

SUMMARY
Cortical spreading depolarizations (CSDs) are increasingly suspected to play an exacerbating role in a range of acute brain injuries, including stroke, possibly through their interactions with cortical blood flow. We use simultaneous wide-field imaging of neural activity and hemodynamics in Thy1-GCaMP6f mice to explore the neurovascular dynamics of CSDs during and following Rose Bengal-mediated photothrombosis. CSDs are observed in all mice as slow-moving waves of GCaMP fluorescence extending far beyond the photothrombotic area. Initial CSDs are accompanied by profound vasoconstriction and leave residual oligemia and ischemia in their wake. Later, CSDs evoke variable responses, from constriction to biphasic to vasodilation. However, CSD-evoked vasoconstriction is found to be more likely during rapid, high-amplitude CSDs in regions with stronger oligemia and ischemia, which, in turn, worsens after each repeated CSD. This feedback loop may explain the variable but potentially devastating effects of CSDs in the context of acute brain injury.
Graphical abstract
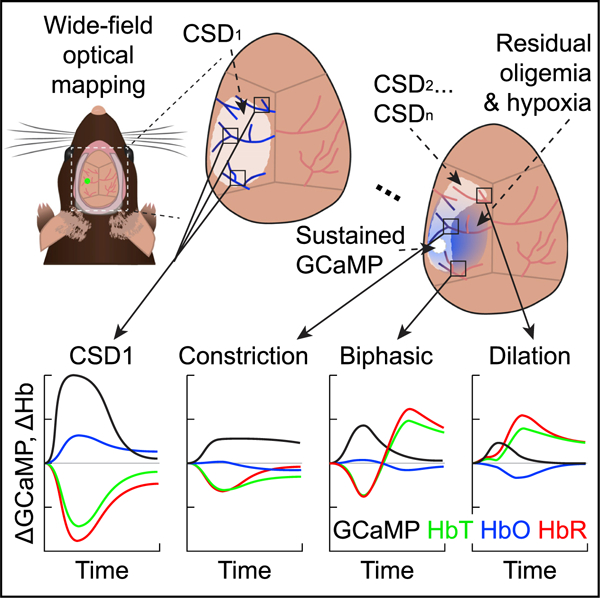
In brief
Zhao et al. use wide-field optical mapping of neuronal and hemodynamic activity in mice, capturing CSDs immediately following photothrombosis. Initial CSDs are accompanied by strong vasoconstriction, leaving persistent oligemia and ischemia. Region-dependent neurovascular responses to subsequent CSDs demonstrate a potential vicious cycle of CSD-dependent damage in acute brain injury.
INTRODUCTION
Cortical spreading depolarizations (CSDs) are known to occur across species and consist of a slow-moving wave of near-complete neuronal depolarization followed by electrical silencing (Fabricius et al., 2006; Koroleva and Bures, 1996; Leão, 1944; Shin et al., 2006; Strong et al., 1983). CSDs have been shown to occur during migraine with aura (Hadjikhani et al., 2001; Lauritzen, 1994; Lauritzen et al., 1983; Major et al., 2020; Olesen et al., 1981; Pietrobon and Moskowitz, 2014) and appear to be relatively benign in otherwise well-perfused, uninjured brains. However, CSDs have also been observed following acute brain injury, such as subarachnoid hemorrhage (Dreier et al., 1998; Hubschmann and Kornhauser, 1980, 1982), traumatic brain injury, and ischemic stroke (Dohmen et al., 2008). Under these pathological conditions, CSDs appear to play a role in injury exacerbation and worsening outcomes (Ayata et al., 2004; Dreier, 2011; Luckl et al., 2009; Murphy et al., 2008; Østergaard et al., 2015; Shin et al., 2006; Strong et al., 2002; Yuzawa et al., 2012).
CSDs represent a massive metabolic challenge to the brain and flood the tissue with excess neurotransmitters, neuromodulators, and vasoactive substances such as potassium ions (Chuquet et al., 2007; Hansen and Zeuthen, 1981; Lothman et al., 1975; Phillips and Nicholson, 1979). By themselves, these dramatic shifts in ionic gradients and depletion of tissue resources could mediate CSD-related brain damage (Hashemi et al., 2009; Nedergaard and Hansen, 1988). However, another potential mechanism for damage is the interaction between CSDs and the brain’s vasculature. While some CSDs are accompanied by local vasodilation or hyperemia, which should improve delivery of fresh oxygen and glucose to the brain, CSDs can also be accompanied by vasoconstriction and hypoperfusion (Dreier et al., 1998). Under pathological conditions in particular, CSDs have been reported to induce severe initial vasoconstriction and spreading ischemia (Dreier et al., 1998; Takano et al., 2007). In many cases, CSDs also result in prolonged (1 to 2 h) oligemia, a reduction in cerebral blood volume and flow within the territory of the CSD (Ayata et al., 2004). All of these neurovascular effects could cause a mismatch between tissue energy demand and supply that could both worsen the impact of, and prevent recovery from, the CSD’s metabolic overload (Ayata et al., 2004; Chang et al., 2010; Somjen, 2001). The neurovascular effects of CSDs could thus be major contributors to their cumulative impact on the injured brain. However, the conditions dictating why some CSDs may be more or less likely to exacerbate persistent damage in the brain are not well understood.
One obstacle to understanding the complexity of CSD responses has been the availability of methods to accurately record both the neural and vascular effects of CSDs simultaneously, particularly in the context of acute brain injury. For example, CSDs are often detected using point-sampling electrophysiology (Ayata et al., 2004; Chang et al., 2010; Fabricius et al., 1995; Lückl et al., 2018; Murphy et al., 2008; Østergaard et al., 2015), where a large negative direct current (DC) potential shift and an absence of spiking indicates massive neuronal depolarization and consequently electrical silencing. However, the lack of spatial resolution can limit visualization of the overall shape or propagation pattern of the neural CSD wave. The presence of a CSD is also sometimes inferred from concomitant changes in blood flow, assessed by methods such as speckle flow imaging (Ayata et al., 2004; Chang et al., 2010; Fabricius et al., 1995; Murphy et al., 2008; Østergaard et al., 2015; Strong et al., 2006; Yuzawa et al., 2012). However, to deduce the spatiotemporal pattern of a CSD using this approach, one must have a priori knowledge of how blood flow changes are coupled to the moving CSD, introducing ambiguity and precluding assessment of the true coupling relationship between cortical hemodynamics and the underlying CSD. As a result, we still have an incomplete view of how CSDs initiate, their spatiotemporal spread, the vascular responses they generate, and their cumulative impact on affected brain regions.
Here, we present the application of in vivo neurovascular imaging in mice to examine the occurrence and properties of CSDs immediately following photothrombosis in a small cortical area. We developed a customized implementation of wide-field optical mapping (WFOM) in an intact, thinned-skull mouse model to enable bilateral cortical imaging of both neural activities via genetically encoded fluorescent calcium indicators expressed in excitatory neurons (Thy1-GCaMP6f) and hemoglobin oxygenation dynamics via diffuse reflectance (Ma et al., 2016a). This approach allows for a high-speed, holistic examination of neurovascular changes across the bilateral cortex in individual mice in real time, enabling the assessment of how CSDs interact with both the zone of primary photothrombotic damage and naive cortex beyond the boundaries of the photothrombosis site.
In all mice, we observed a large CSD radiating from the site of photothrombosis, which was accompanied by profound vasoconstriction. Most mice experienced additional CSDs, which were more variable in amplitude, temporal dynamics, and propagation patterns and were accompanied by neurovascular responses ranging from constrictions to biphasic (a constriction followed by a dilation) to pure dilations. The type of cortical neurovascular response elicited was found to depend on the local amplitude and temporal properties of the impinging CSD, as well as on baseline levels of GCaMP and cortical oligemia, which accumulated in the path of prior CSDs. In particular, cortical regions with hypoxic baseline conditions were more likely to exhibit a vasoconstriction response to a traversing CSD. This pattern creates a potential negative feedback loop in which progressing cortical ischemia and oligemia have an increased likelihood of driving further damage.
These interdependencies, combined with the observed variability in CSD types and trajectories, might explain why, in some cases, the cortex can recover from CSDs without harm, while in others the CSDs could trigger a downward spiral of significantly worsening ischemia and metabolic stress. Importantly, many of these CSD-related effects were observed in regions of the cortex that were far beyond the territory of our initial photothrombotic insult. These results suggest that the CSDs, and their neurovascular consequences, could be major contributors to variability and progression in the pathophysiology of ischemic brain damage.
RESULTS
Visualizing the neural and hemodynamic effects of acute photothrombosis
All experiments were performed in urethane-anesthetized or awake female Thy1-GCaMP6f C57BL/6 transgenic mice (Labat-gest and Tomasi, 2013), who were surgically prepared with a thinned-skull cranial window spanning the bilateral dorsal cortex (Figure 1). These mice express the genetically encoded fluorescent calcium indicator GCaMP in layers 2/3 and 5 excitatory neurons. Increases in the fluorescence of the brain of these mice provide a spatially resolved optical readout of increases in neuronal intracellular calcium and thus neuronal depolarization. Changes in the diffuse reflectance of the cortex can also provide a spatially resolved measurement of hemodynamics, with multi-color imaging capturing the oxygen-dependent absorption properties of hemoglobin, permitting mapping of oxy, deoxy, and total hemoglobin concentrations (HbO, HbR, and HbT, respectively). Changes in [HbT] ( = [HbO] + [HbR]) indicate a change in the concentration of red blood cells present in the tissue, which can generally only be modulated by physical dilation and constriction of local blood vessels. Changes in [HbO] and [HbR] within vessel compartments result from dilations, constrictions, changes in flow, and changes in the rate of oxygen extraction.
Figure 1. Experimental setup for wide-field optical mapping (WFOM) during photothrombosis.
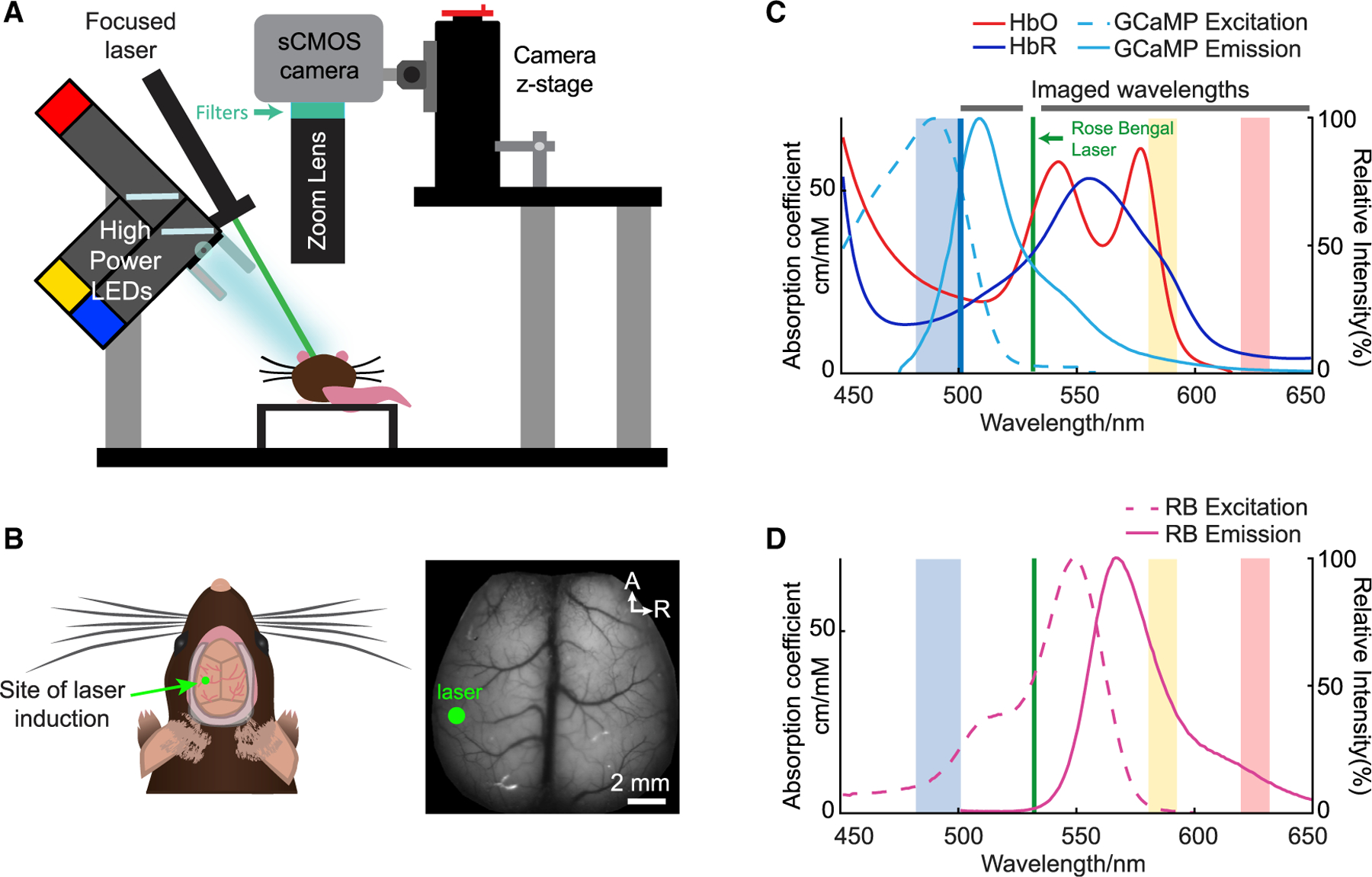
(A) Setup of WFOM imaging system. Blue, amber, and red strobed LEDs sequentially illuminate the cortex, synchronized with frames acquired by an overhead sCMOS camera. Long-pass and notch filters remove blue excitation light as well as contamination from the green laser used for photothrombosis respectively, while passing green GCaMP emissions and amber and red diffuse reflectance to map hemoglobin dynamics.
