

Abstract
An estimated 5 million Americans are living with Alzheimer’s disease (AD), and there is also a significant impact on caregivers, with an additional 16 million Americans providing unpaid care for individuals with AD and other dementias. These numbers are projected to increase in the coming years. While AD is still without a cure, continued research efforts have led to better understanding of pathology and potential risk factors that could be exploited to slow disease progression. A bidirectional relationship between sleep deprivation and AD has been suggested and is well supported by both human and animal studies. Even brief episodes of inadequate sleep have been shown to cause an increase in amyloid-β and tau proteins, both well-established contributors to AD pathology. Sleep deprivation is also the most common consequence of post-traumatic stress disorder (PTSD). Patients with PTSD frequently present with sleep disturbances and also develop dementia at twice the rate of the general population accounting for a disproportionate representation of AD among U.S. Veterans. The goal of this review is to highlight the relationship triad between sleep deprivation, AD, and PTSD as well as their impact on molecular mechanisms driving AD pathology.
Keywords: Alzheimer’s disease, amyloid-β, post-traumatic stress disorder, sleep deprivation, tau
INTRODUCTION
Sleep is a reoccurring period of restorative unconsciousness necessary for normal human function. The cycle of consciousness and unconsciousness is under the control of the circadian clock, a complex system of endocrine and neuroendocrine processes that input ques from the environment to create a sleep-wake rhythm. Significant variability exists in when people sleep and how frequently, termed chronotypes. However, sleep duration less than 5 hours per night corelates with poor cardiovascular health, type 2 diabetes, and obesity across different populations [1, 2]. Individuals with chronotypes far outside the average are considered to have sleep disorders [3]. Requirement for sleep is evolutionarily conserved and is present in invertebrates and vertebrates [4]. Chronic lack of adequate sleep leading to sleep deprivation can cause both systemic and neurological problems ranging from metabolic disruption and obesity to stroke, and impaired learning [5, 6]. Sleep deprivation is associated with neurodegenerative disorders like Alzheimer’s disease (AD) [7] as well as neuropsychiatric disorders such as post-traumatic stress disorder (PTSD) [8], but it remains unclear if sleep deprivation is a cause or a consequence for these disorders.
Quality sleep has been shown recently to be necessary in helping coordinate the clearing of metabolic waste and toxic proteins involved in AD and inadequate sleep may therefore accelerate or cause AD. Non-rapid eye movement (Non-REM) sleep presents on an electroencephalogram (EEG) with low-frequency (less than 4 Hz) oscillations, important in supporting consolidations of new memories and neuronal processing [9–11]. During Non-REM sleep, low frequency oscillations in neuronal activity were found to co-occur with large influx of cerebrospinal fluid (CSF) into the brain and hemodynamic changes [12]. In mice, sleep is associated with clearance of metabolic waste from the CSF which is stronger during low-frequency EEG oscillation associated with Non-REM sleep [13, 14]. Sleep has been found to regulate CSF levels of AD related proteins amyloid-β (Aβ) and tau [15, 16]. Even a single night without sleep can cause accumulation of Aβ in otherwise healthy human brains suggesting a close relationship with sleep disruption and key components of AD pathology [17]. AD is the most common neurodegenerative disorder that disrupts neural circuits, leading to a progressive loss of neurological function and death. Disruption of neural circuitry and neurodegeneration is in part driven by accumulation of “senile plaques” made up of misprocessed Aβ protein and also neurofibrillary tangles consisting of hyperphosphorylated tau. Studies suggest that the incidence of AD is higher in those individuals diagnosed with PTSD [18, 19], a psychiatric disorder triggered by exposure to one or more traumatic events which can lead to a number of neurological and physiological problems, though there have been few studies investigating the factors that may contribute to observed correlation between PTSD and AD. While PTSD patients can present with a number of characteristic symptoms, one of the most common complaints is the presences of sleep disturbances. Large studies suggest that sleep disruption is present in 70–87% of patients with PTSD [20–22]. In this review, we discuss the important role of sleep deprivation in the pathogenesis of AD and as a component of PTSD that may drive the higher incidence of AD in PTSD patient population. We examined primary literature over the past two decades spanning human and animal studies on PTSD, sleep deprivation, and AD, that together implicate sleep deprivation as the likely link between PTSD and increased susceptibility to AD.
