

Abstract
The hippocampus contains a diverse array of inhibitory interneurons that gate information flow through local cortico-hippocampal circuits to regulate memory storage. Although most studies of interneurons have focused on their role in fast synaptic inhibition mediated by GABA release, different classes of interneurons express unique sets of neuropeptides, many of which have been shown to exert powerful effects on neuronal function and memory when applied pharmacologically. However, relatively little is known about whether and how release of endogenous neuropeptides from inhibitory cells contributes to their behavioral role in regulating memory formation. Here we report that vasoactive intestinal peptide (VIP)-expressing interneurons participate in social memory storage by enhancing information transfer from hippocampal CA3 pyramidal neurons to CA2 pyramidal neurons. Notably, this action depends on release of the neuropeptide enkephalin from VIP neurons, causing long-term depression of feedforward inhibition onto CA2 pyramidal cells. Moreover, VIP neuron activity in the CA2 region is increased selectively during exploration of a novel conspecific. Our findings, thus, enhance our appreciation of how GABAergic neurons can regulate synaptic plasticity and mnemonic behavior by demonstrating that such actions can be mediated by release of a specific neuropeptide, rather than through classic fast inhibitory transmission.
Keywords: CA2, social memory, enkephalin, VIP, ITDP
Introduction
Over the past several years, it has become increasingly clear that inhibitory neurons can actively participate in memory storage by regulating the flow of information through distinct neural circuits [1–7]. To date, such studies have focused on the inhibitory or disinhibitory actions of different classes of interneurons that release the inhibitory transmitter GABA at synapses that target, respectively, excitatory pyramidal neurons or other inhibitory interneurons. However, many classes of interneurons are known to co-express with GABA a variety of neuropeptides that often define separate interneuron classes [8]. Although pharmacological studies using injections of neuropeptides or their antagonists indicate that these agents can exert powerful effects to modulate neural excitability, synaptic transmission, and various behaviors [9–12], relatively little is known about whether neuropeptide release from defined classes of hippocampal interneurons under physiological conditions is recruited to mediate the encoding of memory.
Here, we focus on a class of hippocampal interneurons that express the modulatory neuropeptide vasoactive intestinal peptide (VIP neurons). VIP neurons preferentially form inhibitory synapses with other interneurons [13–15] and thereby suppress their firing [16–18], leading to increased overall excitability through the process of disinhibition. A number of studies have recently found that VIP neurons play important roles in regulating a number of different forms of learning and memory, including amygdala-dependent fear memory [5], hippocampal-dependent goal-oriented spatial memory [4] and avoidance behavior [6], and sensory cortical dependent perceptual learning [7]. These studies either explicitly found or implicitly assumed that the VIP neurons acted to regulate memory storage through disinhibition. However, VIP neurons also express neuropeptides, including VIP [19] or the endogenous opioid enkephalin [20]. To date, the role of such modulatory actions of VIP neurons in learning and memory has received little attention.
We explored the potential role of enkephalin release from CA2 VIP interneurons [21] in regulating the activity of CA2 pyramidal neurons and CA2-dependent social memory formation. This study was motivated by the finding that enkephalin was previously implicated in a form of synaptic plasticity involving long-term depression of feedforward inhibition from parvalbumin-expressing interneurons (PV neurons) onto CA2 pyramidal neurons that requires the activation of PV neuron delta opioid receptors (DORs) [22, 23]. This plasticity can be triggered by strong tetanic stimulation of either of the two major sources of excitatory input to CA2 pyramidal neurons – the Schaffer collaterals (SC) from CA3 pyramidal neurons [22] and the entorhinal cortical (EC) perforant path (PP) [24] – or by weak but precisely timed paired activation of the two inputs, a process termed input-timing dependent plasticity or ITDP [23]. Importantly, this form of plasticity has been implicated in CA2-dependent social memory storage [23]. However, neither the source of enkephalin nor its mechanism of release by neural activity during social learning is known. Moreover, as CA2 dysfunction has been implicated in schizophrenia in humans [25, 26] and is thought to contribute to social memory deficits in a mouse model of the 22q11.2 microdeletion [27, 28] — one of the greatest genetic risk factors for schizophrenia —, an enhanced understanding of how endogenous opioids regulate CA2 function will contribute to our understanding of disease mechanisms.
