

Abstract
N-linked glycosylation is a complex, co- and post-translational series of events that connects metabolism to signaling in virtually all cells. Metabolic assembly of N-linked glycans spans multiple cellular compartments, and early N-linked glycan biosynthesis is a central mediator of protein folding and the unfolded protein response. In the brain, N-linked glycosylated proteins participate in a myriad of processes from electrical gradients to neurotransmission. However, it is less clear how perturbations in N-linked glycosylation impact and even potentially drive aspects of neurological disorders. In this review, we discuss our current understanding of the metabolic origins of N-linked glycans in the brain, their role in modulating neuronal function, and how aberrant N-linked glycosylation can drive neurological disorders.
Keywords: N-linked glycosylation, carbohydrate metabolism, Alzheimer’s disease, neurodegeneration, neuroinflammation
The ever-expanding carbohydrate code
Simple sugars, also known as monosaccharides, are one of the foundational groups of biomolecules that comprise a cell. The most well-studied monosaccharide is glucose, an essential substrate for bioenergetics and a carbon source for anabolic synthesis of macromolecules. More than ten unique monosaccharides have been identified thus far that are important for mammalian cellular and organismal physiology [1]. Unlike glucose, the primary role of most monosaccharides is to serve as building blocks for complex carbohydrate molecules such N-linked glycans, O-linked glycans, and proteoglycans that regulate protein structure and cellular functions. Similar to the DNA-centric genetic code where four deoxynucleotides give rise to life’s biodiversity, the carbohydrate code, comprised of unique monosaccharides, is a fundamental element of the protein interactome that leads to organismal complexity (Figure 1). With at least ten identified monosaccharides within a cell and over 1012 possible combinations [2], we are just beginning to unravel the carbohydrate code complexity that impacts organismal physiology and disease pathology.
Figure 1. The carbohydrate code is a critical component of organismal complexity.
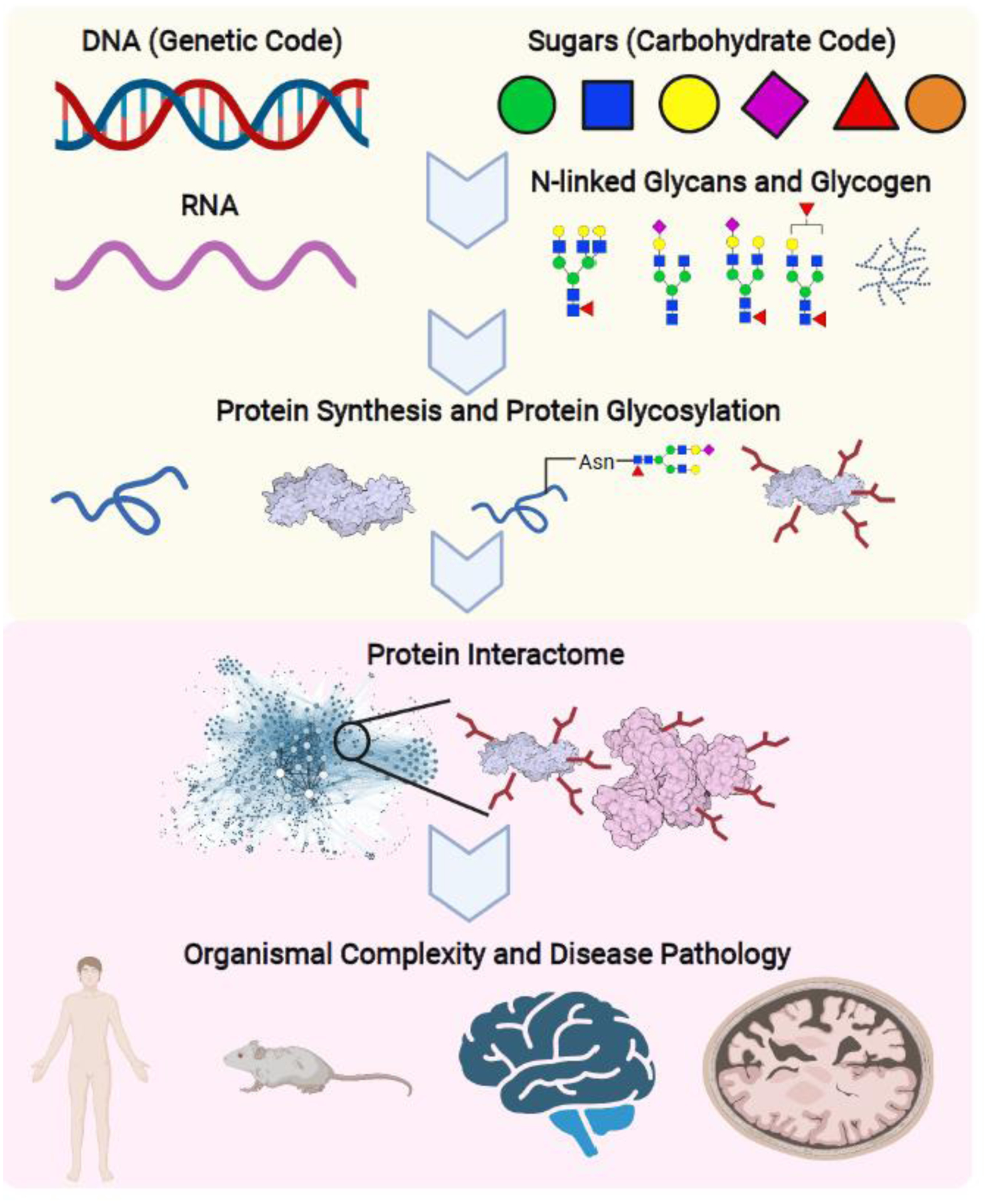
The carbohydrate code operates in parallel and synergistically with the genetic code to boost organismal complexity through the protein interactome. The genetic code is built on four deoxynucleotides that translate to exponential combinations of amino sequences, giving rise to the cellular and organismal proteome. Similarly, unique combinations of monosaccharides, or simple sugars, generate an ever-expanding repertoire of diverse N-linked glycans. This protein modification is both a co- and post-translational event modulating protein activity, trafficking, turnover, and oligomerization. N-linked glycosylation is a foundational event and vital component of the protein interactome that gives rise to organ function and organismal complexity, this holds especially true for the mammalian brain. Thus, N-glycans represent an essential, yet understudied, aspect of biodiversity that impacts disease progression on a systemic and organismal level.
Among the major classes of complex carbohydrates, N-linked glycans are the most diverse both in terms of their structure and functional roles in mammalian cells [3]. N-linked glycosylation is a highly regulated co- and post-translational series of events that modifies cell surface, secreted, and circulating proteins [4]. Synthesis of N-linked glycans is a multi-compartmental process that requires over 700 glycosyltransferases and glycosidases acting in a tightly regulated manner that ultimately determines the glycan profile of a cell. N-glycan synthesis is intimately linked to protein folding and the unfolded protein response (UPR) and many salvage pathways in the lysosome [5] and autophagosome [6]. Further, N-linked glycans participate in a diverse repertoire of cellular processes related to protein structure and function including protein maturation, stability, subcellular localization, enzymatic activity, and protein-protein interaction [7]. Genetic knockout of enzymes in the glycosylation pathway in immortalized cell lines typically only mildly impacts cellular proliferation [8]; however, whole-body knockout of the same enzymes in mice is often embryonic lethal [9]. These parallel experiments highlight the importance of N-linked glycans in cell-cell communication and signaling that are essential during differentiation and embryonic development of multi-cellular organisms. This review focuses on the emerging roles of N-linked glycans in brain physiology and neurological disorders.
Monosaccharides are metabolically channeled in the central nervous system
Glucose, glucosamine, fucose, mannose, galactose, and sialic acid are the basic monosaccharide building blocks for N-linked glycosylation. Within a cell, monosaccharides are biochemically conjugated to a nucleotide to generate sugar-nucleotides that act as carrier molecules (i.e., UDP-N-acetyl glucosamine (UDP-GlcNAc), GDP-fucose, GDP-mannose, UDP-galactose, and CMP-sialic acid) for transport across the endoplasmic reticulum (ER) and/or Golgi membranes (Figure 2). In the central nervous system (CNS), all sugar-nucleotides can be synthesized from glucose, although sugar-nucleotides can also be synthesized from other free monosaccharides that are present in the brain. For example, UDP-GlcNAc can be de novo synthesized from glucose, glucosamine, and fructose. Further, GDP-fucose, GDP-mannose, UDP-galactose can be synthesized from free fucose, mannose, and galactose, respectively. Ultimately, the final glycosylation profile and overall glycan complexity in the brain is greatly impacted by availability of monosaccharide precursors. Although glucose is thought to be the preferred carbon source for their synthesis, cells are able to adapt during glucose deprivation and cellular stress to directly uptake other N-glycan monosaccharides to maintain proper N-linked glycosylation [10]. N-glycan biosynthesis is initiated in the ER where the core glycan structure is assembled through sequential addition of GlcNAc and mannose monomers and transported to the Golgi for maturation [11]. In the Golgi, the glycan core is further modified for complexity. Additional branching is initiated in the cis Golgi, the addition of galactose occurs at medial Golgi, and sialic acid and fucose modifications follow in the trans Golgi and transport vesicles. Together these enzymatic events generate structurally diverse N-linked glycans that are critical for brain function (Figure 2).
Figure 2. Monosaccharide metabolism is metabolically channeled within a cell.