SLEEP DEPRIVATION
Disruption of sleep-wake cycle is frequently caused by work and lifestyle choices [23]. Persistent disruption of sleep-wake cycle can result in chronic sleep deprivation which has been linked to systemic and neurological problems including disruption of normal metabolism, obesity, heart disease, high blood pressure, and stroke [5, 6]. The consequences of sleep deprivation are not simply a result of wear and tear but also due to long lasting hormonal, genetic, and epigenetic changes [5, 6, 24]. Brain is particularly affected by sleep deprivation, resulting in diminished learning and memory even after brief periods of sleep deprivation [25–27]. Even among college athletes sleep deprivation causes deterioration in learning, vigilance, mood, and athletic performance [28]. By altering gene expression and hormone levels, and by disrupting normal metabolic function, sleep deprivation can predispose, cause, or worsen neurodegenerative and neuropsychiatric disorders including AD.
Sleep deprivation and AD
A bidirectional relationship between sleep disruption and AD has been proposed and is supported by mounting evidence, demonstrating that sleep deprivation increases AD related pathology and that increasing AD pathology further causes sleep disruption [29, 30]. Sleep cycles between non-REM and REM sleep stages, several times during a typical night. Non-REM is the longer of the two sleep stages and is further subdivided into 3 successive sub-stages, that occur before the shorter REM period is reached. Non-REM sleep is characterized by low-frequency (less than 4 Hz) oscillations, important in supporting consolidations of new memories and neuronal processing [9–11]. Non-REM sleep is also accompanied by large influx of cerebrospinal fluid (CSF) into the brain and also with hemodynamic changes [12]. In mice, as in humans, diurnal variation in Aβ levels were found to occur, with higher Aβ levels detected in the CSF during waking hours, that decrease following sleep or in case of mice higher levels of Aβ were detected during their active dark phase compared to light phase [15]. Chronic sleep disruption in mice resulted in significantly higher levels of Aβ, that decreased following sleep suggesting that Aβ levels are dependent on wakefulness rather than time of day [15]. Orexin is a molecule involved in regulating wakefulness and is released from hypothalamic neurons promoting wakefulness [31]. Diurnal fluctuations in orexin correspond to Aβ. Brain infusion of orexin in mice resulted in increased Aβ levels, while inhibition of orexin receptors abolished diurnal Aβ variation [15]. Chronic sleep deprivation of APP transgenic mice that develop Aβ plaques, resulted in increased plaque burden, while blocking orexin receptors decreased plaque burden [15]. Sleep duration of less than 5 hours or greater than 11 hours per night have been linked to increased risk for cognitive impairment [32, 33]. Prospective study looking at sleep demonstrated that sleep fragmentation increases the risk of developing AD [7]. AD related pathology is thought to start decades before the onset of symptoms and eventual diagnosis. Identifiable pathological changes in AD include reduction of soluble Aβ42 levels in the CSF 10–15 years before the onset of cognitive symptoms associated with AD [35]. Asymptomatic individuals with reduced soluble Aβ42 levels, which is an indication of Aβ sequestration into insoluble senile plaques, have worse sleep quality compared to their peers with normal Aβ42 levels [36]. These clinical observations were replicated in two different mouse models (APPSWE and APPSWE/PS1DE9) where sleep deprivation accelerated deposition of Aβ into amyloid plaques and enhanced sleep showed decrease in Aβ plaque deposition [15].
Sleep deprivation in PTSD as a risk factor for AD
Individuals with PTSD are reported to have exaggerated cognitive changes with aging, and increased incidence of AD; however, well-controlled studies in this area are relatively few in number. Administrative data from the Veterans Integrated Service Network (VISN) of Department of Veterans Affairs healthcare facilities revealed that the prevalence and incidence of a dementia diagnosis remained nearly two times as high in Veterans with diagnosed PTSD compared to the control group consisting of Veterans without diagnosed PTSD [18]. Another study of Veterans aimed at determining whether PTSD is associated with increased risk for developing dementia reported that during a 7-year follow-up, Veterans with PTSD had a cumulative incident dementia rate of 10.6% whereas those without PTSD had a rate of 6.6%. These results were similar even after those with a history of head injury, substance abuse, or clinical depression were excluded [19]. While PTSD is common among Veterans, it is also present in the general population. A study of 600 older persons revealed that individuals that were categorized as being in the 90th percentile of “distress-proneness” were 2.7 times more likely to develop AD than those not prone to distress (10th percentile). In this study, “distress-proneness” was also associated with overall cognitive decline [37].