(B) Illustration and grayscale image of the bilaterally exposed mouse cortex. Green dot indicates the point of focus of the photothrombosis induction laser.
(C) Spectra of HbO and HbR absorption (left axis) and GCaMP fluorescence excitation and emission (right axis). Faded blue, yellow, and red sections indicate the wavelength bands of the strobed LEDs used during imaging.
(D) Fluorescence excitation and emission spectra of Rose Bengal. Amber and red LEDs were chosen to avoid effects of Rose Bengal absorption on spectroscopic conversion (see also Figure S1) and to allow for strategic removal of green laser contamination.
WFOM can capture changes in both neuronal activity and hemodynamics simultaneously by acquiring interlaced measurements of both fluorescence and multi-spectral reflectance. Raw reflectance measurements were converted to hemodynamics, and raw fluorescence measurements were corrected for hemodynamic cross-talk using methods previously described (Ma et al., 2016a) (see Figures 1 and S1 and STAR Methods). Imaging wavelengths and filters were selected to permit simultaneous photothrombosis via 532-nm laser irradiation of intravascular Rose Bengal. WFOM data were acquired before, during, and up to 2.5 h after photothrombosis, targeting a cortical branch of the middle cerebral artery in 8 mice, denoted as M1–8.
Photothrombosis-induced CSDs occur across all mice
Between 3.7 and 34.1 minutes (on average 18.98 ± 10.2 min; n = 8 mice) after photothrombosis initiation, a wave of increased GCaMP fluorescence was observed to spread radially outward from the induction site in all animals, irrespective of functional or anatomical boundaries (Figure 2A; Video S1). The GCaMP fluorescence signal (Figure 2C) during these events exceeded 50% (n = 8 mice), rising to a peak within 1 minute, and then decreasing back toward baseline in most regions. An additionnal example of neural activity during an initial CSD is shown in Figures S2A–S2C.
Figure 2. Spatial and temporal characterization of neural activity during an initial CSD.
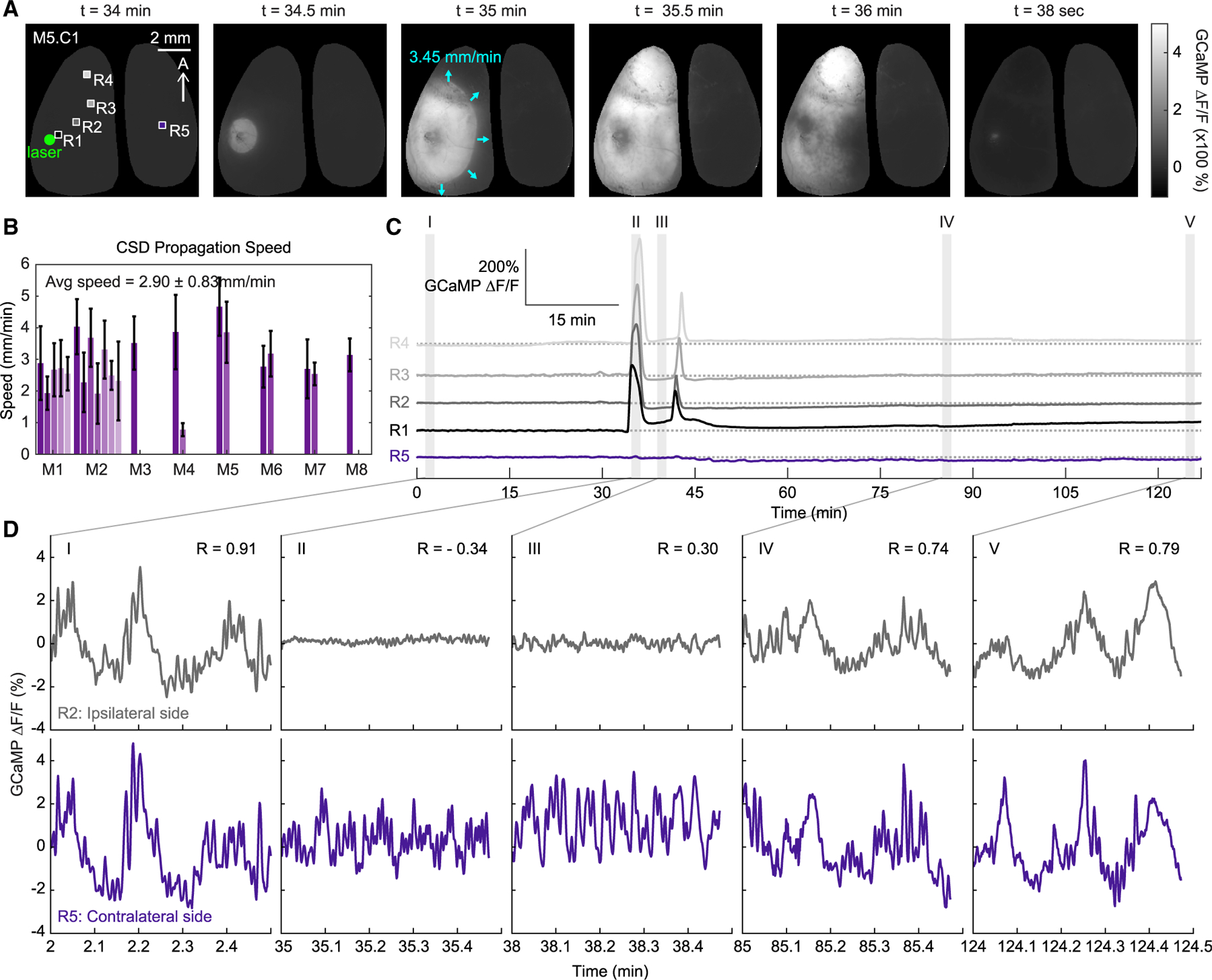
(A) Spatial maps show the propagation of an initial CSD in a representative mouse. Times indicate the amount of time after the laser was turned on (t = 0 minutes). Boxes indicate the regions of interest used for the time courses in this figure.
(B) Average speed of CSD propagation for each CSD across each animal. Error bars show the standard deviation of propagation speeds along the outer edge of each depolarization wave.
(C) Full time courses of GCaMP over more than 2 h of imaging for four regions ipsilateral to the photothrombosis induction site and one contralateral region. Shaded areas show the specific time points along the CSD event highlighted in (D). Note that the first CSD occurs at t = 34.1 minutes and one second CSD occurs at t = 41.2 minutes after initiation of photothrombosis.
(D) 30 s time courses of spontaneous GCaMP activity at each of the shaded time points in (C) for one region (gray) ipsilateral to the photothrombosis site, and one (purple) region on the contralateral side. R values indicate the correlation between the ipsilateral R2 and contralateral R5 time courses. (See also Figure S2 for additional experimental and control examples, and Video S1 for movie of CSD.)
To validate that the spatiotemporal properties of these waves are consistent with prior descriptions of CSDs, we calculated propagation speed for each event across mice. The geodesic distance of each pixel along the wave boundary was assessed in 10 s intervals and averaged for each CSD (Figure 2B). The average speed of initial CSDs was found to be 3.45 ± 0.65 mm/minute, consistent with prior observations of CSDs (Dunn et al., 2001; Farkas et al., 2010; Lauritzen et al., 2011; Leão, 1944). The depolarization wave was largely confined to the ipsilateral cortex, as previously reported (Eikermann-Haerter et al., 2011), although in some mice, a much smaller and delayed GCaMP response on the contralateral side was observed (Figure S2B, inset).
Consistent with prior studies, bilaterally correlated, spontaneous fluctuations in neural activity were seen prior to the CSD (Figure 2DI; R = 0.91) (Ma et al., 2016b). Local fluctuations were abolished at the peak of the CSD, consistent with near-complete depolarization of the labeled neuronal population (see Figure 2D; Video S2). Fluctuations in neural activity in the contralateral cortex appeared altered but continued during the ipsilateral CSD (Figure 2DII). Although spontaneous neuronal activity in the CSD-affected cortex eventually returned, bilateral synchrony remained disrupted up to an hour after the first CSD event (Figure 2DIV). No CSD events or changes in neural fluctuations were observed following laser illumination of saline-injected control mice (Figures S2D–S2F). These findings demonstrate that the effects of the CSD on cortical neuronal activity far outlast the event itself.
Initial CSDs are accompanied by large, spatiotemporally coupled vasoconstrictions
The time courses in Figure 3A′ show the temporal progression of changes in both GCaMP fluorescence and cortical hemodynamics (∆[HbO], ∆[HbR], and ∆[HbT]) beginning when the photothrombosis laser is turned on at t = 0 minutes and ending after the first CSD. A striking feature is the large decrease in cortical [HbT] and [HbO] and the increase in [HbR] occurring concomitantly with the large increase in GCaMP fluorescence. This effect was observed during the initial CSD of every mouse and indicates significant, acute vasoconstriction.
Figure 3. Initial CSD mediates widespread vasoconstriction relative to photothrombosis.
