

Abstract
Circular RNAs are useful entities for various biotechnology applications, such as templating translation and binding or sequestering miRNA and RNA binding proteins. Circular RNA as highly resistant to degradation in cells and are more long-lived than linear RNAs. Here, we describe a method for intracellular trans ligation of RNA transcripts that can generate hybrid circular RNAs. These hybrid circular RNAs comprise two separate RNA that are covalently linked by ligation to form a circular RNA. By incorporating self-cleaving ribozymes at each site of ligation, trans ligation of the transcripts occurs in mammalian cells with no additional material. We provide a protocol for designing and testing trans ligation of transcripts and demonstrate detection of hybrid circular RNAs using fluorescence microscopy.
Keywords: circular RNA, expression system, synthetic biology, RNA ligation
1. INTRODUCTION
Recent studies have shown widespread biogenesis of circular RNAs from human genes [1,2]. These circular RNAs have diverse roles including sequestering specific miRNAs [3], binding to intracellular proteins [4], and possibly acting as templates for translation of proteins [5]. To investigate novel functions of circular RNA and to validate their proposed biological activities, it is important to design circular RNAs that have specified sequences. Additionally, the ability to efficiently express circular RNAs is useful for unveiling potential regulatory roles of a circular RNA. Circular RNAs may also be useful as a general research tool, aside from furthering our understanding of their role in biology.
Circular RNAs are fundamentally more stable than linear RNAs. By definition, circular RNAs lack a 5’ and 3’ end. Therefore, these molecules are completely protected from exoribonuclease activity, which reduces their rate of degradation in cells and lengthens their half-life. While linear mRNAs typically have a half-life counted in hours, circular RNAs have substantially increased half-lives that may be measured in days or weeks [6]. The immune properties of circular RNAs are still debated following disparate results [7,8], yet at a minimum, the lack of 5’ and 3’ ends reduces activation of certain innate immune responses. Innate immune activation is known to occur through MDA5 [9] and RIG-I [10], which detects 5’-triphosphates – entities that cannot exist in a truly circular RNA.
Circular RNA biogenesis typically occurs through an alternative splicing pathway during mRNA intron removal [1]. Through a “backsplicing” process, exonic circular RNAs result from splicing of a downstream splice donor to the splice acceptor of the same exon or of an upstream exon within the same transcript [11]. This pathway generally results in lowly abundant circular RNAs [2]; however, the expression level of individual exonic circular RNAs is not necessarily tied to that of their corresponding linear mRNAs [12,13].
Another biological mechanism of RNA circularization occurs during tRNA maturation. Several human tRNA genes encode an intron in their anticodon loops. These introns are normally removed by a tRNA splicing pathway, which is wholly distinct from the pathway for mRNA splicing. The exons of the tRNA are spliced together by a ubiquitous ligase named RtcB [14–16]. Occasionally, RtcB also ligates the ends of the introns together as well, resulting in a circular RNA designated as a tricRNA (tRNA intronic circular RNA) [6]. Both the backsplicing and tricRNA mechanisms can be incorporated into transgenes in order to express custom circular RNAs [4,6]. Yet, the amount of circular RNA that is expressed by the cell is limited.
A new approach was recently described that results in markedly enhanced levels of circular RNA biogenesis. This approach, termed “Tornado” (Twister-optimized RNA for durable overexpression), causes the overall level of circular RNA expression to be increased by 50-fold compared to the tricRNA approach [17]. The Tornado system involves expressing an RNA of interest flanked by self-cleaving RNA ribozymes. Similarly to the mechanism of tricRNA expression, the ends of the ribozyme-cleaved transcript join together to form a stem that resembles the native tRNA stem during tRNA splicing. Ribozyme cleavage produces a transcript with a 5’-hydroxyl group on the 5’ end and a 2’,3’-cyclic phosphate on the 3’ end. This stem and it’s unique modifications are specifically recognized and ligated by RtcB [17]. Expression of circular RNA by the Tornado system leads to a higher intracellular concentration of circular RNA than the other circular RNA expression systems that were tested. This accumulation is mainly due to the efficiency of the ribozymes, the RtcB ligation step, and the high transcription associated with the RNA Polymerase III promoter.
The abundance of circular RNA that results from Tornado can be the basis of useful and interesting research tools like fluorescent reporters of gene expression. Almost any conceivable functional RNA sequence can be overexpressed using this system. For instance, fluorogenic RNAs are RNA aptamers that bind an otherwise nonfluorescent dye, activating its fluorescence. The field of genetically encoded fluorogenic RNAs began with the “Spinach” and “Broccoli” aptamers [18,19], which have now grown to include numerous other aptamers with different fluorescence properties [20–25]. By circularizing these RNA sequences, the fluorescence signal is much easier to detect in each cell [17]. Occasionally some minor sequence modifications are necessary. Compared to other research tools, such as protein-based fluorescent reporters, these RNAs are advantageously much smaller in molecular weight and in the DNA length when genetically encoded.
The unique ribozyme-dependent mechanism of RNA circularization presents the possibility of performing “trans ligation” reactions in which two different RNAs are ligated together. In one type of trans ligation reaction, two RNAs are ligated to form a longer RNA. In a second type of trans RNA ligation reaction, two RNAs are ligated to each other at both of their ends, resulting in a “hybrid” circular RNA. This hybrid circular comprises two transcripts that are ligated so that each of one transcript’s 5’ and 3’ ends are joined to the opposite ends of the other transcript (Fig 1A).