PTSD and AD have also been studied in animal models. Studies in animals have shown that various stressors can be associated with accelerated development of amyloid plaques [38–41], increased levels of Aβ [42, 43], and tau hyperphosphorylation [44–46]. One study in mice found that a PTSD-like induction chronically elevated levels of Aβ in the CSF, exacerbating ongoing AD pathogenesis [47]. Justice et al. demonstrated that Aβ resulted in hyperexcitation of corticotropin-releasing factor (CRF) neurons and that lowering of Aβ levels attenuated the PTSD-like phenotype. Their data demonstrated that exposure to PTSD-like trauma can drive AD pathogenesis and perturb CRF signaling, important in stress activated responses, thereby enhancing chronic PTSD symptoms and increasing the risk for AD.
PTSD patients can present with a number of psychiatric symptoms, including nightmares and hyperarousal, which invariably impact sleep duration and quality [48]. PTSD and sleep quality deficits are common among combat Veterans. One study of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) Veterans found that 89% were reported as “poor sleepers” according to the Pittsburgh Sleep Quality Index (PSQI) and that sleep quality was worse among Veterans presenting with PTSD symptoms [49].
Neuroimaging studies have long shown that neuronal excitability plays a key role in PTSD pathogenesis [50–52]. One study demonstrated that patients with PTSD exhibited a mean conditioned motor evoked potential amplitude higher than that observed in control groups. They demonstrated that PTSD can give rise to abnormalities in intracortical inhibition which leads to cortical hyperexcitability [53]. Similarly, changes in neuronal excitability may underlie the effects of sleep deficit on AD pathogenesis. In a Drosophila model of AD, Aβ accumulation led to fragmented and reduced sleep, while chronic sleep deprivation led to increased Aβ burden. Moreover, neuronal excitability was found to mimic the effects of reduced sleep on Aβ accumulation. Suppressing