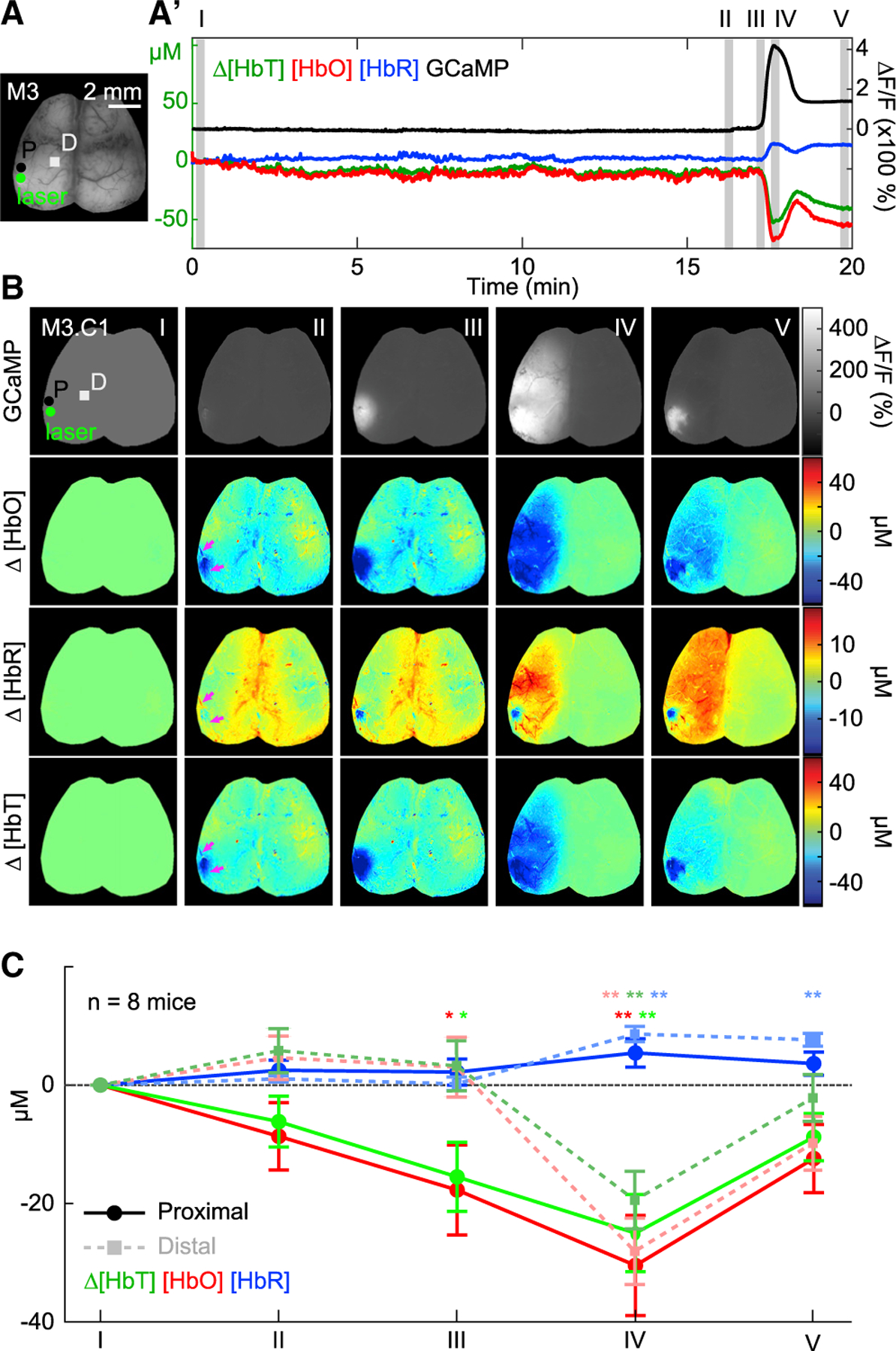
(A and A′) Time courses of fluorescence and hemodynamic changes, starting from laser initiation and ending after the first CSD in one mouse, extracted from the region labeled P in (A).
(B) Maps show GCaMP fluorescence and hemodynamics at five time points: (I) when the laser is turned on, (II) one minute prior to CSD onset, (III) during CSD onset, (IV) peak of CSD, and (V) after recession of CSD when signal gradient returns to zero. Proximal (P) and distal (D) boxes indicate the regions of interest used for hemodynamic analysis in (C).
(C) Average hemodynamic values show a hypoxic trend in regions proximal to the photothrombosis site prior to CSD onset but not in distal regions (epoch II). Once the CSD initiates (epochs III and IV), widespread vasoconstriction, denoted as a decrease in HbT and HbO, and increase in HbR, occurs consistently across all animals. Plot values are expressed as mean ± standard deviation. Significance of hemodynamic deviation from baseline values was determined using Student’s t test (*p < 0.05; **p < 0.01; n = 8 mice).
The increase in GCaMP fluorescence occurs 1.83 s (±0.5 s; n = 8 mice) before changes in diffuse reflectance corresponding to vasoconstriction (Figures S3A and S3B), indicating that vasoconstriction temporally lags the CSD wave. These results confirm that the observed increase in fluorescence is unlikely to be an artifact caused by changes in hemoglobin absorption.
To further confirm the presence of physical vasoconstriction, raw camera images were used to directly measure pial vessel diameters. Pial arteries and veins were identified based on their direction and shape. Cross sections were then used to evaluate their diameter changes during CSD propagation (Figures 4A and 4B). Effects of cortical brightening due to decreased blood flow to the surrounding parenchyma were accounted for by normalizing the cross-section signal to its neighboring non-vessel pixels (Figures S3C–S3E). Consistent with active vasoconstriction, pial arteries in the ipsilateral cortex exhibited a statistically significant decrease in diameter (Student’s t test; **p < 0.001; n = 8 mice), whereas no significant changes in diameter were observed in ipsilateral pial veins or contralateral pial vessels (Figure 4C).
Figure 4. Initial CSDs are accompanied by vessel constriction.
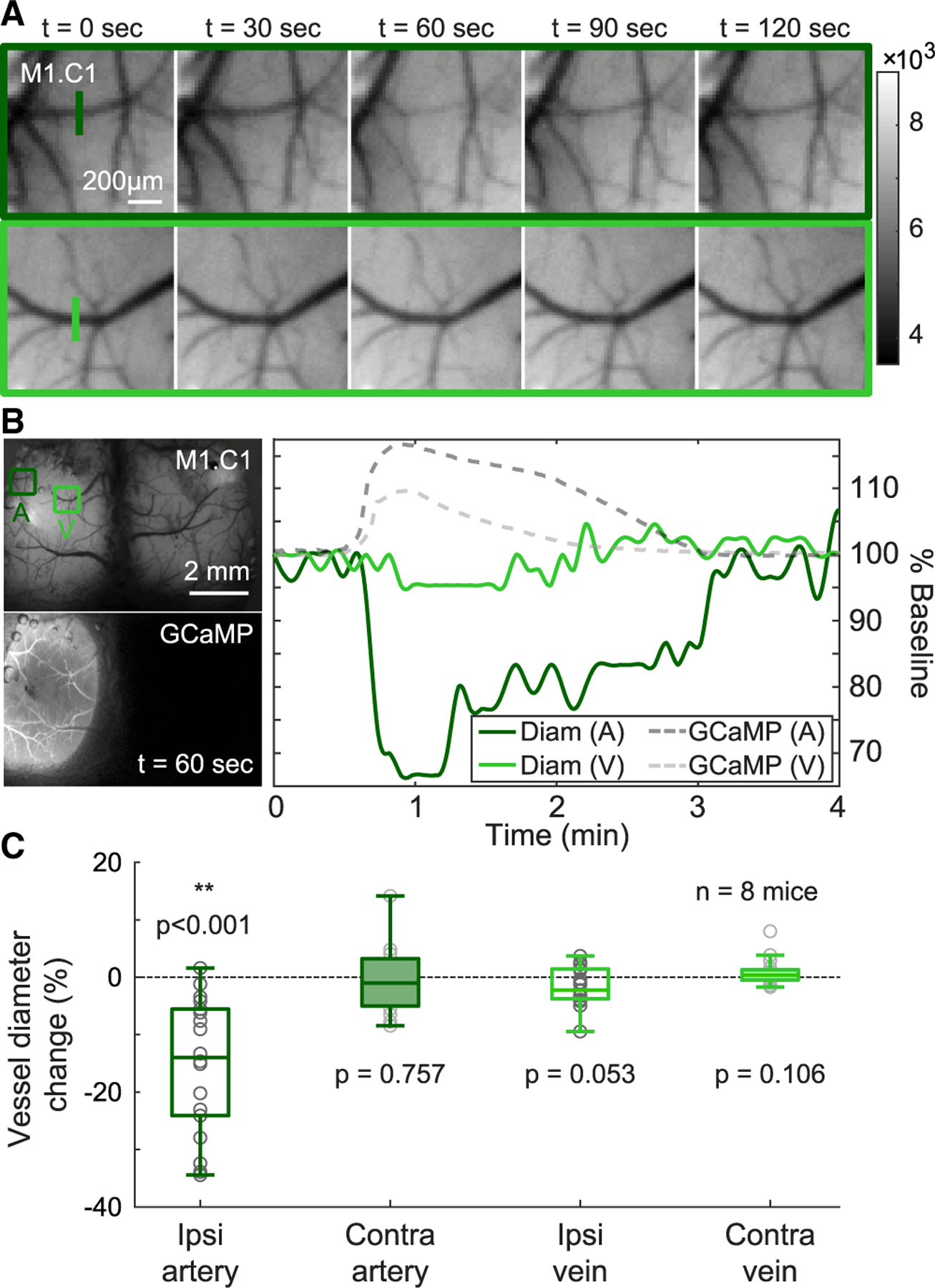
(A) Time-sequence of zoomed-in grayscale maps of a pial artery and vein in the hemisphere ipsilateral to photothrombosis site showing visible arterial constriction (top) and little change in venous diameter (bottom).
(B) Plot shows percentage change in both arterial (A) and venous (V) diameters relative to pre-CSD baseline as a function of time in a representative animal. GCaMP fluorescence shown with dotted gray traces (dark gray for arterial region A, and light gray for venous region V) emphasize that constriction occurs concurrently with the CSD.
(C) Average maximum change in diameters of arteries and veins across the cortex are plotted. Error bars denote standard deviation of the mean. Three arteries and three veins in each hemisphere, totaling twelve vessels, per mouse were evaluated and only ipsilateral arteries exhibit a significant constriction (Student’s t test; **p < 0.001; n = 8 mice). See also Figure S3 and STAR Methods for calculations.
The spatiotemporal evolution of neurovascular changes during the first CSD can be seen in Figure 3B, which shows cortical maps of GCaMP fluorescence, [HbO], [HbR], and [HbT] at the time points indicated in the time-course in Figure 3A′: (I) onset of laser illumination, (II) one minute prior to CSD, (III) during CSD onset, (IV) at the CSD peak, and (V) after recession of CSD. Figure 3C shows average hemodynamic values in cortical regions proximal and distal to where photo-thrombosis was induced across all mice at equivalent time-points (I–V) relative to their first CSD.
The small effect of the photothrombosis itself can be seen at epoch II, indicated by magenta arrows in Figure 3B and quantified for all mice in Figure 3C as a small, local decrease in [HbO] and [HbT] with a small increase in [HbR]. The relatively restricted size of the area affected by primary photothrombosis aligns with previous studies showing the limited effect of single surface vessel occlusion due to the brain’s extensive collateral vascular network (Nishimura et al., 2007; Schaffer et al., 2006). In contrast, upon CSD onset, large decreases in [HbT] and [HbO] (and increases in [HbR]) are observed across both proximal and distal cortical regions (epochs III–IV; Figure 3C). These changes in hemoglobin during the CSD are tightly matched with the leading edge of the neuronal depolarization wave (see Video S1). Together, these results are consistent with “spreading ischemia,” described previously as a spreading depolarization-induced perfusion deficit (Dreier, 2011).
After the CSD, levels of [HbO] and [HbT] remain decreased, while [HbR] remains elevated, characteristic of persistent oligemia or a reduction in local blood volume (epoch V, Figure 3C) (Ayata and Lauritzen, 2015; Ayata et al., 2004). Together, these findings emphasize that CSDs mediate changes in cortical function across the entire cortical hemisphere at a time point that far precedes infarct development (Dreier et al., 2018; Lipton, 1999). Indeed, the very ability to mount a CSD indicates that the tissue is at this moment alive (Dreier et al., 2018). This temporal evolution is paramount as it situates CSDs at a critical time point in which they can potentially shape whether tissue ultimately goes on to recover or die. Furthermore, it emphasizes the complexity of disentangling the consequences mediated by the CSDs from the initial brain injury.
Subsequent CSDs are accompanied by variable hemodynamic responses
While an initial CSD accompanied by vasoconstriction was uniformly observed across all mice, additional CSDs occurred in 6 of the 8 mice. In almost all cases, subsequent CSDs were of lower amplitude than initial ones, as measured by percent change in GCaMP fluorescence (Figures 5A–5D and summarized across mice in Figure S4). There was also significant variability in the spatial spread and hemodynamic response to each of these subsequent CSDs (Figures 2B and 2Di; Video S3).