Here we show that VIP interneurons within or bordering on CA2 provide both the source of enkephalin for inhibitory plasticity and the likely site for integration of the SC and EC inputs. Moreover, we find that enkephalin release from these VIP neurons is necessary for social memory storage. Finally, CA2 VIP neurons show increased activity during social exploration of a novel conspecific unlike during exploration of an object or familiar conspecific. Thus, our study indicates how activity-dependent release of a neuropeptide intrinsic to the hippocampus can be an important mechanism by which a defined class of inhibitory neurons contributes to declarative memory encoding.
Results
VIP and enkephalin expression overlap throughout the CA fields
Given that some VIP interneurons in the hippocampus express enkephalin [20], we explored the hypothesis that these neurons play a key role in both ITDP and social memory. To test this idea, we performed in situ hybridization against the mRNA coding for the enkephalin precursor peptide preproenkephalin (Penk) and the mRNA coding for the vasoactive intestinal peptide (Vip, Table 1). We quantified the density of Penk-expressing neurons in each sub-region of the hippocampus proper (Fig. S1a) and found the highest density of somas in the pyramidal layer of CA1 and CA2, while CA3 showed very few neurons expressing Penk (Fig. S1b). We then quantified the number of cells with overlapping expression (Fig. 1a–b). Penk+ and Vip+ neurons showed extensive colocalization in CA2 (72%, 107 cells, 5 mice; Fig. 1c) and CA1 (67%, 72 cells, 3 mice; Fig. S1c). Interestingly, the dorsal blade of the dentate gyrus showed many cells labelled for Penk but not Vip. We then performed immuno-histochemistry against the ENK and VIP proteins (Fig. 1d–f) and found an even higher number of overlapping cells in CA2 (86%, 54 cells, 3 mice). These results suggest that VIP neurons are the major source of ENK in CA2 and that the Vip-Cre mouse line can be used to target viral expression to ENK+ neurons in CA2. Therefore, we performed local injections of Cre-dependent viral vectors in the CA2 region of Vip-Cre mice, which usually resulted in a relatively restricted pattern of viral expression to VIP neurons in CA2 and neighboring CA3a regions (Fig. 1g). Mice that showed expression outside of these regions or failed to express in CA2 were excluded from the analysis. Because the Vip-Cre mouse line has been reported to have a reduction in Vip mRNA level in the suprachiasmatic nucleus [29], we quantified Vip mRNA level in CA2 using in situ hybridization and observed a 22% decrease in signal (Fig. S1d). Despite this decrease, the Vip-Cre mice had no obvious deficit in ITDP (Fig. 2b) or social memory (Fig. 5). However, to control for any potential effects of the decrease in Vip mRNA, we always compared results using injection of a given viral vector versus a control viral vector (e.g., expressing GFP) in the CA2 region of the Vip-Cre mouse line.