neuronal excitability reduced the effects of sleep deprivation on Aβ accumulation [54]. These results suggest that both neuronal excitability and sleep deprivation exacerbate the accumulation of Aβ and may provide a link between the neuronal excitability and sleep deprivation in PTSD patient populations which can eventually lead to development of AD. We summarize the relationship between PTSD, sleep deprivation, and AD in Fig. 1.
Fig. 1.
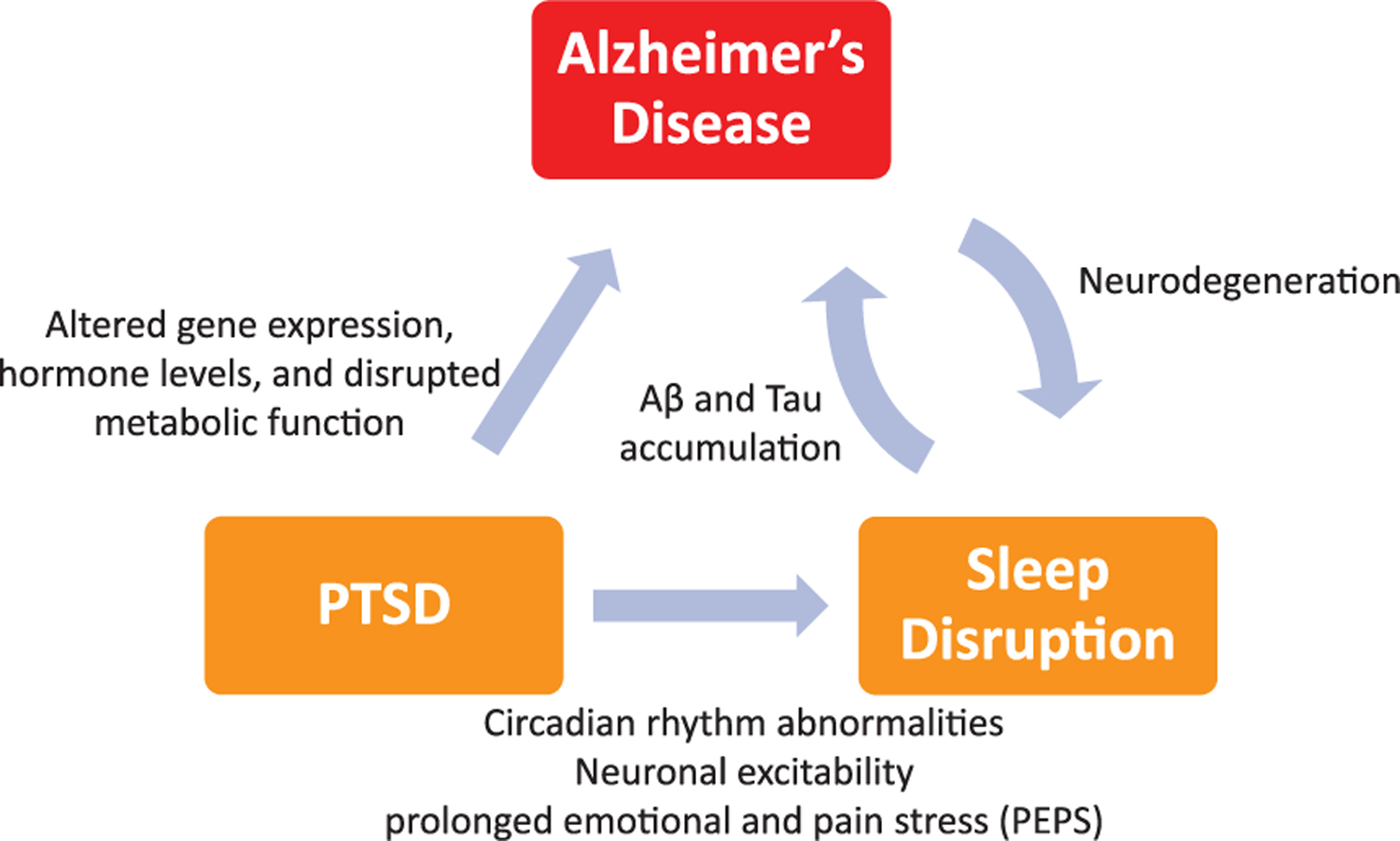
PTSD can cause altered gene expression, altered hormone levels, and disrupted metabolism, thereby creating an environment that can promote amyloid-β (Aβ) aggregation and neurodegeneration associated with Alzheimer’s disease. Sleep deprivation resulting from PTSD can cause a positive feedback loop where sleep deprivation increases likelihood of Alzheimer’s disease and in turn, progression of Alzheimer’s disease can result in further sleep disturbances.
MOLECULAR MECHANISMS OF AD DRIVEN BY SLEEP DEPRIVATION
The hallmark plaques observed in AD brains are the result of an accumulation and inability to clear the protein Aβ, a metabolic waste product. Though the mechanisms by which Aβ is cleared in healthy human brains is not well understood, there is evidence that sleep may play a role in clearance of waste products from the brain including Aβ [13]. Animal models provide further evidence that Aβ levels and sleep quality and duration are tightly linked. In mice, measurement of hippocampal Aβ levels using in vivo microdialysis revealed that Aβ levels were positively correlated with the amount of time spent awake and negatively correlated with the amount of time spent asleep. This negative correlation was even stronger with non-REM sleep [15]. In a human study, even a single unrestricted night of sleep led to a 6% decrease in CSF Aβ levels in cognitively normal middle-aged men and just one night of total sleep deprivation counteracted this decrease, interfering with the physiological morning decrease in Aβ levels [55].
