

Summary
The functional inactivation of tumor suppressor microRNA (miRNA) is closely related to the tumorigenesis of cancer. There are instances where the miRNA and the corresponding target both exist in a cell, but the target gene silencing do not occur as expected. Herein, we found that both miR-506 and its target CDK6 are highly co-expressed in lung cancer cells. Sequence analyses suggested that a miR-506 binding site (1648–1654) and a cis-element (1785–1795) for binding by heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) are evolutionarily conserved and forms a stem structure in the 3′ untranslated region (3′UTR) of CDK6. Furthermore, HNRNPA2B1 can bind to the stem structure to denature it and recruit the RNA helicase DExH-box helicase 9 (DHX9) to the 3′UTR, which ultimately facilitates miRNAs-mediated CDK6 silencing. These results indicate that the cis-element of the 3′UTR of CDK6, where HNRNPA2B1 binds, serves as an RNA switch to regulate miRNAs’ function in cancer cells.
Subject areas: Biological sciences, Biochemistry, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Both miR-506 and its target CDK6 are highly co-expressed in lung cancer
-
•
HNRNPA2B1 facilitates miR-506-mediated CDK6 silence by switching structure in 3′UTR
-
•
HNRNPA2B1 also recruits the DHX9 to the 3′UTR of its targets
-
•
HNRNPA2B1 extensively regulates miRNAs-mediated gene silencing through binding to 3′UTR
Biological sciences; Biochemistry; Molecular biology; Cell biology
Introduction
miRNAs are short ∼22 nt (nucleotides) in length noncoding RNA molecules in eukaryotes (Bartel, 2009), which play critical roles in posttranscriptional gene regulation. Mature miRNAs imperfectly base pair with the complementary sequences in the 3′ untranslated region (3′UTR) of target mRNA, thereby inducing translation suppression or mRNA degradation (Fabian et al., 2010). Suppression of mRNA targets by miRNAs relies on sophisticated steps that are under tight regulation. One of the crucial steps is the miRNA base pairing with a target 3′UTR. Although the 3′UTR contains many potential miRNA seed region binding sites, the target cannot be predicted by the mere presence of miRNA binding sites in the 3′UTR (Pasquinelli, 2012). The suppression efficiency of a miRNA depends on the ability of the RISC to access the target 3′UTR (Agarwal et al., 2015; Bartel, 2009; Grimson et al., 2007; Kertesz et al., 2007). Previous studies found that miRNA binding sites located in the middle of the 3′UTR are less responsive to miRNA-mediated inhibition than the ones located at the end (Agarwal et al., 2015; Betel et al., 2010; Grimson et al., 2007). This suggests that the presence of a complex structure in the middle region of the 3′UTR may act as an obstacle for RISC binding to target mRNAs. The underlying mechanism involved in recruitment and binding of RISC to the structured 3′UTR of the target genes still remains unexplored. Therefore, it is crucial to explore the regulation of RISC interaction with the binding site of target mRNAs so that accuracy in the identification of miRNA targets could be achieved.
In addition to miRNA binding sites, the 3′UTRs also contain evolutionarily conserved cis-elements for RNA binding proteins (RBPs). The cis-elements in 3′UTRs that are bound with RBPs in a sequence-specific manner can play important roles in determining the diverse fates of mRNAs through modulation of mRNA stability, localization, and translation (Matoulkova et al., 2012; Mayr, 2016; Ray et al., 2009; Xu et al., 2017). Moreover, an interplay can exist between RBPs and miRNAs binding at the 3′UTR, which consequently may determine the fate of target mRNA (Elcheva et al., 2009; Kedde et al., 2007, 2010; Kenny et al., 2014; Kundu et al., 2012). Analogous to the riboswitches, which are activated by complexed metabolites (Serganov and Patel, 2012; Winkler and Breaker, 2005), the conformational change of mRNA switches can be dependent on RBP binding in eukaryotes. For instance, Ray et al. found that an RNA switch in the VEGFA 3′UTR is responsive to the stress-stimulated HNRNPL, an RNA binding protein that regulates VEGFA expression in hypoxic conditions (Ray et al., 2009). Similarly, it was reported that PUM1 binding induces a switch of RNA structure that facilitates association with miR-221 and miR-222, thus inducing efficient suppression of p27 expression (Kedde et al., 2010). Understanding the mechanism of RBP function in context of conformational changes in the 3′UTR is therefore desirable not only for miRNA target identification but also for manipulating miRNA for the development of therapies against diseases.
Heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) is an RNA binding protein that belongs to the HNRNP protein family (Martinez-Contreras et al., 2007). HNRNPA2B1 plays key roles in the regulation of RNA metabolism, including mRNA processing (David et al., 2010), miRNA processing (Alarcon et al., 2015), miRNA exosomal sorting (Villarroya-Beltri et al., 2013), mRNA decay (Geissler et al., 2016), and telomere maintenance (Mckay and Cooke, 1992). Recently, it has been reported that HNRNPA2B1 functions in the recognition of pathogenic DNA and the amplification of the innate immune response (Wang et al., 2019a). Previously reported literature evidence indicate that HNRNPA2B1 is overexpressed in many human cancers, including breast cancer, prostate cancer, hepatocellular carcinoma, glioblastomas, and lung cancer (Cui et al., 2010; Golan-Gerstl et al., 2011; Pino et al., 2003; Stockley et al., 2014; Zhou et al., 2001). In addition, a few reports also highlight a critical role for HNRNPA2B1 in the regulation of certain hallmarks of cancer, such as aerobic glycolysis (Clower et al., 2010), cell migration, and proliferation (Tauler et al., 2010). Notably, knockdown of HNRNPA2B1 expression can increase the proliferation rate of lung cancer cells (Tauler et al., 2010). However, the precise regulatory mechanism of HNRNPA2B1 in cell proliferation remains elusive.
Herein, we have found that HNRNPA2B1 can specifically bind to the UUAGG (1785–1795) motif and that motif can base-pair with miR-506 binding site (1648–1654) in the CDK6 3′UTR according to the prediction. HNRNPA2B1 binding resulted in an increase in the linear structure of CDK6 3′UTR, which consequently facilitated RISC binding. Further mechanistic studies revealed that HNRNPA2B1 may recruit the RNA helicase DHX9 to the 3′UTR, whereas its knockdown could relieve CDK6 suppression that was mediated by miR-506 overexpression. Importantly, HNRNPA2B1 was also found to regulate other miRNA-mediated suppression of mRNAs in a similar fashion that miR-506 targets CDK6.
Results
miR-506 induces arrest in the G1 phase of lung cancer cells by targeting the CDK6
Lung cancer is the leading cause of cancer death worldwide due to its poor prognosis and lack of effective therapy (Torre et al., 2016). Thus, it is crucial to dissect different regulatory layers involved in the development and progression of lung cancer. Regulatory RNAs, especially miRNAs, have been reported to play significant roles in cancer development (Calin and Croce, 2006). Therefore, in order to identify miRNAs associated with lung cancer progression and to explore their regulatory role in the early research, we first profiled the expression levels of 500 human miRNAs in lung tumor tissue and noncancerous tissue specimens by using RT-qPCR. Among these, we selected 15 highly deregulated miRNA candidates, which were either overexpressed or underexpressed, for further analysis (Figure S1A). To dissect the biological significance of these aberrantly expressed miRNAs in lung cancer, we synthesized miRNA mimics. Then, corresponding miRNA mimics were transfected into the non-small-cell lung carcinomas (NSCLC) cell line A549, which is one of the most commonly used cell lines in EdU (5-ethynyl-2′-deoxyuridine) cell proliferation assay (Feng et al., 2011; Ren et al., 2014). The effect of the miRNA mimics on cell proliferation was measured via the EdU cell proliferation assay. The results showed that transfection of A549 cells with miR-506 caused a significant inhibition of cell proliferation of lung cancer cells (Figure S1A).
Next, we focused on cell-proliferation-related genes to further investigate targets. First, the 3′UTRs of predicted candidates, including CCND2, CDK2, MAPK1, E2F3, and CDK6, were cloned into a luciferase reporter vector. Then, 3′UTR reporter assays were conducted to test the potential targets of miR-506 in lung cancer cells. It was observed that the reporter luciferase activity of CDK6 3′UTR was obviously suppressed by miR-506 treatment, compared with other candidates (Figures S1B). Therefore, we selected CDK6 as a potential target gene to further study the function and mechanism of miR-506 in lung cancer. We further mutated the putative miR-506 binding site of the CDK6 3′UTR in the reporter vector (Figure 1A). Mutation of the reporter construct led to restoration of the reporter luciferase activity (Figure 1A). Accordingly, transfection of miR-506 mimics induced reduction of CDK6 expression at both the protein level and the mRNA level (Figures 1B and 1C). Consistently, the inhibition of miR-506 by its specific Antagomir (A-506) resulted in a significant increase in CDK6 protein levels (Figure 1C). Furthermore, we examined the effect of miR-506-mediated cell-cycle arrest through flow cytometry analysis. The result showed that transfection with miR-506 mimics and siRNA-CDK6 (si-CDK6) induced cell-cycle arrest at the G1 phase. Meanwhile, cells transfected with the miR-506 Antagomir (A-506) manifested a clear progress to the S phase of the cell cycle (Figure 1D). In addition, an EdU cell proliferation assay was conducted on 95D cells. The percentage of EdU-positive cells in the NC-treated group, the miR-506-treated group, and the miR-506 Antagomir-treated group was 39%, 9%, and greater than 55%, respectively (Figure S1C). Taken together, these results suggested that miR-506 induced cell-cycle arrest in G1 phase and suppressed the proliferation of lung cancer cells by targeting CDK6 expression.
Figure 1.

miR-506 induces G1 phase arrest in lung cancer cells by targeting CDK6 expression
(A) The result of the CDK6 3′UTR reporter assay. A diagram of the wild-type or the mutant luciferase reporter vector encompassing the binding site of miR-506 in the CDK6 3′UTR is shown (right). The bold characters in miR-506 indicate the seed region. The bold italic characters show the binding site of the miR-506 seed region binding site in the CDK6 3′UTR. The luciferase reporter assay was conducted 48 h after transfection of luciferase reporter constructs containing the wild-type or mutated miR-506 binding sites from the CDK6 3′UTR. Data are presented as the mean ± SD (n = 4). ∗p < 0.05, Mann-Whitney U test.
(B) CDK6 mRNA levels were measured by RT-qPCR in 95D cells. Cells were transfected with no target control (NC) and miR-506. Forty-eight hours after transfection, cells were collected for RT-qPCR analysis. mRNA expression was normalized to β-Actin. Data are presented as the mean ± SD (n = 3). ∗p < 0.05, two-tailed Student’s t test.
(C) Western blot analysis of CDK6 protein levels in 95D cells. Cells were transfected with NC, miR-506, and Antagomir (A-506). Forty-eight hours after transfection, cells were harvested for western blot analysis. β-Actin was used as a reference gene for western blot. Band densities were quantified using Image J, and normalized band densities are shown on the bottom. Data are presented as the mean ± SD (n = 3). ∗p < 0.05, two-tailed Student’s t test.