Figure 1:
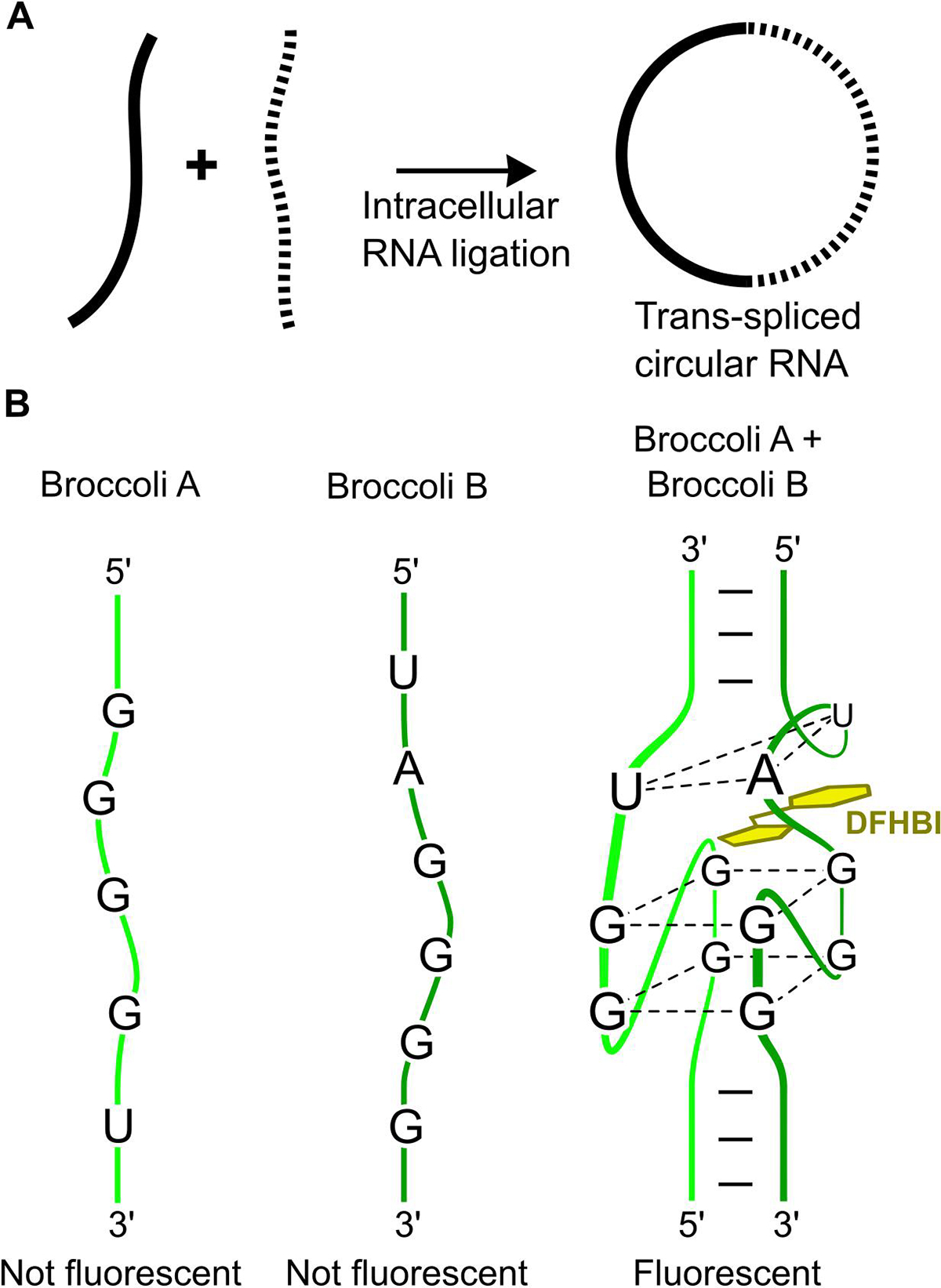
(A) Trans RNA ligation of two RNAs to form a circular RNA. If two transcripts can be trans ligated at both ends, then a circular RNA is formed that contains both sequences. (B) Split Broccoli is only fluorescent when both Broccoli A and Broccoli B are present. Both Broccoli A and Broccoli B are essential to form the DFHBI-binding site (two G-quartets and the U-A-U triplet). A stable and fluorescent RNA is only formed after trans ligating Broccoli A and Broccoli B into a circular RNA.
Trans RNA ligation technology can be used for several applications. First, it could be used to make reporter systems that only fluoresce when two different promoters are active. Each promoter can make one part of the hybrid RNA, which together would produce fluorescence. As an example, this could be used in gene editing applications to identify cells in which two alleles have undergone homologous recombination. This type of approach would reduce the need to test many clones for homologous recombination at both alleles.
Trans RNA ligation can also be used to create long linear RNAs with chemical modifications. Long RNAs are typically made by in vitro transcription using T7 RNA polymerase [26]. However, this method does not allow site-specific incorporation of modified nucleotides, such as N-acetylglucosamine, 2’-O-Me or phosphorothioate modifications. Thus, an RNA with a 3’ ribozyme can be in vitro transcribed, resulting in an RNA product with a 2’,3’ cyclic phosphate on the 3’ end of the RNA. Using RtcB, this RNA can then be ligated with a synthetic chemically modified RNA oligonucleotide with a 5’ hydroxyl. The resulting longer linear RNA would therefore have site-specific chemical modifications. This approach presents an alternative to previous methods that generate long linear RNAs by utilizing traditional T4 RNA ligase or DNAzymes [27,28].
Overall, ligation of RNA molecules together using recombinant RtcB is an attractive way to assemble RNAs from two or more transcripts. Here, we outline the steps needed to design transcripts that can be trans-ligated in mammalian cells or in an in vitro enzymatic reaction, and how to validate ligation and/or circularization of transcripts.
2. CONSIDERATIONS/DISCUSSION
Designing two transcripts that properly undergo RtcB-mediated ligation is primarily about correctly designing the transcripts’ sequences. The sequence requirements for designing a Tornado-based circularizing transcript [17] apply to the sequences of RNAs for trans ligation. If the RNAs are to be expressed and circularized in cells, the RNAs are typically transcribed by RNA polymerase III. Therefore, poly(U) sequences must be avoided throughout the transcript to prevent premature termination. A stretch of four or more uridines in a row is the conventional termination sequence for RNA polymerase III; however, noncanonical termination sequences [29] should also be generally avoided. If possible, we recommend mutating the sequence to eliminate these polymerase termination signals.
The efficiency of RNA ligation by RtcB decreases as length increases. The Tornado system has been used to express a circular RNA that is several hundred nucleotides long [17]. However, when Tornado was used to express circular RNAs as long as 2 kb, a lower efficiency was observed. The same trend is likely to also be true for expressing hybrid circular RNAs. However, the negative effect of length on ligation may be more pronounced since two or more ligations are required from trans ligation. The requirement for multiple ligations would give exonucleases more time to degrade the RNA before circularization renders the RNA resistant to exonucleases.
Another consideration is to design RNAs that can be visualized in cells and in gels. This is important for confirmation that the RNAs are purely synthesized in full-length and for quantification. Detecting circular RNAs can be achieved by including a fluorogenic RNA sequence such as Broccoli [30] in the transcripts. Broccoli contains no termination sequences and can easily be split into two RNA strands. In this way, trans ligation can be visualized. Each portion of split Broccoli (Broccoli A and Broccoli B) is independently non-fluorogenic and only become fluorogenic when both are present and in complex (Fig. 1B). Thus, by including Broccoli A on one transcript and Broccoli B on the other, the trans-ligated circular RNAs can be detected on a gel stained by DFHBI-1T, the cognate fluorophore for Broccoli [18,30].
An important consideration is to ensure that the two RNA fragments hybridize to each other, which will facilitate their ligation. For these two RNAs to be ligated by RtcB, the 5’ sequence of each RNA should hybridize to the 3’ end of the other RNA to form a helical stem. This stem comprises a “ligation site” which resembles the normal tRNA substrate of RtcB. For trans RNA ligation of two RNAs into a circular RNA, two ligation sites are required. Three ligation sites would be required for ligating three RNAs into a circle. Furthermore, trans ligation of two RNAs to form a linear RNA only requires one ligation site.
2.1. Selecting sequences at each ligation site
Trans RNA ligation works best when the sequences at the 5’ and 3’ ends of the RNAs that comprise a ligation site are unique from the sequences that form the other ligation sites. By carefully designing these 5’ and 3’ “ligation sequences,” ligation will occur at the desired ligation sites in a controlled manner. If two ligation sites are similar enough that erroneous hybridization can occur between the wrong pair of 5’ and 3’ ligation sequences, they could become ligated by RtcB, producing an unexpected RNA. Thus, the precise ligation sequences must be designed with minimal potential for hybridization with other ligation sites. The possibility for dsRNA to form G-U base pairs should be considered when designing unique ligation sequences for each ligation site.
Some aspects of the ligation site should not change from site to site. Each 5’ ligation sequence should begin with 5’-AACCATGC-3’ and each 3’ ligation sequence should end with 5’-GCGTGGACTGTAG-3’ (Fig. 2). Where underlined, these two “tRNA intermediate sequences” hybridize to form a short helical stem, with 5’ and 3’ overhangs, mimicking a human tRNATyr splicing intermediate. This is the natural target of RtcB, yet the sequences of this helical stem can be changed to further reduce erroneous hybridization between different ligation sites. Any mutations should preserve dsRNA base pairing in the ligation site (see Fig. 2). Considering the ribozyme cleavage precursor transcript (Fig. 2), these mutations also must be made within only the dsRNA regions of the ribozyme, and so that base pairing within the ribozyme is preserved (see red-outlined nucleotides in Fig. 2 and section 2.2 for more details).