Table 1:
Key resources
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Met/Leu-enkephalin(NOC1/35) antibody produced in mouse | Santa Cruz Biotechnologies | Cat# sc-47705 RRID:AB_2161515 |
| Leu Enkephalin antibody | Fitzgerald Industries International | Cat# 10-L20A RRID: AB_1287677 |
| VIP antibody produced in rabbit | Immunostar | Cat# 20077 RRID:AB_572270 |
| PCP4 antibody produced in rabbit | Sigma-Aldrich | Cat# HPA005792 RRID:AB_1855086 |
| Anti-Reelin/CR-50 antibody produced in mouse | MBL International | Cat# D223–3 RRID:AB_843523 |
| Calbindin 1 antibody produced in rabbit | Abcam | Cat# ab11426 RRID:AB_298031 |
| GFP antibody produced in chicken | AVES Labs | Cat# GFP-1020 RRID:AB_10000240 |
| RFP antibody produced in rabbit | Rockland | Cat# 600-401-379 |
| GFP polyclonal antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A21311 RRID:AB_221477 |
| Streptavidin conjugated to Alexa 488 | Thermo Fisher Scientific | Cat# S32354 RRID:AB_2315383 |
| Streptavidin conjugated to Alexa Fluor 555 | Thermo Fisher Scientific | Cat# S21381 RRID:AB_2307336 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 568 conjugate | Thermo Fisher Scientific | Cat# A11011 RRID:AB_143157 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 633 conjugate | Thermo Fisher Scientific | Cat# A21070 RRID:AB_2535731 |
| Goat anti-mouse IgG (H+L) Secondary Antibody, Alexa Fluor 633 conjugate | Thermo Fisher Scientific | Cat# A21052 RRID:AB_2535719 |
| Goat anti-Mouse IgG1 Secondary Antibody, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | Cat# A21121 RRID:AB_141514 |
| Goat anti-Mouse IgG1 Secondary Antibody, Alexa Fluor 633 conjugate | Thermo Fisher Scientific | Cat# A21126 RRID:AB_2535768 |
| Goat Anti-Chicken IgG (H+L) Secondary Antibody, Alexa Fluor 488 Conjugate | Thermo Fisher Scientific | Cat# A11039 RRID:AB_142924 |
| In situ hybridization probes | ||
| Mm-Vip | ACD Bio | Cat# 415961 |
| GFP-O2-C2 | ACD Bio | Cat# 409011-C2 |
| Mm-Penk-C3 | ACD Bio | Cat# 318761-C3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Colchicine | Tocris | Cat# 13–641-G |
| Naltrindole | Sigma-Aldrich | Cat# N115 |
| CNO | Cayman Chemical | Cat# 16882 |
| CNQX | Cayman Chemical | Cat# 14618 |
| D-AP5 | Cayman Chemical | Cat# 14539 |
| CGP 55845 | Tocris | Cat# 1248 |
