

Abstract
Objective:
Malaria is an ancient disease that still causes more than 200 million of cases 7 with high mortality globally. Identification of new drug targets and development of novel antimalarial drugs with unique mode of action encounter the drug resistance and reduce the mortality by Plasmodium parasites. Actin protein is one of the key proteins in Plasmodium falciparum playing multifarious important roles including transport, cell motility, cell division, and shape determination. This study investigated Actin I as a drug target, in silico screening of diverse molecules through molecular docking was considered. Further, pharmacokinetic parameters of the selected molecules from the docking and interaction studies were planned to propose the lead molecules.b
Methods:
Molecules were selected according to score and protein ligand interaction and selected molecules were subjected for pharmacokinetic studies to investigate important drug parameters.
Results:
The docked molecules were ranked according to the binding score and good interaction pattern was observed with Actin I within top 20 scoring molecules. The selected molecules also had optimum pharmacokinetic parameters.
Conclusion:
The current study provides a set of hit molecules which can be further explored through in vitro and in vivo experiments for the development of potential drugs against malaria, there by encountering drug resistance and establishing Actin I as an important drug target.
Keywords: Actin I, actin, drug resistance, drug, malaria, molecular docking, Plasmodium falciparum
Introduction
Malaria is a life-threatening disease caused by parasites belonging to five species of genus Plasmodium falciparum which are transmitted through the bite of female Anopheles mosquito. In 2017 as per the WHO report, 219 million cases of malaria were reported in 87 countries and the estimated number of deaths was found to be 435,000. Despite such drastic figures there is still no widely used efficacious vaccine available for malaria parasites.[1] Although, antimalarial drugs are available from long back and the popular antimalarial medications used against malarial parasites includes Chloroquine, Chloroguanide (Proguanil), Sulfadoxine/pyrimethamine, Quinine, Mefloquine, Halofantrine, Artemisinin (ART), and Atovaquone. In 21st century, the drug resistance has emerged to almost all classes of antimalarial drugs and therefore a sudden leap has been witnessed in malaria-related mortality, especially in Africa.[1]
Both drug resistance and unavailability of highly effective vaccine makes malaria a highly challenging disease and a great public health burden. The identification of new/novel drug targets as well as development of novel antimalarial drugs with unique mode of action can be the possible answer to drug resistance in malaria and subsequently it can control the mortality due to malaria.[2,3] Being highly studied diseases several potential drug targets have been suggested in research which can be further developed as a drug target in experiments. Actin I is also such kind of drug target in malaria. Recently, our group has discovered that Actin I protein is expressed in almost all stages of P. falciparum which are present in human which justifies its importance in plasmodium lifecycle and its drug target potential.[4] Although in different literature actin is proposed as the drug target in malaria, it is not yet studied for structure based drug design and subsequently not developed as an important drug target.[4,5] The main reasons for the selection of the Actin I in the current study are the Actin I protein is novel drug target which is important in different stages of P. falciparum and can be a probable answer to drug resistance.[4] Although the Actin I protein is conserved in both, there is difference between human and P. falciparum actin protein. In human, three isoforms of actin have been identified, alpha, beta, and gamma whereas in P. falciparum there exists only two isoforms, that is, Actin I and Actin II.[6,7] This protein is an essential part of the parasite motor machinery and responsible for gliding motility of the parasite.[8,9] In nature, there exists six different mammalian actin isoforms which differ from each other by a maximum of 6% of the sequence, and are practically identical across the species. Commonly most actins have the capacity to form long filaments but in P. falciparum the presence of regular actin filaments is uncertain. In P. falciparum Actin I is abundant and expressed throughout the lifecycle of the parasite. In vitro, apicomplexan actins form short, ~100-nm long filaments, which suffer from rapid tread milling.[10,11] Recently, specific antibodies revealed filament-like structures in motile forms of P. falciparum.[12] The P. falciparum Actin I fibers are refined from merozoites and are thereby distinctive for their helical symmetry from canonical actin structures.[13,14]
The most prevalent forms of actin protein are ATP-G-actin and ADP-F-actin. The structural determination of G-actin reveals that it has two lobes separated by a cleft, which is an ATPase fold. The functional form of G-actin exhibits only when the cleft comprises of an ATP or ADP. The F-actin polymer is a filamentous protein which is also referred as microfilament with levorotatory helix structure. The cell creeping process helps actin cytoskeleton to encounter dynamic assembly and disassembly, which further controls central attachment of assembly/disassembly bulge advancement, and contractile fiber association. Moreover cell relocation and adhesion is inhibited by the disturbance of the actin cytoskeleton.[15-17] The assembly and disassembly of local Actin I filament regulates the dynamic formation of lamellipodia. The lamellipodia is slim, sheet-like film distensions of the motile cells. During movement, cells grow the film forward to explore their environment. The branching and elongation are the two examples of actin fiber assembly, which advance the improvement of the actin “work” in the cell distension.[16,18] The nucleation promoting factors such as neuronal Wiskott - Aldrich syndrome Protein (N-WASP) and WASP-family verprolin-homologous protein (WAVE) prohibit the movement of Arp2/3 complex, which are in turn modulated by upstream regulators.[16] The Actin I fibers are organized into two kinds of arrays: Bundles and web like systems. Similarly, the Actin I fibers cross-connecting the proteins help to stabilize and keep up these structures and are divided into two classes: Packaging proteins and web-forming proteins. Actin I plays an important role in the intracellular blood stages of P. falciparum, such as endocytosis and vesicle trafficking,[18,19] ring stage morphology,[20] and spatial positioning of genes in the nucleus.[21] As per the recent studies, the actin elements have appeared to assume significant roles in the inheritance of intracellular organelles, for example, the apicoplast, mitochondria and secretory vesicles during intracellular parasite replication.[21-24] The Actin I association has also been well studied in zoite and asexual blood stages. In this intracellular stage, the parasite experiences a wonderful morphological change during which the parasite embraces a crescent or falci from shape in anticipation for transmission by means of the Anopheles mosquito.[25,26] In Actin I structure the C terminus acquires the shape of a-helix and it interacts with the bottom of the sub domain 1. Actin I protein consist of cleft named hydrophobic cleft between the sub-domains I and III. This cleft is major regulatory site and regarded as hotspot for the protein binding.[7] The binding pocket of Actin I has two parameters, the “mouth” and “phosphate clamp.” The “mouth” is considered as the distance between the Ca atoms of Gln59and Glu207. The distance between the Caatoms of Gly15 and Asp157 makes the “phosphate clamp.” As compared to the canonical actins, in Actin I there is a smaller cleft where large hydrophobic residues have adopted different conformations together withTrp357. Actin I shows significant structural deviations in specific regions which are involved in binding of proteins.[7,27] This may provide opportunities for structure-based drug design targeted at the P. falciparum act in-regulatory boundaries. The importance of Actin I protein is established in most of the stages of parasite. The crucial function of Actin I in asexual, blood, liver, mosquito and sexual stages is reported in the literature which also correlates with our earlier finding of its expression in nearly all stages of P. falciparum.[4] The active site residues of protein consist of amino acids, that is, Asn 17(A), Gly 16(A), Lys 19 (A), Gly 303(A), Glu 215(A), Lys 214(A), Val 160 (A), and Gly 159(A).