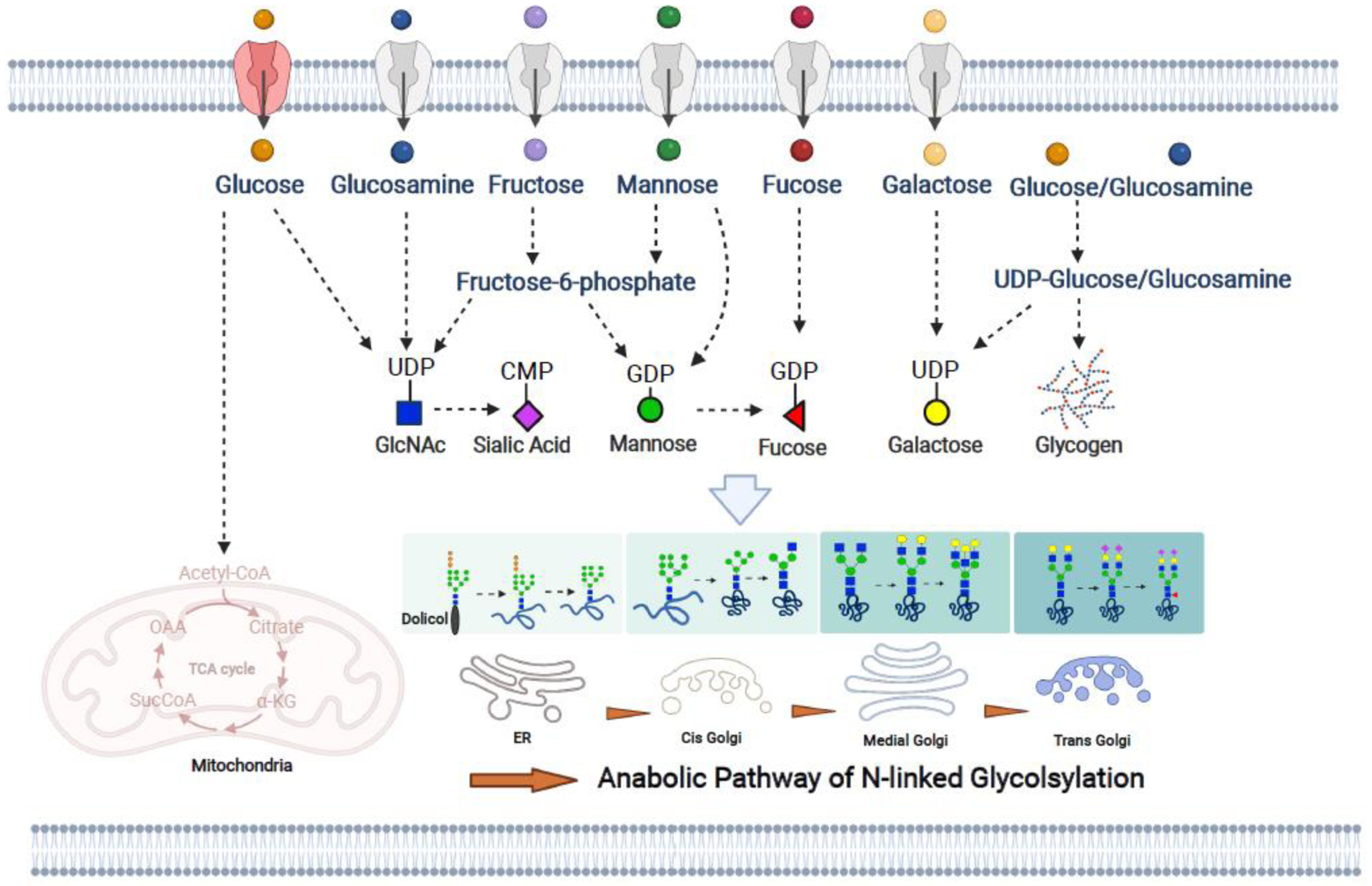
The most common extracellular monosaccharides of the brain are glucose, glucosamine, fructose, mannose, fucose, and galactose, and they can directly enter cells through the GLUT family of membrane transporters. Additionally, monosaccharides can be utilized for the biosynthesis of N-linked glycan substrates also known as sugar-nucleotides that include: UDP-GlcNAc, CMP-Sialic acid, GDP-mannose, UDP-glucose, GDP-fucose, and UDP-galactose. For example, UDP-GlcNAc can be synthesized from glucose, glucosamine, fructose, and mannose based on monosaccharides availability. Brain glycogen provides an additional intracellular pool of glucose and glucosamine. UDP-glucose is the substrate for glycogen but also the substrate for UDP-galactose, further adding to the complexity of sugar-nucleotide metabolism. Sugar-nucleotides are synthesized in the cytoplasm, yet N-linked glycan assembly occurs in the ER and Golgi. Early N-linked glycan biosynthesis occurs in the ER and primarily utilizes GlcNAc and mannose as the basic building blocks. N-linked glycan maturation occurs in the Golgi, additional branching occurs in the cis Golgi, galactose additions occur in the medial Golgi, and fucose and sialic acid are linked in the trans Golgi. Collectively, monosaccharide metabolism and N-linked glycan biosynthesis are complex processes traversing multiple cellular compartments.
Glycogen is another critical complex carbohydrate in the brain (Box 1) [12], and decades of research have highlighted the relationship between glycogen and N-linked glycan biosynthesis [13]. Glycogen can be catabolized into glucose 1-phosphate by glycogen phosphorylase or glucose monomers by acid glucosidase (GAA). Both glucose 1-phosphate and glucose are preferred substrates for the de novo synthesis of sugar-nucleotides. Early reports suggested the possibility that glycogen may contain monosaccharides other than glucose [14,15]. Indeed, a recent study reported that nearly 25% of brain glycogen consists of glucosamine, and glycogen-associated glucosamine represents a substantial substrate pool for the production of UDP-GlcNAc, and subsequently the biosynthesis of N-linked glycans and other complex carbohydrates and glycolipids [16]. Strikingly, glycogen granules are frequently observed localized to the cytosolic side of the ER membrane [17] and a recent study elegantly detailed that the carbohydrate binding enzyme STBD1 is required to anchor glycogen to the ER membrane [18]. During ER stress, glycogen clusters at the ER membrane [18] and is proposed to serve as a rapid supply of substrates to support ER function. Further, genetic approaches that prevent substrate release from glycogen drive a hypoglycosylation phenotype in the brain through simultaneous constriction of glucosamine availability and altered carbon flux through the hexosamine pathway for the de novo synthesis of sugar-nucleotides [16]. Thus, glycogen and N-linked glycans are metabolically channeled both through common substrates and spatial approximation. These studies highlight that glycogen accessibility plays a critical role in maintaining N-glycan metabolic homeostasis in the brain. However, a deeper understanding of substrate availability and cellular compartmentalization is needed to elucidate the complex regulatory pathways that control carbohydrate metabolism during normal brain physiology.
Box 1. Glycogen in brain physiology and neurodegeneration.
Glycogen serves as the primary source of carbohydrate storage in most tissues including liver, heart, and brain. However, the function of brain glycogen extends beyond a simple glucose cache, and includes a central role in maintaining brain homeostasis and higher-order brain functions including memory formation, speech, sensory perception, and cognitive ability [1]. On a cellular level, glycogen is a metabolic substrate for neuronal signaling, ATP production, and GABA biosynthesis [2]. Both neuronal and glial cells possess the machinery to synthesize glycogen, but glycogen is predominately reported in astrocytes in the healthy brain. Recent reports suggest glycogen accumulates within neurons in the degenerative brain leading to bioenergetic imbalance and neurotoxicity [3,4]; however, detailed mechanisms of action remain to be elucidated.
Glycogen storage diseases (GSDs) are a class of disorders defined by the dysregulation of glycogen metabolism [5]. GSDs of the brain share similar neurological defects as congenital disorders of glycosylation (CDGs). For example, germline mutations in UDP-glucose pyrophosphorylase (UGP2) are classified both as a GSD and CDG. UGP2 synthesizes UDP-glucose, and loss-of-function in UGP2 results in CNS accumulation of glycogen granules and aberrant glycosylation that manifests as intractable epilepsy and severe developmental delay in patients [6]. Additionally, CDGs often show acute intracellular glycogen accumulation; in fact, there are documented misdiagnoses of CGDs as GSDs [7, 8]. These clinical phenotypes lead to the hypothesis that glycogen and N-linked glycans are metabolically channeled in the CNS to modulate brain physiology. Early reports alluded to the possibility that pathogenic glycogen aggregates in the brain contain monosaccharides other than glucose [9, 10]. Recently, it was definitively demonstrated that brain glycogen is comprised of 25% glucosamine (GlcN), while liver and muscle glycogen contain <1% GlcN. Cells in the CNS leverage glycogen-associated GlcN as a substantial substrate pool for the production of UDP-GlcNAc, an essential building block for N-glycans [11]. Further, this study demonstrated that modulating glycogen metabolism can affect N-linked glycan biosynthesis. For example, inhibiting glycogenolysis restricts UDP-GlcNAc pools and total N-linked glycan levels in the brain. Conversely, enzymatic release of glycogen monomers through intracerebroventricular (ICV) delivery of recombinant amylase stimulated N-linked glycan biosynthesis [11]. Given the intimate metabolic connection between glycogen and N-link glycosylation in the brain, glycogen represents a potential strategy to mitigate aberrant N-linked glycan phenotypes.
N-linked glycans in brain physiology
N-linked glycans are essential to brain function and neuronal development, highlighted by the fact that nearly all congenital disorders of glycosylation (CDGs) manifest with neurological symptoms such as brain hypotrophy, seizures, cognitive impairment, and delayed intellectual development [19–21]. It is worth noting that CDG patients also suffer from developmental delays and noticeable pathologies in the muscle, liver, and the lung due to peripheral roles of N-linked glycosylation and glycoproteins. Studying the detailed mechanisms of pathogenesis in CDGs is challenging due to the fact that many whole body knockouts of glycosylation genes in mice result in either embryonic lethality or neonatal death [22]. Neurological symptoms in CDGs are not surprising given the pivotal roles of N-linked glycans in modulating neuronal morphology and growth, synaptic plasticity, and basal glia cell homeostasis [23]. There is clearly an increasing appreciation regarding complexity that exists within CNS cell populations. This review focuses on the signaling roles of N-linked glycans in neurons, astrocytes, and microglia, the three major CNS cell types that are implicated in brain function and neurological disorders.