Figure 5. Heterogeneity in vascular responses to successive CSDs.
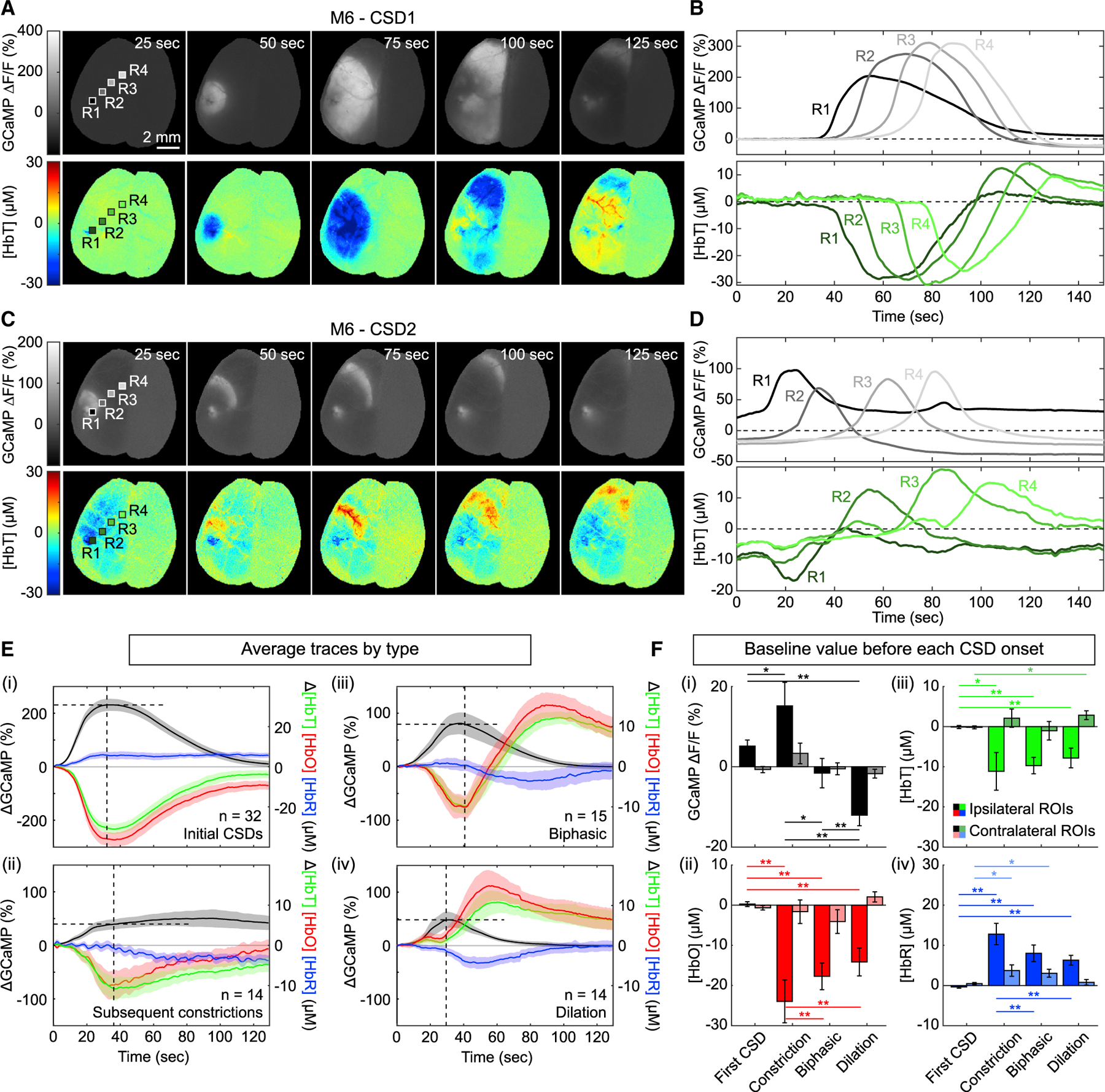
(A and B) Cortical maps (A) and time courses (B) of GCaMP fluorescence (top) and total hemoglobin (bottom) for an initial CSD in one mouse. GCaMP maps highlight the uniform circular pattern of the initial CSD. Total hemoglobin maps and time courses show that all regions across the hemisphere predominantly exhibit intense vasoconstriction in response to an initial CSD.
(C and D) Cortical maps (C) and time courses (D) of GCaMP fluorescence (top) and total hemoglobin (bottom) for a second CSD in the same mouse. In this example, the second CSD propagated outward as a moving arc. Hemoglobin maps and traces show a biphasic hemodynamic response to this CSD. See Video S4 for video of both CSD events and Figure S5 for additional examples.
(E) CSD event-aligned averages of ∆GCaMP, ∆[HbT], ∆[HbO], and ∆[HbR] for all CSDs across all animals. Each trace was subtracted by its respective baseline prior to averaging (see STAR Methods). Traces were categorized into (i) responses to initial CSDs, (ii) subsequent constrictions, (iii) biphasic, and (iv) dilation responses. Shaded area indicates standard error across eight animals (N value indicates the numbers of traces included in each average). Horizontal dotted lines show the peak GCaMP value, while vertical dotted lines show the time of vasodilation onset. On average, the GCaMP peak slightly preceded the onset of vasodilation by 4.15 ± 1.67 s* for the first CSD constriction, 5.67 ± 1.26 s** for biphasic responses, and 0.82 ± 1.00 s for dilation responses (values shown as mean ± SD. Paired t test; *p < 0.05; **p < 0.01).
(F) Bar plots show the averaged non-zeroed starting values for each trace used in (B). Error bars indicate standard error across traces. Saturated colors correspond to ROIs taken from the ipsilateral hemisphere, while faded colors correspond to ROIs taken from the contralateral hemisphere. Paired t tests were used to quantify differences in pre-CSD values of GCaMP ∆F/F, [HbT], [HbO], and [HbR] across first CSDs, constrictive, biphasic, and dilatory vascular responses (Paired t test; *p < 0.05; **p < 0.01).
As an example, Figures 5A–5D compares changes in GCaMP fluorescence and hemodynamics during the first and second CSDs for one mouse. Here, the second CSD propagates as an arc whose wavefront is coincident with an increase in [HbT] (hyperemia) in cortical regions more distant from the site of photothrombosis (Figure 5C; Video S4). Neuronal and hemodynamic time-courses extracted from four different regions of interest (ROIs; extending outward from the site of photothrombosis) reveal a combination of vasoconstriction and vasodilation responses to the passing CSD wave, which itself has a very different spatiotemporal profile to the first CSD wave (Figures 5B and 5D). First and second CSDs observed in two additional mice are shown in Figure S5.
To establish a framework for understanding these widely varying CSDs and neurovascular responses in different mice, we extracted neural and hemodynamic signals from four regions of interest, ranging in distance relative to photothrombosis sites across all animals. To assess neuronal activity in each region, the peak amplitude, onset speed, and duration of increased GCaMP fluorescence for each CSD event were quantified. Region-specific hemodynamic responses to each CSD were classified as either constriction, biphasic (constriction followed by a dilation), or dilation events. Results for all mice are compared in Figure S4. This summary illustrates that a single CSD event can elicit spatially varied neurovascular responses, and that the same cortical region can respond differently over time to successive CSDs.
To compare constriction, biphasic, and dilation response types, all neural and hemodynamic time-courses were aligned to their respective neural CSD onset times, baseline-subtracted relative to the beginning of each CSD, grouped by neurovascular response type, and averaged (Figure 5E). Initial vasoconstriction responses (Figure 5Ei) were averaged separately from subsequent vasoconstrictions (Figure 5Eii), as they have distinct response properties.
These averaged responses reveal that CSD-evoked vasoconstrictions appear to occur in cortical regions where the neuronal depolarization has rapid onset, high amplitude, and/or longer duration as it passes through. All initial CSDs are characterized by this neuronal signature and thus trigger profound vasoconstriction across all animals (Figure 5Ei). Conversely, regions where the CSD has a more gradual onset, reaches a smaller peak, and recovers more quickly are more likely to exhibit some level of vasodilation (Figure 5Eiv).
As a general pattern, we notice that all neurovascular response profiles shown in Figures 5Ei–5Eiv can be explained by the combination of an initial vasoconstriction followed by a vasodilation that begins around the same time at which GCaMP reaches its peak value (i.e., the time at which neural recovery begins, indicated by vertical dotted lines), and peaks around when GCaMP returns to baseline. Careful examination of these inflection points indicates that depolarization recovery precedes the cessation of vasoconstriction by 4.15 ± 1.67 s for the first CSD constriction, 5.67 ± 1.26 s for biphasic responses, and 0.82 ± 1.00 s for dilation responses. This sequence suggests that neuronal recovery is not the result of restored perfusion.
Recognizing this pattern, we see that if a smaller or slower CSD reaches its GCaMP peak quickly, before significant vasoconstriction has occurred, the net hemodynamic response will be a delayed vasodilation (Figure 5Eiv). Meanwhile, both constriction and biphasic events initiate similarly, with vasoconstriction beginning as the GCaMP signal rises (Figures 5Eii and 5Eiii). If GCaMP fluorescence peaks quickly enough, vasodilation results in a hemodynamic overshoot yielding a biphasic response (Figure 5Eiii). However, if GCaMP signals are either very high, as in initial CSDs, or sustained, as in subsequent CSD-induced constrictions, the vasodilation phase does not overcome the hypoperfusion in that region (relative to the pre-CSD baseline level) and hyperemia above baseline is not observed. These findings suggest that a cortical region’s net neurovascular response to a CSD depends on how quickly that region is able to repolarize and recover from the effects of each successive passing CSD.
The patterns above focused on relative changes in neural and hemodynamic signals during a CSD. However, we also noted significant differences in pre-CSD baseline levels for the different CSD neurovascular response types. Figure 5F compares pre-CSD GCaMP and hemodynamic baseline levels for each neurovascular response type in both ipsilateral and contralateral regions. Regions exhibiting vasoconstriction in response to a non-initial CSD show significantly lower levels of initial [HbT] and [HbO] and higher levels of [HbR] relative to pre-initial CSD baseline (Figure 5Fii–5Fiv), while GCaMP fluorescence in these regions was elevated compared to pre-initial CSD baseline (Figure 5Fi). Cortical regions exhibiting biphasic hemodynamic responses to a non-initial CSDs (Figures 5Fii–5Fiv) also showed lower levels of initial [HbT] and [HbO] and higher levels of [HbR], although to a lesser degree than regions that undergo constriction and have relatively unchanged levels of baseline GCaMP fluorescence (Figure 5Fi). Cortical regions exhibiting pure vasodilation (Figure 5Fii–5Fiv) showed the smallest reduction in pre-CSD [HbT] and [HbO] but also had lower GCaMP fluorescence levels than pre-initial CSD levels. For all CSD events, contralateral values were relatively less affected, confirming that the observed differences cannot be accounted for by systemic effects.
These findings indicate that there is heterogeneity in the baseline GCaMP and perfusion levels of the cortex after the initial CSD and that this baseline state is a determining factor in a region’s ability to respond to subsequent CSDs with dilatory, biphasic, or constriction responses.
Spatially heterogeneous sustained effects of subsequent CSDs
The apparent dependence of neurovascular responses to CSDs on baseline cortical conditions prompts the question of how these baseline conditions progress and change with repeated CSD events. WFOM enables spatial mapping of the cumulative effects of CSDs on both neuronal intracellular calcium and cortical hemoglobin levels, thus allowing us to explore this progression.
Figure 6B shows neuronal and hemodynamic time-courses extracted from three regions of interest (indicated in Figure 6A) in a mouse that had 7 CSDs. Figure 6Di depicts the spatiotemporal spread of each successive CSD, while Figure 6Dii–6Dv show the post-CSD levels of GCaMP, [HbT], [HbO], and [HbR] relative to baseline levels before the first CSD. To confirm that these cumulative effects were not due, in part, to prolonged anesthesia or physiological decline, an additional control experiment was performed in an awake mouse, with equivalent analysis shown in Figure S6.