In current study, Actin I protein was taken for molecular docking studies because it is a new potential drug target and relatively not explored as drug target in drug discovery in spite of its conservation in different species of P. falciparum. In P. falciparum intracellular functions of Actin I have been recommended, that is, endocytosis,[20] secretion, and antigenic variation.[18,28,29] Molecular docking is an important tool for drug discovery and can be used to model the interaction between small drug like molecule and a protein at atomic level, which permit us to describe the enactment of small molecules in the binding sites of target proteins as well as to irradiate the biochemical processes. Docking is very useful and one of the most commonly used methods in structure-based drug design, due to its capability to predict the binding-conformation of small molecule ligands to the appropriate target binding site.[4,30-32] In the current study, the library of small molecules is screened against Actin I protein. The top scoring molecules from docking study were subjected for protein-ligand interaction studies and prediction of pharmacokinetics properties. Favorable pharmacokinetics property is also essential parameter for the ligand to be considered in drug discovery process. Therefore, the best molecules according to these three studies can be strongly suggested for further wet lab experiments of drug design.
Materials and Methods
Retrieval of protein and ligand libraries
The crystal structure of Actin I of P. falciparum (PDB ID: 4CBU) was downloaded from protein databank in pdb format [https://www.rcsb.org/structure/4CBU]. For virtual screening organic molecule library from NPACT database consisting of total 1574 molecules (https://zinc.docking.org/browse/catalogs/natural-products) was downloaded from zinc database in mol2 format.
Target protein preparation
The crystal structure of P. falciparum Actin I (PDB ID: 4CBU) is of 1.3 A° resolution with bound ligand ATP and have two chains; chain A and chain G. It was observed that the binding pocket is located in chain-A and chain-G was away from binding pocket. Hence, chain-G was removed through UCSF Chimera before the protein preparation and in further docking studies only chain-A was considered. The bound ligand molecule was also removed before docking. The protein was prepared for docking studies by assigning hydrogen, polaraties, calculating Gasteiger charges to protein structures, and converting protein structures from the pdb format to pdbqt format using Auto-Dock tool1.5.4.
Preparation of ligand libraries for virtual screening
The natural occurring plant-based compound activity target database library was downloaded from ZINC database (https://zinc.docking.org/)in MOL2 format and was further used for performing docking studies. The library was in a single molecular file and all the ligand molecules were extracted from it with the help of in-house developed PERL scripts. Subsequently all the ligand molecules were converted into PDB format from MOL2 format by using open babel with the help of in-house developed PERL scripts. Antimalarial drugs including Chloroquine, Chloroguanide, Quinine, Mefloquine, and Halofantrine were also used as positive control to target ACTIN I and structure of these molecules were taken from PubChem (https://pubchem.ncbi.nlm.nih.gov/).
Molecular docking
All the ligands within the organic molecular library were used to perform molecular docking studies[33,34] against Actin I to find the potential hit molecules for further drug discovery experiments. In the current research for performing the docking studies the version 4.2 of AutoDock was used. AutoDock uses a Lamarckian Genetic Algorithm (LGA) and is based on a semi empirical free energy force field.[35] The docking grid was set manually through visualization of the protein and grid was defined to cover entire binding site of Actin I.
The size of grid was defined as X=54, Y=54 and Z=54 and the coordinates used for docking of ligands library with 4CBU were X=9.175, Y=20.366, and Z=18.583. Also for molecular docking for every molecule ten runs of LGA were performed. The main interacting residues in the binding site were Asn17, Lys19, Tyr338, Lys337, Lys214, Gln215, and other nearby residues. Finally, molecular docking was carried out on target proteins with all ligands from the library using self-developed PERL script, which was used for screening of these large number of ligand molecules one by one [Figure 1]. The top scoring compounds were selected on the basis of binding energy of ligands with the receptor. The lower is the binding energy of ligand with the receptor the more strongly it will bound to the target receptor.
Figure 1.
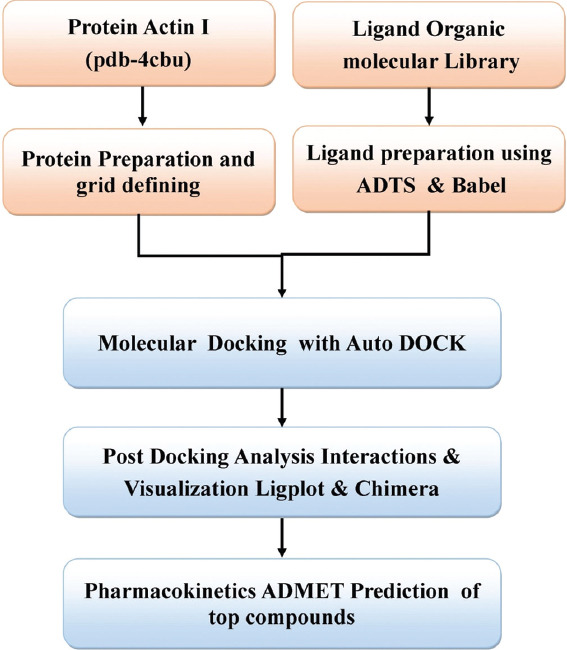
The methodology followed for docking and screening
Interaction study and visualization of docked complex
The UCSF Chimera1.8.1 was used to visualize and analyze docked complexes of ligand and protein.[36] The interaction such as hydrogen bonds and hydrophobic interaction was also analyzed using Ligplot+.[37,38] The generated plots have shown the hydrophobic interaction patterns and hydrogen bonding between the main chain/side-chain atoms of the protein with the ligand.
ADME-toxicity prediction
The pharmacokinetic profile prediction of potential compounds was done using pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) with incorporated prediction of toxicity, metabolism, distribution, absorption, and excretion.[39] Some other properties such as molecular descriptors and drug likeliness properties were also analyzed with the help of pkCSM. The drug likeliness properties were predicted according to the Lipinski Rules of five.[40]
Results
In the present study, considering the importance of Actin I protein as a drug target in P. falciparum molecular docking of actin I of P. falciparum was carried out. The organic molecular library was docked into the active site of Actin I to find the potential ligand molecules which can be used in further drug design experiments and also establish the Actin I as an important drug target in malaria. Pharmacokinetics properties of top ranked molecules were also calculated computationally so that molecules with better pharmacokinetics properties can be further shortlisted for wet lab experiments.