| SR 95531 | Tocris | Cat# 1262 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mus musculus | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J Mus musculus |
Jackson Laboratories | RRID:IMSR_JAX:007914 |
| Viptm1(Cre)Zjh/J Mus musculus | Jackson Laboratories | RRID:IMSR_JAX: 010908 |
| B6.Cg-Tg(Amigo2-Cre)1Sieg/J Mus musculus | Jackson Laboratories | RRID:IMSR JAX:030215 |
| Recombinant DNA | ||
| PX552 | Addgene | Cat# 60958 |
| AAV2/8 U6.shRNAPenk.hSyn.GFP.WPRE | Custom from PX552 | n/a |
| AAV2/8 U6.shRNCtrl.hSyn.GFP.WPRE | Custom from PX552 | n/a |
| AAV2/8 U6.FLEX.shRNAPenk.hSyn.GFP.WPRE | Custom from pUC | n/a |
| AAV2/8 U6.FLEX.shRNACtrl.hSyn.GFP. WPRE | Custom from pUC | n/a |
| AAV2/5 EF1a.DIO.eYFP | UNC vector core | Addgene Cat#27056 |
| AAV2/9 CBA.FLEX.Arch3.0-GFP.WPRE.SV40 | UPenn vector core | Addgene Cat#22222 U Penn Cat# AV-5-PV2432 |
| AAV2/9 CAG.FLEX.eGFP.WPRE.bGH | Addgene | Cat# 51502-AAV9 |
| AAV2/5 EF1a.DIO.hChR2(E123T/T159C)-eYFP | Addgene | Cat# 35509-AAV5 |
| AAV2/5 hSyn.DIO.hM4D(Gi)-mCherry | Addgene | Cat# 44362-AAV5 |
| AAV2/8 hSyn.DIO.hM4D(Gi)-mCherry | Addgene | Cat# 44362-AAV8 |
| AAV2/8 hSyn.DIO.mCherry | Addgene | Cat# 50459-AAV8 |
| AAV2/8 syn.DIO.TVA.2A.GFP.2A.B19G | UNC/Wickersham | Addgene Cat# 52473 |
| Rabies SAD.B19.EnvA.ΔG.mCherry | Salk vector core | Addgene Cat# 32636 |
| AAV2/2 CAG.DIO.ArchT-TdTomato | UNC/Boyden | n/a |
| AAV2/2 CAG.DIO.TdTomato | UNC/Boyden | n/a |
| AAV2/1.syn.FLEX.GCaMP6f.WPRE.SV40 | Addgene | Cat# 100833-AAV1 |
| Software | ||
| AxoGraph | AxoGraph | 1.6.4 |
| PRISM 8 | Graphpad | 8.4.2 (464) |
| Microsoft office Word | Microsoft | 2016 |
| Microsoft office Exel | Microsoft | 2016 |
| Adobe illustrator Illustrator | Adobe | 2020 v24.1 |
| FIDJI | N/A | N/A |
| MATLAB | Mathworks | 2018 |
| Inscopix Data Acquisition & Analysis Software | Inscopix | 1.3.1 |
| CaImAn | Ref [68] | https://github.com/flatironinstitute/CaImAn |
| ANY-maze | Stoelting Co. | 4.99 |
| DeepLabCut | Ref [69] | http://www.mousemotorlab.org/deeplabcut |
Figure 1: VIP and enkephalin expression overlaps in the hippocampal CA2 region.
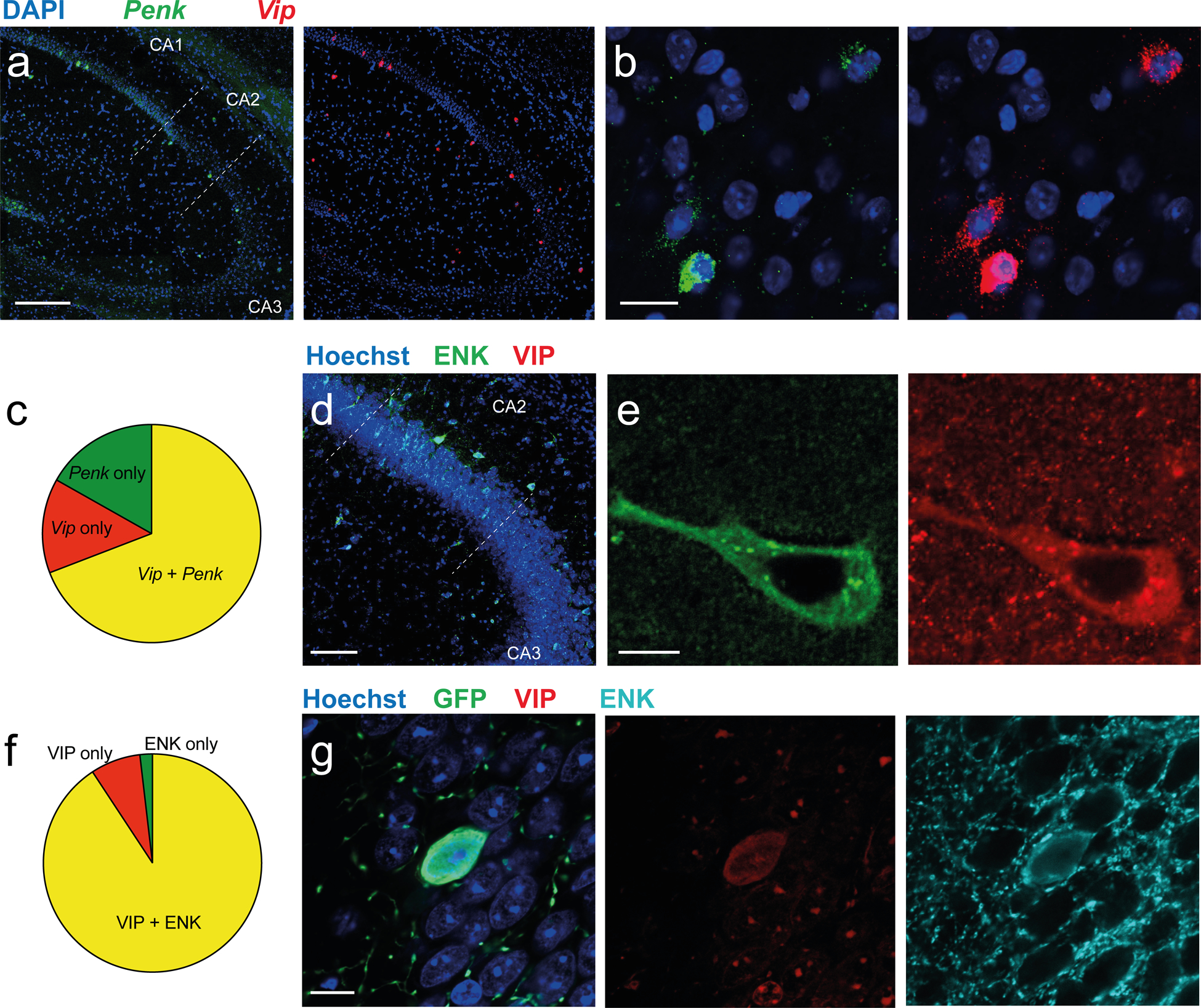