(D) FACS (fluorescence-activated cell sorting) analysis of cell-cycle phases in 95D and H1299 lung cancer cells. The cell was harvested for cell-cycle testing 48 h after transfection of NC, miR-506, miR-506 Antagomir (A-506), and siRNA-CDK6 (si-CDK6).
Both miR-506 and target CDK6 are highly expressed in lung cancer
Our previous work showed that miR-506 was highly expressed in lung cancer tissue samples (Yin et al., 2015). In this study, we further found that there were approximately one-third (26/77) lung cancer patient samples in which both the miR-506 and the CDK6 mRNA levels were elevated as compared with the paired normal tissue (Figure 2A). Furthermore, we analyzed the relationship between different groups and pathological parameters of clinical samples. For disease-free survival (DFS) analysis, the group with upregulated levels of both miR-506 and the CDK6 mRNA showed no significant difference compared with the other group (Figure S2C). The DFS analysis was not statistically significant (p = 0.335), probably because of lacking sufficient samples. For analysis of other pathological parameters of clinical samples, we then associated elevated expression of both miR-506 and the CDK6 mRNA with the patients’ clinicopathological features, such as smoking, gender, age, and death. The analysis revealed that upregulated levels of both miR-506 and the CDK6 mRNA were significantly associated with patient smoking history (p < 0.001). Elevated expression levels of both miR-506 and CDK6 in the lung cancer patients could not correlate with sex (p = 0.063), age (p = 0.091), recurrence (p = 0.173), and death (p = 0.816) (Table 1).
Figure 2.

The expression of miR-506 and CDK6 in lung cancer cells
(A) Detection of miR-506 and CDK6 in the lung cancer patients. The expression levels of miR-506 and CDK6 were tested in lung cancer patients (n = 77). The expression level of each gene in lung cancer tissue was normalized to paired normal tissue. The normalized expression levels of CDK6 and miR-506 were plotted. U6 was used as a reference gene for miR-506. The mRNA expression was normalized to GAPDH.
(B) Detection of miR-506 and CDK6 in the normal lung cell line and lung cancer cell lines. Western blot (left) detection of CDK6 in normal lung cell line MRC5, HLF, and lung cancer cell lines, including A549, 95D, H1975, H460, HCC827, H661, PC9, H23, H358, and H1299. Cells were grown into confluence. Then Cells were harvested for RT-qPCR and western blot analysis. β-Actin was used as a reference gene for western blot. U6 was used as a reference gene for miR-506. The mRNA expression was normalized to β-Actin.
(C) CDK6 protein levels in different lung cancer cells. Lung cancer cells were transfected with no target control (NC) and miR-506. Forty-eight hours after transfection, cells were collected for western blot. β-Actin was used as a reference gene for western blot.
(D) The CCK-8 cell viability assay in lung cancer cell lines. Forty-eight hours after transfection, cell viability was tested with CCK-8 assay. Data are presented as the mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, two-tailed Student’s t test.
Table 1.
Association of miR-506-CDK6 expression with clinicopathologic variables
| Variable | miR-506- and CDK6-elevated patients (%) | Others (%) | p value |
|---|---|---|---|
| Sex | |||
| Male | 19 (42.2) | 26 (57.8) | 0.063 |
| Female | 7 (21.9) | 25 (78.1) | |
| Age (years) | |||
| ≤60 | 10 (25.0) | 30 (75.0) | 0.091 |
| >60 | 16 (43.2) | 21 (57.8) | |
| Smoking history | |||
| No | 11 (20.4) | 43 (79.6) | 0.00014 |
| Yes | 15 (65.2) | 8 (34.8) | |
| Recurrence | |||
| No | 17 (40.5) | 25 (59.5) | 0.173 |
| Yes | 9 (25.7) | 26 (74.3) | |
| Death | |||
| No | 6 (36.4) | 12 (63.6) | 0.816 |
| Yes | 20 (33.3) | 35 (66.7) | |
The group of miR-506, CDK6 elevated based on high expression of both miR-506 and CDK6 in lung cancer patients. p value calculated from Χ2 (Pearson chi square) test.
Apart from this, the expression levels of both miR-506 and its target CDK6 were also found high in lung cancer cell lines, such as 95D, H460, HCC827, PC9, and H1299, compared with lung normal cell line MRC5 (Figure 2B). Interestingly, transfection of miR-506 mimics into 95D, A549, H460, and H1299 lung cancer cells induced more than 15,000-fold increase in the level of miR-506 (Figure S2A). To our surprise, treatment of miR-506 mimic in 95D and A549 cells caused obvious reduction in CDK6 expression at both protein and mRNA levels, but in H460 cell line it failed to do so (Figures 2C and S2B). Consistent with this, the transfection of the corresponding miR-506 mimics led to a significantly reduced cell viability in 95D and A549 cells (Figure 2D). Taken together, these results suggested that the miR-506 underwent a partial loss-of-function in its ability to regulate CDK6 protein expression in lung cancer cells. Identifying the underlying mechanism behind the loss of function ability of miR-506 in lung cancer cells appears to be a research worthy question.
HNRNPA2B1 engages with conserved cis-elements in the CDK6 3′UTR to regulate miR-506-mediated CDK6 expression
In order to find the mechanism involved in the loss of function of miR-506 on CDK6 protein expression, we planned to investigate if there are other factors involved in the miR-506-mediated regulation of CDK6. Considering that the miRNA usually inhibits target exppression through binding to the 3′UTR of target gene, we analyzed the sequence around the miR-506 binding site (1648–1654) in the CDK6 3′UTR. The analysis showed that the sequence of the miR-506 binding site (seed region binding site) on the CDK6 3′UTR harbors an evolutionarily conserved motif, UGCCUUA (1648–1654) (Figure 3A). Interestingly, the sequence located 131 nt downstream of the miR-506 binding site (1648–1654) on the CDK6 3′UTR contains two conserved motifs, UUAGG (1785–1795) (Figure 3A). Previously, it was reported that the UUAGG motif can be recognized by RNA binding protein HNRNP family members, including HNRNPA1, HNRNPA2B1, and HNRNPD (Han et al., 2010). We wondered whether these RBPs can regulate the expression of CDK6 through binding to the UUAGG motif. To test it, we firstly analyzed the function of conserved UUAGG motif through luciferase reporter assay. The reporter construct containing the mutated UUAGG motif showed enhanced luciferase activity compared with the results from the wild-type CDK6 3′UTR reporter (Figure S3A). To identify which protein was binding to the UUAGG motif in the CDK6 3′UTR, an RNA pull-down assay was performed using wild-type CDK6 3′UTR (wild-type) RNA and UUAGG-mutated CDK6 3′UTR (mutant) RNA (Figure 3B). Using silver staining, we found a 36 kD protein band was specifically enriched in the wild-type RNA pull-down sample compared with the mutant RNA sample (Figure 3B). Chromatograph-mass spectrometer (LC-MS) analysis of this enriched band resulted in identification of HNRNPA2B1 (Table S1). Next, the RNA pull-down samples were tested by western blot using a HNRNPA2B1 monoclonal antibody. Western blot results confirmed that HNRNPA2B1 was specifically enriched in the wild-type CDK6 3′UTR RNA pull-down sample (Figure 3C). We wondered whether the CDK6 3′UTR binding protein HNRNPA2B1 regulates CDK6 protein expression. To confirm this, we firstly knocked down HNRNPA2B1 expression by treating cells with synthesized siRNAs targeting HNRNPA2B1 (siRNA-HNRNPA2B1, si-A2B1). The result indicated that the knockdown of HNRNPA2B1 led to an increase in CDK6 expression compared with the NC-treated samples in lung cancer cells (Figure S3B). Furthermore, the overexpression of exogenous HNRNPA2B1 repressed CDK6 protein in HEK293 and 95D cells (Figure S3C). These results suggested that the CDK6 3′UTR binding protein HNRNPA2B1 can suppress CDK6 expression. These results prompted us to speculate that HNRNPA2B1 can regulate miR-506-mediated suppression of CDK6 in 95D cells. To confirm this, we conducted analyses of the mRNA and protein levels of CDK6 in cellular lysates prepared from 95D lung cancer cells after transfection with siRNA-HNRNPA2B1 (si-A2B1) and miR-506 mimics. Results showed that both mRNA and protein levels of CDK6 were reduced in miR-506-mimic-treated cells and significantly increased in siRNA-HNRNPA2B1 (si-A2B1)-treated cells (Figures 3D and 3E). Interestingly, co-transfection of siRNA-HNRNPA2B1 and miR-506 reversed the miR-506-induced suppression of CDK6 at both mRNA (Figure 3D) and protein levels (Figure 3E). We then utilized the CDK6 3′UTR luciferase reporter assay to detect whether HNRNPA2B1 could posttranscriptionally regulate miR-506-mediated CDK6 suppression. Consistent with the data in Figures 3D and 3E, knockdown of HNRNPA2B1 restored the luciferase expression level in the 95D cells that were co-transfected with miR-506 mimics (Figure S3D). Similar results were also observed in HeLa cells after the co-transfection of miR-506 and siRNA-HNRNPA2B1 (si-A2B1) (Figures S3E and S3F). The HNRNPA2B1 binding site mutant reporter vector was co-transfected with miR-506 mimics to test the influence of miR-506 suppression on CDK6 3′UTR. Similarly, the transfection with HNRNPA2B1 binding site mutant plasmid relieved the miR-506-mediated suppression of luciferase activity compared with the WT (Figure 3F). HNRNPA2B1 binding site mutation reduced the inhibitory effect of miR-506 on reporter gene by about 20%, which was less than the miR-506 binding site mutation inducing inhibitory effect (Figure 3F). One likely reason, perhaps, is that the HNRNPA2B1 binding site mutation may change the specific RNA structures in 3′UTR, which, in consequence, partially opens the structure. Besides HNRNPA2B1, other RBPs may involve in the miR-506-mediated repression. Furthermore, the luciferase activity of double mutation vector (containing mutations to both the HNRNPA2B1 and miR-506 binding sites) showed no obvious increase compared with miR-506 binding site mutant vector (Figure 3F). The EdU cell proliferation analysis of 95D cells showed a positive correlation with the CDK6 protein levels (Figure S3G). Altogether, these findings suggested that HNRNPA2B1 modulated the miR-506-mediated suppression of CDK6 in cancer cells.
Figure 3.

HNRNPA2B1 regulates miR-506-mediated inhibition of CDK6
(A) Diagram showing the CDK6 3′UTR evolutionarily conserved sequence analysis of miR-506 and HNRNPs binding site in the 3′UTR. The CDK6 3′UTR harbors two nearby evolutionarily conserved motifs, including UGCCUUA for miR-506 and putative UUAGG for HNRNPs.
(B) RNA pull-down assay identification of CDK6 3′UTR-specific binding proteins. The in vitro transcribed 223 nt wild-type or the mutant (bold italic characters) CDK6 3′UTR RNA was used to perform pull-down assays in 95D cells. Then, the sample was analyzed by silver staining assay and LC-MS. The arrow shows the enriched protein band in the wild-type CDK6 3′UTR RNA pull-down sample.