The increased risk for AD associated with chronic sleep deficit appears to be modulated through multiple molecular pathways of neuronal injury in addition to the effect on Aβ accumulation. Two-month sleep deprivation in mice not only altered the Aβ protein precursor processing but also raised the phosphorylated tau (p-Tau) level and resulted in impairment of cognitive performance compared to non-sleep deprived controls. In addition to increased Aβ42 production and more senile plaques in the cortex and hippocampus, sleep deprivation also caused neuronal mitochondrial damage, caspase cascade activation, and mediated neuronal apoptosis. These changes were long-lasting and were irreversible during a 3-month follow-up under normal conditions [56]. Transgenic mice that were sleep deprived showed significant increase in the insoluble fraction of tau, lower levels of postsynaptic density protein 95, and increased glial fibrillary acidic protein levels [57]. Sleep-deprived mice displayed increased Aβ and p-Tau levels in the cortex and higher circulating levels of the hormone corticosterone, responsible for energy, immune, and stress response regulation, compared to controls [58]. Other animal models of chronic sleep deprivation have shown depletion of glycogen stores and increase in oxidative stress and free radical formation [59], thus emphasizing that multiple molecular pathways are involved in neurodegeneration in response to chronic sleep deficit.
Orexin
Orexin, the neuropeptide that promotes wakefulness, appears to play a significant role in Aβ-mediated neurodegeneration. APP/PS1 transgenic mice, in which the orexin gene was knocked out, display a marked decrease in the amount of Aβ pathology in the brain with an associated increase in sleep time. In contrast, sleep deprivation or increasing wakefulness by rescue of orexinergic neurons in APP/PS1 mice lacking orexin increase the amount of Aβ pathology in the brain [60]. Orexin activation has also been shown to play a role in behavioral fear expression in an animal model [61]. Similarly, another animal study showed that orexin administration impaired fear extinction [62]. These studies suggest that inappropriate excitation of this pathway may account for the fear generalization observed in PTSD. Additionally, altered orexin signaling may lead to sleep disturbance and changes in Aβ pathology.
Alzheimer’s disease, sleep, and the immune system
Sleep deprivation is associated with increased risk of cardiovascular disease, diabetes, hypertension, and obesity with a 45% increase in the risk of a fatal heart attack. These consequences of sleep loss are characterized, in part, by inflammatory processes [63]. Sleep deficit-induced proinflammatory response is considered a risk factor for neurodegenerative diseases such as AD [64]. Sleep appears to have a bidirectional relationship with the immune system and over the past few decades it has become increasingly apparent that sleep is closely intertwined with the immune system. In 1975, Pappenheimer et al. [65] reported isolating a substance which they termed sleep-promoting factor (factor S) and later research identified this substance as a bacterial cell wall peptidoglycan fragment known as muramyl peptide, a pyrogenic cytokine. This substance was shown to induce sleep in non-sleep deprived animals and it also induced inflammatory cytokines [66]. These findings led to further research to delineate the role of sleep, in immune responsiveness and the findings have confirmed a bidirectional relationship between sleep and immune function. It has been found that through neuro–immune interactions, sleep loss alters immune function and immune challenges alter sleep pattern. As more became known about the sleep response to infectious challenge, it became clear that IL-1β and TNF-α are among the inflammatory markers that are involved in the central nervous system regulation of physiological sleep [67].
SUMMARY
Individuals diagnosed with PTSD are at least twice as likely to develop AD than those without PTSD. Sleep deprivation is the most commonly reported consequence of PTSD and has been reported as a major accelerant of AD pathology in patients and in AD animal models. Sleep deprivation appears to affect multiple molecular pathways leading to higher risk for neurodegeneration and therefore interventions aimed at improving sleep patterns will have a much broader impact than medications targeting any single molecular mechanisms. It has been established that neurodegeneration in AD begins years before symptom manifestation presenting an opportunity for intervention prior to onset of symptoms.
ACKNOWLEDGMENTS
We thank Dr. U. Nalla B. Durai for useful discussion and inspiration. This review was supported by the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Rehabilitation/Biomedical Laboratory Research and Development (RX001520, RX003253, BX005015), the Assistant Secretary of Defense for Health Affairs through the Congressionally Directed Gulf War Illness Research Program (W81XWH-16–1–0626), The Bay Pines Foundation, and the Veterans Bio-Medical Research Institute.
The contents do not represent the views of the Department of Veterans Affairs or the United States Government and the opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1378r2).