Docking analysis
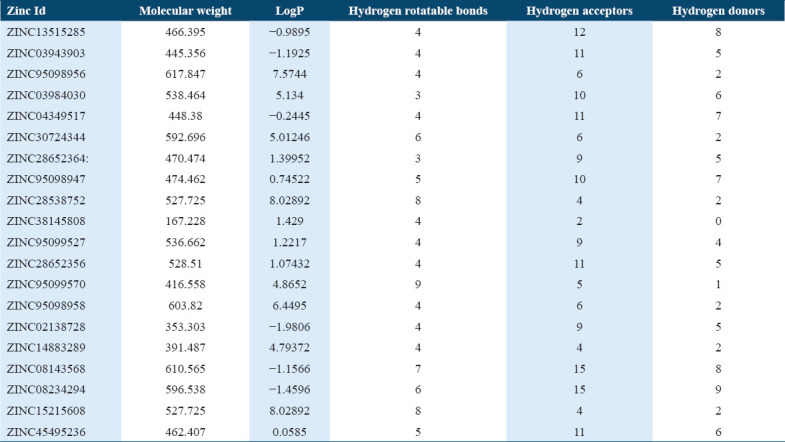
Molecular docking of all the ligands within the library was found to be successful on the basis that all the ligands were docked in the active site of the receptor. The 20 top scoring molecules as per estimated free energy of binding (EFEB) were selected for further investigations. The protein ligand complexes of top scoring molecules were analyzed for interactions, the orientation of the docked compound, and interacting active site residues were visualized. The selected 20 molecules had EFEB in the range of −13.20–−10.51 [Table 1]. The molecule, that is, ZINC30724344 shows least EFEB of −13.20 [Table 1]. The four other top scoring molecules ZINC13515285, ZINC03943903, ZINC95098956, and ZINC03984030 showed the EFEB −12.61, −12.57, −12.37, and −12.23, respectively [Table 1] [Supplementary Figures 1-3]. Despite these molecules the other molecules also had EFEB better than-10 and also show good interaction with the target molecule [Table 1] [Supplementary Table 2] [Supplementary Table 3].
Table 1.
Binding affinity of selected potential molecules
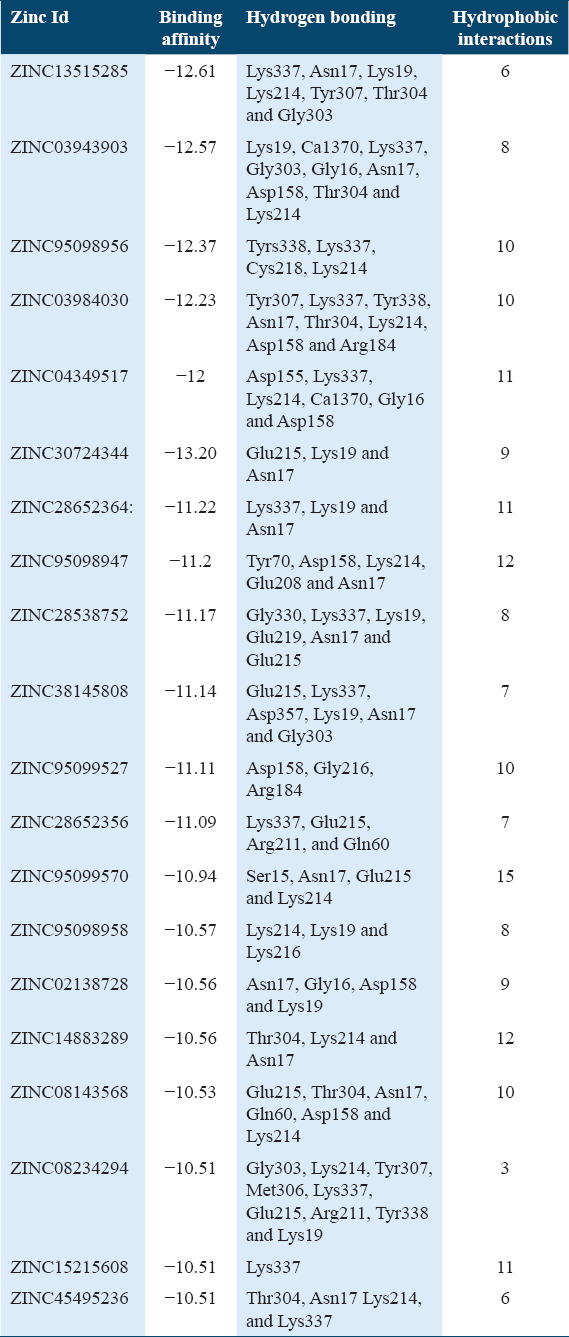
The top scoring molecule, that is, ZINC30724344 strongly interacts with the Glu215, Lys19 and Asn17 residues of the binding cavity [Figure 2] [Table 1]. The second-best scoring molecule as per the EFEB, that is, ZINC13515285 forms hydrogen bonds with the Lys337, Asn17, Lys19, Lys214, Tyr307, Thr304, and Gly303 [Figure 3] [Table 1].
Figure 2.
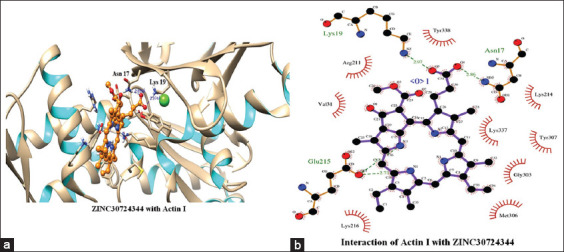
(a and b) Interactions of ZINC30724344 with Actin I
Figure 3.
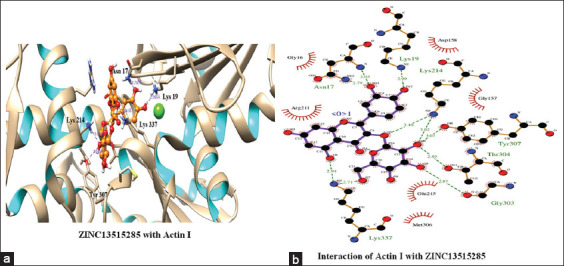
(a and b) Interaction of ZINC13515285 with Actin I
The molecule ZINC03943903 strongly interacts with the Lys19, Ca1370, Lys337, Gly303, Gly16, Asn17, Asp158, Thr304, and Lys214. All other selected molecules also show good patterns of hydrogen bond and hydrophobic interactions [Table 1].