Figure 2:
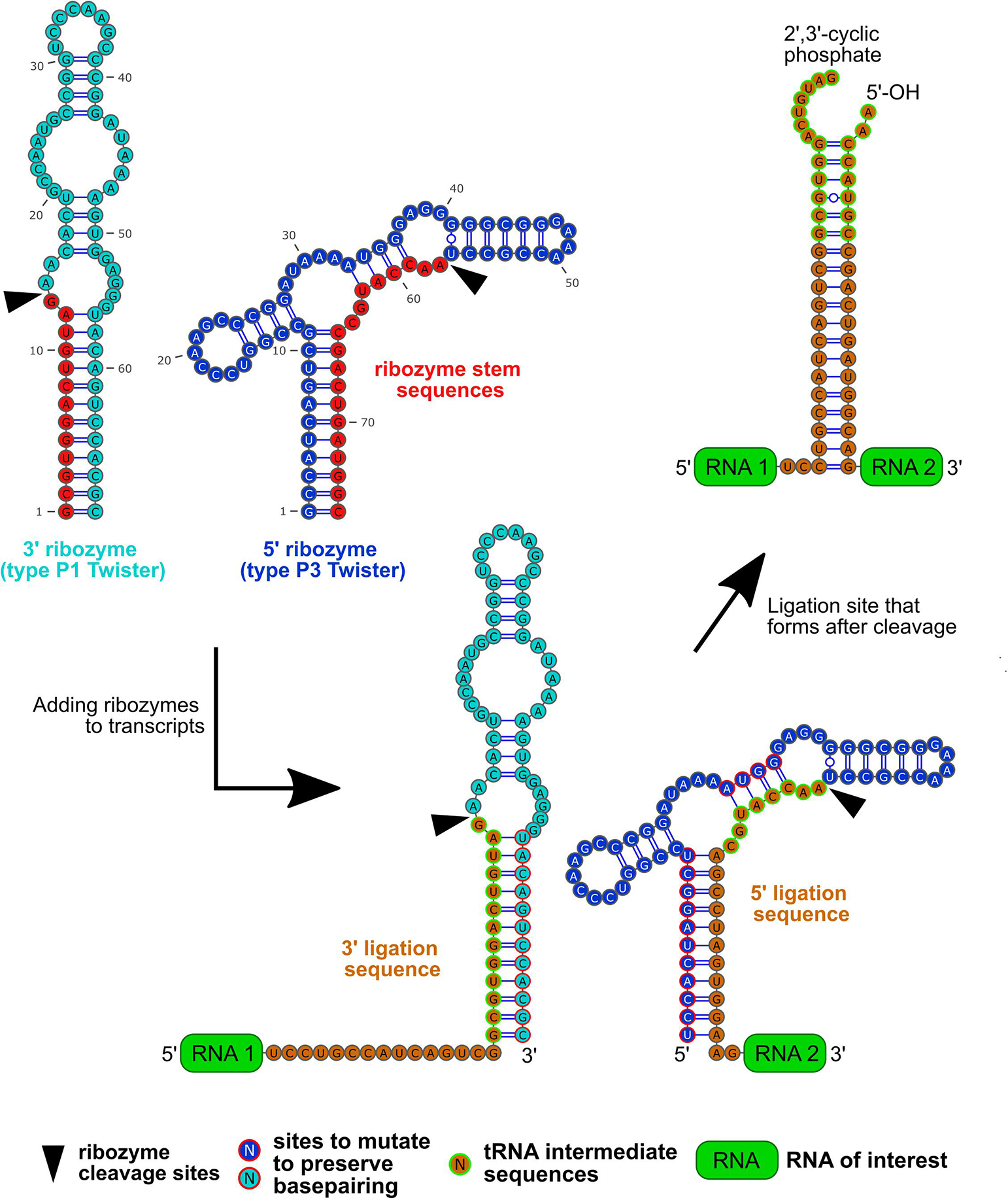
The process for adding type P1 and P3 Twister ribozymes along with the ribozyme sequences are shown. The type P1 Twister ribozyme sequence (cyan) has a ribozyme stem sequence (red) between its cleavage site (black arrow head) and 5’ end. The type P3 Twister ribozyme sequence (blue) has a ribozyme stem sequence that is between its cleavage site and 3’ end.
The ligation sequences (orange), which are unique for each ligation site are designed so that they will not form helices with ligation sequences from other ligation sites. They are also designed to include the 5’ and 3’ tRNA intermediate sequences (orange with green outline).
The type P1 Twister is added to the upstream transcript (labeled “RNA 1” and green) by replacing the ribozyme’s stem sequence with the 3’ ligation sequence. Subsequently, the type P3 ribozyme Twister sequence is added to the downstream transcript (labeled “RNA 2” and green) by replacing its ribozyme stem sequence with the 5’ ligation sequence. If the 5’ tRNA intermediate sequences have been modified to avoid erroneous hybridization, then mutations need to be made that maintain the helices within both ribozymes. The sites to make these mutations (blue or cyan with red outline) are the same for each ligation site, but the exact mutations may differ.
After cleavage of both ribozymes, the 5’ and 3’ ligation sequence form a helix that will contain both tRNA intermediate sequences with 2’,3’-cyclic phosphate and 5’-OH groups. This ligation site can be recognized by RtcB, which ligates the 5’ and 3’ overhangs, leading to trans ligation of the two RNAs of interest.
To promote hybridization of the ligation site, each pair of 5’ and 3’ ligation sequences should include sequences that together form a 10–15 base pair helix (Fig. 2). Helices less than 10 bp might not be thermodynamically stable in cells, and stems longer than 20 bp might become substrates for DICER in cells. In this work, we describe transcripts (Supplementary Table 1) wherein one pair of ligation sequences matches the 15 bp stem-forming sequences from the originally published Tornado system (stem1). The second pair of ligation sequences is compositionally different but forms the same length: a 15 bp stem (stem2). It may be useful to choose the same ligation sequences described here (Supplementary Table 1) because they have already been validated to circularize a single transcript with high efficiency.
When trans ligating RNA transcripts, each transcript will share at least one ligation sites with one other transcript. The 5’ and 3’ ligation sequences at each ligation site must be added to one of the two transcripts at every ligation site. For the transcript at the 3’ end of the ligation site, the 5’ ligation sequence should be inserted just after 5’ tRNA intermediate described above. For the transcript at the 5’ end of the ligation site, the 3’ ligation sequence should be inserted just before 3’ tRNA intermediate. This must be done for every ligation site shared between any two RNA transcripts.
2.2. Adding ribozymes to each transcript
Once the ligation sequences have been added to the appropriate transcripts, ribozyme sequences are added to the DNA template for each transcript. This is necessary so that the encoded RNA transcript will undergo cleavage to form the necessary end modifications (5’-OH and/or 2’,3’-cyclic phosphate). A 5’ end and a 3’ end, and their modifications, must be present at each ligation site for trans RNA ligation by RtcB. Adding each of the two different types of Twister ribozymes to the ends of transcripts occurs in three steps: (1) one type of ribozyme is added to the 3’ end of each transcript with a ligation site at its 3’ end. (2) a second type of ribozymes is added to the 5’ end of each transcript with a ligation site at its 5’ end. (3) mutations are made to each riboyzme on the 5’ ends of transcripts to promote proper ribozyme folding.