(C) Western blot assay detection of the enrichment of HNRNPA2B1 in wild-type or mutant CDK6 3′UTR RNA pull-down samples treated as in (B).
(D) RT-qPCR detection of CDK6 mRNA levels in 95D cells. Cells were transfected with no target control (NC) miR-506, si-HNRBPA2B1 (si-A2B1), and miR-506+ si-A2B1. Forty-eight hours after transfection, the CDK6 mRNA levels were measured with RT-qPCR. The mRNA expression was normalized to β-Actin. Data are presented as the mean ± SD (n = 4). ∗p < 0.05, ∗∗p < 0.01, one-way ANOVA test.
(E) Western blot detection of the CDK6 protein level in 95D cells were treated as in (D). Forty-eight hours after transfection, western blot assays were conducted. β-Actin was used as a reference gene for western blot. Band densities were quantified using ImageJ.
(F) The CDK6 3′UTR dual luciferase reporter assay of HNRNPA2B1 binding sites mutation. 95D cells were co-transfected with NC, miR-506 along with the CDK6 3′UTR wild-type (WT), HNRNPA2B1 binding site mutant (A2B1 bs mutant), and miR-506 binding site mutant (miR-506 bs mutant) reporter vector. Luciferase activity was tested 48 h after transfection. Firefly luciferase activity was normalized to the Renilla luciferase internal control. Data are presented as the mean ± SD (n = 4). ∗p < 0.05, ∗∗p < 0.01, Mann-Whitney U test.
HNRNPA2B1 unwinds CDK6 3′UTR for facilitating AGO2 binding
Given that the miR-506 showed different suppression ability toward CDK6 (Figure 2C), and HNRNPA2B1 can regulate miR-506-mediated suppression of CDK6 (Figures 3D and 3E), we questioned whether HNRNPA2B1 was related to the miR-506-mediated different suppression of CDK6 in these cells. To answer it, we checked the protein level of HNRNPA2B1 in 95D, A549, H460, and H1299 cells. The results showed there was no obvious relationship between HNRNPA2B1 and miR-506-mediated CDK6 suppression in these cells (Figure S4A). Based on these results, we reasoned that the binding of HNRNPA2B1 with the 3′UTR of CDK6 could probably be different in different cell lines. This notion was further confirmed by the RNA pull-down data generated from the lysates of 95D and H460 cells, which showed different CDK6 suppression after transfection with miR-506 mimics (Figure 2C). The CDK6 3′UTR RNA pull-down analysis showed that the enrichment of HNRNPA2B1 in 95D cells was higher than that in H460 cells (Figure S4B). Moreover, the enrichment of HNRNPA2B1 by CDK6 3′UTR was positively related to the miR-506-mediated suppression of CDK6 in 95D lung cancer cells. Next, we focused on the mechanism of HNRNPA2B1 in miR-506-mediated repression of CDK6. Firstly, the interaction between HNRNPA2B1 and the CDK6 3′UTR was confirmed by RIP-PCR (RNA immunoprecipitation PCR). The results showed that the region in the CDK6 3′UTR containing the HNRNPA2B1 binding motif and the miR-506 binding site was enriched in the HNRNPA2B1 immunoprecipitation sample (Figure 4A). The result was consistent with previously reported data (Alarcon et al., 2015), demonstrating that the same region was enriched in HNRNPA2B1 HITS-CLIP-seq (cross-linking immunoprecipitation and high-throughput sequencing) analysis (Figure S4C). To further confirm HNRNPA2B1 binding to the CDK6 3′UTR, confocal microscopy experiments using immunofluorescence staining combined with RNA in situ hybridization were conducted. The CDK6 3′UTR (labeled with red fluorescent dye) and HNRNPA2B1 (labeled with green fluorescent dye) were co-localized (yellow color) in the cytoplasm of 95D lung cancer cells (Figure 4B). Meanwhile, the subcellular localization of HNRNPA2B1 was examined in 95D cell. The western blot results showed that although HNRNPA2B1 was mainly located in the nucleus, part of HNRNPA2B1 was still distributed in the cytoplasm (Figure S4D). These results suggested that the HNRNPA2B1 interacts with CDK6 mRNA in cytoplasm. Subsequently, we investigated how HNRNPA2B1 mechanistically regulates miR-506-mediated suppression of CDK6. First, we analyzed the structure of the CDK6 3′UTR with Mfold software (http://unafold.rna.albany.edu). The predicted CDK6 3′UTR structure showed that the HNRNPA2B1 binding elements can fold into two preferred conformation, including a conformation of base-paired stem structure with the miR-506 binding site (1648–1654) and a conformation of base-paired stem structure with region (1717–1731) (Figure S4E). Notably, a report showed that HNRNPA2B1 can bind with AGG and UAG motifs by forming hydrogen bonds with both base and ribose of the RNA (Wu et al., 2018). This result provided a hint that HNRNPA2B1 can compete with the miR-506 binding site or the region (1717–1731) for the UUAGG motif in the CDK6 3′UTR, which led to release the linear form of the CDK6 3′UTR (Figure 4C). To confirm this hypothesis, we adopted an enzymatic approach to probe the switch of the CDK6 3′UTR. The in vitro folded CDK6 3′UTR RNA was firstly incubated with HNRNPA2B1 and GFP and subsequently treated with RNaseT1. Then, the levels of digested RNA were examined through RT-PCR. The result showed that the efficiency of CDK6 3′UTR digestion increased after incubation with HNRNPA2B1 compared with GFP treatment (Figure 4C). Next, it needs to be explored whether HNRNPA2B1 can enhance RISC accessibility to the CDK6 3′UTR. Thus, we performed AGO2 RIP-PCR to determine the change of AGO2 enrichment in the same region in the CDK6 3′UTR after HNRNPA2B1 knockdown. The enrichment of AGO2 binding to the CDK6 3′UTR was reduced after HNRNPA2B1 knockdown (Figure 4D). Collectively, these results suggested that HNRNPA2B1 can bind to the CDK6 3′UTR and denature the base-paired structure of the 3′UTR, which facilitated the engagement of AGO2 with the CDK6 3′UTR.
Figure 4.

HNRNPA2B1 binding denatures a stem structure in CDK6 3′UTR
(A) The enrichment of HNRNPA2B1 at the CDK6 3′UTR was tested with an exogenous HNRNPA2B1-Flag RIP-PCR assay. RIP of HNRNPA2B1 was performed using a Flag monoclonal antibody. GFP-Flag was used as negative control. (left) RIP-PCR, (right) western blot. Data are presented as the mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, two-tailed Student’s t test.
(B) The combination of RNA FISH and HNRNPA2B1 immunohistochemistry analyses showed co-localization of CDK6 RNA and HNRNPA2B1 in 95D cells. The CDK6 3′UTR was labeled with Cy3-probe (red fluorescence), the HNRNPA2B1 was detected with an HNRNPA2B1 antibody (green fluorescence), and the nucleus was stained with DAPI. The images were captured and analyzed by a Leica Biosystem. The scale bar at the bottom left corner indicates 20 μm.
(C) Enzymatic probing of RNA structural change in the CDK6 3′UTR was performed by RT-PCR (top). The 223 nt CDK6 3′UTRs were synthesized in vitro. Then, the RNA was folded and incubated with exogenous purified GFP-flag or HNRNPA2B1-flag, which was followed by digestion with RNase T1. The digested RNA was tested by RT-PCR (middle). The binding of HNRNPA2B1-flag to the CDK6 3′UTR was confirmed by western blot (upper right).
(D) AGO2 RIP-PCR validation of the interaction between AGO2 and the CDK6 3′UTR after HNRNPA2B1 knockdown in 95D cells. The 95D cells were transfected with NC and siRNA-HNRNPA2B1 (si-A2B1). Forty-eight hours after transfection RIP-PCR was performed using the AGO2 monoclonal antibody. The enrichment of the CDK6 3′UTR in AGO2 immunoprecipitation was determined via RT-qPCR analysis. Data are presented as the mean ± SD (n = 3). Statistically significant differences were determined by Student’s t test. ∗p < 0.05, ∗∗p < 0.01.
HNRNPA2B1 relieves other miRNA-mediated suppression of different genes
In addition to miR-506, we wondered whether HNRNPA2B1 could regulate the functions of other miRNAs. Previously, the CDK6 expression level was reported to be regulated by other miRNAs, such as miR-34a and miR-195 (Sun et al., 2008; Xu et al., 2009). We tested whether HNRNPA2B1 also functions in miR-34a- and miR-195-mediated suppression of CDK6. Accordingly, we assessed the CDK6 3′UTR for these miRNA binding sites and found cis-elements for both miR-34a and miR-195 (Figure S5A). Furthermore, co-transfection of these miRNAs and siRNA-HNRNPA2B1 (si-A2B1) in HEK293 cells resulted in relief from miR-195- and miR-34a-mediated suppression of CDK6 (Figure S5B). In addition, after analyzing previously published HNRNPA2B1 HITS-CLIP-seq data (Alarcon et al., 2015), we found that the 3′UTR of another gene, Aurora Kinase A (AURKA), also contains binding sites of both miR-506 and HNRNPA2B1 (Figure S5C). The 3′UTR of AURKA can be enriched by HNRNPA2B1 through RIP-PCR analysis (Figure S5C). Accordingly, co-transfection of siRNA-HNRNPA2B1 and miR-506 showed that HNRNPA2B1 knockdown can relieve miR-506-mediated suppression of AURKA at both RNA and protein levels (Figures S5D and S5E). Moreover, the reporter assay result with the AURKA 3′UTR also showed that HNRNPA2B1 knockdown relieved miR-506-mediated suppression of AURKA 3′UTR reporter luciferase activity (Figure S5F).
Besides CDK6 and AURKA, we wanted to explore whether other miR-506 target genes are also regulated by HNRNPA2B1. Firstly, we used CLIP data to analyze the enrichment of HNRNPA2B1 in 3′UTR of other target genes of miR-506, including SIRT1, CDK4, FOXQ1, RELA, HMGA2, and YAP1 (Xia et al., 2020; Hossian et al., 2018; Zhang et al., 2015; Yin et al., 2015; Li et al., 2020; Zhu et al., 2019). The analysis revealed that the enrichment of HNRNPA2B1 in 3′UTR of SIRT1, CDK4, FOXQ1, and RELA was comparatively less than that of the 3′UTR of HMGA2 and YAP1 (Figure S5G). Therefore, in order to detect the impact of HNRNPA2B1 on miR-506-mediated target gene suppression, we co-transfected siRNA-HNRNPA2B1 (si-A2B1) and miR-506 in 95D cells. The results showed that HNRNPA2B1 KD could not relieve the inhibition of CDK4 and RELA protein mediated by miR-506 treatment (Figure S5H). The following reporter gene detection results also verified that HNRNPA2B1 KD could not restore the luciferase activity of CDK4 and RELA 3′UTR reporter vectors (Figure S5I).
We next wondered whether the binding site of HNRNPA2B1 in CDK6 3′UTR can function as cis-elements. To answer this, we cloned the binding site of HNRNPA2B1 into the 3′UTR reporter vector of SIRT1, CDK4, and FOXQ1 (Figure S5J). The insertion of the binding site of HNRNPA2B1 could promote the inhibitory effect of miR-506 on the target gene (Figure S5J). These results suggested that the HNRNPA2B1 can regulate miR-506-mediated suppression of mRNAs through specifically binding to the 3′UTR of target genes.