N-linked glycans mediate neuronal signaling
CNS controlled processes, such as memory consolidation and formation, social behavior, and decision making, are physiological outputs of neuronal homeostasis and signaling. While other CNS cell types influence these processes, ultimately, neuronal signaling and metabolism dictate brain function. Neuronal signaling is a complex set of molecular processes that involves maintaining resting membrane potential through Na+/K+ ATPase, axon firing through voltage gated ion channels (VGICs), synaptic vesicle loading and trafficking, neurotransmitter binding, and subsequent signaling cascades at the synaptic cleft. N-linked glycans are observed on virtually all proteins involved in modulating or refining the molecular processes that comprises neuronal signaling (Figure 3). As expected, not all N-linked glycan modifications are critical for proper neuronal signaling and some are dispensable [24]. Yet some dramatically impact neuronal activity. Indeed, sialylated N-glycan species on VGICs are one of the most potent modulators of VGIC activity [25], and the addition of charged sialylated N-linked glycans drastically increases the rate of axon firing compared to other N-glycan species [26]. Several proton pump and neurotransmitter antiporters located in the synaptic vesicles are also sialylated glycoproteins [27,28]. N-linked glycan modification at multiple asparagine residues is required for trafficking of synaptic vesicle protein isoform 2 (SV2) for the formation of synaptic vesicles at the synaptic clef [23,29]. Additionally, N-linked glycosylation is essential for stable surface expression of metabotropic glutamate receptor 7 (mGlu7), a key receptor modulating excitatory synaptic transmission in neurons [30]. Importantly, glucose metabolism supports bioenergetics but also provides substrates for N-linked glycosylation, and dysregulation in glucose metabolism directly impacts both N-glycan structural diversity and the extent to which neuronal proteins are N-glycosylated [31,32]. Thus, N-linked glycans couple metabolism to signaling within a neuron, and proper control of N-linked glycosylation is critical for neuronal function.
Figure 3. N-linked glycans modulate neuronal activity.
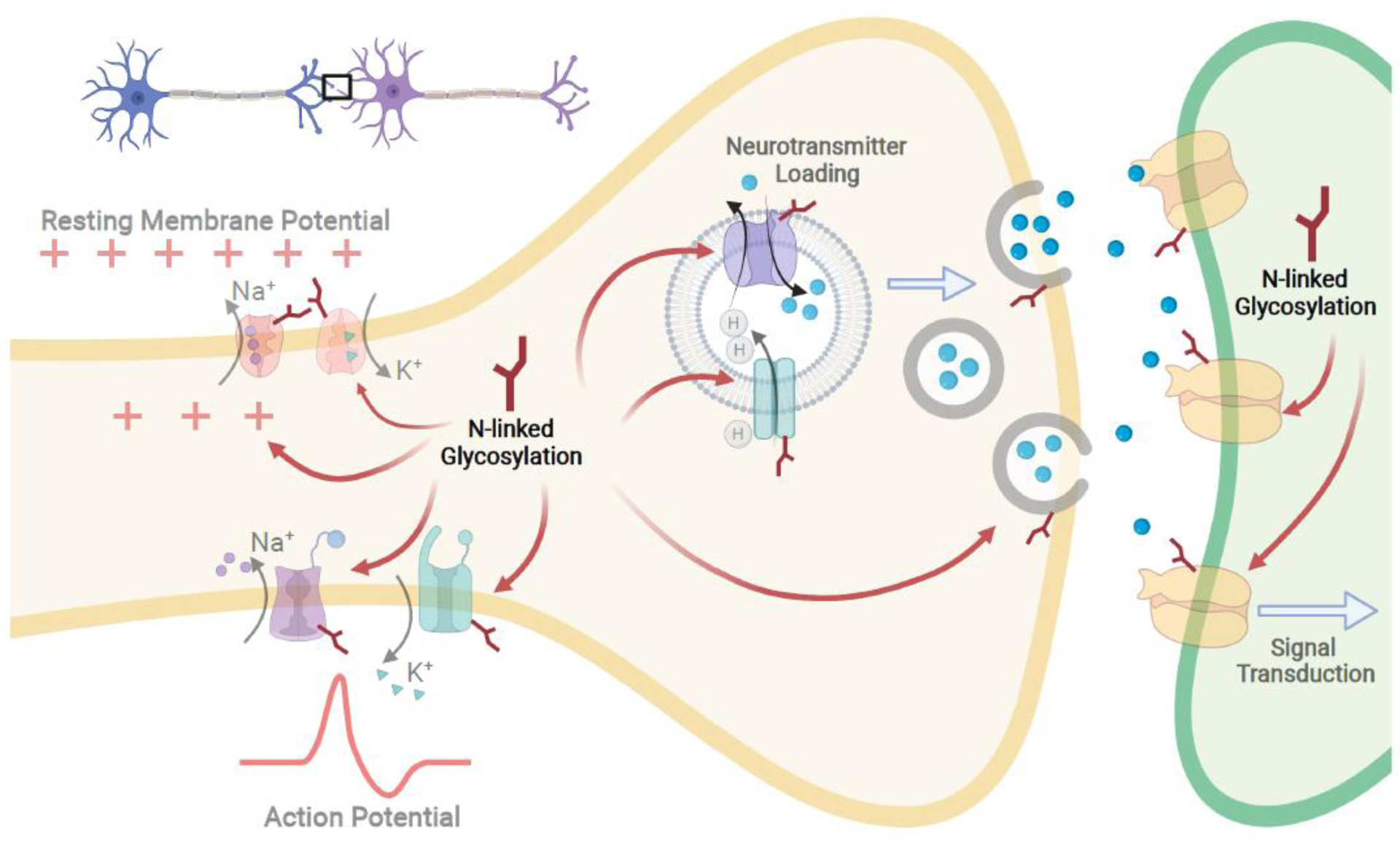
Neuronal processes translate into brain function through synaptic transmission. These processes include maintaining resting membrane potential, axon firing through action potentials, neurotransmitter loading in vesicles and their release, and ultimately neurotransmitter and receptor binding leading to signal transduction. All of these processes are regulated or fine-tuned by N-linked glycosylation. Thus far, Na+/K+ ATPases, voltage gated ion channels (VGICs), and antiporters on synaptic vesicles and neurotransmitter receptors are all reported glycoproteins, and N-linked glycosylation assists in their folding and trafficking, but also modulate their activity. Therefore, proper control of N-linked glycosylation is critical for neuronal function.
N-linked glycans in resting glial cell homeostasis
Astrocytes and microglia are also important cell populations within the CNS, and recent advances in our knowledge of astrocyte biology have elevated our understanding of astrocytes from supportive cells to a central modulator of neuronal signaling and brain physiology [33]. Unlike neurons, astrocytes arise from the glial progenitors and adapt to different morphology according to the brain regions where they are located [34]. The emerging roles of astrocytes in brain function have expanded to include maintaining neuronal survival, synaptogenesis, neurotransmitter uptake and recycling, calcium signaling, and blood-brain barrier (BBB) integrity [35]. N-linked glycosylation plays critical roles in each of these processes. For example, astrocytes are crucial for glutamate homeostasis by maintain cellular uptake and recycling. The excitatory amino acid transporter (EAAT) family of excitatory glutamate transporters are responsible for transporting internalized glutamate in astrocytes and regulating glutamatergic signaling. EAAT1 and EAAT2 are both N-glycosylated and decreased N-glycosylation is associated with retention of EAAT1/2 in the ER and decreased trafficking to the plasma membrane, which is required for glutamate transport [36]. These data suggest a potential critical role for glycosylation in glial-specific glutamate cycling that remains to be elucidated. Further, astrocytes are a vital component of the BBB, which is a specialized and selective boundary comprised of endothelial cells and pericytes that protects the CNS. In mice, glycosylation of Dentin matrix protein 1 (Dmp1) is critical for astrocyte maturation and BBB formation both in vitro and in vivo [37]. Astrocytes and microglia express a multitude of cell surface lectins, including galectins, sialic acid-binding immunoglobulin-type lectins (Siglecs), mannose-binding lectins (MBLs), and other glycan binding proteins. These lectin receptors are critical for modulating pro- and anti-inflammatory signaling in astrocytes and microglia. Siglecs are a class of lectins containing membrane receptors responsible for anti-inflammatory responses in resting microglia by recognizing terminal sialic acid N-glycan residues [38]. Moreover, Siglecs themselves rely on N-linked glycosylation for trafficking to the membrane [39]. Despite these observations, the regulatory roles of N-linked glycans in resting microglia is less clear. However, given the increasing evidence demonstrating the crucial roles microglia play in brain function, this area warrants further investigation.
N-linked glycans and neurological disorders
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are devastating age-related neurodegenerative disorders that are characterized by substantial neuronal loss, neuroinflammation, and aggregate pathologies. Recent proteomic and glycoproteomic analyses of CSF from AD patients revealed global changes in complex carbohydrate metabolism [40] and N-linked glycosylation [41], alluding to aberrant N-glycan metabolism as a potential biomarker in AD progression. Both AD and PD share similar key pathologies such as accumulation of extracellular amyloid plaques containing amyloid beta (Aβ) aggregates, hyperphosphorylated tau aggregates called neurofibrillary tangles (NFT), and α-synuclein aggregates [42,43]. Recent reports reveal that AD and PD protein aggregates are in fact glycoprotein aggregates, highlighting defects in glycosylation during neurodegeneration [43,44].