REFERENCES
- 1.Schmid SM, Hallschmid M, Schultes B (2015) The metabolic burden of sleep loss. Lancet Diabetes Endocrinol 3, 52–62. [DOI] [PubMed] [Google Scholar]
- 2.Cappuccio FP, Taggart FM, Kandala NB, Currie A, Peile E, Stranges S, Miller MA (2008) Meta-analysis of short sleep duration and obesity in children and adults. Sleep 31, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dagan Y (2002) Circadian rhythm sleep disorders (CRSD). Sleep Med Rev 6, 45–54. [DOI] [PubMed] [Google Scholar]
- 4.Miyazaki S, Liu CY, Hayashi Y (2017) Sleep in vertebrate and invertebrate animals, and insights into the function and evolution of sleep. Neurosci Res 118, 3–12. [DOI] [PubMed] [Google Scholar]
- 5.Skuladottir GV, Nilsson EK, Mwinyi J, Schioth HB (2016) One-night sleep deprivation induces changes in the DNA methylation and serum activity indices of stearoyl-CoA desaturase in young healthy men. Lipids Health Dis 15, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Javaheri S, Zhao YY, Punjabi NM, Quan SF, Gottlieb DJ, Redline S (2018) Slow-wave sleep is associated with incident hypertension: The Sleep Heart Health Study. Sleep 41, zsx179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA (2013) Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert KS, Kark SM, Gehrman P, Bogdanova Y (2015) Sleep disturbances, TBI and PTSD: Implications for treatment and recovery. Clin Psychol Rev 40, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diekelmann S, Born J (2010) The memory function of sleep. Nat Rev Neurosci 11, 114–126. [DOI] [PubMed] [Google Scholar]
- 10.Marshall L, Helgadottir H, Molle M, Born J (2006) Boosting slow oscillations during sleep potentiates memory. Nature 444, 610–613. [DOI] [PubMed] [Google Scholar]
- 11.Van Someren EJ, Van Der Werf YD, Roelfsema PR, Mansvelder HD, da Silva FH (2011) Slow brain oscillations of sleep, resting state, and vigilance. Prog Brain Res 193, 3–15. [DOI] [PubMed] [Google Scholar]
- 12.Fultz NE, Bonmassar G, Setsompop K, Stickgold RA, Rosen BR, Polimeni JR, Lewis LD (2019) Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 366, 628–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (2013) Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hablitz LM, Vinitsky HS, Sun Q, Staeger FF, Sigurdsson B, Mortensen KN, Lilius TO, Nedergaard M (2019) Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv 5, eaav5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (2009) Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM (2019) The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, Lindgren E, Ramirez V, Zehra A, Freeman C, Miller G, Manza P, Srivastava T, De Santi S, Tomasi D, Benveniste H, Volkow ND (2018) beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad SciUSA 115, 4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qureshi SU, Kimbrell T, Pyne JM, Magruder KM, Hudson TJ, Petersen NJ, Yu HJ, Schulz PE, Kunik ME (2010) Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. J Am Geriatr Soc 58, 1627–1633. [DOI] [PubMed] [Google Scholar]
- 19.Yaffe K, Vittinghoff E, Lindquist K, Barnes D, Covinsky KE, Neylan T, Kluse M, Marmar C (2010) Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry 67, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leskin GA, Woodward SH, Young HE, Sheikh JI (2002) Effects of comorbid diagnoses on sleep disturbance in PTSD. J Psychiatr Res 36, 449–452. [DOI] [PubMed] [Google Scholar]
- 21.Ohayon MM, Shapiro CM (2000) Sleep disturbances and psychiatric disorders associated with posttraumatic stress disorder in the general population. Compr Psychiatry 41, 469–478. [DOI] [PubMed] [Google Scholar]
- 22.Maher MJ, Rego SA, Asnis GM (2006) Sleep disturbances in patients with post-traumatic stress disorder: Epidemiology, impact and approaches to management. CNS Drugs 20, 567–590. [DOI] [PubMed] [Google Scholar]
- 23.Boivin DB, Boudreau P (2014) Impacts of shift work on sleep and circadian rhythms. Pathol Biol (Paris) 62, 292–301. [DOI] [PubMed] [Google Scholar]