The first step of adding ribozymes to the transcripts is to add a ribozyme to the 3’ end of each transcript that has a ligation site at its 3’ end. A type P1 Twister ribozyme [31] should be used for each 3’ end of these transcripts because this ribozyme specifically cleaves at a site near its 5’ end (Fig. 2). Thus, only a small sequence of the ribozyme is left on the RNA of interest. This “ribozyme stem sequence” exists between the cleavage site and the 5’ end of the P1 Twister ribozyme and forms an RNA duplex with another sequence in the ribozyme (Fig. 2). Each of these transcripts is merged with the type P1 Twister sequence by replacing the ribozyme stem sequence with the 3’ tRNA intermediate sequence and 3’ ligation sequence. This ensures that the 3’ tRNA intermediate sequence is at the 3’ end of the transcript after ribozyme cleavage.
The second step is to add a ribozyme to the 5’ end of each transcripts with a ligation site at its 5’ end. For 5’ ends, type P3 Twisters [32] are appropriate to add because they self-cleave near the ribozyme’s 3’ end. Again, this leaves a small fragment on the RNA of interest. For type P3 Twister, the ribozyme stem sequence is between its cleavage site and the 3’ end of the ribozyme (Fig. 2). Each of these transcripts are merged with the 3’ end of the P3 Twister sequence. This is done in such a way that the 5’ tRNA intermediate sequence and the 5’ ligation sequence replace the ribozyme stem sequence (Fig. 2).
The third step is to make mutations to each transcript containing a type P3 Twister, so that this ribozyme folds properly, promoting rapid cleavage. After merging type P3 Twister to the 5’ ends of transcripts in step two, crucial base pairs in the ribozyme structure are disrupted, which must be fixed by mutating each instance of this ribozyme. Mutations must be made to the sequence at the very beginning of the ribozyme that normally forms a duplex with the ribozyme stem sequence (Fig. 2).
If the tRNA intermediate sequences has been changed for any ligation site (see section 2.1), then additional compensatory mutations should be made to the ribozymes (Fig. 2). Compensatory mutations must be made to the middle of the type P3 Twisters that preserve base pairing with the 5’ tRNA intermediate sequence (see red-outlined nucleotides in Fig. 2). Compensatory mutations to the 3’ end of the type P1 Twister must be made that preserve base pairing with the 3’ ligation tRNA intermediate sequence (see red-outlined nucleotides in Fig. 2). Together, these mutations promote rapid cleavage of the ribozymes by promoting the consensus secondary structures.
Once the ribozymes have been added to the ligation sites for each transcript, it will be possible for RtcB to trans ligate the RNAs (Fig. 3). Each transcript can be encoded in a T7 promoter-driven template for in vitro transcription (kits supplied by Lucigen cat# ASF3507) or a U6 promoter-driven plasmid for mammalian expression. Trans ligation of RNAs is possible in vitro using recombinant E. coli RtcB (supplied by New England Biolabs), while a protocol for trans ligation in mammalian cells is described here.
Figure 3:
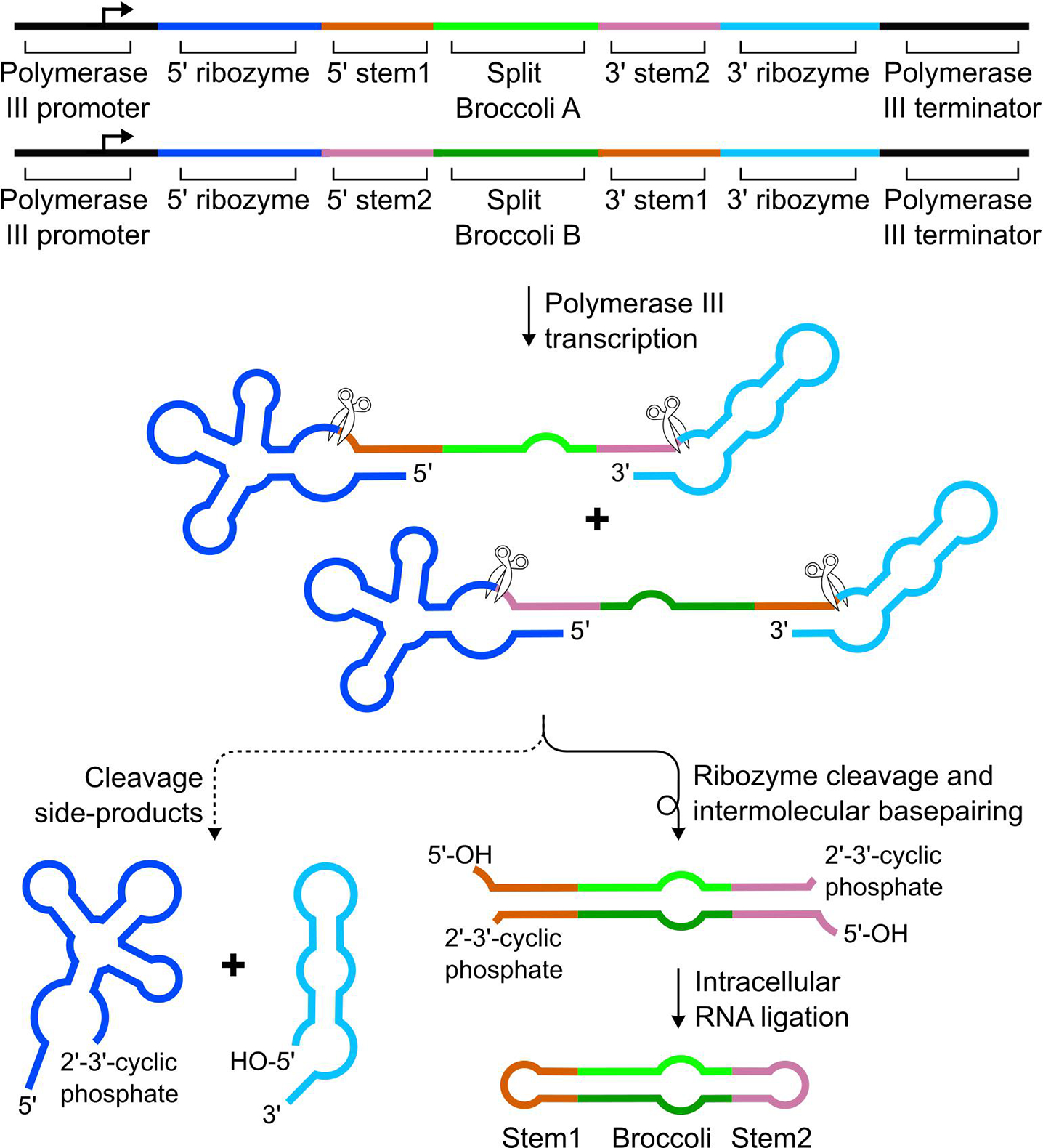
The overall scheme for trans ligation of two transcripts into circular Brococli is shown. The sequence for Broccoli A is flanked by a 5’ ligation sequence from the stem1 ligation site and a 3’ ligation sequence from the stem2 ligation site. Broccoli B is flanked by a 5’ ligation sequence from the stem2 ligation site and a 3’ ligation sequence from the stem1 ligation. After transcription by RNA polymerase III, then cleavage by both ribozymes, the stem1 and stem2 ligation sequences in each transcript base pair to form helices containing 2’,3’-cyclic phosphate and 5’-OH modifications. RtcB recognizes these unique modifications, leading to ligation at both ligation sites. A stable Broccoli sequence is formed that can bind DFHBI and fluoresce.