The physical interaction between HNRNPA2B1 and DHX9
It was reported that HNRNPA2B1 interacts with other functional partners such as DiGeorge syndrome critical region gene 8 (DGCR8) to mediate miRNA processing (Alarcon et al., 2015) and TANK binding kinase 1 (TBK1) to initiate type I interferon (IFN-I) expression (Wang et al., 2019a). We wondered whether there were other factors involved in the function of HNRNPA2B1 in 3′UTR regulation. In the RNA pull-down assay, proteins enriched in sample of wild-type 3′UTR were analyzed by LC-MS. The results showed that DHX9 (∼130 kD) was one of the proteins with a high score identified by LC-MS analysis (Table S2). Previously, it was reported that DHX9 is a key cofactor of RISC (Robb and Rana, 2007) and plays roles in regulation of CDK6 expression in hepatocellular carcinoma (HCC) (Wang et al., 2019b). Therefore, we hypothesized that HNRNPA2B1 can recruit DHX9 to the CDK6 3′UTR. To confirm this, we first examined the DHX9 protein level in wild-type and UUAGG-motif-mutated CDK6 3′UTR RNA pull-down samples by western blot. The results showed that the DHX9 protein level was higher in wild-type CDK6 3′UTR sample than it was in the UUAGG-mutated sample (Figure 5A). To test the interaction between HNRNPA2B1 and DHX9, we then performed co-immunoprecipitation (co-IP) experiments and found that HNRNPA2B1 and DHX9 can mutually bind to each other (Figure 5B). In addition, AGO2 was also found to interact with DHX9 (Figure 5B). To further investigate whether the interaction between HNRNPA2B1 and DHX9 is RNA dependent, we performed co-IP using samples treated with RNase. Interestingly, the DHX9 co-IP with HNRNPA2B1 is slightly reduced in the presence of RNase (Figure 5C). HNRNPA2B1 is also co-precipitated with DHX9 in RNase-treated co-IP samples (Figure 5D). This suggests that HNRNPA2B1 and DHX9 can interact with each other in both an RNA-dependent and RNA-independent manner.
Figure 5.

The interaction between HNRNPA2B1 and DHX9
(A) The CDK6 3′UTR RNA pull-down assay. Western blot identification of DHX9 and HNRNPA2B1 enriched by CDK6 3′UTR RNA with HNRNPA2B1 binding site wild-type or mutant sample in 95D cells. The pull-down RNA was tested with RT-PCR (bottom).
(B) Co-immunoprecipitation (co-IP) detection of the interaction among HNRNPA2B1, DHX9, and AGO2 in 95D lung cancer cells. co-IP was performed using endogenous DHX9, AGO2, and HNRNPA2B1 anti-body. Immunoprecipitate (IP), immunoblotting (IB).
(C) In vivo interaction between DHX9 and HNRNPA2B1 was tested by co-IP in 95D cells. Cellular lysates were immunoprecipitated with an HNRNPA2B1 antibody in samples treated with RNaseA/T1. HNRNPA2B1 and DHX9 were detected by western blot. Immunoprecipitate (IP), immunoblotting (IB).
(D) Same as (C), but reciprocal immunoprecipitation was performed. In this case, endogenous DHX9 was immunoprecipitated, and its interaction with HNRNPA2B1 was detected by western blotting under similar conditions. Immunoprecipitate (IP), immunoblotting (IB).
Next, we explored the effect of the interaction between HNRNPA2B1 and DHX9 on CDK6 inhibition. First, the individual silencing of HNRNPA2B1 and DHX9 in lung cancer cells resulted in an increase in the expression level of CDK6 protein (Figure S6A). In addition, the simultaneous inhibition of DHX9 and HNRNPA2B1 resulted in the increase of CDK6 level, which was similar in extent to that of HNRNPA2B1 KD alone (Figure S6A). Then, we overexpressed the HNRNPA2B1 in lung cancer cells. The results showed that the HNRNPA2B1 overexpression can reduce the CDK6 protein level (Figure S6B). Interestingly, the KD of DHX9 was able to restore the CDK6 protein level suppressed by HNRNPA2B1 overexpression (Figure S6C). These findings highlighted that the role of HNRNPA2B1 suppression on CDK6 was dependent on DHX9. Collectively, these data described earlier indicate that HNRNPA2B1 may recruit RISC to the CDK6 3′UTR through directly binding with DHX9.
HNRNPA2B1 regulates miRNA-mediated gene silencing pathways
In addition to CDK6, we speculated that HNRNPA2B1 and DHX9 could colocalize to the 3′UTR of other target mRNAs to regulate AGO2-mediated gene suppression. By analyzing HNRNPA2B1 HITS-CLIP data (Alarcon et al., 2015) and DHX9 FLASH data (fast ligation of RNA after some sort of affinity purification for high-throughput sequencing) (Aktas et al., 2017), we found that the HNRNPA2B1 binding sites are enriched in the 3′UTRs of 2,167 genes (Figure 6A). Similarly, the 3′UTRs of 284 genes were bound by DHX9 (Figure 6A). Notably, HNRNPA2B1 and DHX9 binding sites co-localized in the 3′UTR of 117 genes (Figure 6A), including BCL2 like 2 (BCL2L2), G protein nucleolar 3 like (GNL3L), mitochondrial antiviral signaling protein (MAVS), and von Hippel-Lindau (VHL) (Figure 6B). Moreover, the RIP-PCR results validated that HNRNPA2B1, DHX9, and AGO2 can bind to the 3′UTR of BCL2L2, GNL3L, MAVS, and VHL (Figure 6C). The enrichment of AGO2 and DHX9 at the BCL2L2, GNL3L, MAVS, and VHL 3′UTRs was significantly reduced after knocking down HNRNPA2B1 (Figure 6D). In addition, we tested the RNA-binding ability of AGO2 by PAR-CLIP assay, which showed that the RNA-binding signal of AGO2 protein was reduced after knocking down of HNRNPA2B1 (Figure S7A). These results suggested that the enrichment of AGO2 and DHX9 at the 3′UTRs of BCL2L2, GNL3L, MAVS, and VHL was dependent on HNRNPA2B1 binding to the 3′UTRs. Consistent with the reduced binding of the 3′UTR by AGO2 and DHX9, the 3′UTR RNA levels of BCL2L2, GNL3L, MAVS, and VHL were increased after siRNA-HNRNPA2B1 treatment (Figure S7B). Moreover, overexpression of HNRNPA2B1 could repress VHL and BCL2L2 expression at the protein level (Figure S7C). Given the previously reported literature evidence that BCL2L2 is a target of human miR-195 (Qu et al., 2015), we analyzed the HNRNPA2B1 HITS-CLIP data (Alarcon et al., 2015) as well as the miRNA target predication data (Figure S7D). From the analysis, we observed that the binding motifs of both miR-195 and HNRNPA2B1 were found together in the 3′UTR of BCL2L2 (Figure S7D). Interestingly, RNA structure prediction showed that the strand containing the HNRNPA2B1 binding cis-elements formed a stem-loop structure with the strand containing miR-195 binding sites (Figure S7E). Next, we tested whether HNRNPA2B1 and DHX9 regulate the miR-195-mediated suppression of BCL2L2. The result showed that HNRNPA2B1 and DHX9 knockdown relieved miR-195-mediated BCL2L2 suppression at both the mRNA and protein levels (Figure S7F). These findings indicated that HNRNPA2B1 can regulate the binding of DHX9 and AGO2 to the target 3′UTR, facilitating miRNA-mediated target suppression.
Figure 6.

HNRNPA2B1 modulates the enrichment of AGO2 and DHX9 to target genes
(A) Venn diagram comparing the number of 3′UTRs enriched by HNRNPA2B1 HITS-CLIP and DHX9 FLASH. The enrichment of 3′UTRs by HNRNPA2B1 was analyzed on the basis of HITS-CLIP data (Alarcon et al., 2015), and the 3′UTR binding by DHX9 was obtained from the FLASH data (Aktas et al., 2017).
(B) Integrative Genomics Viewer (IGV) snapshot depicting the HNRNPA2B1 (HIST-CLIP-seq data) and the endogenous DHX9 (FLASH data) binding regions. Co-localization of HNRNPA2B1 and DHX9 in 3′UTRs is shown by rectangles.
(C) Detection of the enrichment of BCL2L2, GNL3L VHL, and MAVS 3′UTR by AGO2, HNRNPA2B1, and DHX9 RIP-PCR in 95D cells. Data are presented as the mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, two-tailed Student’s t test.
(D) RIP-PCR validation of the enrichment of AGO2 (left) and DHX9 (right) at BCL2L2, GNL3L, VHL, and MAVS 3′UTR after HNRNPA2B1 knockdown in 95D cells. Cells were transfected with siRNA-HNRNPA2B1 (si-A2B1). After forty-eight hours of transfection, cells were harvested for AGO2 and DHX9 RIP-PCR. The RIP-PCR results are shown (top), and the efficiency of RIP and HNRNPA2B1 knockdown was tested by western blot (bottom). Data are presented as the mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, two-tailed Student’s t test.
Discussion
RBPs play crucial roles in RNA metabolism. Previous studies found that RBP binding sites and miRNA binding sites are enriched in the 3′UTR of mRNAs, suggesting that both RBPs and miRNAs play a role in gene posttranscriptional regulation (Matoulkova et al., 2012; Mayr, 2016). In addition, studies of RBPs identification systemically found that many proteins that associated with poly(A)+ RNA were members of the HNRNP family RBPs (Bao et al., 2018; Castello et al., 2012). It was reported that the HNRNPA2B1 and HNRNPA1 took part in a widespread sequence-specific mRNA decay pathway via mRNA deadenylation, probably mediated by the CCR4–NOT complex (Geissler et al., 2016). This suggests that a wide variety of posttranscriptional events might be regulated by HNRNPs acting on 3′UTRs. miRNAs play key role in gene posttranscriptional regulation mainly through binding to 3′UTRs of target genes. The effectiveness of miRNA-mediated suppression of target genes is dependent on the accessibility of the RISC to the seed region binding sites in 3′UTRs, particularly in long 3′UTRs (Agarwal et al., 2015; Grimson et al., 2007). However, the underlying mechanism regulating miRNA accessibility to target 3′UTRs is largely unknown. The availability of linear miRNA binding sites in the 3′UTR is a preliminary and key step for RISC engagement with a target, especially for the target containing long 3′UTR. The length of the CDK6 3′UTR is more than 10 kilobases (kb). Many reports found that CDK6 expression was regulated by kinds of trans-factors, such as miRNAs, RBPs, and cis-elements at the posttranscriptional level (Hossian et al., 2018; Liu et al., 2014; Sun et al., 2008; Konishi et al., 2015; Wang et al., 2019b). Our findings revealed that the conserved cis-elements of miR-506 (UGCCUUA) and HNRNPA2B1 binding sites (UUAGG) can form a stem structure in the CDK6 3′UTR. The RNA-binding protein HNRNPA2B1 binds to the UUAGG motif, and thereof denatures the stem structure, leading to release of the linear conformation for switching “on” the miR-506-mediated suppression of CDK6 protein expression. Notably, research on the HNRNPA2B1-RNA structure showed that HNRNPA2B1 can form hydrogen bonds with bases in the AGG, UAG motif (Wu et al., 2018). Considering our data in light of this report, it is conceivable that HNRNPA2B1 denatures a paired RNA structure through competitive hydrogen bond formation in CDK6 3′UTRs, thus inducing RNA structure switching and activation of miRNA-mediated target suppression (Figures 4C and S4E). In addition, HNRNPA2B1 interacts with the RNA helicase DHX9, which can recruit AGO2 to the 3′UTR of target genes. Based on these observations, we suggest a model wherein HNRNPA2B1 triggers miRNA function by inducing a structural switch in the 3′UTR and, thereby recruiting DHX9 to the target 3′UTR. Accordingly, the RISC can access the 3′UTR and suppress target gene expression (Figure 7).