- 24.Gaine ME, Chatterjee S, Abel T (2018) Sleep deprivation and the epigenome. Front Neural Circuits 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prince TM, Wimmer M, Choi J, Havekes R, Aton S, Abel T (2014) Sleep deprivation during a specific 3-hour time window post-training impairs hippocampal synaptic plasticity and memory. Neurobiol Learn Mem 109, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Havekes R, Vecsey CG, Abel T (2012) The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell Signal 24, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, Bruinenberg VM, Poplawski SG, Day JP, Aton SJ, Radwanska K, Meerlo P, Houslay MD, Baillie GS, Abel T (2016) Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife 5, e13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolin DJ (2019) Sleep deprivation and its contribution to mood and performance deterioration in college athletes. Curr Sports Med Rep 18, 305–310. [DOI] [PubMed] [Google Scholar]
- 29.Ju YE, Lucey BP, Holtzman DM (2014) Sleep and Alzheimer disease pathology–a bidirectional relationship. Nat Rev Neurol 10, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daulatzai MA (2015) “Boomerang neuropathology” of late-onset Alzheimer’s disease is shrouded in harmful “BDDS”: Breathing, diet, drinking, and sleep during aging. Neurotox Res 28, 55–93. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S (2001) Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci 14, 1075–1081. [DOI] [PubMed] [Google Scholar]
- 32.Tworoger SS, Lee S, Schernhammer ES, Grodstein F (2006) The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis Assoc Disord 20, 41–48. [DOI] [PubMed] [Google Scholar]
- 33.Faubel R, Lopez-Garcia E, Guallar-Castillon P, Graciani A, Banegas JR, Rodriguez-Artalejo F (2009) Usual sleep duration and cognitive function in older adults in Spain. J Sleep Res 18, 427–435. [DOI] [PubMed] [Google Scholar]
- 34.Webb WB (1982) Sleep in older persons: Sleep structures of 50- to 60-year-old men and women. J Gerontol 37, 581–586. [DOI] [PubMed] [Google Scholar]
- 35.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH (2011) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association work-groups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM (2013) Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA (2006) Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology 27, 143–153. [DOI] [PubMed] [Google Scholar]
- 38.Devi L, Alldred MJ, Ginsberg SD, Ohno M (2010) Sex- and brain region-specific acceleration of β-amyloidogenesis following behavioral stress in a mouse model of Alzheimer’s disease. Mol Brain 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL (2009) Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem 108, 165–175. [DOI] [PubMed] [Google Scholar]
- 40.Ni Y, Zhao X, Bao G, Zou L, Teng L, Wang Z, Song M, Xiong J, Bai Y, Pei G (2006) Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat Med 12, 1390–1396. [DOI] [PubMed] [Google Scholar]
- 41.Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG (2004) Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 127, 601–609. [DOI] [PubMed] [Google Scholar]
- 42.Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong WN, Martin B, Mattson MP (2012) 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol Aging 33, 830.e831–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM (2007) Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad SciUSA 104, 10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rissman RA, Lee KF, Vale W, Sawchenko PE (2007) Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci 27, 6552–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VM, Trojanowski JQ (2011) Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J Neurosci 31, 14436–14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rissman RA, Staup MA, Lee AR, Justice NJ, Rice KC, Vale W, Sawchenko PE (2012) Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc Natl Acad Sci USA 109, 6277–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Justice NJ, Huang L, Tian JB, Cole A, Pruski M, Hunt AJ Jr., Flores R, Zhu MX, Arenkiel BR, Zheng H (2015) Posttraumatic stress disorder-like induction elevates beta-amyloid levels, which directly activates corticotropin-releasing factor neurons to exacerbate stress responses. J Neurosci 35, 2612–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khazaie H, Ghadami MR, Masoudi M (2016) Sleep disturbances in veterans with chronic war-induced PTSD. J Inj Violence Res 8, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plumb TR, Peachey JT, Zelman DC (2014) Sleep disturbance is common among service members and veterans of Operations Enduring Freedom and Iraqi Freedom. Psychol Serv 11, 209–219. [DOI] [PubMed] [Google Scholar]