3. MATERIALS
- Cloning reagents and equipment
- SalI (New England Biolabs, cat# R0138)
- XbaI (New England Biolabs, cat# R0145)
- Standard cloning reagents, for example
- Taq polymerase (New England Biolabs, cat# E5000)
- Deoxynucleotide mix (dNTPs) (Sigma Aldrich, cat# D7295)
- Quick Ligase kit (New England Biolabs, cat# M200)
- Agarose (Sigma Aldrich, cat# D7295)
- 50x TAE buffer
- 2 M tris base (Sigma Aldrich, cat# 648310)
- 1 M glacial acetic acid (Sigma Aldrich, cat# A6283)
- 0.05 M EDTA, pH 8.0 (Sigma Aldrich, cat# EDS-100G)
- PCR thermocycler
- agarose gel casting and running apparatus
- LB agar (Thermo Fisher, cat# 22700025)
- LB broth base (Thermo Fisher, cat# 12780052)
- Carbenicillin (Sigma Aldrich, cat# C1389)
- Bacterial incubator
- RNA expression
- HEK293T cells (ATCC, cat# CRL-3216)
- Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher, cat# 11054001)
- Penicillin/Streptomycin (Thermo Fisher, cat# 15140122)
- Glutamax (Thermo Fisher, cat# 35050061)
- FBS (Thermo Fisher, cat# 26140)
- TrypLE (Thermo Fisher, cat# 12563011)
- PBS (Thermo Fisher, cat# 70011044)
- FuGENE HD (Promega, cat# E2311)
- Trizol LS (Thermo Fisher, cat# 10296010)
- Trizol LS contains phenol and chloroform, which present health and safety hazards. Please refer to the Safety Data Sheet for appropriate use.
- Isopropanol (Sigma Aldrich, cat# I9516)
- Tissue culture hood and incubator
- Page gel
- Low Range ssRNA ladder (New England Biolabs, cat# N0364S)
- Novex TBE-Urea sample buffer (Thermo Fisher, cat# LC6876)
- 10x TBE
- 1 M tris base (Sigma Aldrich, cat# 648310)
- 1 M boric acid (Sigma Aldrich, cat# B6768)
- 0.02 M EDTA, pH 8.0 (Sigma Aldrich, cat# EDS-100G)
- Novex TBE-Urea gels (Thermo Fisher, cat# EC6875)
- or custom PAGE gels (optional)
- Novex gel cassettes (Thermo Fisher, cat# NC2010)
- Gel cassette combs (Thermo Fisher, cat# NC3010)
- Acrylamide/Bis-acrylamide (29:1) (Sigma Aldrich, cat# A2792)
- Ammonium persulfate (Sigma Aldrich, cat# A3678)
- TEMED (Sigma Aldrich, cat# T9281)
- Urea (Sigma Aldrich, cat# U5378)
- PAGE gel running apparatus
- PCR thermocycler or 80 °C heat block
- Broccoli staining solution
- 40 mM HEPES-K pH 7.4 (Sigma Aldrich, cat# 54457)
- 100 mM KCl (Sigma Aldrich, cat# P9541)
- 1 mM MgCl2 (Sigma Aldrich, cat# M8266)
- 10 μM DFHBI, DFHBI-1T or DFHBI-BI (Lucerna Technologies cat# 400, 410, or 600)
- SYBR TM Gold Nucleic Acid Gel Stain (Invitrogen cat# S11494)
- Fluorescence gel imager
- Live cell circular RNA detection
- Fluorescence microscope with EGFP filter set
- FluoroBrite medium (Thermo Fisher, cat# A1896701)
- DFHBI, DFHBI-1T or DFHBI-BI (Lucerna Technologies, cat# 400, 410, or 600)
4. METHODS
4.1. Cloning hybrid circular constructs
Plasmids expressing each transcript to be trans ligated are derived from a pAV vector with a U6 promoter driving expression. The transcript contains modified 5’ and 3’ ribozymes (Fig. 2) flanking the RNA of interest. The RNA of interest is flanked by different ligation sequences. The insert comprising the ribozymes, the ligation sequences, and the RNA of interest is prepared by PCR with a 5’ SalI site and a 3’ XbaI site. Constructs can be generated by ligating inserts into the pAV plasmid at the SalI and XbaI sites using standard cloning techniques.
4.2. RNA expression and collection
HEK293T cells are cultured in growth media (DMEM supplemented with 1x Penicillin-Streptomycin, 1x Glutamax and 10% fetal bovine serum) in a tissue culture incubator that maintains 5% CO2 and 37 °C. All tissue culture work is conducted in a biosafety cabinet.
Plasmids encoding RNA transcripts that will be trans ligated are co-transfected into cells using FuGENE HD transfection reagent according to the manufacturer’s protocol. Plasmids encoding circular Brococli from one transcript are transfected in the same way. The day before transfection, cells are seeded onto 12-well plates.
- Three days after transfection, the cells are collected and the cellular RNA is harvested. A single set of transfected cells can be used for analysis of total cellular RNA and imaging trans ligated in live cells. If the cells expressing the circular RNA will be imaged (optional), first prepare imaging dishes.
- (optional) Coat a sterile 24-well glass-bottom dish with 0.5 mL poly-D-lysine in each well for 3–24 hours. Then, wash the dishes twice with 0.5 mL of sterile PBS in each well and allow to dry in the tissue culture hood.
- Next, the total cellular RNA is harvested from the cells. First, each well of cells is trypsinized by washing briefly with sterile PBS and placing in the tissue culture incubator for 5 min. To quench trypsin, add 0.5 mL of growth medium and dislodge cells from the plate. If these cells will be imaged for trans RNA ligation (optional), then the trypsinized cells will need to be seeded onto the imaging dish at this step as follows below. To collect total RNA from the trypsinized cells, they are then centrifuged in 15 mL conical tubes for 5 minutes at 300 g. Next, aspirate supernatant from the tubes and resuspend cells in 250 μL PBS and transfer to Eppendorf tubes. Next, a volume of 750 μL of Trizol LS is added to this suspension outside of the biosafety cabinet, ideally in a chemical fume hood, and inverted multiple times. The mixture of Trizol LS and cells can be stored at −20 °C until ready to isolate total cellular RNA by following the manufacturer’s protocol.
- (optional): To prepare cells for imaging trans ligated RNA, they are seeded onto imaging dishes. The cell density of each tube of trypsinized cells can be counted using a haemocytometer. Plate 80,000 cells for each condition into a well of the dried imaging plate (~5 × 104 cells/cm2), adding growth media to 0.5 mL total in each well. Cells are then cultured in the tissue culture incubator. Imaging proceeds 24 hours later from section 4.4.
4.3. Hybrid circular RNA analysis and detection
- First, total cellular RNA is separated on denaturing polyacrylamide gels. 6% acrylamide gels efficiently resolve circular RNAs that are longer than 250 nt, while 10% acrylamide gels are more appropriate for smaller circular RNAs. Denaturing PAGE gels containing 6–8 M urea in TBE can be prepared from acrylamide (optional) or they can be purchased already polymerized in cassettes.
- (optional) To prepare polyacrylamide gels, gather gel cassettes and carefully washed and dried gel combs. For two 10% polyacrylamide mini-gels combine 5 mL of 30% acrylamide/bis-acrylamide (29:1) with 1.5 mL 10x TBE, 7.2 g urea, and water to a total volume of 14.7 mL. Then add 250 μL 10% APS, and mix, then add 12.5 μL TEMED and quickly mix, then add the mixture into the gel cassettes with a serological pipette, avoiding formation of bubbles in the solution. Then add combs to the cassettes. Gels will polymerize within 30 minutes.