Perturbed N-linked glycosylation of amyloid precursor protein (APP), the precursor to Aβ peptide, results in decreased secretion and microsomal localization of APP [45], and disruption of neuronal iron homeostasis [46]. Specifically, APP exhibits altered glycosylation in AD patient brain samples, and, importantly, APP sialylation increases its secretion and Aβ plaque development [44]. Likewise, aggregation of tau, a microtubule-assembly protein that functions to stabilize microtubules, also exhibits aberrant glycosylation in both AD and PD [43]. Additionally, α-synuclein, a neuronal protein involved in synaptic vesicle trafficking, is a major component of Lewy body aggregates in PD and is found in plaques of AD brains [43]. Extracellular N-linked glycans are key regulators of α-synuclein neuronal uptake [47], and they promote clearance of pathogenic α-synuclein aggregates [48]. Collectively, these findings suggest a fundamental shift in N-glycan metabolism defects during AD and PD that warrant further investigation. For example, how does glucose hypometabolism, a key clinical hallmark of AD, impact N-linked glycan biosynthesis. Additionally, studies have shown autophagic/lysosomal pathways that are required for salvage and recycling of surface N-linked glycans are dysfunctional in AD [49,50]. How do perturbations in these processes impact the overall N-linked glycosylation landscape in neurological disorders? Dissecting these molecular events will aid in the understanding of the precise contributions of aberrant N-linked glycosylation to neurodegeneration. Further basic and preclinical investigations are needed to elucidate these answers.
Epilepsy is a major neurological disorder that manifests as myoclonic jerks and/or seizures that are the result of neuronal hyperactivity [51]. Nearly 30% of epilepsy patients do not respond to anti-epileptic drugs and suffer from refractory epilepsy. For more than 70% of refractory patients, the ketogenic diet is an effective treatment option, suggesting that there is a bioenergetic component of the disease [52]. Interestingly, the N-linked glycan profiles of epilepsy patients change during ketogenic diet, highlighting N-linked glycosylation as one of the many processes altered during ketosis [53]. The link between N-linked glycosylation and epilepsy is further supported by SLC35A2-CDG that presents predominantly with neurological features and has been described as an early onset epileptic encephalopathy [54,55]. SLC35A2 encodes the X-linked UDP-galactose transporter that shuttles UDP-galactose from the cytosol into the Golgi. Using deep sequencing, mutations in SLC35A2 were identified in 13 patients with non-lesional focal epilepsy. Further, patient samples had reduced galactose and increased GlcNAc glycan structures [56]. Importantly, investigation of oral supplementation with D-galactose for 18 weeks in a small cohort of pediatric SLC35A2 patients demonstrated decreased epileptic events, as well as general growth and development improvements [55]. These data suggest N-glycosylation defects alone are major contributors to seizures, and supplementation of restricted glycan monomers can rescue aspects of the disease.
N-linked glycosylation impacts neuronal cell fate through the unfolded protein response
Healthy brain function is the organismal manifestation of neuronal homeostasis and signaling. Similarly, the development of neurological disorders is a direct result of neuronal dysfunction. Recent advances in our knowledge of neuronal function place N-linked glycosylation and the unfolded protein response (UPR) at the center of neuronal cell fate [57–59]. Early N-linked glycosylation is a central mediator of the UPR, a complex set of signaling events that control diverse cellular processes. The UPR is a physiological process that is initiated in the ER and is linked to nuclear, lysosomal, and mitochondrial salvage pathways to recycle misfolded proteins, and it is crucial for neuronal survival (Figure 4). The UPR can be activated by internal and external stimuli including ischemia, oxidative stress, starvation, and calcium imbalance [60]. During protein synthesis, polypeptide chains are first glycosylated and then disulfide bonds form within the polypeptide as the protein folds into its tertiary structure [61]. If misfolding occurs, glucose regulated protein 78 (GRP78) binds to misfolded glycopeptides and triggers a sequence of events the results in the UPR. Binding of GRP78 to misfolded glycoproteins initiates either the refolding process or endoplasmic-reticulum-associated degradation (ERAD) that shunts glycoproteins to the proteasome, lysosome or autophagosome for recycling of misfolded proteins to their metabolic constituents [62].
Figure 4. The unfolded protein response (UPR) and ER stress are at the crossroads of neuronal cell survival and cell death.
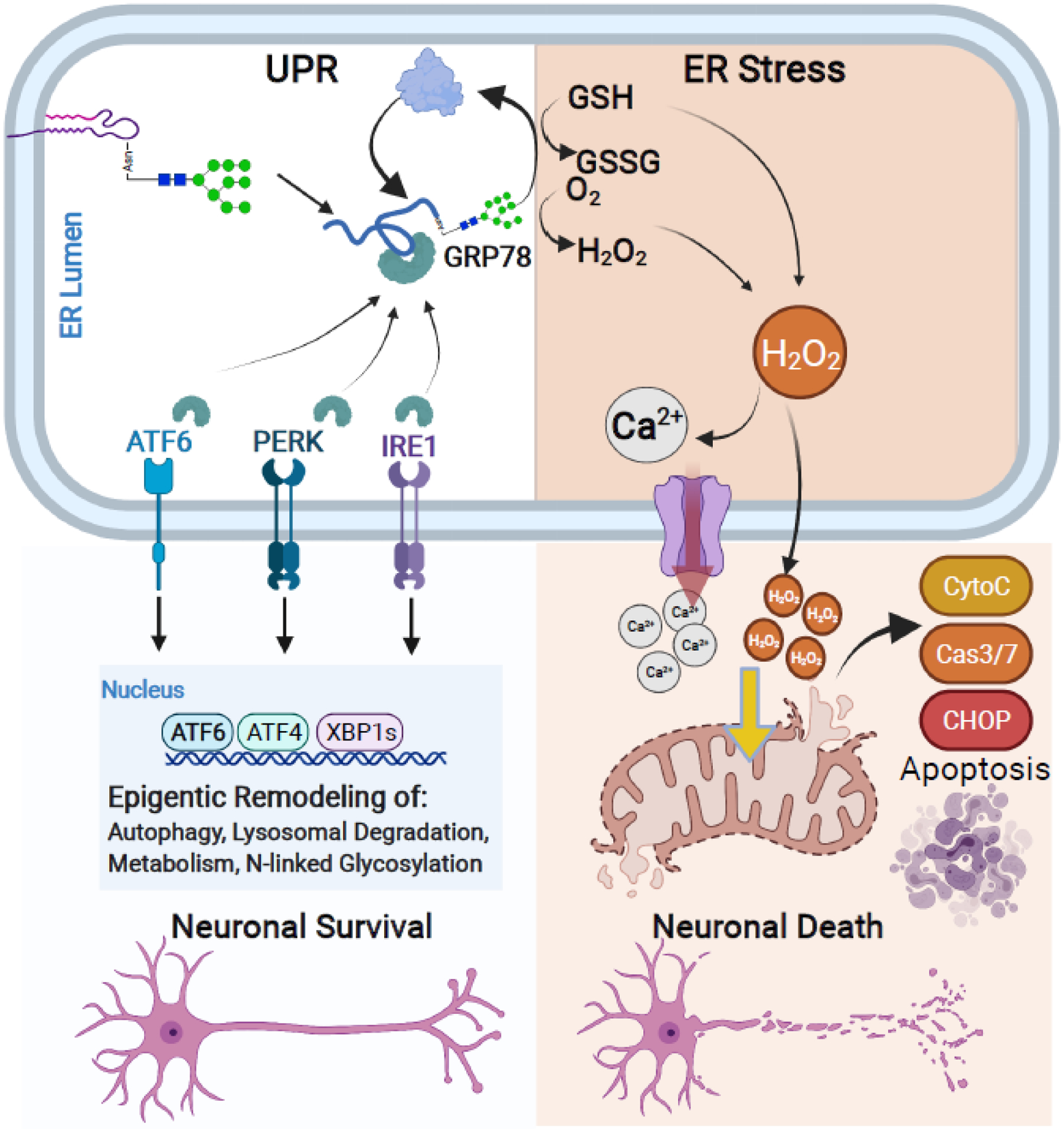
Early N-linked glycan biosynthesis is a central mediator of the UPR pathway. The UPR leads to protein refolding or salvage and activation of pro-survival pathways in neurons. However, if accumulation of unfolded proteins or prolonged stress exceeds capacity, the UPR leads to ER stress that triggers mitochondrial apoptotic signaling cascades and neuronal cell death. The UPR and pro-survival pathways (Left): Accumulation of misfolded proteins recruits available GRP78 and sequesters GRP78 away from ER membrane signaling transducers: ATF6, PERK, and IRE1. Binding of GRP78 to misfolded glycoproteins leads to protein refolding or salvage through lysosomal and proteasomal degradation. Dissociation of GRP78 with ATF6, PERK, and IRE1 also leads to their activation and subsequent signaling cascades for epigenetic remodeling of neurons. This process promotes neuronal cell survival by upregulating genes involved in the autophagy, lysosomal processing, metabolism, and N-linked glycosylation as a protective process against cellular stress. ER stress occurs when misfolded proteins or stress exceeds the UPR capacity (Right): If the UPR fails to re-establish cellular homeostasis, excess utilization and depletion of GSH and production of H2O2 during the refolding process leads to the ectopic release of Ca2+ and H2O2 from the ER into the cytosol. Excess of either cytosolic Ca2+ and H2O2 initiates mitochondrial-driven pro-apoptotic pathways including release of Cytochrome C and Caspase 3/7 activation and signaling cascade, ultimately inducing neuronal cell death.