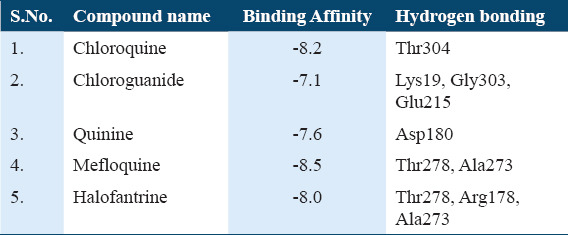
Among the five antimalarial drugs which were used for control docking Mefloquine exhibit the best estimated free energy of −8.5 kcal/mol with Actin I and interact strongly with Thr278, Ala273 [Supplementary Table 1]. The other drugs which were used as positive controls, that is, Chloroquine, Chloroguanide, Quinine, and Halofantrine show the EFEB −8.2, −7.1, −7.6, and −8.0, respectively [Supplementary Table 1].
Molecular parameters
The drug molecules which are having molecular weight less than 500Da can be easily transported, diffused and absorbed as compared to bigger molecules.[41] The physicochemical properties were calculated and the molecular weight of 10 hit molecules out of 20 was less than 500 Da. Most of the molecules [Table 1] showed log P < 5 [Table 1]. The 13 out of 20 selected hit molecules have less than 5 hydrogen bond donors and also have less than 10 hydrogen bond acceptors [Table 2].
Table 2.
Physicochemical properties of potential compounds
ADMET properties
The selected 20 top scoring molecules were subjected to ADMET properties prediction. In ADMET, the absorption properties such as intestinal solubility (% absorbed), water solubility (log mol/L), and skin permeability (logKp) values were calculated. The other properties such as Ames test, BBB penetrability, and Caco2 (cell line of human colorectal adenocarcinoma cells) permeability was included under ADMET. For human intestinal absorption, the compounds which have value less than 30% are poorly absorbed [41,42] Out of the 20 selected hit molecules only three molecules depicted the intestinal absorption value less than 30% and other molecules displayed good absorption potential in human intestine [Table 3]. The selected top 20 scoring molecules also showed good skin permeability values as compared to standard value (−2.5 logKp) [41] and only few can penetrate to Caco2 as compared to (>0.90 as per pkCSM) standard value [Tables 3 and 4].
Table 3.
Absorption profile of potential compounds
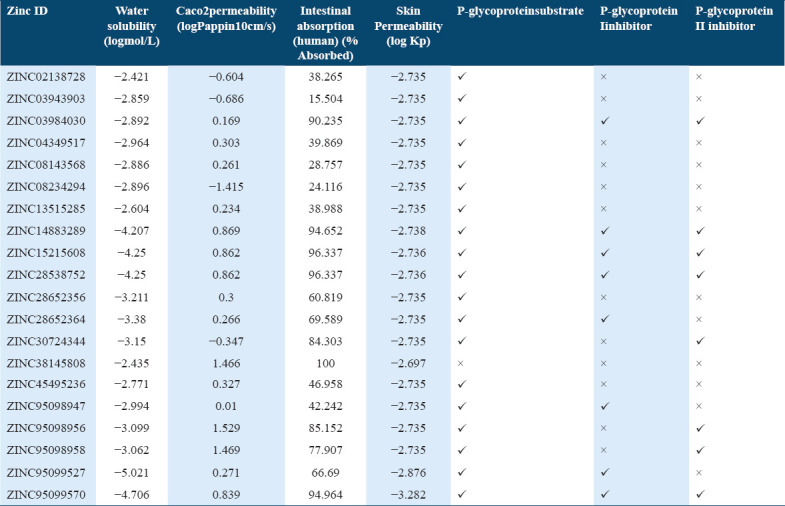
Table 4.
Distribution/excretion profile of compounds
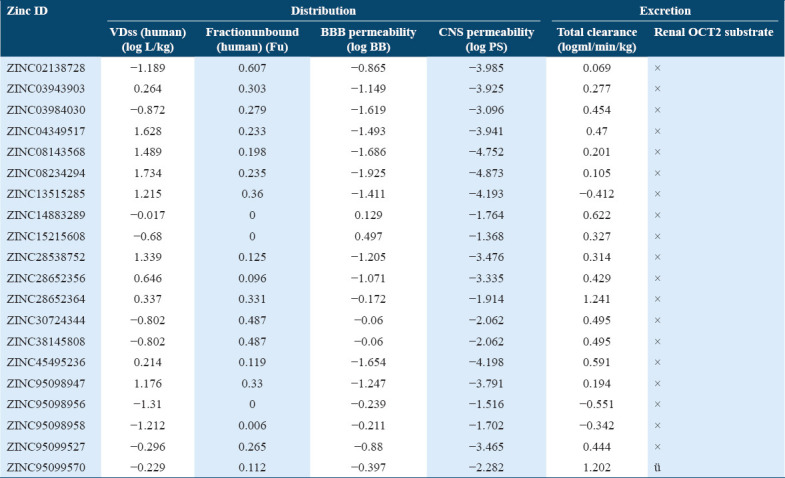
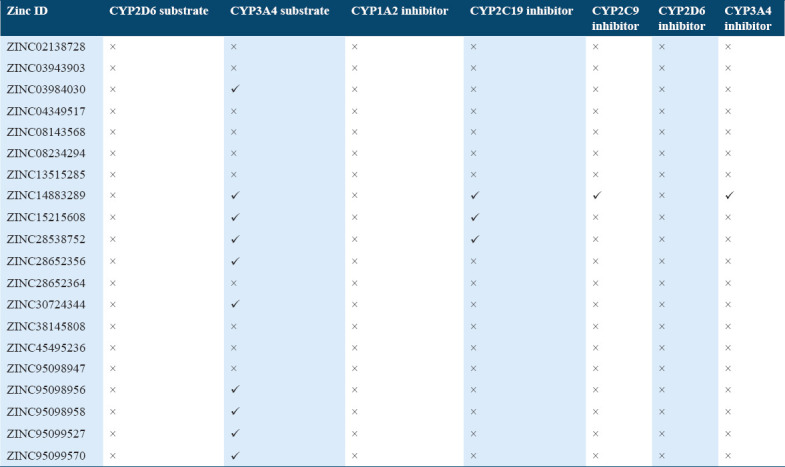
The human microsomal P450s aromatase catalyze the metabolism of a wide variety of compounds including drugs and xenobiotic. In ADMET under metabolism profile, the top scoring molecules were tested whether they were acting as an inhibitor or substrate for different isoforms of cytochrome P450 as it is important for drug metabolism. Many drugs can be inactivated by cytochrome P450 whereas some can be activated also. The inhibitors of cytochrome P450 can affect metabolism of drugs. Hence, from the predicted values, it was observed that the ten molecules out of 20 can acts as a substrate for one of the isoforms of P450, that is, CYP3A4 and none of the molecule acts as a substrate for other isoforms, that is, CYP2D6. The predictive values of the selected hit compounds suggest that only few compounds were acting as inhibitors for CYP3A4, CYP2C19, CYP2C9 and CYP3A4 isoforms of the cytochrome [Table 5].