a-b. In situ hybridization images of dorsal hippocampal slices (a) and CA2 (b) showing staining for Penk and Vip mRNAs. c. Quantification of the fraction of neurons positive for Penk alone (green), for Vip alone (red) and for both Penk and Vip (yellow) across all layers of the CA2 region. d-e. Immunohistochemistry images of dorsal hippocampal slices with antibodies against ENK (Santa Cruz antibody) and VIP. f. Quantification of the fraction of hippocampal cells positive for ENK alone, VIP alone, and co-labeled for both ENK (Fitzgerald antibody) and VIP in the CA2 region. g. Immunohistochemistry for ENK and VIP in the CA2 region of a Vip-Cre mouse injected in dorsal CA2 with AAV2/5 EF1a.DIO.eYFP. Scale bars: a 200 μm, b 20 μm, d 100 μm, e and g 10 μm.
Figure 2: VIP neurons are necessary and sufficient to induce CA2 ITDP through DOR activation.

a. Vip-Cre mice injected in dorsal CA2 with AAV2/9 CBA.FLEX.Arch3.0-GFP.WPRE.SV40 to express Arch-3.0-GFP or with AAV2/9 CAG.FLEX.eGFP.WPRE to express eGFP as a control (top). Image shows immunohistochemistry in acute hippocampal slice labelling a VIP neuron expressing GFP (green) next to a CA2 PN filled with biocytin during a patch-clamp recording (red). b. Average normalized SC-evoked PSP amplitude in CA2 PNs before and after electrical pairing of EC and SC inputs to induce ITDP in presence of continuous illumination with yellow light (which activates Arch3.0). The PSP showed a significant progressive potentiation following pairing, indicative of ITDP, in slices in which GFP was expressed in VIP neurons (black). In contrast, ITDP was absent in slices expressing Arch3.0 (red). Inset shows example PSPs before and after pairing. c. Vip-Cre mice injected in dorsal CA2 with AAV2/5 EF1a.DIO.hChR2(E123T/T159C)-eYFP (top). Immunohistochemistry of acute hippocampal slice showing labelling of VIP neurons expressing GFP next to a recorded CA2 PN filled with biocytin (bottom). d. Average normalized SC PSPs in CA2 PNs evoked by electrical stimulation before and after photostimulation of VIP neurons expressing ChR2, with or without 0.1 μM naltrindole in bath. Photostimulation of VIP neurons was sufficient to enhance the PSP, suggestive of ITDP, in the absence (red) but not presence (black) of naltrindole. Inset shows example PSPs before and after photostimulation. e. Photostimulation of VIP neurons expressing ChR2 induces long-term depression of SC-evoked IPSC recorded from CA2 PNs voltage-clamped at +10 mV (same viral injection than panel d). Scale bars: a 100 μm, c 200 μm, b and d 50 ms / 2 mV, e 100 ms / 100 pA.