After removing the comb and tape from the PAGE gel, assemble the gel in the gel running apparatus and fill the top reservoir with 1x TBE buffer and the bottom reservoir with at least 3 cm of buffer. Then pre-run the gel for 30 minutes at 300 V.
Meanwhile prepare 1–5 μg of each total RNA sample in denaturing PAGE loading buffer in a total volume of 10–20 μL, depending on the maximum load volume of the wells of the gel comb. Also prepare 500 ng of the ladder with the denaturing PAGE loading buffer in the same total volume. Next, heat all samples and the ladder to 80 °C for 10 minutes using a PCR thermocycler heat block. Once samples have cooled to room temperature and the gel has finished pre-running, clear any residual urea from the wells by rinsing with TBE buffer using a p1000 pipette or syringe. Then, load samples and ladder into the wells and run at 250 V for 30 minutes or until the lower dye front (bromophenol blue) has reached the bottom of the gel. RNAs longer than 250 nt many require longer run times for optimal separation.
The next step is to visualize the fluorescent RNAs in the gel. While the gel is running, prepare the Broccoli staining solution. Once the gel has finished running, allow it to cool briefly. Then, remove the gel from the cassette and carefully transfer the gel to a plastic tray or the inverted lid of a pipette tip box. Rinse the gel with 10 mL of deionized water or enough to cover the gel. Incubate on an orbital shaker for 5 min and repeat with fresh deionized water 2–3 times. Then, add the fluorescent gel stain and incubate for another 20 minutes. This stain can be stored at 4 °C and reused 3–5 times. Remove the stain and image the gel image with Alexa488 settings (or equivalent settings using approximately 470 nm excitation and 530 nm emission) to collect the fluorescence image. The visualized bands from this setting correspond to RNAs containing Broccoli RNA.
Next, image the total cellular RNA and the ladder bands of the gel using SYBR Gold Nucleic Acid Gel stain. To do so, rinse the gel 3× 5 min with deionized water, then combine 1.5 μL of concentrated SYBR Gold with 15 mL water and add to the gel. Incubate on an orbital shaker for 10 minutes and then image the gel using SYBR Gold settings (or equivalent settings with approximately 302 nm excitation and 590 nm excitation). The bands of the ladder and the total cellular RNA should be visible.
4.4. Detection of trans ligated RNA in live cells
To image Broccoli-containing circular RNAs in live cells, the cells are first transfected, then plated onto imaging plates as described in step 2. The cells should be imaged the day after seeding onto imaging plates.
30 minutes before imaging cells, the growth medium is aspirated and replaced with FluoroBrite medium containing 40 μM DFHBI-1T or BI. Use a fluorescent microscope that is compatible with EGFP imaging. The filter set used for Broccoli detection is a standard EGFP filter set (470 nm excitation and 525 nm emission).
If using DFHBI-1T, Broccoli can quickly photobleach by cis-trans isomerization. Therefore, it is advisable that the field of view is selected before opening the fluorescence light source shutter. If this shutter is opened, wait 20–30 seconds before collecting another image. This time period allows the photobleached Broccoli to recover fluorescence due to exchange of fluorescence-incompetent trans-DFHBI-1T with fluorescence-competent cis-DFHBI-1T from the cytosol. Excitation light source intensity levels can vary by microscope. To reduce the rate photobleaching, excitation should be as low as possible while fluorescence is still detected.
4.5. Sample results
Previously, we have described a pair of ligation sequences that is efficiently ligated by RtcB (stem1) [17]. For proper trans ligation of two RNAs in a circular RNA, two ligation sites are needed. The sequences at these ligation sites must be different in order to avoid erroneous hybridization of transcripts from different ligation sites. Furthermore, these ligation sites must become ligated in cells with similar efficiency. If one ligation site is less efficient than another, there may be a bias towards ligating RNAs at only one ligation site rather than at both sites, thus producing more linear and less circular RNA. Thus, it is important to follow the steps outlined in section 2.1 and assess the relative efficiency of each ligation site separately.
To test the previously described ligation sequences (stem1) alongside a new pair of ligation sequences (stem2), we compared the ability of each pair to circularize Broccoli-containing transcripts (Fig. 4A). The ligation sequences in each transcript are flanked by ribozymes (Supplementary Table 1), as described for the Tornado system [17], leading to ligation of circular Broccoli from a single transcript (“single-transcript circular Broccoli”). Both ligation sites generated a bright circular Broccoli band when measuring DFHBI-1T-depending fluorescence of total cellular RNA on a gel (Fig. 4A). This indicates that both ligation sequences form equally efficient ligation sites and substrates of intracellular RtcB.
Figure 4:
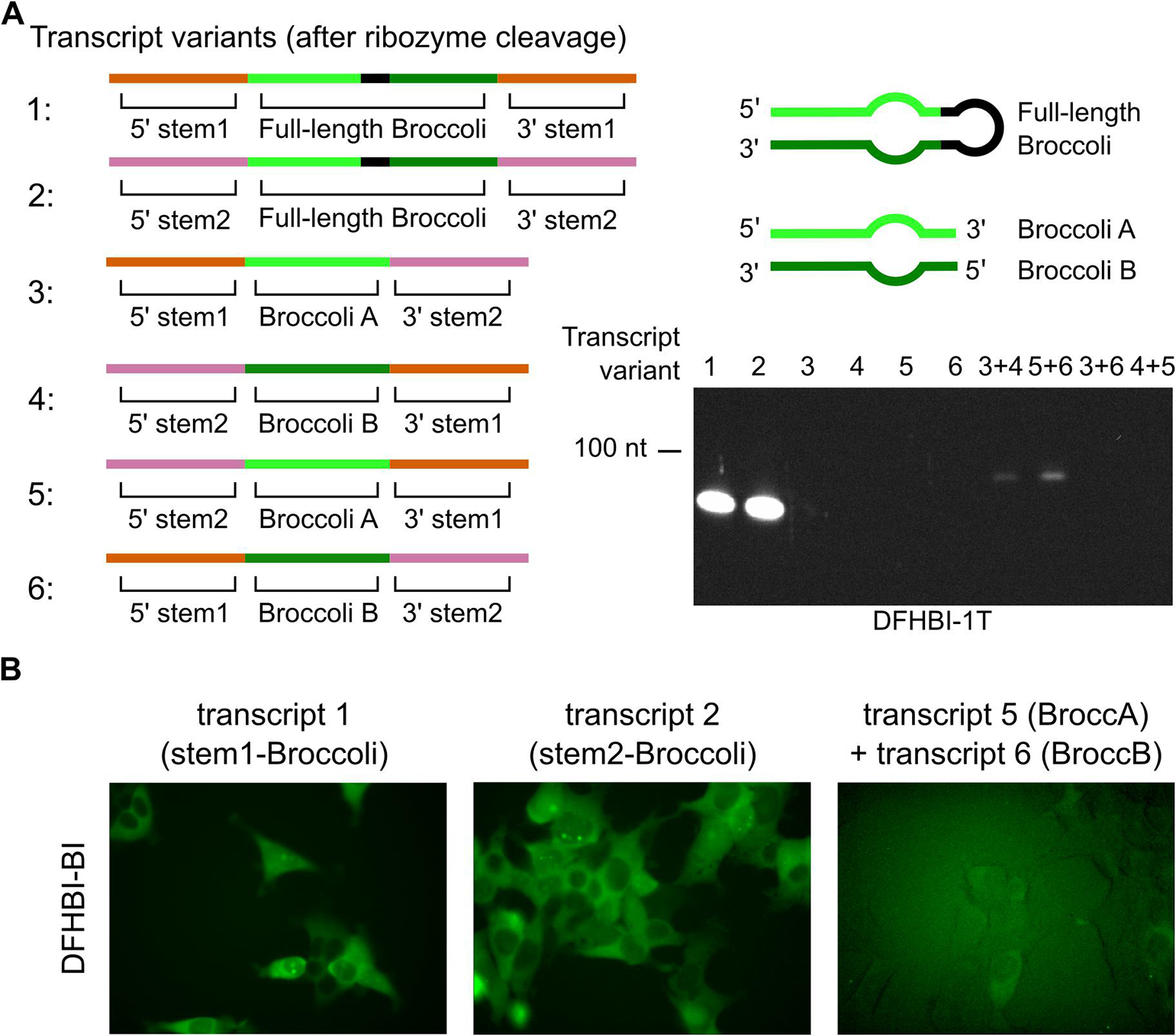
(A) Multiple transcripts were designed with the aim of validating trans RNA ligation into circular Broccoli in cells. Transcripts containing full-length Broccoli and only the stem1 or stem2 ligation site (transcript 1 and 2, respectively) are efficiently circularized. Transcripts 3, 4, 5, and 6 contain Broccoli A (transcripts 3 and 5) or Broccoli B (transcripts 4 and 6) and do not form fluorescent species on their own. Co-transfection of constructs containing Broccoli A and Broccoli B transcripts leads to a circular Broccoli signal only when the stem1 and stem2 ligation sequences match after ribozyme cleavage, according to Figure 3 (transcript 3 matches with 4 and transcript 5 matches with 6). (B) Live cell fluorescence of circular Broccoli expression or trans ligated circular Broccoli. Circular Broccoli is detected in HEK293T cells expressing either circular Broccoli control (both transcript 1 and 2). Trans ligated circular Broccoli is detected in cells co-transfected for expression of transcript 5 and 6. Brightness levels of these images have been adjusted to improve visibility of fluorescence.