Under physiological conditions GRP78 also binds to and inactivates three well-characterized ER membrane proteins and UPR sensors: protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Binding of misfolded glycopeptides sequesters GRP78 away from all three ER membrane proteins and initiates signaling cascades that lead to epigenetic changes and alter cellular metabolism, bioenergetics, and salvage pathways to adapt to extracellular pressure. Thus, all three signaling cascades protect neurons from cell death. For example, ectopic expression of ATF6 in forebrain neurons activates pro-survival pathways such as increased autophagy, increased apoptosis inhibition through B cell lymphoma 2 (BCL2), improved neuronal survival, and improved behavior functions [63]. Interestingly, ATF6 itself is glycosylated, and its glycosylation status modulates its activity as a transcription factor [64]. Further, activation of X-Box binding protein 1 (XBP1) during the UPR also leads to global changes in expression of genes involved in N-linked glycosylation [65]. Together, these events suggest a feed forward loop that remodels the cellular N-linked glycome as an adaptive process against cellular stress.
The goal of UPR activation is to reduce cellular stress and restore homeostasis. However, if the UPR fails to re-establish cellular homeostasis, it can lead to ER stress that amplifies cellular dysfunction and death through redox and calcium toxicity [66]. When protein misfolding occurs, the ER initiates refolding mechanisms of glycopeptides through the calnexin-calreticulin cycle and ER oxidoreductin 1 (ERO1)/protein disulfide isomerase (PDI) to direct disulfide bond formation (Box 2). The first gatekeeping enzyme of this process is calnexin, which only recognizes glycosylated peptides. Specifically, the addition of a single carbohydrate monomer on N-linked glycans is required for both calnexin activity and to initiate protein refolding [67,68]. Both calnexin and ERO1 are ER glycoproteins, and N-linked glycosylation is critical for their trafficking to the ER lumen and their enzymatic activities [69].
Box 2. The calnexin/calreticulin cycle in the UPR and neurodegeneration.
During early glycoprotein synthesis, N-glycans participate in protein folding via the calnexin/calreticulin (CNX/CRT) cycle [12]. Both CNX and CRT contain lectin binding domains that recognize and bind the early N-linked glycan moiety on newly synthesized glycoproteins. The collective actions of CNX/CRT allow the trimming of N-linked glycans as needed while the protein adopts its native confirmation, and prevents proteins from exiting the ER prematurely [13]. CNX/CRT is also a strong buffering system for ER luminal Ca2+, and critical for compartmentalized Ca2+ control. Glycoprotein bound-CNX/CRT acts as a chaperone and recruits oxidoreductases including ER oxidoreductin 1 (ERO1) and protein disulfide isomerase (PDI) to catalyze disulfide bond formation [13]. ERO1/PDI enzymatic actions require the antioxidant glutathione (GSH) and produce H2O2. Thus, the disulfide bond formation process is a major contributor of intracellular reactive oxygen species [14]. It is worth noting that CNX/CRT cycle share the common substrate, UDP-glucose, with glycogen metabolism. In response to excess unfolded proteins, the CNX/CRT cycle functions as a vital check point to mitigate accumulation of misfolded proteins in the ER and initiate the refolding process [15]. In addition, misfolded polypeptide chains form transient disulfide bonds with ERp57, a PDI family protein to stabilize its interaction with the CNX/CRT complex and prevent protein aggregation [16]. Finally, CRT acts as a Ca2+ buffering chaperone in the ER to maintain intracellular Ca2+ homeostasis and prevent Ca2+ induced apoptotic events [17]. In the event that misfolded proteins exceed the ER refolding machinery capacity, the unfolded protein response (UPR) leads to ER stress and initiates apoptosis in one of two ways. 1) CNX/CRT loses its Ca2+ buffering activity and results in ectopic Ca2+ release into the cytoplasm. 2) The actions of ERO1/PDI to form disulfate bonds in the protein refolding process simultaneously deplete GSH while producing H2O2. The combined actions of cytoplasmic Ca2+ and excess H2O2 can lead to apoptosis and ultimately neuronal cell death.
Attempts to correct misfolded glycoproteins generates increased oxidative stress by one of two mechanisms. First, the formation of disulfide bonds utilizes the reduced antioxidant glutathione (GSH) as the electron acceptor and produces oxidized glutathione (GSSG) [70]. Second, the biochemical reactions of ERO1 and PDI require oxygen as electron acceptors and they produce hydrogen peroxide, a form of reactive oxygen species [71]. When the number of misfolded glycopeptides overwhelms the refolding capacity and distorts the redox balance within the ER, hydrogen peroxide can enter the cytoplasm. The ER represents the largest storage of intracellular calcium and acts as a cytoplasmic calcium buffer in physiological and adverse conditions [72]. Increased hydrogen peroxide can trigger aberrant calcium release from the ER [73,74]. Ectopic levels of both cytoplasmic hydrogen peroxide and calcium lead to neuronal cell death by initiating mitochondrial-driven apoptotic pathways that include c/enhancer-binding protein homologous protein (CHOP) inhibition of BCL2, cytochrome C release, and caspase 3/7 activation [75,76]. Collectively, early N-linked glycosylation events in the ER are critical for modulating the UPR and sufficiently clearing misfolded glycoproteins. While detailed N-glycan changes in neurological disorders remain to be elucidated, ER stress has been highlighted as a key molecular process involved in AD disease progression (Box 3). Thus, N-linked glycosylation could sit at the crossroads of neuronal survival and neuronal cell death (Figure 4).
Box 3: ER stress in AD. [18–23].
The unfolded protein response (UPR) is one of the key regulatory processes for protein homeostasis (i.e. proteostasis) through activation of ERAD or autophagic/lysosomal pathways. Perturbed proteostasis leads to protein aggregation and ER stress. AD is clinically classified by loss of cognitive function and accumulation of protein aggregates known as Aβ or NFT. Indeed, multiple signaling proteins in the ER stress pathway are altered in human AD brain specimens when compared to matched normal controls [24]. For example, increased GRP78 has been observed in human brain regions affected by AD [25] and IRE1α phosphorylation directly correlates with Braak staging in AD patients [26]. Further analysis in AD experimental models raised the interesting hypothesis that only a subset of neurons are affected by ER stress [27]. Thus far, analyses of human tissue and multiple AD mouse models with different genetic drivers all share the common phenotypes of ER stress and neurodegenerations [24]. This correlation raises the interesting question whether ER stress is a driver or simply resultant of the disease. Recent studies using both genetic and pharmacological interventions have revealed causal relationship between ER stress and AD. For example, siRNA against ATF4, a transcription factor downstream of PERK activation, provided neuroprotection and restored nearly all choline acetyltransferase positive neurons in the PSEN1/APP mouse AD mouse model [28]. Further, pharmacological inhibition of PERK restored neuronal protein synthesis and prevented further neuronal loss in the rTg4510 tauopathy AD mouse model [29]. It is important to note that ER stress does not occur exclusively in neurons. ER stress in glial cells directly affects JAK/STAT and NF-κB pathways [30, 31], both pathways provide signaling cascades for microglia activation and astrogliosis [31]. Collectively, ER stress could act as a double-edged sword that drive neuronal cell death and activate neuroinflammation during AD.
N-linked glycosylation modulates neuroinflammation
Recent reports have highlighted neuroinflammation as a key driver of many neurodegenerative disorders including AD, PD, and epilepsy. More specifically, chronic exposure to reactive astrocytes and microglial activation leads to neuronal cell death. Neuronal cell death is initiated by acute reactive oxygen species (nitric oxide and hydrogen peroxide), cathepsin B, calcium, and tumor necrosis factor (TNF) being released from reactive astrocytes and activated microglia. These signaling molecules trigger neuronal cell death, followed by clearance of presumably dead neurons by complement-directed phagocytosis [77,78]. N-glycosylation is intimately involved in all of these processes during neuroinflammation (Figure 5). Specifically, lectin receptors are essential for proinflammatory microglia activation. For example, MBLs can bind to multiple glycan monomers including mannose, fucose, and GlcNAc and play a proinflammatory role in microglial activation and phagocytosis [79]. Further, the glycoprotein binding protein galectin-3 is required for astrocyte and microglial proliferation through the Janus kinase/signal transducers and activator of transcription (JAK/STAT) pathway and boosts IL-6 production [80]. Recent studies have suggested that MBL levels are negatively associated with poor prognosis in major neurological disorders [79]. It is also interesting to note that while microglia express surface Siglec receptors that recognize sialic acid on other CNS cells, microglia themselves also express sialic acid-containing N-liked glycans on the surface [81,82]. Intriguingly, the microglia surface sialic acid-containing glycans modulate inflammatory and phagocytic activates through the complement receptor or the Toll-like receptor signaling pathways [81,82]. N-linked glycosylation and microglia homeostasis are very active areas of research due to renewed interest in the roles of activated microglia in AD.
Figure 5. N-linked glycans are essential mediators of neuroinflammation.