Table 5.
Metabolism profile of potential compounds
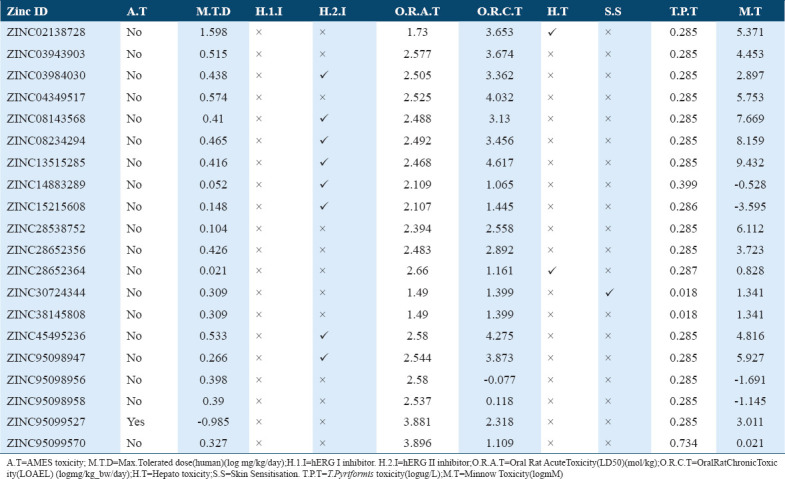
The toxicity and mutagenicity of the selected molecules were depicted with the help of Ames test data, only single molecule, that is, ZINC95099527 displayed the positive results and remaining 19 molecules were note found to be toxic according to Ames test data [Table 6]. Furthermore, for a given molecule maximum recommended tolerated dose (MRTD) ≤0.477 was considered as low and MRTD high if greater than 0.477 as per pkCSM, although most of selected molecules show low MRTD [Table 6].
Table 6.
Toxicity profile of the potential compounds
Discussion
Malaria is still a major global health problem, although antimalarial drugs are available drug resistance has emerged thereby increasing morbidity and mortality rate. The research for the development of efficient licensed malaria vaccine is still not over despite many rigorous efforts have been made in the last decade. It was previously suggested in the literature that the proteins such as actin, tubulin, and histone are involved in the structural assembly of the pathogen.[43,44] Hence, the potential new drug target Actin I is one of the most important structural proteins which is found in P. falciparum and plays important role in its motility apparatus, sexual, asexual and blood stages of the organism.[39] It is expressed in all human stages and little is known about its potential as a drug target. In the current repertoire, we have considered library of 1574 natural organic molecules of diverse nature which were docked with Actin I protein of P. falciparum employ in one of the most important docking software.[6] AutoDock 4.2. The selected molecules were further subjected to their pharmacokinetic studies to make them potentially promising drug candidates.[45] These approaches were widely utilized to design drugs against a number of pathologies such as cardiovascular diseases and cancer.[46-48] The current study is the first report to explore the potential of receptor protein Actin I as an efficient drug target in structure-based drug design.
The prospect of virtual screening techniques was employed to screen the 20 top scoring molecules on the basis of their binding affinity. Although EFEB of ligand protein interaction is relative score and direct correlation of this score is not studied in different docking studies, EFEB of selected ligands in current study was better than the ligands found to be active in recent in silico and in vitro studies conducted by our group in falcipains 2 and 3 of P. falciparum.[49] In addition, when the docked complexes of these selected hit molecules were subjected to post docking analysis, they displayed good interaction patterns (hydrogen as well as hydrophobic interactions). The compound with zinc ID ZINC30724344 was the top scoring molecule and also depicted good interactions indicating a potent promising candidate. Similar trend was also discovered for other remaining 19 molecules which were also prioritized as hit molecules displaying good binding affinities and interaction patterns [Table 1]. Furthermore, the 20 top scoring molecules interact strongly with the target protein forming number of hydrogen bonds [Table 1] [Figures 2 and 3]. In most of these top scoring molecules Lys337, Asn17, and Lys19 were found to be most common interacting residues forming hydrogen bonding. The top scoring molecules exhibit better binding affinity in terms of EFEB as compared to the antimalarial drugs which were used as positive control [Supplementary Table 1].
Further, these top scoring molecules were subjected to ADMET prediction and physicochemical analysis to ensure drug likeness of hit molecules as part of Lipinski’s rule of five. The ADMET properties are important criteria because significant number of molecules failed in the clinical trial due to poor ADMET properties.[50] The top scoring molecule from docking studies ZINC30724344 exhibited molecular weight of 592.696 Da, logP of5.01246, HBA of 6 and HBD value as 2. Furthermore, this hit molecule was non-toxic as per Ames test data and displayed good intestinal absorption (84.303%) and skin permeability (logKp-2.735) values. The molecular weight and logP were observed to be slightly higher as per the standard rules, but natural origin and all other parameters justified that the above potential hit molecule can be developed against malaria which can serve as a roadmap for perusing drug discovery process. The remaining molecules having zinc ID ZINC38145808, ZINC95099570, ZINC14883289, and ZINC02138728also showed high binding affinities and these molecules also justified the different essential parameters to be considered as hit molecules for the drug discovery [Table 1] [Figures 2 and 3]. Although current results are very promising, computational studies require further wet lab experimental verification including target validation. Moreover, current findings warrants that these selected hit molecules can be subjected to further wet lab experimentations for the efficacy of hits compound in vitro and in vivo studies against plasmodium and subsequently modification accordingly so that these molecules can be used as potential hit molecules for anti-malarial drug discovery.
Conclusion
First time virtual screening methodology was implemented successfully for Actin I of P. falciparum using structure-based drug design. Library of diverse molecules was screened to find potential hit molecules on the basis of EFEB. Protein ligand interaction and ADMET properties of top selected scoring molecules were found to be favorable. Among the selected top scoring molecules, the five molecules, that is, ZINC30724344, ZINC13515285, ZINC03943903, ZINC95098956, and ZINC03984030 looks more prominent as a potential candidate for further drug development. The current findings provide a suitable starting point for further in vitro and in vivo analyses to exploit Actin I as an optimal drug target which can encounter drug resistance.
Authors’ Declaration Statements
Ethics approval and consent to participate
Not applicable (as no human subject was used in the study)
Consent for publication
Not applicable (as no human subject was used in the study)
Availability of data and material
The data used in this study are available and will be provided by the corresponding author on a reasonable request.
Competing interests
None declared.