Next, we designed transcripts that would generate a strong fluorescent signal only when trans ligated into a circular RNA. To detect whether the RNAs were trans ligated, the Broccoli sequence was split into two halves, and one of each half was added to each of the two RNA transcripts (Supplementary Table 1). Only when the 5’ half (Broccoli A) is ligated to the 3’ half (Broccoli B) will fluorescence be observed. There are two RNA helix regions of Broccoli and each is an ideal ligation site. In the manner described in sections 2.1 and 2.2, we added each ligation sequence (stem1 or stem2) to a different ligation site of split Broccoli (Supplementary Table 1). The result was four different transcripts (transcripts 3–6), comprised of two transcripts with Broccoli A and two with Broccoli B, none of which produced fluorescence when transcribed alone (Fig 4A). Although, linear Broccoli RNAs are unstable in cells and would be difficult to fluorescently detect [6,17], RNA half-life measurements using actinomycin D can indicate whether such an RNA is linear or circular.
Next, we tested whether these split-Broccoli RNAs are trans ligated by co-transfecting pairs of plasmids that encode a Broccoli A and a Broccoli B transcript. For pairs of transcripts where both ligation sites are able to form as in Figure 3 (transcripts 3+4, and 5+6), Broccoli signal was detected (Fig. 4A). Slightly more Broccoli signal was present when stem1 was added to the ligation site downstream of Broccoli A and stem2 downstream of Broccoli B (transcript 5+6), as compared to the alternative arrangement (transcript 3+4), as in Figure 4A. These bands can be confirmed as circular RNAs by a half-life measurement [6] or an exonuclease treatment [33]; however, the gel mobility of the RNA band is too rapid for it to be the linear precursor to circularization. The transcript pairs that are not able to form ligation sites (transcripts 3+6 and 4+5) did not produce RNAs with observable Broccoli fluorescence (Fig. 4A).
Compared to single-transcript circular Broccoli, the amount of trans ligated circular Broccoli was greatly diminished. This drop in efficiency could stem from multiple factors. Primarily, trans ligation requires formation of the ligation site from two transcripts. The rate of this second-order reaction relies on the concentration of two RNAs, reducing the efficiency of ligation site formation relative to ligation sites formed by a single transcript. Based on previous measurements of circular Broccoli levels at 20 μM in cells [17], we estimate that the level of trans-ligated circular Broccoli is as high at 400 nM (Fig. 4A).
Next, we tested whether the expression level of trans ligated Broccoli was high enough to detect its fluorescence in live cells. We performed fluorescence microscopy on the same HEK293T cells used for PAGE detection experiments. Trans ligated circular Broccoli signal was detected by microscopy and appeared to have similar localization as expression of single-transcript circular Broccoli expression (Fig. 4B). All cells expressing only half of this hybrid RNA produced no fluorescence signal during microscopy (not shown). Thus, a trans-ligated circular RNA reporter can be used in mammalian cells in a way that depends on expression of two transcripts.
5. CONCLUSIONS
The Tornado expression system can be modified to generate trans ligated circular RNAs, leading to many potential applications. Firstly, trans ligation may be useful when generating a hybrid circular RNA as a reporter in cell lines. While trans ligated circular RNAs do not accumulate in cells as highly as single-transcript circular RNA, the level of trans ligated circular RNA is still sufficient to use as a fluorescent reporter in live cells.
Secondly, trans RNA ligation using purified bacterial RtcB could potentially enable RNA syntheses that are difficult by traditional RNA synthesis or by in vitro transcription alone. Currently, synthesis of RNAs with site-specific chemical modifications is limited to short RNAs, but methods for ligating small RNAs together into longer modified linear RNAs do exist. These methods rely on either a third oligonucleotide “splint” that templates ligation by T4 RNA ligase or on a DNA enzyme [28] that ligates 3’-OH and 5’-triphosphates with specific end sequences. By contrast, the scheme for trans ligation by RtcB produces the required phosphorylation ends intrinsically, and only the two template oligonucleotides are required.
We expect that trans ligation of RNAs by purified bacteria RtcB outside of cells is much more efficient than ligation by mammalian RtcB inside cells. Highly improved RNA ligation would be achieved by using optimal conditions for RtcB and concentrations of cations and RNA transcripts that promote hybridization. In theory, this system for trans ligation of RNAs will efficiently produce long RNA molecules with modifications to the nucleobase or backbone that are otherwise costly and difficult to synthesis as a single transcript.
A third potential application of trans RNA ligation by RtcB is for generating complex libraries of RNA sequences. This could be beneficial when diverse mutants of protein will be screened during protein evolution or selection. While we have shown here that two transcripts can be ligated into one RNA, it is possible to extend this methodology to three or more transcripts. Conceivably, one could ligate three or more RNA libraries together, generating mRNA libraries with variable length, depending on which transcripts were ligated at each site. Differences in the order by which each transcript was ligated would also increase the complexity of the libraries in this application.
Overall, the straightforward steps required to design hybrid linear or circular RNAs have been described here and can be replicated for nearly any RNA of interest.
Supplementary Material
HIGHLIGHTS.
RNA transcripts are trans ligated by endogenous RNA ligases
Multiple sites of ligation between RNAs can generate ligated circular RNAs
Fluorescence detection of ligated circular RNAs is possible in live mammalian cells
FUNDING
This work was supported by NIH grant R35NS111631 to S.R.J.