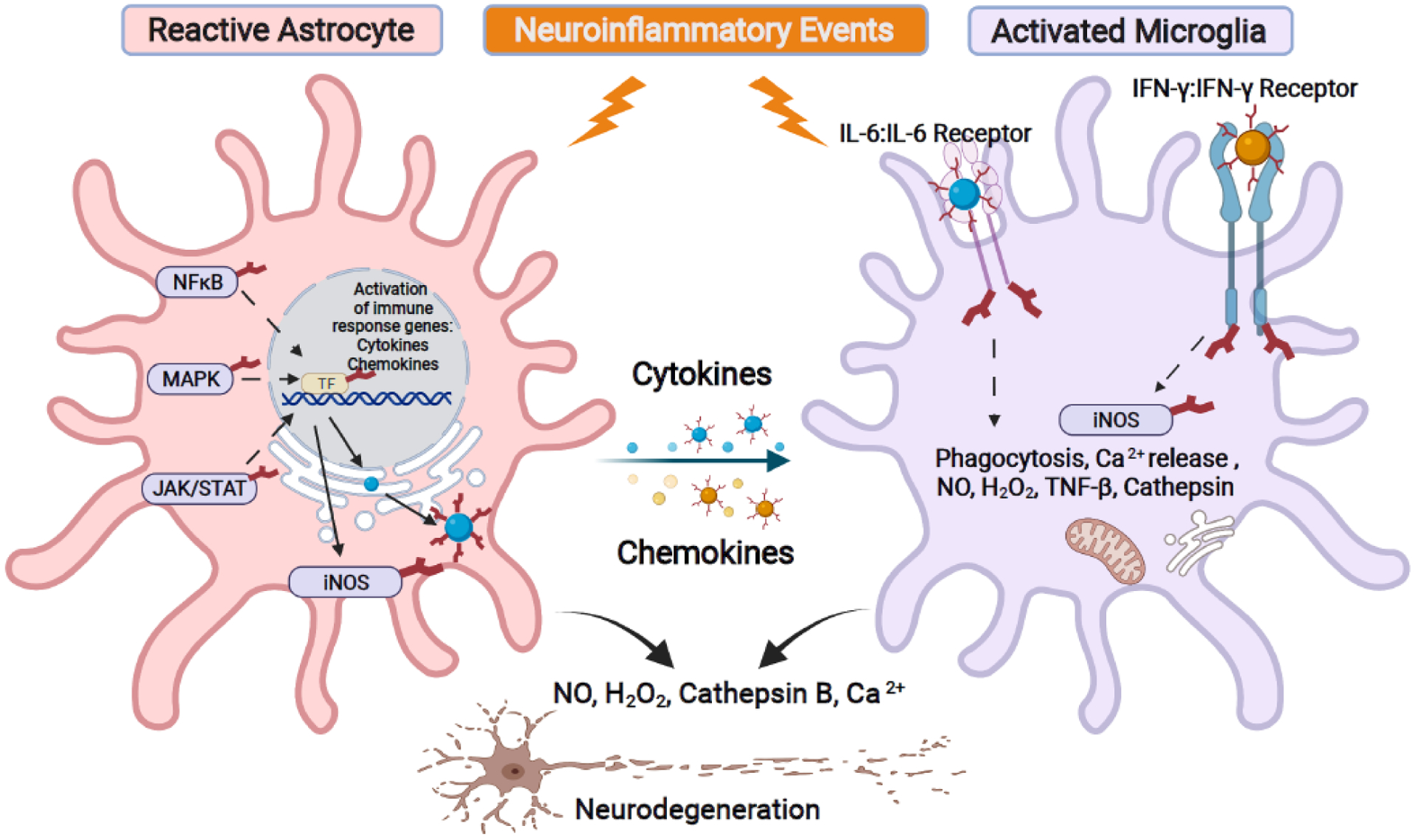
Neuroinflammation is one of the major drivers of neurogenerative diseases. Glial cells such as astrocytes and microglia contribute to this process by producing pro-inflammatory cytokines/chemokines to recruit and mount an immune response. They also generate reactive oxygen species and activate phagocytosis to clear cellular debris. Pro-inflammatory triggers activate signaling cascades such as NFκB, MAPK, and JAK/STAT pathways that initiate cytokine, chemokine, and nitric oxide synthase (iNOS) production in astrocytes. N-linked glycosylation is a modifier of NFκB, MAPK, JAK/STAT pathway components as well as participates in the production, trafficking, and interaction of cytokines/chemokines/iNOS. Released cytokines and chemokines can recruit and activate microglia by binding to their corresponding cell surface receptors. Cytokine ligand and receptor binding (such as IL-6 and IFN-γ) require N-linked glycosylation for the signaling transduction events that lead to expression of iNOS, intracellular release of Ca2+, NO, H2O2, cathepsin, and the action of phagocytosis. Collectively, N-linked glycans are an integral component of these neuroinflammatory pathways, especially in driving the release of pro-apoptotic factors that lead to neuronal cell death.
During early phases of the neuroinflammatory response, the primary role of reactive astrocytes is the production of cytokines and chemokines to recruit additional immune cells, including microglia, to the site of action [78]. Many signaling pathways regulate production of cytokines and chemokines. Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFκB), mitogen-activated protein kinase (MAPK), and JAK/STAT all initiate signaling cascades that lead to the epigenetic production of cytokine and chemokine transcripts. Interestingly, all three signaling pathways require glycosylation in one or more components to fine-tune pathway activity [83]. Cytokines/chemokines are extracellular proteins, and their maturation and trafficking are controlled through the ER-Golgi organelle compartments. Therefore, it is not surprising that cytokines and chemokines and their cell surface receptors are glycosylated, and their glycosylation is important for both receptor-ligand interaction as well as protein trafficking and release. For example, both interleukin 6 (IL6) and interferon gamma (IFN-y) and their respective microglia cell surface receptors contain multiple glycosylation sites [84,85]. N-linked glycan status on both ligand and receptor modulate signaling dynamics within activated microglia, contributing to its pro-apoptotic responses that could drive nitric oxide production, ectopic calcium release, cathepsin release, and complemental cascades that lead to phagocytosis. Many of these pro-apoptotic factors are known inducers of neuronal cell death in neurological disorders. Of note, both nitric oxide synthase (iNO2) and C3 complement cascade activities are modulated by N-linked glycosylation [86,87]. However, the exact regulatory dynamics within these enzymes and signaling cascades by N-linked glycans within the context of chronic inflammation during neurological disorders remain to be elucidated.
Concluding Remarks
Decades of research have drastically increased our knowledge in the essential roles of N-linked glycans in signaling, enzyme activity, and cellular trafficking. However, significant knowledge gaps remain at the system level regarding how N-linked glycosylation modulates the protein interactome, both at the cellular level on deciding cellular fate and function, and on an organismal level on driving whole-body physiology. These gaps are especially true in the case of the human brain, an organ with incredible complexity that gives rise to human memory, cognition, and behavior. N-linked glycans are an essential component of neuronal function and modulate a myriad of neuronal processes from membrane potential to neurotransmitter vesicle release. N-linked glycosylation is also a central mediator of the UPR, a critical decision point for neuronal survival or death. These important functions highlight why aberrant N-linked glycosylation is implicated in major neurological disorders. N-linked glycosylation represents a complex set of pathways that span three cellular compartments and involves over 700 enzymes. Therefore, clinical targeting of N-linked glycosylation should be carefully considered and designed, as preventing the initiation of N-linked glycosylation could have myriad unwanted side effects. Nevertheless, N-linked glycosylation pathways represent an exciting therapeutic opportunity in neurological disorders that has yet to be explored, and treatments should be carefully tailored to the specific N-linked glycan species perturbed in each unique neurological disorder.
Outstanding questions.
How do brain cells coordinate de novo synthesis of monosaccharides versus exogenous uptake for subsequent sugar-nucleotide synthesis?
How do neurons balance/regulate bioenergetics and N-linked glycan biosynthesis, two facets of glucose metabolism?
What are the unique classes of N-linked glycans over- and under-represented in neurological disorders?
Are aberrant N-linked glycans displayed on neurons or protein aggregates a chronic driver of neuroinflammation in neurological disorders?
Does targeting glycogen accumulation represent a potential therapeutic strategy to modulate defective glycosylation in neurodegenerative diseases?
Highlights:
At least ten unique monosaccharides are present in cells of the central nervous system, and they provide an essential repertoire of oligosaccharides critical for brain function.
Monosaccharide and sugar-nucleotide biosynthesis exhibit metabolic plasticity and are channeled through multiple substrates.
N-linked glycans impact nearly all neuronal functions, including maintenance of resting membrane potential, axon firing, and synaptic vesicle release.
N-linked glycosylation is a central mediator of the unfolded protein response that determines neuronal cell fate.
Cytokines, nitric oxide synthase, and other protein/enzymes involved in the innate immune response are N-linked glycosylated, suggesting a central role for N-linked glycans in neuroinflammation.
Acknowledgments
We would like to thank Drs. Vander Kooi and Charles J. Waechter as well as members of the Gentry and Sun laboratories for vigorous discussions regarding the work.
Funding
This Review was supported by National Institute of Health (NIH) grants R01 AG066653, St Baldrick’s Career Development Award, V-Scholar Grant, Rally Foundation Independent Investigator Grant to R.C.S., R35 NS116824 and P01 NS097197 to M.S.G., and L.R.C was supported by NIH/NCI training grant T32CA165990.
Glossary
- α-synuclein
a neuronal protein that controls synaptic vesicle trafficking and neurotransmitter release.
- Alzheimer’s disease
a progressive neurodegenerative disorder that causes dementia, memory loss, and loss of cognitive function.
- Amyloid beta (Aβ)
the peptide derived from amyloid precursor protein that is the major component of amyloid plaques that accumulate in the brains of Alzheimer’s disease patients. Axon firing: the traveling of an action potential or nerve impulse down an axon to the synaptic cleft for neuronal cell communication.
- Carbohydrate code
the diverse range of complex carbohydrates generated by limitless combinations of individual monosaccharides.
- Congenital disorders of glycosylation (CDGs)
an umbrella term for a group of over 130 rare genetic disorders resulting in defects in glycosylation.
- Glycoproteins
proteins that contain covalent attachment of glycans to amino acid side chains.
- Lectins
highly specific carbohydrate-binding proteins.
- N-linked glycans/glycosylation
the attachment of oligosaccharides (N-linked glycans) to asparagine residues on newly synthesized proteins.