Funding Statement
This research received no specific grant from any funding agency
Authors’ contributions
The research “Conceptualization, V.J. and S.C.; Methodology, V.G.; Software, B.S. and V.G.; Validation, T.P., and B.S.; Formal Analysis, B.S. and V.G.; Data Curation, T.P. and B.S.; Writing – Original Draft Preparation, S.C. and V.G.; Writing – Review & Editing, V.J., T.P., B.S. and S.C.; Visualization, V.G. and B.S.; Supervision, V.J. and T.P.; Project Administration, V.J.;
Acknowledgement
Authors would like to acknowledge Shoolini University, Solan Himachal Pradesh, India.
ORCID link of the submitting author: https://orcid.org/0000-0001-8068-0672
Supplementary Data
Supplementary Table 1.
Docking of Actin I with different Antimalarial drugs:
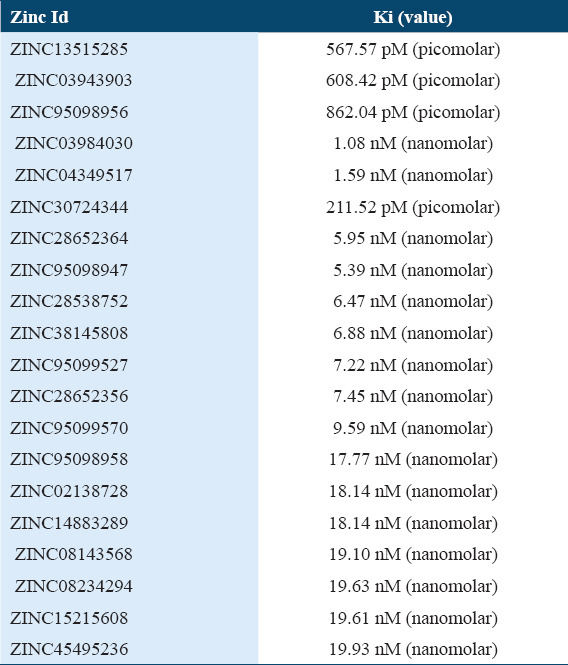
Supplementary Table 2.
Top 20 molecules ki (Values)
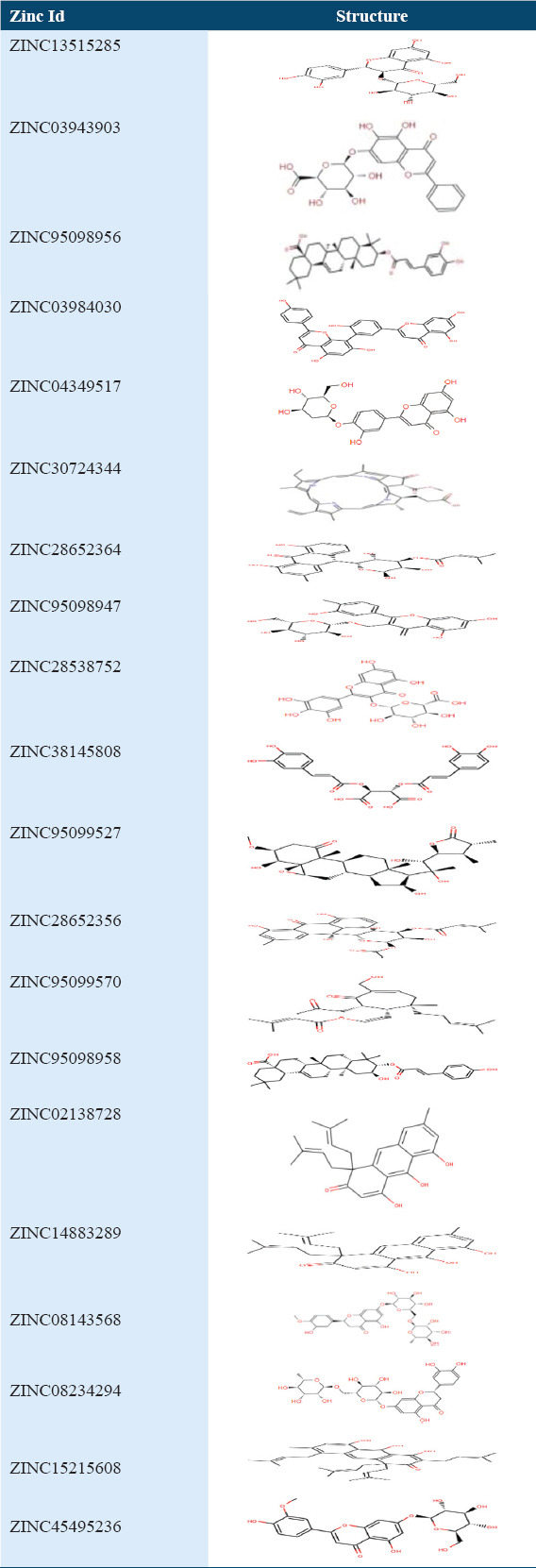
Supplementary Table 3.
Top 20 molecules structures
Supplementary Figure 1.
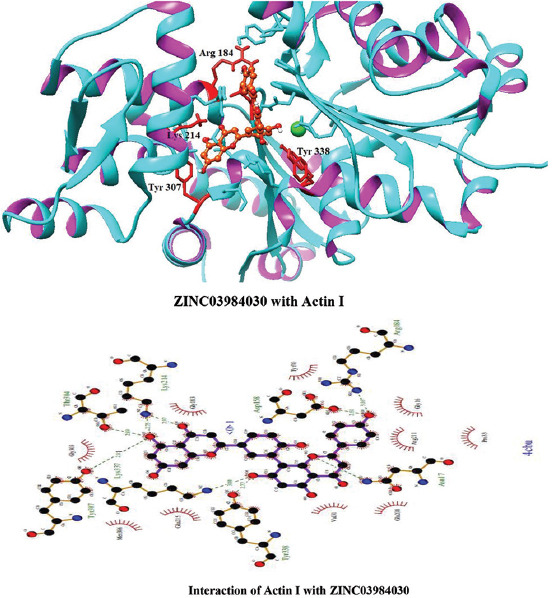
Interactions of ZINC03984030 with Actin I
Supplementary Figure 2.
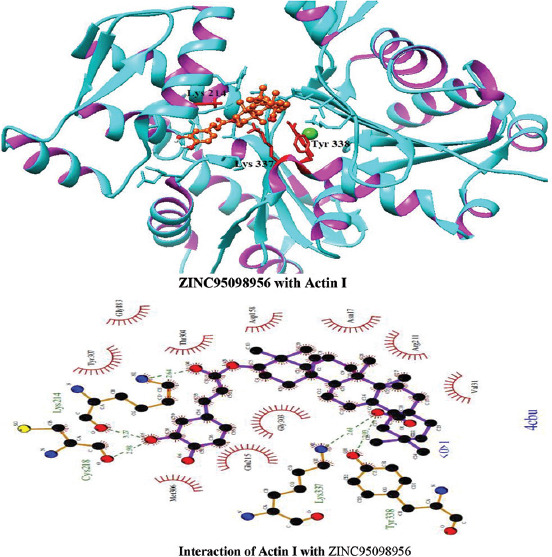
Interactions of ZINC95098956 with Actin I
Supplementary Figure 3.