Figure 6. Characterization of the cumulative effects of successive CSDs.
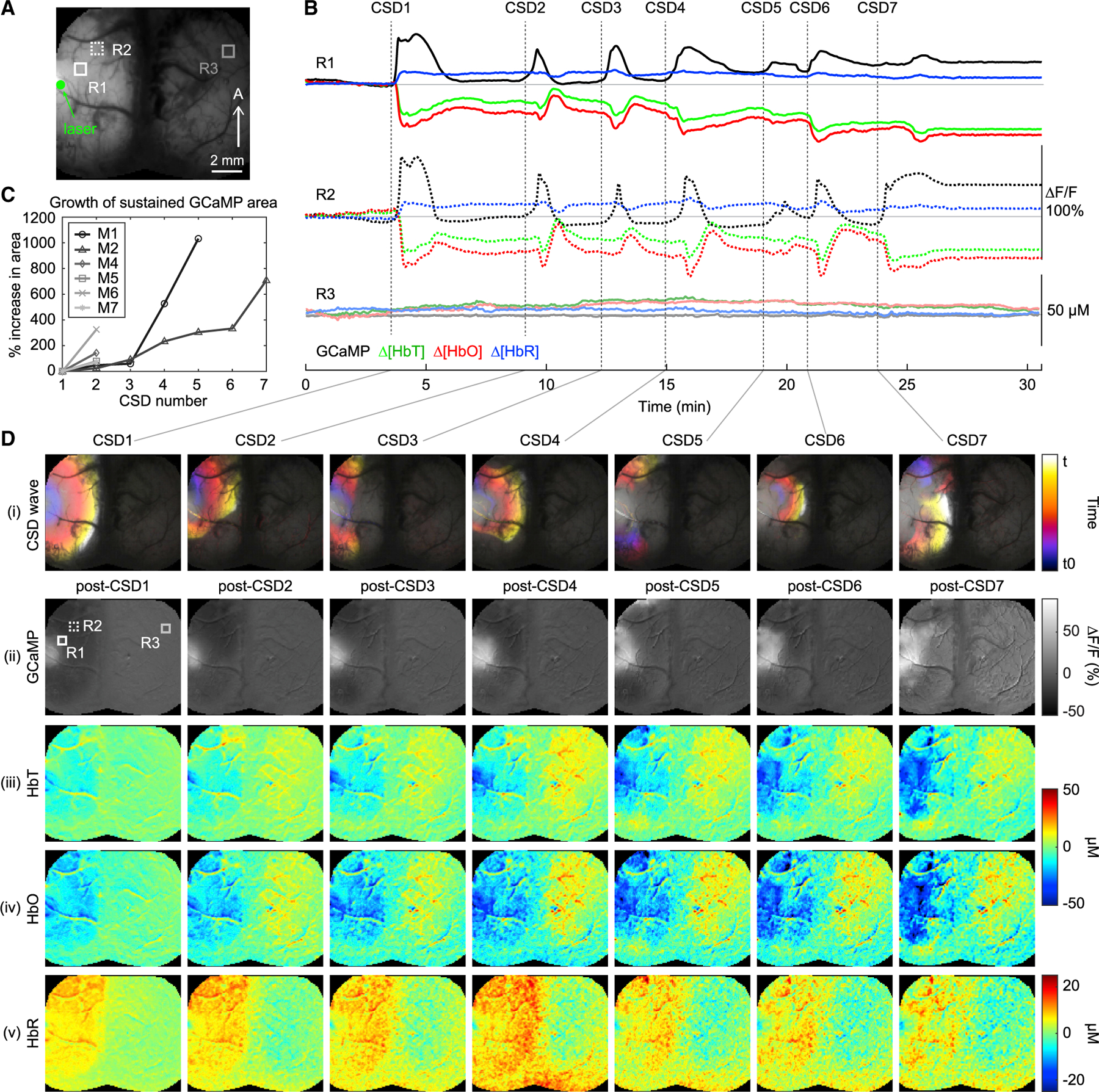
(A) Grayscale cortical image shows the ROIs used for the time courses in (B) and their relative locations to the laser focus.
(B) GCaMP ∆F/F, [HbT], [HbO], and [HbR] traces extracted from two ipsilateral regions, R1 and 2, and one contralateral region, R3, indicated by boxed ROIs in the raw cortical image in (A).
(C) Plot shows percentage increase in area of the region with sustained GCaMP fluorescence (relative to post-CSD1) following successive CSDs in each mouse. See STAR Methods for details on how percentage growth was calculated.
(D) (i) Cortical maps color-coded by time, showing the propagation dynamics of each CSD for an animal that experienced 7 CSDs (denoted as CSD1-CSD7, see STAR METHODS). See also Video S3 for full movies of these events. (ii-v) Cortical maps showing post-CSD levels of GCaMP ∆F/F, [HbT], [HbO], and [HbR] respectively 5 s after the completion of each CSD event. The spatial maps highlight the growing areas of elevated GCaMP fluorescence and accompanying hypoperfusion after each successive CSD.
One striking cumulative effect seen across mice was a central region of sustained GCaMP fluorescence, which gradually increased in area with each additional CSD (Figures 6C, 6Dii, and S6Cii; Video S3). This sustained level of high GCaMP fluorescence likely indicates an inability of the neuronal tissue to restore its prior transmembrane ion gradients and membrane potential. The stepwise growth of the sustained GCaMP fluorescence area mirrors that observed in the growing central region of decreased [HbT] and thus oligemia (Figures 6Diii and S6Ciii). While gradually increasing ischemia might be expected as a result of photothrombosis, this distinct spatial pattern of decreased [HbT] suggests that regions of sustained depolarization may also be experiencing microvascular vasospasm. During subsequent CSDs, these regions typically showed minimal changes in both GCaMP fluorescence and hemoglobin concentration and thus were not included in the analysis shown in Figures 5E and 5F (and are denoted as X in Figure S4).
Beyond these core changes, cumulative changes were also observed in more distant cortical regions. As noted already in Figure 3C′, we see clear evidence of progressive post-CSD oligemia, which is a well-documented effect of CSDs (Ayata, 2013; Ayata et al., 2004). The post-CSD [HbT] maps after the initial CSD show that the shape of this area of oligemia and hypoxia closely matches the domain of the first CSD (Figures 6Diii and S6Ciii). With each successive CSD, these areas around the photothrombotic core show gradually decreasing [HbO] and increasing [HbR] consistent with worsening ischemia, with the different spatial pattern of each CSD (Figure 6Di) introducing more regional heterogeneity (Figure 6Div–6Dv). Of note, the reduction in both HbT and HbO and the increase in HbR due to photothrombosis (epoch II; Figure 3C) is relatively mild compared to the post-CSD oligemia (Figure 6D; CSD7). These data suggest that CSDs drive expanding ischemia in the acutely injured brain at an early time-point that precedes neuronal death.
Post-CSD decreases in baseline GCaMP fluorescence are also observed in more distant regions (also visible in Figures 5A, 5C, and S5) and overlap with regions of oligemia and cortical ischemia. This decrease may represent a change in baseline neural activity in response to decreased energy availability. Analysis in Figure 5F suggests that these regions might be expected to better tolerate subsequent CSDs via vasodilatory responses. Notably, with additional CSDs, these darker regions become less prominent as the central bright core region spreads, tending toward the high baseline GCaMP condition prone to CSD-induced vasoconstriction (Figure 5F) and thus exacerbation of metabolic tissue damage.
We note also that the propagation trajectories of successive CSDs vary in shape and spread non-uniformly (Figure 5DI; Video S3). This additional heterogeneity could be influenced by the ability of cortical tissue at the wavefront to generate neuronal depolarization or may reflect the inability of the wave to travel uniformly through tissue that is heterogeneously accumulating damage (Ayata and Lauritzen, 2015).
The time-courses in Figure 6B underscore how all of these heterogeneous factors impact the regional dependence of the cortical response to each CSD. Although the strong initial CSD induces uniform vasoconstriction, subsequent CSDs are accompanied by a range of vascular responses that ultimately progress back to pure vasoconstriction, oligemia, ischemia, and elevated GCaMP. The same sequence was observed in an awake mouse, confirming that this progression is not the result of the type, duration, or level of anesthesia. The type of neurovascular response generated clearly depends heavily on the pre-CSD baseline and the temporal shape of each CSD, which in turn depends on the trajectory of damage of a given cortical region based on the spatial pattern and amplitude of prior CSDs.
DISCUSSION
CSDs have emerged as common pathophysiological events in a number of diseases, including ischemic stroke, traumatic brain injury, and subarachnoid hemorrhage (Dohmen et al., 2008; Dreier et al., 1998; Hadjikhani et al., 2001; Hubschmann and Kornhauser, 1980, 1982; Lauritzen, 1994; Pietrobon and Moskowitz, 2014). This phenomenon, which was first thought to be an experimental oddity (Leão, 1944), is now recognized as a potentially major contributor to the progression of tissue damage in human brain injury (Dohmen et al., 2008; Dreier et al., 2006; Hartings et al., 2009). Although CSDs associated with migraine with aura are often considered benign, we also note that migraine with aura has been identified as an independent risk factor for ischemic stroke (Etminan et al., 2005). While extensive studies using in vivo imaging and electrophysiological tools have significantly increased our understanding of CSDs (Ayata et al., 2004; Balbi et al., 2021; Bauer et al., 2014; Østergaard et al., 2015; Strong et al., 2006), they have thus far failed to capture a comprehensive, bilateral view of both neural activity and hemodynamics, and the spatiotemporal evolution of CSDs at the moment of and after the onset of acute brain injury. Reliance on hemodynamic-only imaging, invasive techniques, and inconsistent experimental paradigms have resulted in conflicting findings across different studies that have been challenging to reconcile (Ayata and Lauritzen, 2015).
Understanding variability in neurovascular responses to CSD
A wide range of different neurovascular responses to CSDs have been reported across species and disease states. Many studies have observed a primarily hyperemic or vasodilatory response in rats but a vasoconstriction response in mice (Ayata et al., 2004; Shin et al., 2006). However, under pathological conditions, CSDs can be accompanied by varying levels of vasoconstriction in both rodents and humans (Ayata, 2013). The widely varying hemodynamic responses to CSDs observed across rodent studies and patient cohorts has been difficult to explain, leading many to question whether and how CSDs ultimately influence clinical outcomes (Ayata and Lauritzen, 2015).
In this study, we observed a high-amplitude CSD in every mouse following onset of photothrombosis. Each initial CSD was accompanied by a spatially matched but temporally lagged strong vasoconstriction whose amplitude and range far outstripped the small and localized hemodynamic changes caused by the photothrombosis itself. Subsequent CSDs occurred in most mice and were much more variable in amplitude, spatial spread, and duration. These subsequent CSDs were accompanied by more widely varying hemodynamic responses, from constrictions to biphasic and dilatory responses.
By analyzing these responses, we recognized that in cases where we observed vasodilation in mice, the CSD was small, had a slow onset, or baseline perfusion levels were relatively high and baseline GCaMP levels were reduced. Biphasic vascular responses were observed under conditions in which the CSD was relatively larger and more rapid, but the depolarization was not sustained. CSDs of higher amplitude, rapid onset, sustained depolarization, and those occurring in cortex with decreased [HbT], elevated [HbR] and high baseline GCaMP levels drove vasoconstriction with no hyperemia. Our analysis confirmed that the same CSD could cause different hemodynamic responses in different regions of the cortex, while the same area of cortex can show different hemodynamic responses to different CSDs. These findings lead us to conclude that the temporal shape of the hemodynamic response to a given CSD is resource- and rate-dependent, and that the distinction between whether a net CSD response is vasoconstrictive or vasodilatory is dependent on the dynamic properties of the CSD relative to the underlying metabolic landscape. This tissue-state dependence may provide a more unified explanation for the widely varying hemodynamic responses to CSD seen in prior animal and clinical studies. For example, this model is consistent with clinical observations of CSD-induced vasoconstriction in aneurysmal subarachnoid hemorrhages (aSAHs) being correlated with CSDs of a longer duration and greater frequency. In addition, while vasoconstriction responses to CSDs have been seen in malignant cerebral infarction (MCI), cortical regions further from the infarct core are more likely to show CSD-induced hyperemia (Dreier et al., 2006, 2009).