- Neurodegeneration
the progressive loss of function and ultimately cell death of neurons, which is a driver of many neurodegenerative diseases.
- Neurofibrillary tangles (NFT)
insoluble twisted fibers found in brain cells that consist of aggregates of hyperphosphorylated tau protein.
- Neuroinflammation
activation of the central nervous system’s innate immune system in response to an inflammatory challenge.
- Synaptic cleft
the physical boundary or junction between two neurons.
- Synaptic transmission
the biological process by which neurons communicate with target neurons across the synaptic cleft. Neurotransmitters released from presynaptic neurons bind to and react with receptors on postsynaptic neurons to propagate neuronal signaling.
- Synaptic vesicle
secretory vesicles formed in the Golgi that transport neurotransmitters to the synaptic cleft to be released.
- Voltage gated ion channels (VGICs)
transmembrane proteins that play important roles in electrical signaling of cells. VGICs form ion channels that become activated in response to changes in electrical membrane potential.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests
R.C.S., and M.S.G. are consultants for Maze Therapeutics, M.S.G. is a consultant for Enable Therapeutics, Glut1-Deficiency Syndrome Foundation, and Chelsea’s Hope. M.S.G., and R.C.S., are founders of Atterogen, LLC.
References
- 1.Werz DB et al. (2007) Exploring the structural diversity of mammalian carbohydrates (“glycospace”) by statistical databank analysis. ACS chemical biology 2, 685–691 [DOI] [PubMed] [Google Scholar]
- 2.Laine RA (1994) A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10 (12) structures for a reducing hexasaccharide: the Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 4, 759–767 [DOI] [PubMed] [Google Scholar]
- 3.Narimatsu Y et al. (2019) An Atlas of Human Glycosylation Pathways Enables Display of the Human Glycome by Gene Engineered Cells. Mol Cell 75, 394–407.e395. 10.1016/j.molcel.2019.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drake RR et al. (2017) MALDI Mass Spectrometry Imaging of N-Linked Glycans in Cancer Tissues. Adv Cancer Res 134, 85–116. 10.1016/bs.acr.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winchester B (2005) Lysosomal metabolism of glycoproteins. Glycobiology 15, 1r–15r. 10.1093/glycob/cwi041 [DOI] [PubMed] [Google Scholar]
- 6.Fahie K and Zachara NE (2016) Molecular Functions of Glycoconjugates in Autophagy. J Mol Biol 428, 3305–3324. 10.1016/j.jmb.2016.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarz F and Aebi M (2011) Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol 21, 576–582. 10.1016/j.sbi.2011.08.005 [DOI] [PubMed] [Google Scholar]
- 8.Berger RP et al. (2016) Glycosylation and stem cells: Regulatory roles and application of iPSCs in the study of glycosylation-related disorders. Bioessays 38, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balakrishnan B et al. (2019) A novel phosphoglucomutase-deficient mouse model reveals aberrant glycosylation and early embryonic lethality. Journal of inherited metabolic disease 42, 998–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho-Cruz P et al. (2018) Cellular glycosylation senses metabolic changes and modulates cell plasticity during epithelial to mesenchymal transition. Dev Dyn 247, 481–491. 10.1002/dvdy.24553 [DOI] [PubMed] [Google Scholar]
- 11.Reily C et al. (2019) Glycosylation in health and disease. Nat Rev Nephrol 15, 346–366. 10.1038/s41581-019-0129-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brewer MK and Gentry MS (2019) Brain Glycogen Structure and Its Associated Proteins: Past, Present and Future. Adv Neurobiol 23, 17–81. 10.1007/978-3-030-27480-1_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley P et al. (2017) N-glycans. Essentials of Glycobiology [Internet]. 3rd edition, [Google Scholar]
- 14.Sakai M et al. (1969) Studies of corpora amylacea. I. Isolation and preliminary characterization by chemical and histochemical techniques. Arch Neurol 21, 526–544. 10.1001/archneur.1969.00480170098011 [DOI] [PubMed] [Google Scholar]
- 15.Stam FC and Roukema PA (1973) Histochemical and biochemical aspects of corpora amylacea. Acta Neuropathol 25, 95–102. 10.1007/bf00687554 [DOI] [PubMed] [Google Scholar]
- 16.Sun RC et al. (2021) Brain glycogen serves as a critical glucosamine cache required for protein glycosylation. Cell Metab. 10.1016/j.cmet.2021.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandl J and Bánhegyi G (2018) The ER - Glycogen Particle - Phagophore Triangle: A Hub Connecting Glycogenolysis and Glycophagy? Pathol Oncol Res 24, 821–826. 10.1007/s12253-018-0446-0 [DOI] [PubMed] [Google Scholar]
- 18.Lytridou AA et al. (2020) Stbd1 promotes glycogen clustering during endoplasmic reticulum stress and supports survival of mouse myoblasts. J Cell Sci 133. 10.1242/jcs.244855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeken J (2013) Congenital disorders of glycosylation. Handb Clin Neurol 113, 1737–1743. 10.1016/b978-0-444-59565-2.00044-7 [DOI] [PubMed] [Google Scholar]
- 20.Ng BG and Freeze HH (2018) Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet 34, 466–476. 10.1016/j.tig.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paprocka J et al. (2021) Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci 11. 10.3390/brainsci11010088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanley P (2016) What Have We Learned from Glycosyltransferase Knockouts in Mice? J Mol Biol 428, 3166–3182. 10.1016/j.jmb.2016.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott H and Panin VM (2014) N-glycosylation in regulation of the nervous system. Adv Neurobiol 9, 367–394. 10.1007/978-1-4939-1154-7_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon SE and Chapman ER (2012) Glycosylation is dispensable for sorting of synaptotagmin 1 but is critical for targeting of SV2 and synaptophysin to recycling synaptic vesicles. J Biol Chem 287, 35658–35668. 10.1074/jbc.M112.398883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazniewska J and Weiss N (2017) Glycosylation of voltage-gated calcium channels in health and disease. Biochimica et Biophysica Acta (BBA)-Biomembranes 1859, 662–668 [DOI] [PubMed] [Google Scholar]
- 26.Kruger LC and Isom LL (2016) Voltage-gated Na+ channels: not just for conduction. Cold Spring Harbor perspectives in biology 8, a029264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boll I et al. (2020) Synaptic transmission induces site-specific changes in sialylation on N-linked glycoproteins in rat nerve terminals. BioRxiv, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boll I et al. (2020) Depolarization-dependent induction of site-specific changes in sialylation on N-linked glycoproteins in rat nerve terminals. Molecular & Cellular Proteomics 19, 1418–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stout KA et al. (2019) The Synaptic Vesicle Glycoprotein 2: Structure, Function, and Disease Relevance. ACS Chem Neurosci 10, 3927–3938. 10.1021/acschemneuro.9b00351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park DH et al. (2020) N-linked glycosylation of the mGlu7 receptor regulates the forward trafficking and transsynaptic interaction with Elfn1. Faseb j 34, 14977–14996. 10.1096/fj.202001544R [DOI] [PubMed] [Google Scholar]
- 31.Sun RC et al. (2021) Brain glycogen serves as a critical glucosamine cache required for protein glycosylation. Cell Metabolism, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reily C et al. (2019) Glycosylation in health and disease. Nature Reviews Nephrology 15, 346–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price BR et al. (2021) Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Research Reviews, 101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matyash V and Kettenmann H (2010) Heterogeneity in astrocyte morphology and physiology. Brain research reviews 63, 2–10 [DOI] [PubMed] [Google Scholar]
- 35.Hussaini SM and Jang MH (2018) New roles for old glue: astrocyte function in synaptic plasticity and neurological disorders. International Neurourology Journal 22, S106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauer D et al. (2010) Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophrenia research 117, 92–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jing B et al. (2018) Glycosylation of dentin matrix protein 1 is a novel key element for astrocyte maturation and BBB integrity. Protein & cell 9, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith BA and Bertozzi CR (2021) The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nature Reviews Drug Discovery, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X et al. (2020) N-glycosylation of Siglec-15 decreases its lysosome-dependent degradation and promotes its transportation to the cell membrane. Biochem Biophys Res Commun 533, 77–82. 10.1016/j.bbrc.2020.08.111 [DOI] [PubMed] [Google Scholar]
- 40.Johnson EC et al. (2020) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Z et al. (2021) In-depth Site-specific Analysis of N-glycoproteome in Human Cerebrospinal Fluid and Glycosylation Landscape Changes in Alzheimer’s Disease. Mol Cell Proteomics 20, 100081. 10.1016/j.mcpro.2021.100081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lane CA et al. (2018) Alzheimer’s disease. European Journal of Neurology 25, 59–70. 10.1111/ene.13439 [DOI] [PubMed] [Google Scholar]
- 43.Xie A et al. (2014) Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int 2014, 648740. 10.1155/2014/648740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakagawa K et al. (2006) Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J Neurochem 96, 924–933. 10.1111/j.1471-4159.2005.03595.x [DOI] [PubMed] [Google Scholar]
- 45.Boix CP et al. (2020) Amyloid precursor protein glycosylation is altered in the brain of patients with Alzheimer’s disease. Alzheimers Res Ther 12, 96. 10.1186/s13195-020-00664-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsatsanis A et al. (2019) Post Translational Modulation of β-Amyloid Precursor Protein Trafficking to the Cell Surface Alters Neuronal Iron Homeostasis. Neurochem Res 44, 1367–1374. 10.1007/s11064-019-02747-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birol M et al. (2019) Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. PLoS Biol 17, e3000318. 10.1371/journal.pbio.3000318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo JL and Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20, 130–138. 10.1038/nm.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whyte LS et al. (2017) Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer’s disease? Journal of neurochemistry 140, 703–717 [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y et al. (2017) The role of ubiquitin proteasomal system and autophagy-lysosome pathway in Alzheimer’s disease. Reviews in the Neurosciences 28, 861–868 [DOI] [PubMed] [Google Scholar]