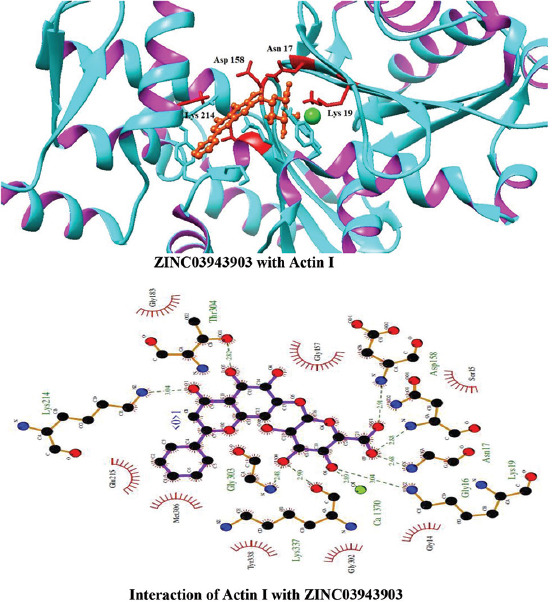
Interactions of ZINC03943903 with Actin I
References
- 1.White NJ. Antimalarial drug resistance. J Clin Investig. 2004;113:1084–92. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Misra H, Mehta D, Mehta BK, Jain DC. Extraction of artemisinin, an active antimalarial phytopharmaceutical from dried leaves of Artemisia annua L., using microwaves and a validated HPTLC-visible method for its quantitative determination. Chromatogr Res Int. 2014;2014:361405. [Google Scholar]
- 3.Pinheiro L, Feitosa LM, Silveira FF, Boechat N. Current antimalarial therapies and advances in the development of semi-synthetic artemisinin derivatives. Anais Acad Bras Ciênc. 2018;90:1251–71. doi: 10.1590/0001-3765201820170830. [DOI] [PubMed] [Google Scholar]
- 4.Guleria V, Jaiswal V. Comparative transcriptome analysis of different stages of Plasmodium falciparum to explore vaccine and drug candidates. Genomics. 2020;112:796–804. doi: 10.1016/j.ygeno.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Vahokoski J, Bhargav SP, Desfosses A, Andreadaki M, Kumpula EP, Martinez SM, et al. Structural differences explain diverse functions of Plasmodium actins. PLoS Pathog. 2014;10:e1004091. doi: 10.1371/journal.ppat.1004091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wesseling JG, Smits MA, Schoenmakers JG. Extremely diverged actin proteins in Plasmodium falciparum. Mol Biochem Parasitol. 1988;30:143–53. doi: 10.1016/0166-6851(88)90107-7. [DOI] [PubMed] [Google Scholar]
- 7.Dominguez R. Actin-binding proteins a unifying hypothesis. Trends Biochem Sci. 2004;29:572–8. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Sibley LD. Invasion of vertebrate cells by Toxoplasma gondii. Trends Cell Biol. 1995;5:129–32. doi: 10.1016/s0962-8924(00)88964-3. [DOI] [PubMed] [Google Scholar]
- 9.Meissner M, Ferguson DJ, Frischknecht F. Invasion factors of apicomplexan parasites:Essential or redundant? Curr Opin Microbiol. 2013;16:438–44. doi: 10.1016/j.mib.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Wetzel D, Håkansson S, Hu K, Roos D, Sibley LD. Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol Biol Cell. 2003;14:396–406. doi: 10.1091/mbc.E02-08-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma B, Hussain T, Khan MA, Jaiswal V. Exploring AT2R and its polymorphism in different diseases:An approach to develop AT2R as a drug target beyond hypertension. Curr Drug Targets. 2021 doi: 10.2174/1389450122666210806125919. [DOI] [PubMed] [Google Scholar]
- 12.Siden-Kiamos I, Louis C, Matuschewski K. Evidence for filamentous actin in ookinetes of a malarial parasite. Mol Biochem Parasitol. 2012;181:186–9. doi: 10.1016/j.molbiopara.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Kalhor HR, Niewmierzycka A, Faull KF, Yao X, Grade S, Clarke S, et al. A highly conserved 3-methylhistidine modification is absent in yeast actin. Arch Biochem Biophys. 1999;370:105–11. doi: 10.1006/abbi.1999.1370. [DOI] [PubMed] [Google Scholar]
- 14.Schmitz S, Schaap IA, Kleinjung J, Harder S, Grainger M, Calder L, et al. Malaria parasite actin polymerization and filament structure. J Biol Chem. 2010;285:36577–585. doi: 10.1074/jbc.M110.142638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerthoffer WT. Migration of airway smooth muscle cells. Proc Am Thoracic Soc. 2008;5:97–105. doi: 10.1513/pats.200704-051VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–12. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cleary RA, Wang R, Waqar O, Singer HA, Tang DD. Role of c-Abl tyrosine kinase in smooth muscle cell migration. Am J Physiol Cell Physiol. 2014;306:C753–61. doi: 10.1152/ajpcell.00327.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Wang D, Johnson AD, Papp AC, Sadée W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–24. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- 19.Lazarus MD, Schneider TG, Taraschi TF. A new model for hemoglobin ingestion and transport by the human malaria parasite Plasmodium falciparum. J Cell Sci. 2008;121:1937–49. doi: 10.1242/jcs.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smythe WA, Joiner KA, Hoppe HC. Actin is required for endocytic trafficking in the malaria parasite Plasmodium falciparum. Cell Microbiol. 2008;10:452–64. doi: 10.1111/j.1462-5822.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Huang Y, Zhang Y, Fang X, Claes A, Duchateau M, et al. A critical role of perinuclear filamentous actin in spatial repositioning and mutually exclusive expression of virulence genes in malaria parasites. Cell Host Microb. 2011;10:451–63. doi: 10.1016/j.chom.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andenmatten N, Egarter S, Jackson AJ, Jullien N, Herman JP, et al. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat Methods. 2013;10:125–7. doi: 10.1038/nmeth.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacot D, Daher W, Soldati-Favre D. Toxoplasma gondii myosin F, an essential motor for centrosomes positioning and apicoplast inheritance. EMBO J. 2013;32:1702–16. doi: 10.1038/emboj.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller C, Klages N, Jacot D, Santos JM, Cabrera A, Gilberger TW, et al. The Toxoplasma protein ARO mediates the apical positioning of rhoptry organelles, a prerequisite for host cell invasion. Cell Host Microb. 2013;13:289–301. doi: 10.1016/j.chom.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Sinden R. Gametocytogenesis of Plasmodium falciparum in vitro:An electron microscopic study. Parasitology. 1982;84:1–11. doi: 10.1017/s003118200005160x. [DOI] [PubMed] [Google Scholar]