- 50.Shaw ME, Strother SC, McFarlane AC, Morris P, Anderson J, Clark CR, Egan GF (2002) Abnormal functional connectivity in posttraumatic stress disorder. Neuroimage 15, 661–674. [DOI] [PubMed] [Google Scholar]
- 51.Rauch SL, van der Kolk BA, Fisler RE, Alpert NM, Orr SP, Savage CR, Fischman AJ, Jenike MA, Pitman RK (1996) A symptom provocation study of posttraumatic stress disorder using positron emission tomography and script-driven imagery. Arch Gen Psychiatry 53, 380–387. [DOI] [PubMed] [Google Scholar]
- 52.Shin LM, Kosslyn SM, McNally RJ, Alpert NM, Thompson WL, Rauch SL, Macklin ML, Pitman RK (1997) Visual imagery and perception in posttraumatic stress disorder. A positron emission tomographic investigation. Arch Gen Psychiatry 54, 233–241. [DOI] [PubMed] [Google Scholar]
- 53.Centonze D, Palmieri MG, Boffa L, Pierantozzi M, Stanzione P, Brusa L, Marciani M, Siracusano A, Bernardi G, Caramia M (2005) Cortical hyperexcitability in post-traumatic stress disorder secondary to minor accidental head trauma: A neurophysiologic study. J Psychiatry Neurosci 30, 127–132. [PMC free article] [PubMed] [Google Scholar]
- 54.Tabuchi M, Lone SR, Liu S, Liu Q, Zhang J, Spira AP, Wu MN (2015) Sleep interacts with Abeta to modulate intrinsic neuronal excitability. Curr Biol 25, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA (2014) Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: A randomized clinical trial. JAMA Neurol 71, 971–977. [DOI] [PubMed] [Google Scholar]
- 56.Qiu H, Zhong R, Liu H, Zhang F, Li S, Le W (2015) Chronic sleep deprivation exacerbates learning-memory disability and Alzheimer’s disease-like pathologies in AbetaPP(swe)/PS1(DeltaE9) mice. J Alzheimers Dis 50, 669–685. [DOI] [PubMed] [Google Scholar]
- 57.Di Meco A, Joshi YB, Pratico D (2014) Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging 35, 1813–1820. [DOI] [PubMed] [Google Scholar]
- 58.Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP (2013) Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Abeta and pTau in a mouse model of Alzheimer’s disease. Brain Res 1529, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McEwen BS (2006) Sleep deprivation as a neurobiologic and physiologic stressor: Allostasis and allostatic load. Metabolism 55, S20–23. [DOI] [PubMed] [Google Scholar]
- 60.Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, Heda A, Snider BJ, Li M, Yanagisawa M, de Lecea L, Holtzman DM (2014) Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med 211, 2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soya S, Takahashi TM, McHugh TJ, Maejima T, Herlitze S, Abe M, Sakimura K, Sakurai T (2017) Orexin modulates behavioral fear expression through the locus coeruleus. Nat Commun 8, 1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flores Á, Valls-Comamala V, Costa G, Saravia R, Maldonado R, Berrendero F (2014) The hypocretin/orexin system mediates the extinction of fear memories. Neuropsychopharmacology 39, 2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Imeri L, Opp MR (2009) How (and why) the immune system makes us sleep. Nat Rev Neurosci 10, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hurtado-Alvarado G, Pavon L, Castillo-Garcia SA, Hernandez ME, Dominguez-Salazar E, Velazquez-Moctezuma J, Gomez-Gonzalez B (2013) Sleep loss as a factor to induce cellular and molecular inflammatory variations. Clin Dev Immunol 2013, 801341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pappenheimer JR, Koski G, Fencl V, Karnovsky ML, Krueger J (1975) Extraction of sleep-promoting factor S from cerebrospinal fluid and from brains of sleep-deprived animals. J Neurophysiol 38, 1299–1311. [DOI] [PubMed] [Google Scholar]
- 66.Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L (1984) Sleep-promoting effects of endogenous pyrogen (interleukin-1). Am J Physiol 246, R994–999. [DOI] [PubMed] [Google Scholar]
- 67.Mullington JM, Simpson NS, Meier-Ewert HK, Haack M (2010) Sleep loss and inflammation. Best Pract Res Clin Endocrinol Metab 24, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]