The combination of neurovascular mechanisms responsible for the constriction and dilation components of the CSD hemodynamic response and how they might be modulated by cortical state remain uncertain (Ayata and Lauritzen, 2015). However, we note that other major neuronal events such as interictal discharges and seizures elicit relatively normal hyperemia responses (Montgomery et al., 2020), suggesting the involvement of potentially CSD-specific mechanisms in driving vasoconstrictions. As such, CSD-evoked vasoconstriction may not represent a simple “inversion” of normal neurovascular coupling but rather demonstrate the net dominance of a vasoconstrictory mechanism for a given CSD and cortical condition (Hartings et al., 2017; Hinzman et al., 2014).
Our results may also help to explain the wide variability in CSD responses observed in studies that have used open cranial windows or the insertion of invasive electrodes or probes. Since CSDs can be induced in the cortex with a simple pin prick, they are thus likely to be evoked during surgical preparation or electrode insertion. Our WFOM method captured both neuronal GCaMP and hemodynamics through a thinned-skull preparation and enabled continuous imaging before, during, and after initiation of photothrombosis. With this minimally invasive approach, we observed marked differences between hemodynamic responses to the first CSD and subsequent CSDs, as well as persistent changes in neural activity and baseline [HbT] following the first CSD. Therefore, as previously proposed (Chang et al., 2010; Tomida et al., 1989), studies using invasive preparations may have been unable to capture the full evolution of CSDs and cumulative tissue damage starting from naive tissue, making the diversity of vascular responses observed across different experimental paradigms more challenging to reconcile.
A further potential contributor to varying neurovascular responses seen in prior CSD studies may have been the use of KCl for non-pathological CSD induction (Ayata et al., 2004; Chen et al., 2006). While high levels of potassium ions can induce CSDs, potentially modeling high potassium levels caused by ischemia (Dreier and Reiffurth, 2015), potassium levels can also independently and differentially modulate the cortical vasculature in a concentration dependent manner (Longden et al., 2017; Chang et al., 2010). Excessive application of KCl to the pial surface could thus contribute to or alter the neurovascular response to CSDs generated in this way (Chang et al., 2010). Species-dependent effects could reflect the differential sensitivity of cerebral vessels in rat and mouse to elevated extracellular potassium concentrations (Ayata et al., 2004).
Cumulative and spatially heterogeneous effects of CSDs
Consistent with prior studies, we observed persistent cortical oligemia and ischemia as well as alterations in baseline neural activity for over an hour after passage of a CSD. With each additional CSD, we observed worsening oligemia and ischemia across an area that far outreached the relatively small territory of the photothrombosis. These CSD-evoked, cumulative changes provide a mechanism for the heterogeneous shifts in baseline perfusion, ischemia, and GCaMP levels that we observed to shape the cortical hemodynamic response to subsequent CSDs. Thus, if the neurovascular response to a given CSD depends on the baseline hemodynamic and metabolic state of the cortex through which it is passing, the persistent impact of prior CSDs has the potential to create a vicious circle of accumulating damage, where oligemia and ischemia worsen further with repeated waves of vasoconstriction (Ayata, 2013; Dreier and Reiffurth, 2015).
Recording from the onset of photothrombosis onward also allowed us to observe a distinct core region of persistently high GCaMP fluorescence remaining after the first CSD. This core region appeared to serve as the starting point for subsequent CSDs and increased stepwise in area with successive CSDs, extending beyond the territory of the initial photothrombotic area. This “bright” region may represent irreversible CSD-evoked damage wherein neurons exhibiting sustained, elevated levels of GCaMP are unable to recover because of inadequate metabolic capacity, and which if left unresolved may lead to cell death (Hartings et al., 2003; Shin et al., 2006). These regions with sustained GCaMP levels also co-localized with strong decreases in local [HbT] and reduced vascular reactivity, suggestive of vasospasm, which might further exacerbate metabolic stress and could plausibly play a role in obstacles to clinical stroke management, such as impaired drug delivery and reperfusion failure after clot removal.
In addition to ischemic effects, cytotoxic edema associated with CSDs can also lead to blood-brain barrier disruption and vasogenic edema, which can further contribute to disturbed tissue perfusion in these boundary regions, exacerbating tissue excitotoxicity, inflammation, and neuronal injury (Dreier et al., 2018; Gursoy-Ozdemir et al., 2004; Simard et al., 2007). In clinical stroke, the ischemic penumbra is characterized by hypoper-fused, electrophysiologically silent tissue surrounding the ischemic core that is viable but at risk of infarction (Astrup et al., 1977) and has thus has been the focus of clinical research and stroke therapeutic development (Lo, 2008; Ramos-Cabrer et al., 2011). Our results strongly support that CSDs could be a major catalyst in penumbral expansion, propagating electrical and biological damage beyond the primary region of impaired perfusion caused by the primary ischemic injury (Lauritzen et al., 2011).
Imaging both neuronal and hemodynamic activity is crucial for understanding CSD variability
Data presented here were acquired with a customized implementation of WFOM, which simultaneously captured intracellular calcium dynamics in excitatory neurons (via GCaMP fluorescence) and cortical hemodynamics across the entire dorsal cortex during acute photothrombosis. A marked benefit of GCaMP imaging over point electrophysiology is the ability to non-invasively analyze both the amplitude and spatiotemporal trajectories of each individual CSD across almost the entire cortex. This feature permitted us to observe the widely varying, spatially dependent amplitudes, speeds, and spatial spreads of successive CSDs that appear to influence the tissue’s resulting neurovascular response. Although visualization of CSDs using GCaMP fluorescence has been previously performed (Balbi et al., 2017, 2021), our simultaneous visualization of hemodynamics permitted spatiotemporally precise comparisons of neurovascular responses to multiple spontaneously occurring CSDs. WFOM also enabled quantification of progressive changes in baseline oligemia and cortical ischemia, which provided a more comprehensive depiction of interdependencies between the CSD-induced vascular response and heterogeneity in the underlying cortical hemodynamic state. Our GCaMP observations also underscore the potential ambiguity of differentiating the effects of photothrombotic occlusion and CSDs from measurements of cortical blood flow alone. With only hemodynamic readouts, temporal delays and spatially dependent differences in neurovascular responses to CSDs will obscure the true amplitude, propagation pattern, and residual effect of the neuronal CSD wave.
Limitations of the study
Limitations of wide-field neuronal GCaMP imaging include the range of different cellular factors that can alter intracellular calcium levels. Under normal physiological conditions, increases in GCaMP fluorescence correlate with action potentials (Chen et al., 2013), but subthreshold activity and subtle changes in voltage gating will not be represented. However, GCaMP fluorescence can also report changes in the calcium homeostasis of neurons, which could be influenced by other drivers of calcium influx such as neuronal cell death. The large negative DC potential shift observed during CSDs is known to correlate with a decrease in extracellular calcium (Dreier, 2011; Hansen and Zeuthen, 1981; Windmüller et al., 2005), which in turn correlates with an increase in neuronal intracellular calcium (Gniel and Martin, 2010). It is thus likely that a component of the elevated GCaMP fluorescence observed during CSDs corresponds to this effect. Further studies are thus needed to fully interpret the neuronal GCaMP signals observed in this study, particularly the residual increases and decreases in baseline GCaMP fluorescence observed after each CSD, which could represent a combination of changes in neuronal firing and pathological increases or decreases in neuronal calcium homeostasis.
CSDs are a complex phenomenon involving interactions between various cell types at different stages of cortical injury (Dreier and Reiffurth, 2015). Additional studies could thus include expressing GCaMP (or contrasting genetically encoded calcium indicators) in other cell types, including other sub-populations of excitatory neurons, interneurons, astrocytes, and microglial cells, in addition to dye-based assessment of blood-brain barrier integrity; all of which could reveal many additional components of the effects of CSDs on the pathophysiology of brain injury. A further valuable addition would be the comparison between WFOM observations during the acute phases of photothrombosis and contrast observable in diffusion and perfusion magnetic resonance imaging (MRI). Such a comparison could potentially reveal clinical biomarkers for CSD-related damage that could better guide clinical decision making (Motta et al., 2015. While the size of our cohort was limited, the fact that CSDs were observed in every single mouse suggests that CSDs are far more common in patients with acute brain injury than currently appreciated.
The aim of the present study was to investigate the spatiotemporal neurovascular dynamics of CSDs in the acute period after a photothrombosis insult. Further studies are needed to explore the longitudinal consequences of the cumulative impact of multiple CSDs during recovery. Although the majority of mice in this study were imaged under urethane anesthesia, our results in awake, freely behaving mice emphasize the potential to use WFOM to investigate how changes in the acute period after brain injury influence or predict longitudinal changes in tissue viability, neuronal activity, and hemodynamic response. Of particular interest is whether regions of persistently increased GCaMP fluorescence represent cortical tissue that is susceptible to cellular death if timely therapeutic intervention is not provided.
Finally, we note that our study only included female mice to reduce sex-dependent variability. Female mice are generally underrepresented in stroke research, and there exist many sex differences in the occurrence and outcomes of stroke in humans (Dotson and Offner, 2017). Further studies are thus needed to confirm whether there are sex differences in the properties of CSDs identified here, and how such differences might play a role in stroke outcomes and potential therapeutic targets.
Summary
This study leveraged in vivo WFOM in transgenic mice to provide important insights into the neurovascular properties of CSDs in the injured cortex. Our results demonstrated that the amplitude, timing, and spatiotemporal patterns of CSDs, and their neurovascular responses, can be highly variable. Importantly, successive CSDs were observed to drive heterogeneous tissue damage far beyond the region of initial insult, altering the baseline perfusion and metabolic state, which in turn shaped the hemodynamic response to subsequent CSDs. The tendency of cortical regions with lower baseline perfusion to elicit vasoconstrictive hemodynamic responses to subsequent CSDs supports a potentially vicious cycle that could significantly exacerbate cortical damage. These results suggest that in the setting of ischemic stroke, CSDs could be a significant contributor to the progression of metabolic stress and tissue damage even after the restoration of blood flow. The highly variable and cumulative effects of CSDs that we observed may also explain why minor brain insults, regardless of size or location, can result in significant deficits and poor outcomes in certain patients, while leading to minimal damage in others.
These results strongly support the idea that CSDs could be a major contributor to secondary progression of tissue damage during and after acute brain injury, emphasizing CSDs as a potential therapeutic target. More rigorous clinical evaluation of CSD occurrence and properties could provide an improved foundation for personalized treatment strategies to inhibit secondary progression in acute brain injury.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests and for reagents and resources should be directed to the Lead Contact, Elizabeth Hillman (eh2245@columbia.edu).
Materials availability
No new or unique reagents were generated in this study.
Data and code availability
The data shown in this study will be shared by the lead contact upon reasonable request.
Analysis code used for this study are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request. Please direct all requests to Elizabeth Hillman (eh2245@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal procedures were reviewed and approved by the Columbia University Institutional Animal Care and Use Committee (IACUC). Adult female Thy1-GCaMP6f mice (Jackson Labs; C57BL/6J-Tg(Thy1-GCaMP6f)GP5.17Dkim/J) were weaned at postnatal day 21 and housed with littermates in temperature and humidity controlled facilities with 12-hour light/dark cycles and ad libitum food and water.
