

Abstract
The first gapless, telomere-to-telomere sequence of a human autosome, chromosome 8, is complete. Sequencing and assembly of the corresponding centromere in the chimpanzee, orangutan and macaque reveals details of its rapid evolution over the past 25 million years.
The problem
The first draft sequence of the human genome was released more than 20 years ago. However, the limitations of available technologies meant that parts of our genetic code — including regions corresponding to centromeres, telomeres and duplicated gene families — could not be fully sequenced. This information gap has limited our understanding of the regions’ organization, genetic variation, evolution and roles in complex biological processes that are required for life. It has also impeded advances in the discovery, diagnosis and treatment of human disease. Scientists have therefore been working towards filling in the gaps in the current draft of the human reference genome sequence (GRCh38).
The solution
Advances in long-read sequencing technologies have enabled the resolution of complex, repeat-rich regions from native DNA1. For example, ultra-long sequence reads from Oxford Nanopore Technologies (ONT) and high-fidelity (HiFi) data from Pacific Biosciences (PacBio) have facilitated the accurate assembly of complex structural variants2–4, segmental duplications5–7, centromeres6,8 and the complete human X chromosome9. Many of these assemblies used DNA from complete hydatidiform mole (CHM) cell lines (for example, CHM13 and CHM1). These have no maternal genome (but a duplicated paternal one), which avoids the need to assemble both haplotypes of a diploid genome.
We developed an assembly method that leverages the strengths of ultra-long reads (>100 kilobases) from ONT and HiFi long reads (~15–25 kb) from PacBio to resolve all five gaps in the DNA sequence of human chromosome 8 from CHM13 cells (Fig. 1). Specifically, the ONT reads were used to build a sequence scaffold that was subsequently replaced with PacBio continuous DNA sequences. The result is a complete, highly accurate (>99.99%) telomere-to-telomere sequence assembly of human chromosome 8. Our assembly contains more than 3.5 million bases that were missing from GRCh38 and map to the centromere, two segmental duplications and both telomeres. We also identified 12 new protein-coding genes, and generated a chromosome-wide DNA methylation map.
Figure 1 |. Resolving the gaps in human chromosome 8.
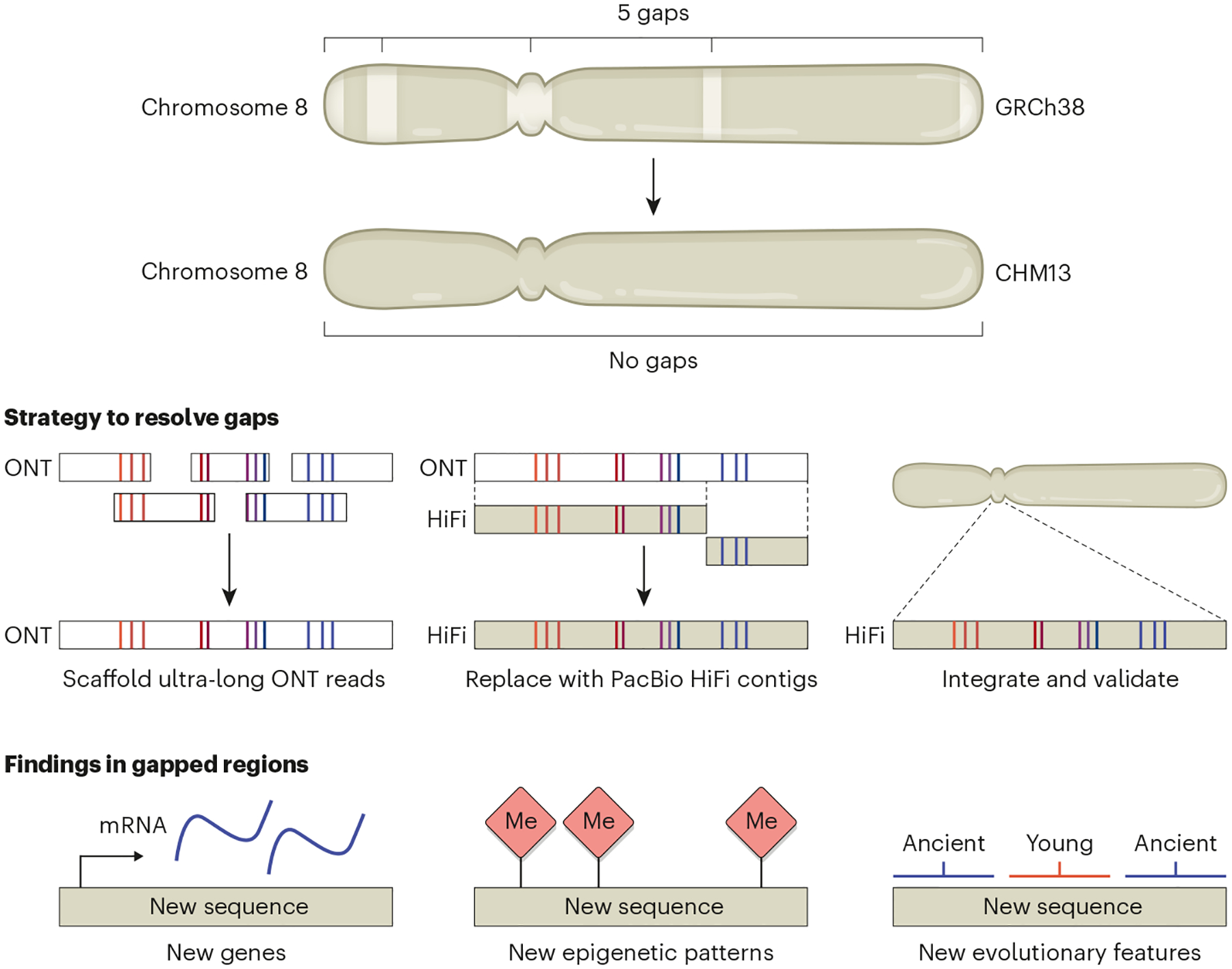
The five gaps present in chromosome 8 in the current human reference genome sequence (GRCh38) were resolved using CHM13 cells, which have a single haploid equivalent of the human genome. Ultra-long reads from Oxford Nanopore Technologies (ONT) were assembled into a sequence scaffold. High-fidelity (HiFi) reads from Pacific Biosciences (PacBio) were then used to generate continuous DNA sequences that replaced the ONT-based scaffold. The HiFi assemblies were integrated into a previously generated assembly of CHM13 chromosome 8 (ref. 4) and validated. Closure of these gaps allowed new genes, epigenetic patterns (such as DNA methylation (red diamonds)) and evolutionary features to be identified in previously unseen regions of the human chromosome 8 sequence.
The completion of the chromosome 8 centromere sequence provides insight into the biology of the region. We found that the centromeric α-satellite higher-order repeat (HOR) array (that is, units of repeating DNA that are arranged in tandem to form a larger repeating structure) is methylated, except for a small (73 kb) region that coincides with the location of nucleosomes containing the centromeric histone CENP-A. This region probably represents the site of the kinetochore, a macromolecular structure that ensures the accurate segregation of chromosomes during cell division.