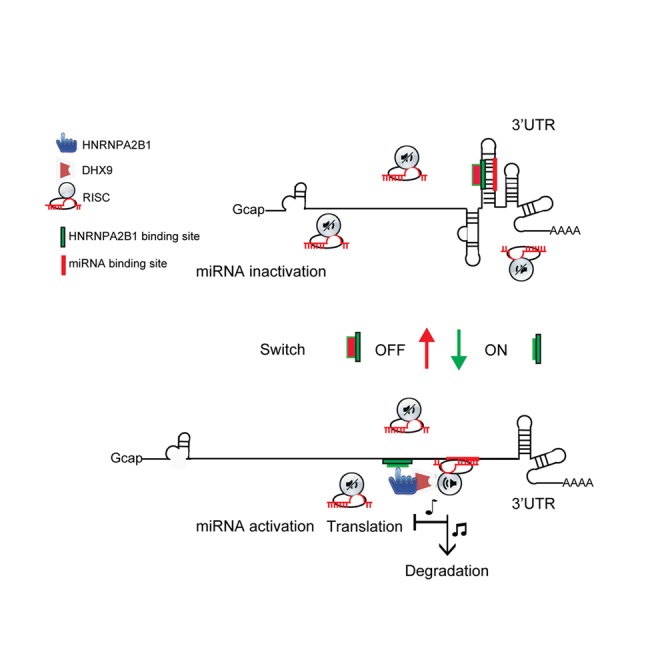
Figure 7.

The proposed model is as follows
Two cis-elements are located in the 3′UTR of CDK6 mRNA; one is the HNRNPA2B1 binding site, and the other is the miR-506 binding site. The sequences of these two elements are complements of each other and form a stem structure that prevents miR-506 from recruiting the RISC to the 3′ UTR of CDK6 mRNA (the OFF status).Upon cytoplasmic HNRNPA2B1 binding, the stem structure is denatured, which releases the miR-506 cis-element, leading to the recruitment of the miR-506-RISC complex for specific gene silencing (the ON status). These results indicate that a cis-element, the HNRNPA2B1 binding site, in the 3′ UTR of CDK6 mRNA serves as a conserved RNA switch.
Generally, the expression level of tumor suppressor miRNAs is downregulated in most patients and cancer cell lines (Macfarlane and Murphy, 2010; Garzon et al., 2010). However, there were few reports that indicate that the miRNAs are also upregulated in part of patients and cancer cell lines. Examples include miR-34 in NSCLC (Wiggins et al., 2010), miR-195 in hepatocellular carcinoma (HCC) (Xu et al., 2009), and miR-449a and miR-192 in NSCLC (Feng et al., 2011; Ren et al., 2014). This suggests that loss of function for tumor-suppressive miRNAs may exist during tumorigenesis. In addition to the aforementioned studies, we herein found that both miR-506 and CDK6 are highly expressed in approximately one-third lung cancer patients. Our result further suggested one of the adaptive mechanisms that lung cancer cells normally use in resisting high expression levels of tumor-suppressive miR-506 was mediated through modulation of the miRNA(s) binding site accessibility on target(s) 3′UTR. To this context, HNRNPA2B1 functions as a key factor for switching “on” the suppression function of miR-506 against CDK6 in cancer cells. Hence, during searching for miRNA targets and miRNAs-based therapies, the interaction between RBPs and the 3′UTR should be considered to achieve a higher efficiency of miRNA suppression. Previous reports indicate that the HNRNPA2B1 is upregulated in many cancer types and correlates with worse prognosis (Clower et al., 2010; Cui et al., 2010; Golan-Gerstl et al., 2011; Pino et al., 2003; Stockley et al., 2014; Zhou et al., 2001). Given this fact, it is possible that the cancer cells may come up with adaptation to inhibit tumor-suppressive function of HNRNPA2B1. In this research, we found that the enrichment of HNRNPA2B1 by 3′UTR of CDK6 was higher in 95D cells than that in H460 cells (Figure S4B). And, KD of HNRNPA2B1 was able to relive the miR-506-mediated CDK6 suppression (Figures 3D and 3E). These results indicate that the reduced binding of HNRNPA2B1 to CDK6 3′UTR may cause a decrease in miR-506-mediated CDK6 suppression, which eventually may increase both miR-506 and CDK6 mRNA levels in lung cancer patients and cell lines (Figures 2A and 2B). In order to completely understand the functions of miRNAs and RBPs, it is worth to explore the up-stream regulatory pathways that control RBPs binding to the 3′UTRs of different transcripts in specific signaling pathways.
DHX9 is a ubiquitously expressed RecA domain RNA helicase. Structural and biochemical studies have reported that the RecA domain of RNA helicase directly interacted with the sugar-phosphate backbone of RNA, indicating that the RNA helicase lacks the ability to bind to a specific RNA motif (Ozgur et al., 2015). However, recent studies have shown that the RNA helicase can be enriched at specific sites of RNA (Aktas et al., 2017; Sloan and Bohnsack, 2018). Given that helicases directly interact with the sugar-phosphate backbone of RNA, these findings raise the possibility that RNA helicase preference for specific RNA sequences may be conferred by cofactors, which bind to both specific RNA sequences and helicases. Our research showed that HNRNPA2B1 and DHX9 co-localized at the 3′UTR of target mRNA (Figures 5A, 6B, and 6C). Further, HNRNPA2B1 knockdown attenuated the binding of DHX9 to the specific mRNA 3′UTR (Figure 6D). In a recent study, it was found that the DHX9 can specifically bind to CDK6 3′UTR, leading to degradation of CDK6 in HCC (Wang et al., 2019b), and HNRNPA2B1 can recognize specific AGG, UAG motif (Wu et al., 2018). To this end, our results suggested that the RNA binding protein HNRNPA2B1 can function as a bridge between a specific RNA motif and an RNA helicase.
Limitations of the study
The complex secondary and tertiary structures of RNA molecules play key roles in its cellular functions. The observations of 3′UTR structure alteration mediated by HNRNPA2B1 are interesting, but the paper fails short to use an orthogonal approach to disclose the widespread RNA structure modulation of HNRNPA2B1, in particular with respect to modulating miRNA-mediated repression. Meanwhile, considering that RNA helicase can unwind the double-stranded RNA structure in an ATP-dependent manner (Jankowsky, 2011) and the RNA helicase DHX9 can progressively unwind RNA duplexes (Koh et al., 2014), it also would be interesting to unmask the landscape of DHX9 RNA helicase activity in 3′UTR structure remodeling following DHX9 recruitment by HNRNPA2B1.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-AGO2 | Abcam | Cat# ab186733; RRID:AB_2713978 |
| anti-BCL2L2 | Proteintech | Cat# 16026-1-AP; RRID: AB_2827646 |
| anti-CDK4 | Proteintech | Cat# 11026-1-AP; RRID:AB_2078702 |
| anti-NFkappaB p65 (RELA) | Santa Cruz | Cat# sc-8008; RRID:AB_628017 |
| anti- Histone H3 | Santa Cruz | Cat# sc-10809; RRID:AB_2115276 |
| anti-CDK6 | Santa Cruz | Cat# sc-177; RRID:AB_631225 |
| anti-HNRNPA2B1 | Santa Cruz | Cat# sc-53531; RRID:AB_2248245 |
| anti-DHX9 | Santa Cruz | Cat# sc-137232; RRID:AB_2261698 |
| anti-ACTB | Santa Cruz | Cat# sc-47778; RRID:AB_626632 |
| anti-VHL | Santa Cruz | Cat# sc-135657; AB_2215955 |
| anti-AURKA | Santa Cruz | Cat# sc-56881; RRID:AB_667838 |
| anti-RELA(p65) | Santa Cruz | Cat# sc-8008; RRID:AB_628017 |
| anti-HNRNPL | Santa Cruz | Cat# sc-32317; RRID:AB_627736 |
| anti-Flag | Santa Cruz | Cat# sc-807; RRID:AB_675756 |
| Anti-rabbit IgG, HRP-linked Antibody | Santa Cruz | Cat# sc-2004; RRID:AB_631746 |
| Anti-mouse IgG, HRP-linked Antibody | Santa Cruz | Cat# sc-2005; RRID:AB_631736 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Antibiotic (Penicillin/Streptomycin) | Thermo Fisher | Cat# 15140122 |
| NP40 Thermo Scientific | Thermo Fisher | Cat# 85124 |
| Lipofectamine2000 | Thermo Fisher | Cat# 11668019 |
| PBS | Thermo Fisher | C10010500BT |
| Trypsin | Thermo Fisher | Cat# 15400054 |
| DMEM | Thermo Fisher | Cat# C11995500BT |
| PRIM1640 | Thermo Fisher | Cat# C11875500BT |
| RNase inhibitors | Thermo Fisher | Cat# EO0381 |
| Bis-TrisNuPage 4-12% gel | Thermo Fisher | Cat# NP0335BOX |
| TRIzol reagent | Thermo Fisher | Cat# 15596026 |
| GlycoBlue coprecipitant | Thermo Fisher | Cat# AM9516 |
| SuperScript III reverse transcriptase | Thermo Fisher | Cat# 18080044 |
| Pre-staining protein marker 10 to 180kDa | Thermo Fisher | Cat# 26617 |
| 20X SSC BUFFER | Thermo Fisher | Cat# AM9763 |
| HEPES (1 M) | Thermo Fisher | Cat# 15630080 |
| TRIS-HCL PH 7.5 1M | Thermo Fisher | Cat# 15567027 |
| 1 M MAGNESIUM CHLORIDE | Thermo Fisher | Cat# AM9530G |
| 2 M KCL | Thermo Fisher | Cat# AM9640G |
| 10X TBE | Thermo Fisher | Cat# AM9865 |
| RNASE T1 | Thermo Fisher | Cat# EN0542 |
| RNase A/T1 | Thermo Fisher | Cat# EN0551 |
| EDTA | Thermo Fisher | Cat# AM9260 |
| RNase-free DNase I | Thermo Fisher | Cat# 18068015 |
| streptavidin Dynabeads beads C1 | Thermo Fisher | Cat# 65002 |
| Proteinase K Invitrogen | Thermo Fisher | Cat# 25530049 |
| Flag antibody coated M2 beads | Sigma Aldrich | Cat# M8823 |
| paraformaldehyde (PFA) | Sigma Aldrich | Cat# 158127 |
| Triton X-100 | Sigma Aldrich | Cat# T8787 |
| TWEEN 20 | Sigma-Aldrich | Cat# P9416 |
| DMSO | Sigma Aldrich | Cat# D2650 |
| NaCl | Sigma Aldrich | Cat# S5150-1L |
| KCl | Sigma Aldrich | Cat# P9333 |
| Protease inhibitor cocktail | Sigma Aldrich | Cat# P8340 |
| 20xSSC Buffer Sigma Aldrich | Sigma Aldrich | Cat# 93017 |
| glycine | Sigma Aldrich | Cat# G8898 |
| Glycerol | Sigma Aldrich | Cat# G5516-500ML |
| Tris-EDTA 0.5M pH 8.0 | Sigma Aldrich | Cat# 93283-500ML |
| 2-Mercaptoethanol | Sigma Aldrich | Cat#M6250 |
| Biotin RNA Labeling Mix | Roche | Cat# 11685597910 |
| Yeast tRNA | Roche | Cat# 10109495001 |
| Fetal Bovine Serum | PAN Biotech | Cat# ST30-3302 |
| Moloney Murine Leukemia Virus Reverse Transcriptase | Promega | Cat# M1705 |
| PVDF membrane | Millipore | Cat# ISEQ00010 |
| Propidium Iodide | Beyotime | Cat# ST511 |
| Critical Commercial Assays | ||
| Cell Light EdU DNA imaging Kit | RiboBio | Cat# C10310-3 |
| Cell Counting Kit-8 (CCK-8) assay kit | Dojindo | Cat# CK04 |