Footnotes
CONFLICTS OF INTEREST
S.R.J. is the co-founder of Lucerna Technologies and has equity in this company. Lucerna has licensed technology related to Spinach and other RNA-fluorophore complexes. S.R.J. and J.L.L. are founders of Chimerna Therapeutics and have equity in this company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE, Circular RNAs are abundant, conserved, and associated with ALU repeats, RNA. 19 (2013) 141–157. 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO, Cell-type specific features of circular RNA expression., PLoS Genet. 9 (2013) e1003777. 10.1371/journal.pgen.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J, MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA, EMBO J. 30 (2011) 4414–4422. 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J, Natural RNA circles function as efficient microRNA sponges., Nature. 495 (2013) 384–8. 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- [5].Chen CY, Sarnow P, Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs., Science. 268 (1995) 415–7. 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- [6].Lu Z, Filonov GS, Noto JJ, Schmidt CA, Hatkevich TL, Wen Y, Jaffrey SR, Matera AG, Metazoan tRNA introns generate stable circular RNAs in vivo, RNA. 21 (2015) 1554–1565. 10.1261/rna.052944.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wesselhoeft RA, Kowalski PS, Anderson DG, Engineering circular RNA for potent and stable translation in eukaryotic cells, Nat. Commun. 9 (2018). 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, Chang HY, Sensing Self and Foreign Circular RNAs by Intron Identity, Mol. Cell. 67 (2017) 228–238.e5. 10.1016/j.molcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S, Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5, J. Exp. Med. 205 (2008) 1601–1610. 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G, 5′-Triphosphate RNA is the ligand for RIG-I, Science (80-. ). 314 (2006) 994–997. 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- [11].Cocquerelle C, Mascrez B, Hétuin D, Bailleul B, Mis-splicing yields circular RNA molecules., FASEB J. 7 (1993) 155–160. 10.1096/fasebj.7.1.7678559. [DOI] [PubMed] [Google Scholar]
- [12].Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ, The RNA Binding Protein Quaking Regulates Formation of circRNAs, Cell. 160 (2015) 1125–1134. 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- [13].Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, Herzog M, Schreyer L, Papavasileiou P, Ivanov A, Öhman M, Refojo D, Kadener S, Rajewsky N, Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed, Mol. Cell. (2015) 870–885. 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- [14].Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lührmann R, Söll D, Martinez J, HSPC117 is the essential subunit of a human tRNA splicing ligase complex., Science. 331 (2011) 760–4. 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- [15].Tanaka N, Shuman S, RtcB is the RNA ligase component of an Escherichia coli RNA repair operon, J. Biol. Chem. 286 (2011) 7727–7731. 10.1074/jbc.C111.219022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Englert M, Sheppard K, Aslanian A, Yates JR, Soll D, Archaeal 3’-phosphate RNA splicing ligase characterization identifies the missing component in tRNA maturation, Proc. Natl. Acad. Sci. 108 (2011) 1290–1295. 10.1073/pnas.1018307108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Litke JL, Jaffrey SR, Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts, Nat. Biotechnol. 37 (2019) 667–675. 10.1038/s41587-019-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Paige JS, Wu KY, Jaffrey SR, RNA mimics of green fluorescent protein., Science. 333 (2011) 642–646. 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Filonov GS, Moon JD, Svensen N, Jaffrey SR, Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution [SUPPLEMENT], J. Am. Chem. Soc. 136 (2014) 16299–16308. 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sunbul M, Jäschke A, Contact-mediated quenching for RNA imaging in bacteria with a fluorophore-binding aptamer, Angew. Chemie - Int. Ed. 52 (2013) 13401–13404. 10.1002/anie.201306622. [DOI] [PubMed] [Google Scholar]
- [21].Dolgosheina EV, Jeng SCY, Panchapakesan SSS, Cojocaru R, Chen PSK, Wilson PD, Hawkins N, Wiggins PA, Unrau PJ, RNA Mango aptamer-fluorophore: A bright, high-affinity complex for RNA labeling and tracking, ACS Chem. Biol. 9 (2014) 2412–2420. 10.1021/cb500499x. [DOI] [PubMed] [Google Scholar]
- [22].Song W, Filonov GS, Kim H, Hirsch M, Li X, Moon JD, Jaffrey SR, Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex, Nat. Chem. Biol. 13 (2017) 1187–1194. 10.1038/nchembio.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Braselmann E, Wierzba AJ, Polaski JT, Chromiński M, Holmes ZE, Hung ST, Batan D, Wheeler JR, Parker R, Jimenez R, Gryko D, Batey RT, Palmer AE, A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells, Nat. Chem. Biol. 14 (2018) 964–971. 10.1038/s41589-018-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bouhedda F, Fam KT, Collot M, Autour A, Marzi S, Klymchenko A, Ryckelynck M, A dimerization-based fluorogenic dye-aptamer module for RNA imaging in live cells, Nat. Chem. Biol. (2019). 10.1038/s41589-019-0381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen X, Zhang D, Su N, Bao B, Xie X, Zuo F, Yang L, Wang H, Jiang L, Lin Q, Fang M, Li N, Chen Z, Bao C, Zhao Y, Zhu L, Loscalzo J, Yang Y, Biology S, Engineering B, Technology I, Hua X, Chen Z, Bao C, Xu J, Du W, Zhang L, Zhao Y, Zhu L, Loscalzo J, Yang Y, Visualizing RNA Dynamics in Live Cells with Bright and Stable Fluorescent RNA Mimics, Nat. Biotechnol. 37 (2019) 1–64. 10.1038/s41587-019-0249-1. [DOI] [PubMed] [Google Scholar]
- [26].Sousa R, Patra D, Lafer EM, Model for the mechanism of bacteriophage T7 RNAP transcription initiation and termination, J. Mol. Biol. 224 (1992) 319–334. 10.1016/0022-2836(92)90997-X. [DOI] [PubMed] [Google Scholar]
- [27].Purtha WE, Coppins RL, Smalley MK, Silverman SK, General deoxyribozyme-catalyzed synthesis of native 3′−5′ RNA linkages, J. Am. Chem. Soc. 127 (2005) 13124–13125. 10.1021/ja0533702. [DOI] [PubMed] [Google Scholar]
- [28].Scheitl CPM, Lange S, Höbartner C, New deoxyribozymes for the native ligation of RNA, Molecules. 25 (2020) 1–14. 10.3390/molecules25163650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Orioli A, Pascali C, Quartararo J, Diebel KW, Praz V, Romascano D, Percudani R, Van Dyk LF, Hernandez N, Teichmann M, Dieci G, Widespread occurrence of noncanonical transcription termination by human RNA polymerase III, Nucleic Acids Res. 39 (2011) 5499–5512. 10.1093/nar/gkr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Filonov GS, Kam CW, Song W, Jaffrey SR, In-Gel Imaging of RNA Processing Using Broccoli Reveals Optimal Aptamer Expression Strategies, Chem. Biol. 22 (2015) 649–660. 10.1016/j.chembiol.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu Y, Wilson TJ, a McPhee S, Lilley DMJ, Crystal structure and mechanistic investigation of the twister ribozyme., Nat. Chem. Biol. 7 (2014) 1–7. 10.1038/nchembio.1587. [DOI] [PubMed] [Google Scholar]
- [32].Roth A, Weinberg Z, Chen AGY, Kim PB, Ames TD, Breaker RR, A widespread self-cleaving ribozyme class is revealed by bioinformatics, Nat. Chem. Biol. 10 (2014) 56–60. 10.1038/nchembio.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A, Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing, Nucleic Acids Res. 34 (2006). 10.1093/nar/gkl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.