METHOD DETAILS
Animal preparation and photothrombosis model
Thy1-GCaMP6f C57BL/6 transgenic mice express the genetically-encoded intracellular calcium indicator GCaMP throughout excitatory neurons of layers 2/3 and 5 (Chen et al., 2012). Increases in intracellular calcium cause an increase in the intensity of fluorescence that can be measured as an indicator of neural activity. Here, adult female Thy1-GCaMP6f C57BL/6 mice (4 months to 1 year old) were anesthetized with 1.5 mg/kg urethane and placed in a mouse stereotax (Kopf) on an electrical heating pad, where the rectal temperature was maintained at 36.5 ± 0.5°C. The skin and connective tissue over the surface of the brain were removed, and the entire bilateral dorsal surface of the skull was thinned to translucency and covered with a layer of cyanoacrylate glue (Figure 1B). A custom laser-cut headplate was then glued to the skull in order to fix the mouse into the imaging rig. Female mice were used to reduce the impact of sex-dependent variability, although the limited research has thus far shown no sex differences with respect to vascular responses to CSDs (Yuzawa et al., 2012).
A photothrombotic model was used following initial baseline imaging (Labat-gest and Tomasi, 2013). Rose Bengal (Sigma), a light-activated photothrombotic agent was administered via intraperitoneal injection (10 uL/g at 15 mg/mL). A green (532 nm, 3–5 mW at the skull) laser was then focused onto a branch of the middle cerebral artery (MCA) supplying the somatosensory cortex. Functional imaging began before laser induction and continued for 2.5 hours after laser induction. Imaging continued during Rose Bengal administration and throughout laser-activated photolysis lasting 20 minutes, after which the laser was turned off and imaging continued.
Wide-field optical mapping (WFOM)
Neuronal activity and hemodynamics were captured using WFOM of GCaMP fluorescence and hemoglobin absorption (Bouchard et al., 2009; Ma et al., 2016a) (Figure 1A). The cortex was illuminated with consecutively strobed blue (Thorlabs, M490L4), amber (Thorlabs, M595L3) and red (Thorlabs, M625L3) LEDs, with bandpass filters (Thorlabs MF460/60, Semrock FF02–586/15–25, and Semrock FF01–623/24–25 respectively). A Semrock FF01–496/LP-25 long-pass filter and Thorlabs NF533–17 notch filter were used to block blue excitation light in order to image green GCaMP fluorescence, and to remove contamination from the green laser, respectively (Figure 1C). These filters were placed between the imaging lens (Nikon Micro-Nikkor 60 mm F/2.8) and an Andor Zyla sCMOS camera. LEDs were strobed in sequence, synchronized using the frame exposure signal from the camera to collect interleaved images of the cortex at each wavelength in turn. In some experiments, an Andor iXon 897 EMCCD camera was used, achieving equivalent results to the Zyla.
Illumination wavelengths were carefully chosen to enable efficient excitation of GCaMP, in addition to illumination with focused 532 nm laser light for photothrombosis, and imaging of oxy- and deoxy-hemoglobin (HbO and HbR) concentrations without contamination from the absorption cross-section of Rose Bengal (Figure 1D). Baseline resting state activity was collected in sequential 60 s trials. Imaging data was acquired for one hour prior to photothrombosis and 2.5 hours thereafter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Conversion and correction of WFOM data
As shown in Figure 1C, cortical images acquired under amber illumination are targeting an isosbestic point in the hemoglobin spectrum, yielding sensitivity primarily to oxygenation-independent changes (i.e., total hemoglobin, HbT). Red illumination provides images that are primarily sensitive to changes in deoxy-hemoglobin. Combining these two measurements permits estimation of changes in the concentrations of HbO, HbR, and HbT, where HbT is the sum of HbO and HbR. Spectral conversion was performed as described previously using the modified Beer-Lambert law with Monte Carlo-derived wavelength-dependent pathlength factors (Bouchard et al., 2009; Hillman, 2007; Ma et al., 2016a). Fluorescence measurements acquired during blue light illumination of the cortex correspond to GCaMP fluorescence, but are multiplied by a spatiotemporal factor corresponding to changes in both oxy- and deoxy-hemoglobin absorption that affect both the 488 nm excitation and 530 nm emission of GCaMP (Figure 1C). A correction for this hemodynamic contamination was derived from the hemodynamic reflectance measurements and applied to the fluorescence signal to yield a corrected DF/F as described in (Ma et al., 2016a). Validation described in the following section and presented in Figure S1 demonstrates that spectral conversion, the presence of Rose Bengal, and the hemodynamic correction of GCaMP fluorescence did not affect our observations or conclusions. For Video S2, spontaneous fluctuations in neural activity during the CSD was obtained by further removing the large CSD signal. After converting raw signals to GCaMP fluorescence, as described above, the data was then temporally smoothed with a 5 s sliding window and subtracted from the original signal on a pixel-by-pixel basis.
Verifying minimal effect of Rose Bengal on GCaMP signal correction
As shown in Figure 1, the absorption of Rose Bengal overlaps with the absorption of HbO and HbR, and therefore could contribute to absorption crosstalk on GCaMP fluorescence. Standard correction methods for hemodynamic absorption crosstalk have been previously described (Ma et al., 2016a). For imaging trials acquired following Rose Bengal injection, we examined the effect of the dye on GCaMP fluorescence calculations. This was done by modifying the absorption coefficient used for hemodynamic cross-talk corrections by introducing a Rose Bengal concentration-dependent term:
where μa,λ is the absorption coefficient, and ξλ and [X] are the wavelength-dependent extinction coefficient and concentration for each absorber, respectively. While we cannot measure the exact concentration of Rose Bengal, we assume that it will affect the overall amber reflectance and therefore the calculated [HbT] by a factor, RB = 0.17. This factor was estimated based on the injected amount of dye and the assumption that an average mouse has a blood volume that is about 7% of its body weight (McClure, 1999). Thus, the equation above can be rewritten as follows:
where [HbT] = [HbO]+[HbR]. A comparison of the GCaMP signal with and without correction for Rose Bengal is shown in Figure S1. For the purposes of this comparison, we assumed no dye clearance in order to determine the maximum effect Rose Bengal could have on fluorescence data over time. Even under this assumption, Rose Bengal is seen to have minimal effect on fluorescence.
Data analysis
In order to determine the speed of propagation of the CSD, we calculated the geodesic distance of each pixel along the leading edge of the wave relative to the centroid point of CSD onset. Distances were assessed at 10 s intervals and averaged for each interval. Standard deviation of the propagation speed shows the variability in speed around the CSD. To ensure that the increase in fluorescence signal was not due to hemodynamic cross-talk, we determined the time lag between onset times of reflectance and fluorescence channels (Figure S3A-B‘). Onset time was calculated as the time at which the first derivative of a signal is at a maximum. The time lag between channels was calculated as the difference in time between when the first derivative of the reflectance channels and fluorescence channel each reach their respective maxima.
To determine changes in hemodynamics due to photothrombosis versus CSD, two regions were selected, one immediately around the site of the laser focus (labeled P) and one more distal region (labeled D) that lay in the path of the initial CSD propagation (Figure 3B). Specific epochs of interest were chosen as follows: [I] after the induction laser is turned on, [II] one minute prior to CSD onset, [III] during CSD onset, [IV] peak of CSD, and [V] after recession of CSD as indicated by when the gradient of the GCaMP signal returns to zero. At each time point, neural and hemodynamic values were obtained by averaging all pixels within each ROI (P and D) over 500 msec (Figure 3C). Student’s t tests were used to evaluate whether signal changes at each time point differed significantly from baseline in all 8 mice.
To confirm physical constriction, we evaluated vessel diameter by plotting pixel intensities of vessel cross sections from the raw signal and calculating the full width half max (Figure S3C-E“). Vessel diameter values were calculated per frame over the course of a CSD. Changes in intensity were normalized to pixels in the surrounding parenchyma for each vessel in order to control for any widespread changes in tissue reflectance, and to ensure that changes in signal were truly due to diameter changes. To determine whether the change in vessel diameter was significant (Figure 4C), Student’s t tests were used where zero mean indicated no diameter change.
For analysis of subsequent CSDs, four regions were chosen ranging in distance from the photothrombotic core (R1–4 in Figures 5, S4, and S5). Time courses were extracted by averaging over each region and smoothed using a moving average with a window size of 1 s. Amplitude was calculated as the peak fluorescence in that region over the course of each CSD. Duration was calculated as the amount of time in which GCaMP fluorescence exceeded 50% of peak amplitude. Onset time was determined to be when the GCaMP signal exceeded 10% of the peak amplitude and onset speed was calculated as the average rate of fluorescence increase from onset time until the GCaMP signal reached 80% of its peak amplitude. These metrics are summarized in Figure S4. To extract baseline values prior to each CSD, the entire imaging sequence was concatenated and average values for GCaMP, HbT, HbO, and HbR over the 500ms prior to each CSD onset were calculated (Figure 5F). To obtain the event-triggered response traces in Figure 5E, the GCaMP and HbT signals from each CSD were aligned to their onset times. Each trace was then subtracted by its respective baseline value, as calculated in Figure 5F. Finally, all traces were grouped and averaged according to their response categories. Paired t tests were used to compare all category pairings.
To determine growth of the persistently depolarized core after each CSD, grayscale maps of GCaMP fluorescence were obtained after each CSD and thresholded for all pixels greater than 10% ∆F/F (Figure 6C). Maps were binarized so that only pixels above threshold were given a value of 1. Any single pixels or clusters of less than 5 pixels in the binarized image were excluded. Percent change in area was then calculated as the increase in the number of pixels with suprathreshold GCaMP fluorescence relative to post-CSD1. To show the CSD dynamics encoded by time in Figure 6D(i), we first isolated the propagation of only the leading edge of the CSD wave by subtracting each frame of the event from one 20 frames prior. We then applied an existing Temporal-Color_Code plugin in ImageJ (Schindelin et al., 2012; Schindelin et al., 2015) to the 3D matrix to color-code each event in time.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Rose Bengal | Millipore-Sigma | 330000 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J-Tg(Thy1-GCaMP6f) GP5.17Dkim/J | Jackson Laboratory | 025393 |
|
| ||
| Software and algorithms | ||
|
| ||
| MATLAB 2016a – 2019b | MathWorks | https://www.mathworks.com/products/matlab.html |
| FIJI/ImageJ | NIH | https://imagej.net/Fiji |
| Modified Beer-Lambert Conversion Excitation-Emission Hemodynamic Correction | Ma et al., 2016a | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5003860/ |
| Temporal-Color Code ImageJ Plugin | GitHub | https://github.com/fiji/fiji/blob/master/plugins/Scripts/Image/Hyperstacks/Temporal-Color_Code.ijm |
Highlights.