| ECL western blotting Kit (, Thermo Scientific Pierce) | Thermo Fisher | Cat# 32106 |
| SYBR Green Supermix | Promega | Cat# S2062, |
| Dual-Glo luciferase assay kit | Promega | Cat# E1980 |
| T7 transcript KIT | TOYOBO | Cat# TSK-101B |
| KOD plus neo KIT | TOYOBO | Cat# KOD-401S |
| Biological samples | ||
| Patient-derived samples | The First Affiliated Hospital of Guangzhou Medical University (Ren et al. 2014) | N/A |
| Experimental Models: Cell Lines | ||
| NCI-HCC827 | ATCC | Cat# CRL-2868 |
| A549 | ATCC | Cat# CCL-185 |
| NCI-H661 | ATCC | Cat# HTB-183 |
| NCI-H1299 | ATCC | Cat# CRL-5803 |
| NCI-H358 | ATCC | Cat# CRL-5807 |
| NCI-H1975 | ATCC | Cat# CRL-5908 |
| NCI-H23 | ATCC | Cat# CRL-5800 |
| HCC827 | ATCC | Cat# CRL-2868 |
| MRC-5 | ATCC | Cat# CCL-171 |
| HLF | ATCC | Cat# CCL-153 |
| HeLa | ATCC | Cat# CCL-2 |
| HEK-293 | ATCC | Cat# CRL-1573 |
| 95D | This paper | N/A |
| PC9 | This paper | N/A |
| Oligonucleotides | ||
| No target control (NC) | RiboBio | Cat# siN0000001-1-5 |
| hsa-miR-506-3p mimic | RiboBio | Cat# miR10002878-1-5 |
| hsa-miR-34a-5p mimic | RiboBio | Cat# miR10000255-1-5 |
| hsa-miR-195-5p mimic | RiboBio | Cat# miR10000461-1-5 |
| si-h-CDK6_001 | RiboBio | Cat# siB084281913 |
| si-h-CDK6_002 | RiboBio | Cat# siB084281932 |
| si-h-CDK6_003 | RiboBio | Cat# siB084281948 |
| si-h-HNRNPA2B1_001 | RiboBio | Cat# siB160409043647 |
| si-h-HNRNPA2B1_002 | RiboBio | Cat# siB160409043801 |
| si-h-HNRNPA2B1_003 | RiboBio | Cat# siB160706015613 |
| si-h-DHX9_001 | RiboBio | Cat# stB0006120A |
| si-h-DHX9_002 | RiboBio | Cat# stB0006120B |
| si-h-DHX9_003 | RiboBio | Cat# stB0006120B |
| CDK6 FISH Cy3 probes, see Table S3 | Generay biotechnology | N/A |
| Primers for RT-qPCR, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pENTER-HNRNPA2B1-Flag | Vigene Biosciences | Cat# CH865056 |
| pENTER-GFP-Flag | This paper | N/A |
| pET28a-GFP | This paper | N/A |
| pET28a-HNRNPA2B1 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Bio-RAD CFX Manager | BIO-RAD | http://www.bio-rad.com/en-us/product/cfx-manager-software?tab=Download |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/int/software-cameras.html |
| Accuri C6 Plus | BD Biosciences | http://www.bdbiosciences.com/us/instruments/research/cell-analyzers/bd accuri/m/1294932/overview |
| MACS2 (v2.1.0) | GitHub | https://github.com/taoliu/MACS/ |
| ChIPSeeker (v1.14.2) | Bioconductor | http://www.bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| FlowJo Software | FlowJo | https://www.flowjo.com/ |
Resource availability
Lead contact
Further information and requests for reagents will be fulfilled by the lead contact Menghui Yin (yin_menghui@gibh.ac.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Tissue samples collection
Lung cancer tissues and matched non-cancerous lung tissues were collected from The First Affiliated Hospital of Guangzhou Medical University (Guangzhou, China). 77 lung cancer tissues and respective non-cancer lung tissues were obtained in 2008 with informed consent and Institutional Review Board permission. All clinical and biological data were available for the samples. Samples were immediately snap-frozen in liquid nitrogen and stored in liquid nitrogen until use.
Cell lines and cell culture
Lung cancer cell lines A549, H1299, H460, H358, H661, H23, H1975, HCC827, HLF and MRC5 were purchased from the American Type Culture Collection (ATCC). HeLa and HEK293 cells were purchased from ATCC. The lung cancer cell line 95D and PC9 were a kind gift from Prof. Duanqing Pei’s lab at GIBH. The cell lines were tested for potential mycoplasma contamination and were confirmed to be mycoplasma negative. HeLa and HEK293 cells were cultured in DMEM supplemented with 10% foetal bovine serum (FBS), 100 ng/ml streptomycin and 100 U/ml penicillin, 1x Glutamax and Normocin at 37°C with 5% CO2 in a cell culture incubator. A549, H1299, H460, H358, H661, H23, H1975, HCC827, PC9 and 95D cells were cultured in PRIM1640 medium supplemented with 10% FBS, 100 ng/ml streptomycin and 100 U/ml penicillin, 1x Glutamax and Normocin at 37°C with 5% CO2.
siRNA and miRNA mimics
siRNAs targeting HNRNPA2B1 (si-A2B1), DHX9 (si-DHX9), CDK6 (si-CDK6) and No target control (NC) were provided by Guangzhou RiboBio. miRNAs mimics, including miR-506, miR-34 and miR-195 were provided by Guangzhou RiboBio.
Method details
RNA interference in cells
The cells were seeded at 60,000 cells per well in a six-well plate 24 hours before transfection. The siRNA transfection (50 nM, mixture of three individual siRNA) and co-transfection of siRNA and miRNA mimics (50 nM) were performed using Lipofectamine 2000 (11668019, Thermo Fisher) according to the manufacturer’s protocol. Forty-eight hours after transfection, cells were collected for further analysis, including analysis of RNA and protein expression levels. Ten μg of GFP-Flag or HNRNPA2B1-Flagplasmids were transfected using Lipofectamine 2000 (11668019, Thermo Fisher) according to the manufacturer’s protocol. Cells were harvested for protein analysis and immunoprecipitation (IP) experiments forty-eight hours after plasmid transfection.
RT-qPCR for mRNA and miRNA
Total RNA of treated cell were extracted with trizol methods. In cDNA syntheses step, 2 ug of total RNA was used for first strand cDNA synthesis using cDNA Moloney Murine Leukemia Virus Reverse Transcriptase (M1705, Promega). 1:20 dilution of cDNA was used to perform RT-qPCR analysis using SYBR Green Supermix (LS2062, Promega) on the Bio-Rad CFX96 Real-Time PCR system. Beta-Actin RNA was used as a loading control and fold change was calculated using the 2(−ΔΔ) method. All primers used for RT-qPCR are listed in Table S3.
Western blot (WB)
Cells were lysed in RIPA lysis buffer containing 1x protease cocktail and 1mM DTT. Protein quantification was measured using BCA Protein Assay. 20μg of total protein from each sample was loaded on a 4–12% Bis-TrisNuPage gel (NP0335BOX, Thermo Fisher) and transferred to 0.22 μM PVDF membrane (ISEQ00010, Millipore) at condition of 10 volt, time 60 minutes. Membrane was blocked in 5% non-fat milk for 1 hour at room temperature. After blocking, blots were probed with specific primary antibody overnight at 4°C. After 3 times washes with TBST, their corresponding secondary antibodies were incubated for 2 hours each. After washing secondary antibodies with TBST for 3 times, membranes were blotted under standard conditions using ECL western blotting Kit (32106, Thermo Scientific Pierce). Antibodies used in this study are: HNRNPA2B1 (sc-374053, Santacruz), AGO2 (ab186733, Abcam) and DHX9 (sc-137232, Santa Cruz), CDK6 (sc-177, Santa Cruz), and Beta-Actin (sc-47778, Santa Cruz). β-Actin was used as a reference gene for each western blot. Band densities were quantified using ImageJ.
Luciferase activity assay
Cells transfected with NC, miR-506, miR-506 Antagomir (A-506) or siRNA were transfected with 200ng of the psi-Check2-CDK6 3′UTR-Fluc plasmid. Forty-eight hours after transfection. Luciferase assays were performed using the Dual-Glo Luciferase activity system according to the manufacturer’s protocol. In brief, Forty-eight hours after transfection, luciferase activity was detected using the Dual-Glo luciferase assay kit (E1980, Promega). Luminescence intensity was read with a Microplate Luminometer according to the Promega protocol. Each experiment was performed with three technical replicates, and three biological replicates were carried out for each sample.
5-Ethynyl-2′-deoxyuridine (EdU) proliferation assay
The EdU proliferation assay was used to test lung cancer cell proliferation according to the Kit protocol (Cell Light EdU DNA imaging Kit, Guangzhou RiboBio, China). In brief, A549 and 95D cells were transfected with miRNA mimics in 96-well plates. Forty-eight hours after transfection, 50 mM 5-ethynyl-2′-deoxyuridine (EdU) was added and the cells were cultured for an additional 2 hours. The cells were then stained according to the Kit protocol: discard the EdU medium mixture, wash cells with PBS, add 4% paraformaldehyde (PFA) to fix cells at room temperature for 30 minutes, wash with glycine (2 mg/ml) for 5 minutes in a shaker, add 0.2% Trion X-100 for 10 minutes, wash with PBS for two times, add click reaction buffer (Tris–HCl, pH 8.5, 100mM; CuSO4, 1mM; Apollo 550 fluorescent azide,100 mM; ascorbic acid, 100mM) for 30 minutes while protecting from light, wash with 0.5% Triton X-100 for three times, stain with DAPI (0.5 mg/L) for 30 minutes at room temperature, wash with 0.5% Triton X-100 for five times, and finally add 150 ul PBS. Images were taken and analyzed using High Content Imaging Pathway 855 (BD, USA). EdU positive cells were calculated with EdU% = (EdU-add-in cells/Hoechst stained cells)∗ 100%.