We also reconstructed the evolution of the ape chromosome 8 centromere over the past 25 million years by sequencing and assembling the orthologous centromeres in the chimpanzee, orangutan and macaque. Our data show that α-satellite HOR structures evolved after great apes diverged from Old World monkeys. In addition, our comparison of human centromere chromosome 8 haplotypes (that is, of those inherited from a single parent) shows that there is greater variation in the centromere than in other regions of the genome, and that the centromere mutates two- to fourfold faster than the rest of the genome.
Future directions
The first telomere-to-telomere assembly of a human autosome sets the stage for a complete map of a haploid human genome. The Telomere-to-Telomere (T2T) Consortium is currently leading an effort to do this and is likely to reveal hundreds of millions of new bases, aiding the study of sequence composition, variation, function and evolution in the human genome. The next step will be to generate complete genomes from normal diploid cells. This will require the development of technologies and computational tools that can completely phase and assemble both maternal and paternal haplotypes across complex structural variant regions of the genome. Such an effort is being led by the Human Pangenome Reference Consortium (HPRC) and the Human Genome Structural Variation Consortium (HGSVC)4. The ability to sequence and assemble diploid genomes could pave the way to the production of complete genomic maps for patients, and to the use of such maps in tailoring treatments to the genetic variant underlying a disease.
EXPERT OPINION
“As well as presenting a blueprint for a method to produce finished human chromosome assemblies, the authors demonstrate the importance of gap-free sequences. The exciting key example of this study is the presentation of the entire chromosome 8 centromere, including the methylation status of the α-satellite HOR arrays, because this predicts the likely position of the functional kinetochore.” Nils Stein, Leibniz Institute of Plant Genetics and Crop Plant Research, Seeland, Germany.
BEHIND THE PAPER
We have been fortunate to work with an amazing team of scientists as part of the T2T Consortium, HPRC and HGSVC. These consortia bring together some of the most creative minds in genome sequencing and assembly. In addition to advances in long-read sequencing technology, the proposal 15 years ago that complete hydatidiform mole DNA (which is devoid of allelic variation2) be used to finish sequencing the human genome has been key. When we first visualized the sequence organization and evolutionary layers of the chromosome 8 centromere, we were excited to realize that there are many features of the centromere, such as DNA methylation patterns and chromatin organization, that were previously unknown. This has opened up new areas in the study of centromere biology and evolution.
Footnotes
Competing interests
The authors declare no competing interests.
REFERENCES
- 1.Logsdon GA, Vollger MR & Eichler EE Nature. Rev. Genet 21, 597–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaisson MJP et al. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain M et al. Nature Biotechnol. 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ebert P et al. Science 372, eabf7117 (2021).33632895 [Google Scholar]
- 5.Vollger MR et al. Ann. Hum. Genet 84, 125–140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nurk S et al. Genome Res. 10.1101/gr.263566.120 (2020). [DOI] [Google Scholar]
- 7.Cheng H, Concepcion GT, Feng X, Zhang H & Li H Nature Methods 18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bzikadze AV & Pevzner PA Nature Biotechnol. 38, 1309–1316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miga KH et al. Nature 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]