Pan-cortical imaging of neural activity in mice reveals CSDs after photothrombosis
Simultaneous hemodynamic imaging reveals varying vascular responses to each CSD
Successive CSDs induce worsening oligemia and ischemia in distal cortical areas
Vascular response dependence on baseline cortical state may exacerbate damage
ACKNOWLEDGMENTS
Funding for this work was provided by National Institutes of Health grants R01NS063226, R01NS076628, RF1MH114276 (E.M.C.H.), and U19NS104649 (D.C.); Toshiba America Medical Systems/RSNA Research Resident Grant RR1312 (D.C.); NIH (NCATS) UL2 RR024156 (E.M.C.H.); NSF CAREER 0954796 (E.M.C.H.); Columbia’s Medical Scientist Training Program (M.C.T.), and funding from the David MC Ju Foundation. We also acknowledge the contributions and support of Carla Kim, Angela Liu, Evelyn Ramirez, Aleksandr Rayshubskiy, David Chung, and Matthew Bouchard. We thank Randall Marshall, Dritan Agalliu, and Franck Polleux for helpful discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109794.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Astrup J, Symon L, Branston NM, and Lassen NA (1977). Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 8, 51–57. [DOI] [PubMed] [Google Scholar]
- Ayata C. (2013). Spreading depression and neurovascular coupling. Stroke 44 (6, Suppl 1), S87–S89. [DOI] [PubMed] [Google Scholar]
- Ayata C, and Lauritzen M. (2015). Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev 95, 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, and Moskowitz MA (2004). Pronounced hypoperfusion during spreading depression in mouse cortex. J. Cereb. Blood Flow Metab 24, 1172–1182. [DOI] [PubMed] [Google Scholar]
- Balbi M, Vanni MP, Silasi G, Sekino Y, Bolanos L, LeDue JM, and Murphy TH (2017). Targeted ischemic stroke induction and mesoscopic imaging assessment of blood flow and ischemic depolarization in awake mice. Neurophotonics 4, 035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbi M, Xiao D, Jativa Vega M, Hu H, Vanni MP, Bernier LP, LeDue J, MacVicar B, and Murphy TH (2021). Gamma frequency activation of inhibitory neurons in the acute phase after stroke attenuates vascular and behavioral dysfunction. Cell Rep. 34, 108696. [DOI] [PubMed] [Google Scholar]
- Bauer AQ, Kraft AW, Wright PW, Snyder AZ, Lee JM, and Culver JP (2014). Optical imaging of disrupted functional connectivity following ischemic stroke in mice. Neuroimage 99, 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MB, Chen BR, Burgess SA, and Hillman EM (2009). Ultrafast multispectral optical imaging of cortical oxygenation, blood flow, and intracellular calcium dynamics. Opt. Express 17, 15670–15678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, and Brennan KC (2010). Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain 133, 996–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Feng Z, Li P, Jacques SL, Zeng S, and Luo Q. (2006). In vivo optical reflectance imaging of spreading depression waves in rat brain with and without focal cerebral ischemia. J. Biomed. Opt 11, 34002. [DOI] [PubMed] [Google Scholar]
- Chen Q, Cichon J, Wang W, Qiu L, Lee SJ, Campbell NR, Destefino N, Goard MJ, Fu Z, Yasuda R, et al. (2012). Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron 76, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, and Nimchinsky EA (2007). High-resolution in vivo imaging of the neurovascular unit during spreading depression. J. Neurosci 27, 4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, and Graf R; Co-Operative Study of Brain Injury Depolarisations (COSBID) (2008). Spreading depolarizations occur in human ischemic stroke with high incidence. Ann. Neurol 63, 720–728. [DOI] [PubMed] [Google Scholar]
- Dotson AL, and Offner H. (2017). Sex differences in the immune response to experimental stroke: Implications for translational research. J. Neurosci. Res 95, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med 17, 439–447. [DOI] [PubMed] [Google Scholar]
- Dreier JP, and Reiffurth C. (2015). The stroke-migraine depolarization continuum. Neuron 86, 902–922. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Körner K, Ebert N, Görner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhäupl KM, et al. (1998). Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J. Cereb. Blood Flow Metab 18, 978–990. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, et al. (2006). Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 129, 3224–3237. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, et al. ; COSBID study group (2009). Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132, 1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Lemale CL, Kola V, Friedman A, and Schoknecht K. (2018). Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 134 (Pt B), 189–207. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Bolay H, Moskowitz MA, and Boas DA (2001). Dynamic imaging of cerebral blood flow using laser speckle. J. Cereb. Blood Flow Metab 21, 195–201. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, et al. (2011). Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J. Neurosci 31, 5755–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etminan M, Takkouche B, Isorna FC, and Samii A. (2005). Risk of ischaemic stroke in people with migraine: systematic review and meta-analysis of observational studies. BMJ 330, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius M, Akgoren N, and Lauritzen M. (1995). Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am. J. Physiol 269, H23–H29. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, and Lauritzen M. (2006). Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 129, 778–790. [DOI] [PubMed] [Google Scholar]
- Farkas E, Bari F, and Obrenovitch TP (2010). Multi-modal imaging of anoxic depolarization and hemodynamic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage 51, 734–742. [DOI] [PubMed] [Google Scholar]
- Gniel HM, and Martin RL (2010). Changes in membrane potential and the intracellular calcium concentration during CSD and OGD in layer V and layer II/III mouse cortical neurons. J. Neurophysiol 104, 3203–3212. [DOI] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, and Moskowitz MA (2004). Cortical spreading depression activates and upregulates MMP-9. J. Clin. Invest 113, 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, et al. (2001). Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 98, 4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, and Zeuthen T. (1981). Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol. Scand 113, 437–445. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Rolli ML, Lu XC, and Tortella FC (2003). Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J. Neurosci 23, 11602–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier JP, Mazzeo AT, Tortella FC, and Bullock MR; Co-Operative Study of Brain Injury Depolarizations (2009). Spreading depolarizations and late secondary insults after traumatic brain injury. J. Neurotrauma 26, 1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, Andrew RD, Boutelle MG, Brennan KC, Carlson AP, et al. (2017). The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J. Cereb. Blood Flow Metab 37, 1571–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemi P, Bhatia R, Nakamura H, Dreier JP, Graf R, Strong AJ, and Boutelle MG (2009). Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leão’s spreading depression. J. Cereb. Blood Flow Metab 29, 166–175. [DOI] [PubMed] [Google Scholar]
- Hillman EM (2007). Optical brain imaging in vivo: techniques and applications from animal to man. J. Biomed. Opt 12, 051402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Andaluz N, Shutter LA, Okonkwo DO, Pahl C, Strong AJ, Dreier JP, and Hartings JA (2014). Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 137, 2960–2972. [DOI] [PubMed] [Google Scholar]
- Hubschmann OR, and Kornhauser D. (1980). Cortical cellular response in acute subarachnoid hemorrhage. J. Neurosurg 52, 456–462. [DOI] [PubMed] [Google Scholar]
- Hubschmann OR, and Kornhauser D. (1982). Effect of subarachnoid hemorrhage on the extracellular microenvironment. J. Neurosurg 56, 216–221. [DOI] [PubMed] [Google Scholar]
- Koroleva VI, and Bures J. (1996). The use of spreading depression waves for acute and long-term monitoring of the penumbra zone of focal ischemic damage in rats. Proc. Natl. Acad. Sci. USA 93, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labat-gest V, and Tomasi S. (2013). Photothrombotic ischemia: a minimally invasive and reproducible photochemical cortical lesion model for mouse stroke studies. J. Vis. Exp 76, 50370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M. (1994). Pathophysiology of the migraine aura. The spreading depression theory. Brain 117, 199–210. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Olsen TS, Lassen NA, and Paulson OB (1983). Regulation of regional cerebral blood flow during and between migraine attacks. Ann. Neurol 14, 569–572. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, and Strong AJ (2011). Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab 31, 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AAP (1944). Spreading depression of activity in the cerebral cortex. J. Physiol 7, 359–390. [Google Scholar]
- Lipton P. (1999). Ischemic cell death in brain neurons. Physiol. Rev 79, 1431–1568. [DOI] [PubMed] [Google Scholar]
- Lo EH (2008). A new penumbra: transitioning from injury into repair after stroke. Nat. Med 14, 497–500. [DOI] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki N, Brayden JE, Hill-Eubanks D, and Nelson MT (2017). Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci 20, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman E, Lamanna J, Cordingley G, Rosenthal M, and Somjen G. (1975). Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res 88, 15–36. [DOI] [PubMed] [Google Scholar]
- Luckl J, Zhou C, Durduran T, Yodh AG, and Greenberg JH (2009). Characterization of periinfarct flow transients with laser speckle and Doppler after middle cerebral artery occlusion in the rat. J. Neurosci. Res 87, 1219–1229. [DOI] [PubMed] [Google Scholar]
- Lückl J, Lemale CL, Kola V, Horst V, Khojasteh U, Oliveira-Ferreira AI, Major S, Winkler MKL, Kang EJ, Schoknecht K, et al. (2018). The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain 141, 1734–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Shaik MA, Kim SH, Kozberg MG, Thibodeaux DN, Zhao HT, Yu H, and Hillman EM (2016a). Wide-field optical mapping of neural activity and brain haemodynamics: considerations and novel approaches. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Shaik MA, Kozberg MG, Kim SH, Portes JP, Timerman D, and Hillman EMC (2016b). Resting-state hemodynamics are spatiotemporally coupled to synchronized and symmetric neural activity in excitatory neurons. Proc. Natl. Acad. Sci. USA 113, E8463–E8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major S, Huo S, Lemale CL, Siebert E, Milakara D, Woitzik J, Gertz K, and Dreier JP (2020). Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury. Geroscience 42, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure DE (1999). Clinical pathology and sample collection in the laboratory rodent. Vet. Clin. North Am. Exot. Anim. Pr 2, 565–590, vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery MK, Kim SH, Dovas A, Zhao HT, Goldberg AR, Xu W, Yagielski AJ, Cambareri MK, Patel KB, Mela A, et al. (2020). Glioma-Induced Alterations in Neuronal Activity and Neurovascular Coupling during Disease Progression. Cell Rep. 31, 107500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta M, Ramadan A, Hillis AE, Gottesman RF, and Leigh R. (2015). Diffusion-perfusion mismatch: an opportunity for improvement in cortical function. Front. Neurol 5, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, and Liu R. (2008). Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J. Neurosci 28, 1756–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, and Hansen AJ (1988). Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449, 395–398. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Schaffer CB, Friedman B, Lyden PD, and Kleinfeld D. (2007). Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc. Natl. Acad. Sci. USA 104, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Larsen B, and Lauritzen M. (1981). Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann. Neurol 9, 344–352. [DOI] [PubMed] [Google Scholar]
- Østergaard L, Dreier JP, Hadjikhani N, Jespersen SN, Dirnagl U, and Dalkara T. (2015). Neurovascular coupling during cortical spreading depolarization and -depression. Stroke 46, 1392–1401. [DOI] [PubMed] [Google Scholar]
- Phillips JM, and Nicholson C. (1979). Anion permeability in spreading depression investigated with ion-sensitive microelectrodes. Brain Res 173, 567–571. [DOI] [PubMed] [Google Scholar]
- Pietrobon D, and Moskowitz MA (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci 15, 379–393. [DOI] [PubMed] [Google Scholar]
- Ramos-Cabrer P, Campos F, Sobrino T, and Castillo J. (2011). Targeting the ischemic penumbra. Stroke 42 (1, Suppl), S7–S11. [DOI] [PubMed] [Google Scholar]
- Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF, Lyden PD, and Kleinfeld D. (2006). Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol 4, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC, and Eliceiri KW (2015). The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev 82, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, and Ayata C. (2006). Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J. Cereb. Blood Flow Metab 26, 1018–1030. [DOI] [PubMed] [Google Scholar]
- Simard JM, Kent TA, Chen M, Tarasov KV, and Gerzanich V. (2007). Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG (2001). Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev 81, 1065–1096. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Venables GS, and Gibson G. (1983). The cortical ischaemic penumbra associated with occlusion of the middle cerebral artery in the cat: 1. Topography of changes in blood flow, potassium ion activity, and EEG. J. Cereb. Blood Flow Metab 3, 86–96. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, Parkin MC, and Lauritzen M. (2002). Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 33, 2738–2743. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Bezzina EL, Anderson PJ, Boutelle MG, Hopwood SE, and Dunn AK (2006). Evaluation of laser speckle flowmetry for imaging cortical perfusion in experimental stroke studies: quantitation of perfusion and detection of peri-infarct depolarisations. J. Cereb. Blood Flow Metab 26, 645–653. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, and Nedergaard M. (2007). Cortical spreading depression causes and coincides with tissue hypoxia. Nat. Neurosci 10, 754–762. [DOI] [PubMed] [Google Scholar]
- Tomida S, Wagner HG, Klatzo I, and Nowak TS Jr. (1989). Effect of acute electrode placement on regional CBF in the gerbil: a comparison of blood flow measured by hydrogen clearance, [3H]nicotine, and [14C]iodoantipyrine techniques. J. Cereb. Blood Flow Metab 9, 79–86. [DOI] [PubMed] [Google Scholar]
- Windmüller O, Lindauer U, Foddis M, Einhäupl KM, Dirnagl U, Heinemann U, and Dreier JP (2005). Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain 128, 2042–2051. [DOI] [PubMed] [Google Scholar]
- Yuzawa I, Sakadžić S, Srinivasan VJ, Shin HK, Eikermann-Haerter K, Boas DA, and Ayata C. (2012). Cortical spreading depression impairs oxygen delivery and metabolism in mice. J. Cereb. Blood Flow Metab 32, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data shown in this study will be shared by the lead contact upon reasonable request.
Analysis code used for this study are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request. Please direct all requests to Elizabeth Hillman (eh2245@columbia.edu).