Cell cycle analysis
H1299 and 95D cells were seeded in a six-well plate and miRNA mimics or siRNA transfections were performed as described above. Forty-eight hours after siRNA transfection, cells were gently washed once in PBS, making sure that no cells were discarded during media removal or washing. After trypsinization, cells were fixed in 66% ice-cold ethanol at 4°C for at least two hours. Propidiumiodide (PI) DNA stained cells were analyized with fluorescence-activated cell sorting on a BD Accuri C6 Plus flow cytometer. Percentage of cells in each cell cycle phase was calculated using volume of respective peak (2N: G0/G1; 2N-4N: S; > 4N: G2/M).
Cell counting Kit-8 assay kit
Cell Counting Kit-8 (CCK-8) assay kit (Dojindo, Kumamoto, Japan) was used to evaluate the viability of lung cancer cell lines after treatment with miR-506 mimics and NC. Cells were transfected with miRNA mimics or siRNAs in 96-well plates. A final concentration of 50 nM mimics was used. At 72 h after transfection, cell viability was measured according to manufacturer’s instructions.
RNA FISH combined with immunofluorescence
The culture of 95D cells took place on coverslips, and then the cells were fixed with 4% paraformaldehyde (PFA). The PFA was subsequently quenched by incubating with 25 mM glycine in PBS for 15 minutes. Cells were then washed twice with PBS and permeabilized with 0.5% (v/v) Triton X-100/PBS for 5 minutes at room temperature, which was followed by three washes with PBS and incubation with prehybridization solution 2xSCC, 10% (v/v) formamide (Sigma) in PBS for 5 minutes at room temperature. Coverslips were then placed cell-side down to ensure that samples were completely immersed in 50 mL of hybridization solution 2 x SSC, 10% (v/v) formamide, 10% (w/v) dextran sulfate, 0.5% (w/v) BSA in PBS containing 250 nM CDK6 3′UTR or negative control (LacZ) Cy3-labelled FISH probes (Generay biotechnology) and HNRNPA2B1 antibodies (diluted at 1/300, Santa Cruz) inside of a humidified Petri dish for 4 hours at 37°C. After hybridization, coverslips were placed back into a 12-well plate, and washing was performed in prehybridization solution. Then, coverslips were incubated with a secondary antibody (goat anti-rabbit-Alexa488, Life Technologies) for 30 minutes at 37°C. Next, the cells were washed again in prehybridization solution for 30 minutes at room temperature. The prehybridization solution was washed three times with PBS for 5 minutes each time. Cells were stained with DAPI (0.5 mg/L), and coverslips were mounted on glass slides and imaged using a Leica Biosystem.
Enzymatic probing of RNA structural change
In vitro transcribed CDK6 3′UTR RNA (10 μg) was folded and incubated with GFP or HNRNPA2B1 at 37 °C for 15 minutes. Then, the RNA was digested with 0.005 U/ml RNaseT1 at 37°C for 15 minutes. The RNA was extracted using TRIzol reagent (15596026, Thermo Fisher), and then the small RNA fraction was enriched using GlycoBlue coprecipitant (AM9516, Thermo Fisher). The RNA was used to perform RT-PCR. The sequences of the RT primers are listed in Table S3.
Co-immunoprecipitation
Cells were lysed with lysis buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1mM EDTA, 0.5% TritonX-100, and protease inhibitors). The lysate were homogenized using a syringe with a 0.4 mm diameter needle and centrifuged at 12000g speed for 10 minutes at 4°C. Supernatants were incubated with HNRNPA2B1 antibody (sc-374053, Santacruz), AGO2 antibody (ab186733, Abcam) and DHX9 antibody (sc-137232, Santacruz) for 2 hours. The protein G beads (10004D, Thermo Fisher) were added to precipitate target protein complex with antibody Then immunoprecipitates were washed twice with low-salt wash buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 0.02%NP-40) and high-salt wash buffer (50 mM Tris-HCl pH7.4, 300 mM NaCl, 0.02%NP-40), respectively. For RNase treatment, samples were treated with 1/1000 RNase A/T1 (EN0551, Thermo Fisher) for 15 minutes at 37°C. After co-IP, samples were tested with western blot using the following antibodies: HNRNPA2B1 antibody (sc-374053, Santacruz), AGO2 antibody (ab186733, Abcam) and DHX9 antibody (sc-137232, Santacruz).
RNA pull-down assay
The CDK6 3′UTR wild type and HNRNPA2B1 binding site mutant sequences were transcribed in vitro with a biotin RNA Labeling Mix (11685597910, Roche) using T7 transcript KIT (TSK-101B, TOYOBO). The in vitro-transcribed RNAs were treated with RNase-free DNase I (18068015, Thermo Fisher) and purified with TRIzol reagent (1559602, Thermo Fisher). One milligram of whole-cell lysate from 95D cells was incubated with 10 pM of folded transcripts for 1 hour at 4°C; complexes were isolated with streptavidin Dynabeads beads C1 (65002, Thermo Fisher). The RNA present in the pull-down fractions was analysed by RT-PCR analysis, and the protein was assessed by western blot analysis.
RNA immune-precipitation (RIP) PCR
For exogenous expression proteins RIP assay, pENTER-GFP-Flag and pENTER-HNRNPA2B1-Flag were transfected into 95D cells. Forty-eight hours after transfection, 2 × 107cells were used to perform RIP experiments using a Flag antibody coated M2 beads (M8823, Sigma). In brief, cells were harvested and suspended with lysis buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.0, 0.5% NP-40, and 1 mM DTT) supplemented with RNase inhibitors (EO0381, Thermo Fisher) and protease inhibitors (P8340, Sigma). Samples were homogenized using a syringe with a 0.4 mm diameter needle and centrifuged at 12000g for 10 minutes at 4°C. The lysate supernatants were incubated with M2 beads for 2 hours at 4°C. The beads were washed five times with NT2 buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1mM MgCl2, and 0.05% NP-40 supplemented with 1 mM DTT and 2 mM EDTA) supplemented with RNase inhibitors (EO0381, Thermo Fisher) and protease inhibitors (P8340, Sigma). Afterwards, the RNA–protein complexes were digested with proteinase K at 55°C for 1h. RNA was purified using TRIzol and analyzed by RT-qPCR. Detailed primers sequence information is provided in Table S3.
For endogenous expression proteins RIP assay, the cells were incubated with lysis buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.0, 0.5% NP-40, and 1 mM DTT) supplemented with RNase inhibitors (EO0381, Thermo Fisher) and protease inhibitors (P8340, Sigma). Then the 50ul protein G beads bound with 5ug HNRNPA2B1 antibody (sc-374053, Santacruz), AGO2 antibody (ab186733, Abcam) and DHX9 antibody (sc-137232, Santacruz), and IgG were added to the cell lysis. The samples were incubated on a rotator at 4°C for 2 hours. Then the beads were washed with NT2 buffer supplemented with RNase inhibitor and protease inhibitor five times at 4°C (5 minutes each wash). Afterwards, the RNA–protein complexes were digested with proteinase K at 55°C for 1 hour. RNA was purified using TRIzol and analyzed by RT-qPCR. Detailed primers information is provided in Table S3.
Quantification and statistical analysis
Western blot analysis
For densitometric western blot analysis, the intensity of each immunoreactive band was analyzed by ImageJ and normalized to the optical density of the corresponding Beta-Actin bands.
Statistical analysis
In most cases, datas obtained from three biological replicates were analyzed, unless indicated otherwise in the Figure legends. Comparisons of multiple groups were performed using 1-way ANOVA followed by Tukey’s multiple comparisons test. Comparisons between 2 groups were performed using the student’s t test. All statistical calculations are included in the relevant figure legends. P values less than 0.05 were accepted as significant, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Bioinformatics analysis
For CLIP-seq and FLASH-seq analysis, aligned reads were obtained from GSE89276 and GSE70061. Peaks were called using MACS2 (v2.1.0) with the settings “--keep-dup 1 -q 0.05 –nomodel” and were annotated using ChIPSeeker (v1.14.2). Peaks were counted if reads were localized in the 3′UTR of mRNA genes.
Acknowledgments
We thank Dr Jingsong Liu for help and suggestion in manuscript revision. We thank Shahzina Kanwal and Muzammal Hussain for assistance in manuscript modification. We thank reading and commenting the manuscript by Dr XichenBao. This work was supported by grants from National Key Research and Development Program (2017YFA0504104); National Natural Science Foundation of China (81871881 and 81401909 to M.Y., 91440115 to B.Z.); National Science and Technology Major Project [2018ZX09733006 to B.Z.]; Science and Technology Program of Guangzhou, China. Grant [No. 201904010464]; and Youth grant from State Key Laboratory of Respiratory Disease (to M. Y. [SKLRD-QN-201718] and C.L. [SKLRD-QN-201905]).
Author contributions
M.Y. and B.Z. conceptualized and supervised the study. M.Y. designed the experiments. M.Y., C.L., and M.C. conducted most of the experiments and data analysis. M.C. mainly completed the experiment of revisions. W.X. and J.F. carried out statistics and data analysis. M.Y., Y.L., and B.Z. wrote the manuscript with input from all other authors.
Declaration of interests
The authors report no competing interests.
Inclusion and diversity
We worked to ensure diversity in experimental samples through the selection of the cell lines. We worked to ensure diversity in experimental samples through the selection of the genomic datasets.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103345.
Contributor Information
Menghui Yin, Email: yin_menghui@gibh.ac.cn.
Biliang Zhang, Email: zhang_biliang@gibh.ac.cn.
Supplemental information
Data and code availability
The datas of proteins MS analysis can be found in Tables S1 and S2.