- 51.Lignani G et al. (2020) Homeostatic plasticity in epilepsy. Frontiers in Cellular Neuroscience 14, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Andrea Meira I et al. (2019) Ketogenic diet and epilepsy: what we know so far. Frontiers in neuroscience 13, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okuda T (2019) Data set for characterization of the glycosylation status of hepatic glycoproteins in mice fed a low-carbohydrate ketogenic diet. Data in brief 27, 104604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quelhas D et al. (2021) SLC35A2-CDG: Novel variant and review. Molecular Genetics and Metabolism Reports 26, 100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Witters P et al. (2020) Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genetics in Medicine 22, 1102–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sim NS et al. (2018) Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurology Genetics 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hetz C and Papa FR (2018) The unfolded protein response and cell fate control. Molecular cell 69, 169–181 [DOI] [PubMed] [Google Scholar]
- 58.Bartoszewska S and Collawn JF (2020) Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cellular & molecular biology letters 25, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z et al. (2019) Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox biology 25, 101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hetz C and Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nature Reviews Neurology 13, 477–491 [DOI] [PubMed] [Google Scholar]
- 61.Ellgaard L et al. (2016) Co-and post-translational protein folding in the ER. Traffic 17, 615–638 [DOI] [PubMed] [Google Scholar]
- 62.Ibrahim IM et al. (2019) GRP78: A cell’s response to stress. Life sciences 226, 156–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu Z et al. (2017) Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. Journal of Cerebral Blood Flow & Metabolism 37, 1069–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang H et al. (2020) ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. Iscience 23, 100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong MY et al. (2018) XBP1s activation can globally remodel N-glycan structure distribution patterns. Proceedings of the National Academy of Sciences 115, E10089–E10098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhattarai KR et al. (2021) The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med 53, 151–167. 10.1038/s12276-021-00560-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kozlov G and Gehring K (2020) Calnexin cycle–structural features of the ER chaperone system. The FEBS journal 287, 4322–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang H et al. (2019) N-glycan-calnexin interactions in human factor VII secretion and deficiency. The international journal of biochemistry & cell biology 113, 67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fan F et al. (2020) Identification of N-glycosylation sites on AtERO1 and AtERO2 using a transient expression system. Biochemical and Biophysical Research Communications 533, 481–485 [DOI] [PubMed] [Google Scholar]
- 70.Shergalis AG et al. (2020) Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacology & therapeutics 210, 107525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee YM et al. (2021) The redox language in neurodegenerative diseases: oxidative post-translational modifications by hydrogen peroxide. Cell Death & Disease 12, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wegierski T and Kuznicki J (2018) Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 74, 102–111 [DOI] [PubMed] [Google Scholar]
- 73.Ostrowski TD et al. (2017) H2O2 augments cytosolic calcium in nucleus tractus solitarii neurons via multiple voltage-gated calcium channels. American Journal of Physiology-Cell Physiology 312, C651–C662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng Y and Shen X (2005) H2O2 directly activates inositol 1, 4, 5-trisphosphate receptors in endothelial cells. Redox Report 10, 29–36 [DOI] [PubMed] [Google Scholar]
- 75.Dong Y et al. (2017) N-methyl-d-aspartate receptor-mediated calcium overload and endoplasmic reticulum stress are involved in interleukin-1beta-induced neuronal apoptosis in rat hippocampus. Journal of neuroimmunology 307, 7–13 [DOI] [PubMed] [Google Scholar]
- 76.Andreyev A et al. (2018) Calcium uptake and cytochrome c release from normal and ischemic brain mitochondria. Neurochemistry international 117, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarlus H and Heneka MT (2017) Microglia in Alzheimer’s disease. The Journal of clinical investigation 127, 3240–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giovannoni F and Quintana FJ (2020) The role of astrocytes in CNS inflammation. Trends in Immunology, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Siew JJ and Chern Y (2018) Microglial lectins in health and neurological diseases. Frontiers in molecular neuroscience 11, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sirko S et al. (2015) Astrocyte reactivity after brain injury—: the role of galectins 1 and 3. Glia 63, 2340–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allendorf DH et al. (2020) Lipopolysaccharide activates microglia via neuraminidase 1 desialylation of Toll-like Receptor 4. Journal of neurochemistry 155, 403–416 [DOI] [PubMed] [Google Scholar]
- 82.Allendorf DH et al. (2020) Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 68, 989–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doud EH et al. (2020) NF-κB Signaling Is Regulated by Fucosylation in Metastatic Breast Cancer Cells. Biomedicines 8, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Riethmueller S et al. (2017) Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS biology 15, e2000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blouin CM et al. (2016) Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell 166, 920–934 [DOI] [PubMed] [Google Scholar]
- 86.Yan J et al. (2020) N-Glycosylation at Asn695 might suppress inducible nitric oxide synthase activity by disturbing electron transfer. Acta Biochimica et Biophysica Sinica 52, 1360–1372 [DOI] [PubMed] [Google Scholar]
- 87.Ritchie GE et al. (2002) Glycosylation and the complement system. Chemical reviews 102, 305–320 [DOI] [PubMed] [Google Scholar]
References
- 1.Rich LR et al. (2019) The Role of Brain Glycogen in Supporting Physiological Function. Front Neurosci 13, 1176. 10.3389/fnins.2019.01176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki A et al. (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. 10.1016/j.cell.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rai A et al. (2018) Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis 9, 201. 10.1038/s41419-017-0190-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robitaille Y et al. (1980) A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal ageing. Brain 103, 315–336. 10.1093/brain/103.2.315 [DOI] [PubMed] [Google Scholar]
- 5.Zhou Z et al. (2019) Antibody-Mediated Enzyme Therapeutics and Applications in Glycogen Storage Diseases. Trends Mol Med 25, 1094–1109. 10.1016/j.molmed.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perenthaler E et al. (2020) Loss of UGP2 in brain leads to a severe epileptic encephalopathy, emphasizing that bi-allelic isoform-specific start-loss mutations of essential genes can cause genetic diseases. Acta Neuropathol 139, 415–442. 10.1007/s00401-019-02109-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi R et al. (2015) Application of whole exome sequencing to a rare inherited metabolic disease with neurological and gastrointestinal manifestations: a congenital disorder of glycosylation mimicking glycogen storage disease. Clin Chim Acta 444, 50–53. 10.1016/j.cca.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 8.Vega AI et al. (2016) Molecular diagnosis of glycogen storage disease and disorders with overlapping clinical symptoms by massive parallel sequencing. Genet Med 18, 1037–1043. 10.1038/gim.2015.217 [DOI] [PubMed] [Google Scholar]
- 9.Sakai M et al. (1969) Studies of corpora amylacea. I. Isolation and preliminary characterization by chemical and histochemical techniques. Arch Neurol 21, 526–544. 10.1001/archneur.1969.00480170098011 [DOI] [PubMed] [Google Scholar]
- 10.Stam FC and Roukema PA (1973) Histochemical and biochemical aspects of corpora amylacea. Acta Neuropathol 25, 95–102. 10.1007/bf00687554 [DOI] [PubMed] [Google Scholar]
- 11.Sun RC et al. (2021) Brain glycogen serves as a critical glucosamine cache required for protein glycosylation. Cell Metab. 10.1016/j.cmet.2021.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ihara Y et al. (2020) Calnexin/Calreticulin and Assays Related to N-Glycoprotein Folding In Vitro. Methods Mol Biol 2132, 295–308. 10.1007/978-1-0716-0430-4_29 [DOI] [PubMed] [Google Scholar]
- 13.Kozlov G and Gehring K (2020) Calnexin cycle - structural features of the ER chaperone system. Febs j 287, 4322–4340. 10.1111/febs.15330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shergalis AG et al. (2020) Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol Ther 210, 107525. 10.1016/j.pharmthera.2020.107525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q et al. (2015) Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 20, 13689–13704. 10.3390/molecules200813689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hebert DN and Molinari M (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev 87, 1377–1408. 10.1152/physrev.00050.2006 [DOI] [PubMed] [Google Scholar]
- 17.Michalak M et al. (2009) Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J 417, 651–666. 10.1042/bj20081847 [DOI] [PubMed] [Google Scholar]
- 18.Sprenkle NT et al. (2017) Endoplasmic reticulum stress and inflammation in the central nervous system. Molecular neurodegeneration 12, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louessard M et al. (2017) Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death & Differentiation 24, 1518–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarvani C et al. (2017) Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacological research 119, 412–421 [DOI] [PubMed] [Google Scholar]
- 21.Remondelli P and Renna M (2017) The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Frontiers in molecular neuroscience 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uddin MS et al. (2020) Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Molecular neurobiology 57, 2902–2919 [DOI] [PubMed] [Google Scholar]
- 23.Ghemrawi R and Khair M (2020) Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. International journal of molecular sciences 21, 6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hetz C and Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nature Reviews Neurology 13, 477–491 [DOI] [PubMed] [Google Scholar]
- 25.Hoozemans JJ et al. (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. The American journal of pathology 174, 1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duran-Aniotz C et al. (2017) IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta neuropathologica 134, 489–506 [DOI] [PubMed] [Google Scholar]
- 27.Saxena S and Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71, 35–48 [DOI] [PubMed] [Google Scholar]
- 28.Baleriola J et al. (2014) Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radford H et al. (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta neuropathologica 130, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Y and Li F (2016) Endoplasmic reticulum stress in brain ischemia. International Journal of Neuroscience 126, 681–691 [DOI] [PubMed] [Google Scholar]
- 31.Yan Z et al. (2018) Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clinical Immunology 189, 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]