- 26.Dixon MW, Dearnley MK, Hanssen E, Gilberger T, Tilley L. Shape-shifting gametocytes:How and why does P falciparum go banana-shaped? Trends Parasitol. 2012;28:471–8. doi: 10.1016/j.pt.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Kursula I, Kursula P, Ganter M, Panjikar S, Matuschewski K, Schüler H. Structural basis for parasite-specific functions of the divergent profilin of Plasmodium falciparum. Structure. 2008;16:1638–48. doi: 10.1016/j.str.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Singh S, More KR, Chitnis CE. Role of calcineurin and actin dynamics in regulated secretion of microneme proteins in Plasmodium falciparum merozoites during erythrocyte invasion. Cell Microbiol. 2014;16:50–63. doi: 10.1111/cmi.12177. [DOI] [PubMed] [Google Scholar]
- 29.Volz JC, Bártfai R, Petter M, Langer C, Josling GA, Tsuboi T, et al. PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe. 2012;11:7–18. doi: 10.1016/j.chom.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Alam A, Jaiswal V, Akhtar S, Jayashree BS, Dhar KL. Isolation of isoflavones from Iris Kashmiriana Baker as potential anti proliferative agents targeting NF-kappaB. Phytochemistry. 2017;136:70–80. doi: 10.1016/j.phytochem.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Chauhan S, Jaiswal V, Attri C, Seth A. Random mutagenesis of thermophilic xylanase for enhanced stability and efficiency validated through molecular docking. Recent Patents Biotechnol. 2020;14:5–15. doi: 10.2174/1872208313666190719152056. [DOI] [PubMed] [Google Scholar]
- 32.Khah S, Goyal R, Chabba A, Jaiswal V, Sharma G, Naushad M. Certain 4-iminoflavones derivatives:Synthesis, docking studies, antiasthmatic and antimicrobial agents. Asian J Chem. 2016;28:1639–42. [Google Scholar]
- 33.Ashraf TM, Mahmood T, Hassan M, Afzal S, Rafique H, Afzal K, et al. Dexibuprofen amide derivatives as potential anticancer agents:Synthesis, in silico docking, bioevaluation, and molecular dynamic simulation. Drug Des Dev Ther. 2019;13:1643. doi: 10.2147/DDDT.S178595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma B, Jaiswal V, Khan MA. In silico approach for exploring the role of AT1R polymorphism on its function, structure and drug interactions. Curr Comput Aided Drug Des. 2020;2020:23113709. doi: 10.2174/1573409916666201023113709. [DOI] [PubMed] [Google Scholar]
- 35.Kalliokoski T, Salo HS, Lahtela-Kakkonen M, Poso A. The effect of ligand-based tautomer and protomer prediction on structure-based virtual screening. J Chem Inf Mod. 2009;49:2742–8. doi: 10.1021/ci900364w. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Kashyap M, Jaiswal V, Farooq U. Prediction and analysis of promiscuous T cell-epitopes derived from the vaccine candidate antigens of Leishmania donovani binding to MHC class-II alleles using in silico approach. Infect Genet Evol. 2017;53:107–15. doi: 10.1016/j.meegid.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 38.Sharma A, Kaur D, Gupta A, Jaiswal V. Application and Analysis of Hyperspectal Imaging. In:2019 5th International Conference on Signal Processing, Computing and Control (ISPCC) 2019 [Google Scholar]
- 39.Pires DE, Blundell TL, Ascher DB. pkCSM:Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–72. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipinski CA. Lead-and drug-like compounds:The rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–41. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Srimai V, Ramesh M, Parameshwar KS, Parthasarathy T. Computer-aided design of selective cytochrome P450 inhibitors and docking studies of alkyl resorcinol derivatives. Med Chem Res. 2013;22:5314–23. [Google Scholar]
- 42.Hassan M, Ashraf Z, Abbas Q, Raza H, Seo SY. Exploration of novel human tyrosinase inhibitors by molecular modeling, docking and simulation studies. Interdiscip Sci. 2018;10:68–80. doi: 10.1007/s12539-016-0171-x. [DOI] [PubMed] [Google Scholar]
- 43.Samant M, Chadha N, Tiwari AK, Hasija Y. In silico designing and analysis of inhibitors against target protein identified through host-pathogen protein interactions in malaria. Int J Med Chem. 2016;2016:2741038. doi: 10.1155/2016/2741038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saleem S, Shaukat F, Gul A, Arooj M, Malik A. Potential role of amino acids in pathogenesis of schizophrenia. Int J Health Sci. 2017;11:63. [PMC free article] [PubMed] [Google Scholar]
- 45.Jazayeri A, Dias JM, Marshall FH. From G protein-coupled receptor structure resolution to rational drug design. J Biol Chem. 2015;290:19489–95. doi: 10.1074/jbc.R115.668251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frédérick R, Robert S, Charlier C, de Ruyck J, Wouters J, Pirotte B, et al. 3, 6-Disubstituted coumarins as mechanism-based inhibitors of thrombin and factor Xa. J Med Chem. 2005;48:7592–603. doi: 10.1021/jm050448g. [DOI] [PubMed] [Google Scholar]
- 47.Ahire V, Das D, Mishra KP, Kulkarni GR, Ackland L. Inhibition of the p53 Y220C mutant by 1-hydroxy-2-methylanthraquinone derivatives:A novel strategy for cancer therapy. J Environ Pathol Toxicol Oncol. 2016;35:355–64. doi: 10.1615/JEnvironPatholToxicolOncol.2016012256. [DOI] [PubMed] [Google Scholar]
- 48.Rasheed Z. Green tea bioactive polyphenol epigallocatechin-3-o-gallate in osteoarthritis:Current status and future perspectives. Int J Health Sci (Qassim) 2016;10:5–8. [PubMed] [Google Scholar]
- 49.Rana D, Kalamuddin M, Sundriyal S, Jaiswal V, Sharma G, Sarma KD, et al. Identification of antimalarial leads with dual falcipain-2 and falcipain-3 inhibitory activity. Bioorg Med Chem. 2020;28:115155. doi: 10.1016/j.bmc.2019.115155. [DOI] [PubMed] [Google Scholar]
- 50.Lagorce D, Douguet D, Miteva MA, Villoutreix BO. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci Rep. 2017;7:1–15. doi: 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in this study are available and will be provided by the corresponding author on a reasonable request.