This study did not generate codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas T., Ilik I.A., Maticzka D., Bhardwaj V., Rodrigues C.P., Mittler G., Manke T., Backofen R., Akhtar A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 2017;544:115. doi: 10.1038/nature21715. [DOI] [PubMed] [Google Scholar]
- Alarcon C.R., Goodarzi H., Lee H., Liu X.H., Tavazoie S., Tavazoie S.F. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X.C., Guo X.P., Yin M.H., Tariq M., Lai Y.W., Kanwal S., Zhou J.J., Li N., Lv Y., Pulido-Quetglas C., Wang X.W. Capturing the interactome of newly transcribed RNA. Nat. Methods. 2018;15:213. doi: 10.1038/nmeth.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin G.A., Croce C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Clower C.V., Chatterjee D., Wang Z.X., Cantley L.C., Heiden M.G.V., Krainer A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. U. S. A. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.Q., Wu F., Sun Y.L., Fan G.C., wang Q.M. Up-regulation and subcellular localization of hnRNP A2/B1 in the development of hepatocellular carcinoma. BMC Cancer. 2010;10:356. doi: 10.1186/1471-2407-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C.J., Chen M., Assanah M., Canoll P., Manley J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–U114. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcheva I., Goswami S., Noubissi F.K., Spiegelman V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell. 2009;35:240–246. doi: 10.1016/j.molcel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Feng S.P., Cong S.J., Zhang X., Bao X.C., Wang W., Li H.P., Wang Z., Wang G.X., Xu J.Z., Du B.W., et al. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucleic Acids Res. 2011;39:6669–6678. doi: 10.1093/nar/gkr232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R., Marcucci G., Croce C.M. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler R., Simkin A., Floss D., Patel R., Fogarty E.A., Scheller J., Grimson A. A widespread sequence-specific mRNA decay pathway mediated by hnRNPs A1 and A2/B1. Genes Dev. 2016;30:1070–1085. doi: 10.1101/gad.277392.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan-Gerstl R., Cohen M., Shilo A., Suh S.S., Bakacs A., Coppola L., Karni R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71:4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- Grimson A., Farh K.K.H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.P., Tang Y.H., Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem. J. 2010;430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- Hossian A., Sajib M.S., Tullar P.E., Mikelis C.M., Mattheolabakis G. Multipronged activity of combinatorial miR-143 and miR-506 inhibits lung cancer cell cycle progression and angiogenesis in vitro. Sci. Rep. 2018;8:10495. doi: 10.1038/s41598-018-28872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem. Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedde M., Strasser M.J., Boldajipour B., Vrielink J.A.F.O., Le Sage C., Nagel R., Voorhoeve P.M., van Duijse J., Orom U.A., Lund A.H., et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell. 2007;131:1273–1286. doi: 10.1016/j.cell.2007.11.034. [DOI] [PubMed] [Google Scholar]
- Kedde M., van Kouwenhove M., Zwart W., Vrielink J.A.F.O., Elkon R., Agami R. A Pumilio-induced RNA structure switch in p27-3 ' UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- Kenny P.J., Zhou H.J., Kim M., Skariah G., Khetani R.S., Drnevich J., Arcila M.L., Kosik K.S., Ceman S. MOV10 and FMRP regulate AGO2 association with MicroRNA recognition elements. Cell Rep. 2014;9:1729–1741. doi: 10.1016/j.celrep.2014.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Koh H.R., Xing L., Kleiman L., Myong S. Repetitive RNA unwinding by RNA helicase A facilitates RNA annealing. Nucleic Acids Res. 2014;42:8556–8564. doi: 10.1093/nar/gku523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H., Fujiya M., Ueno N., Moriichi K., Sasajima J., Ikuta K., Tanabe H., Tanaka H., Kohgo Y. microRNA-26a and -584 inhibit the colorectal cancer progression through inhibition of the binding of hnRNP A1-CDK6 mRNA. Biochem. Biophys. Res. Commun. 2015;467:847–852. doi: 10.1016/j.bbrc.2015.10.055. [DOI] [PubMed] [Google Scholar]
- Kundu P., Fabian M.R., Sonenberg N., Bhattacharyya S.N., Filipowicz W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res. 2012;40:5088–5100. doi: 10.1093/nar/gks148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Wei H.T., Zhang H.F., Xu F., Che G.W. Circ_100565 promotes proliferation, migration and invasion in non-small cell lung cancer through upregulating HMGA2 via sponging miR-506-3p. Cancer Cell Int. 2020;20:160. doi: 10.1186/s12935-020-01241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Sun Y., Ji P., Li X., Cogdell D., Yang D., Parker Kerrigan B.C., Shmulevich I., Chen K., Sood A.K., et al. MiR-506 suppresses proliferation and induces senescence by directly targeting the CDK4/6-FOXM1 axis in ovarian cancer. J. Pathol. 2014;233:308–318. doi: 10.1002/path.4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane L.A., Murphy P.R. MicroRNA: biogenesis, function and role in cancer. Curr. Genomics. 2010;11:537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R., Cloutier P., Shkreta L., Fisette J.F., Revil T., Chabot B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- Matoulkova E., Michalova E., Vojtesek B., Hrstka R. The role of the 3 ' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. Rna Biol. 2012;9:563–576. doi: 10.4161/rna.20231. [DOI] [PubMed] [Google Scholar]
- Mayr C. Evolution and biological roles of alternative 3 ' UTRs. Trends Cell Biol. 2016;26:227–237. doi: 10.1016/j.tcb.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckay S.J., Cooke H. Hnrnp A2/B1 binds specifically to single stranded vertebrate telomeric repeat Ttagggn. Nucleic Acids Res. 1992;20:6461–6464. doi: 10.1093/nar/20.24.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S., Buchwald G., Falk S., Chakrabarti S., Prabu J.R., Conti E. The conformational plasticity of eukaryotic RNA-dependent ATPases. Febs J. 2015;282:850–863. doi: 10.1111/febs.13198. [DOI] [PubMed] [Google Scholar]
- Pasquinelli A.E. Non-coding RNA MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012;13:271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- Pino I., Pio R., Toledo G., Zabalegui N., Vicent S., Rey N., Lozano M.D., Torre W., Garcia-Foncillas J., Montuenga L.M. Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer. 2003;41:131–143. doi: 10.1016/s0169-5002(03)00193-4. [DOI] [PubMed] [Google Scholar]
- Qu J., Zhao L., Zhang P.Z., Wang J., Xu N., Mi W.J., Jiang X.W., Zhang C.M., Qu J. MicroRNA-195 chemosensitizes colon cancer cells to the chemotherapeutic drug doxorubicin by targeting the first binding site of BCL2L2 mRNA. J. Cell Physiol. 2015;230:535–545. doi: 10.1002/jcp.24366. [DOI] [PubMed] [Google Scholar]
- Ray P.S., Jia J., Yao P., Majumder M., Hatzoglou M., Fox P.L. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.S., Yin M.H., Zhang X., Wang Z., Feng S.P., Wang G.X., Luo Y.J., Liang P.Z., Yang X.Q., He J.X., Zhang B.L. Tumor-suppressive microRNA-449a induces growth arrest and senescence by targeting E2F3 in human lung cancer cells. Cancer Lett. 2014;344:195–203. doi: 10.1016/j.canlet.2013.10.031. [DOI] [PubMed] [Google Scholar]
- Robb G.B., Rana T.M. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol. Cell. 2007;26:523–537. doi: 10.1016/j.molcel.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Serganov A., Patel D.J. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu. Rev. Biophys. 2012;41:343–370. doi: 10.1146/annurev-biophys-101211-113224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan K.E., Bohnsack M.T. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem. Sci. 2018;43:237–250. doi: 10.1016/j.tibs.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Stockley J., Villasevil M.E.M., Nixon C., Ahmad I., Leung H.Y., Rajan P. The RNA-binding protein hnRNPA2 regulates beta-catenin protein expression and is overexpressed in prostate cancer. RNA Biol. 2014;11:755–765. doi: 10.4161/rna.28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F., Fu H.J., Liu Q., Tie Y., Zhu J., Xing R.Y., Sun Z.X., Zheng X.F. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. Febs Lett. 2008;582:1564–1568. doi: 10.1016/j.febslet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- Tauler J., ZUDAIRE E., Liu H.T., Shih J., Mulshine J.L. hnRNP A2/B1 modulates epithelial-mesenchymal transition in lung cancer cell lines. Cancer Res. 2010;70:7137–7147. doi: 10.1158/0008-5472.CAN-10-0860. [DOI] [PubMed] [Google Scholar]
- Torre L.A., Siegel R.L., Jemal A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- Villarroya-Beltri C., Gutierrez-Vazquez C., Sanchez-Cabo F., Perez-Hernandez D., Vazquez J., Martin-Cofreces N., Martinez-Herrera D.J., Pascual-Montano A., Mittelbrunn M., Sanchez-Madrid F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013;4:1–10. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wen M.Y., CAO X.T. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. 2019;365:656. doi: 10.1126/science.aav0758. [DOI] [PubMed] [Google Scholar]
- Wang Y.L., Liu J.Y., Yang J.E., Yu X.M., Chen Z.L., Chen Y.J., Kuang M., Zhu Y., Zhuang S.M. Lnc-UCID promotes G1/S transition and hepatoma growth by preventing DHX9-mediated CDK6 down-regulation. Hepatology. 2019;70:259–275. doi: 10.1002/hep.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins J.F., Ruffino L., Kelnar K., Omotola M., Patrawala L., Brown D., Bader A.G. Development of a lung cancer therapeutic based on the tumor suppressor MicroRNA-34. Cancer Res. 2010;70:5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler W.C., Breaker R.R. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- Wu B.X., Su S.C., Patil D.P., Liu H.H., Gan J.H., Jaffrey S.R., Ma J.B. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 2018;9:1–12. doi: 10.1038/s41467-017-02770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X.Y., Yu Y.J., Ye F., Peng G.Y., Li Y.J., Zhou X.M. MicroRNA-506-3p inhibits proliferation and promotes apoptosis in ovarian cancer cell via targeting SIRT1/AKT/FOXO3a signaling pathway. Neoplasma. 2020;67:344–353. doi: 10.4149/neo_2020_190517N441. [DOI] [PubMed] [Google Scholar]
- Xu T., Zhu Y., Xiong Y., Ge Y.Y., Yun J.P., Zhuang S.M. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology. 2009;50:113–121. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- Xu W.L., Gong F.C., Zhang T., Chi B.R., Wang J.Y. RNA-binding protein Dnd1 inhibits epithelial-mesenchymal transition and cancer stem cell-related traits on hepatocellular carcinoma cells. Biotechnol. Lett. 2017;39:1359–1367. doi: 10.1007/s10529-017-2375-5. [DOI] [PubMed] [Google Scholar]
- Yin M., Ren X., Zhang X., Luo Y., Wang G., Huang K., Feng S., Bao X., Huang K., He X., et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappa B p65 to evoke reactive oxygen species generation and p53 activation. Oncogene. 2015;34:691–703. doi: 10.1038/onc.2013.597. [DOI] [PubMed] [Google Scholar]
- Zhang Z.C., Ma J., Luan G., Kang L., Su Y.H., He Y.N., Luan F. MiR-506 suppresses tumor proliferation and invasion by targeting FOXQ1 in nasopharyngeal carcinoma. PLoS One. 2015;10:e0122851. doi: 10.1371/journal.pone.0122851. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhou J., Allred D.C., Avis I., Martinez A., Vos M.D., Smith L., Treston A.M., Mulshine J.L. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 2001;66:217–224. doi: 10.1023/a:1010631915831. [DOI] [PubMed] [Google Scholar]
- Zhu J.F., tao L.Q., Jin L.T. MicroRNA-506-3p reverses gefitinib resistance in non-small cell lung cancer by targeting Yes-associated protein 1. Mol. Med. Rep. 2019;19:1331–1339. doi: 10.3892/mmr.2018.9710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datas of proteins MS analysis can be found in Tables S1 and S2.
This study did not generate codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.