Enkephalin release from VIP INs in CA2 induces ITDP
To determine whether VIP neurons are required for ITDP, we took an optogenetic approach using local injections of a Cre-dependent AAV expressing the Archaerhodopsin3.0 [30] tagged with GFP (Arch3.0-GFP) or GFP only (control) in the CA2 region of Vip-Cre mice (Fig. 2a). We prepared acute hippocampal slices 2–3 weeks after virus injection and obtained whole-cell patch-clamp recordings from CA2 pyramidal neurons (PNs) (Fig. 2a) to monitor synaptic responses to electrical stimulation of the SC inputs. After a 5-min baseline recording, we used a standard ITDP induction protocol [23, 31] in which an electrical stimulus to PP inputs was followed 20 ms later by a stimulus to SC inputs, with the pairings repeated at 1 Hz for 90 s. The CA2 region was illuminated with 590 nm light to inhibit VIP neurons, starting 2 s before the pairing protocol and then throughout the 90 s induction period. In control slices, the ITDP induction protocol increased the amplitude of the CA2 PSP elicited by SC stimulation to 218 ± 38% of baseline after 30 min (n = 6; Wilcoxon signed rank test, p = 0.03), similar to our previous observations [23]. In contrast, in slices from animals in which VIP neurons in CA2 expressed Arch-GFP, the normal enhancement in PSP amplitude in response to the ITDP induction protocol was completely blocked (PSP = 98 ± 11% of baseline, n = 6; Wilcoxon signed rank test, p = 0.7; Mann-Whiney test compared to control slices, p = 0.004; Fig. 2b). These data show that the induction of ITDP requires VIP neuron to be active during pairing. This is in stark contrast to our previous findings that Arch-mediated inhibition of either CA2 pyramidal neurons or PV interneurons during the pairing protocol did not inhibit ITDP [23].
Is activation of VIP neurons in CA2 sufficient to induce ITDP? To explore this question, we used optogenetics to selectively activate VIP neurons by injecting in CA2 of Vip-Cre mice a Cre-dependent AAV expressing Channelrhodopsin-2 tagged with YFP (ChR2-YFP; Fig. 2c). We first asked whether optogenetic stimulation was indeed able to activate VIP neurons. To examine this question, we took advantage of the fact that the VIP neurons preferentially inhibit other interneurons [13], thereby reducing feedforward inhibition of pyramidal neurons. Thus, activation of VIP neurons during stimulation of feedforward inhibitory inputs should decrease the inhibitory postsynaptic current (IPSC) recorded in a CA2 PN. We obtained whole-cell recordings from CA2 PNs in hippocampal slices from Vip-Cre mice expressing ChR2 in VIP neurons. We elicited IPSCs in CA2 PNs using direct electrical stimulation of local inhibitory neurons using a stimulating electrode located in stratum pyramidale, with blockers of excitatory transmission in the bath. Electrical stimulation by itself induced a large IPSC in the PNs, as previously described [23, 32]. The IPSC was significantly reduced when we applied a train of 10, 1-ms blue-light pulses at 20 Hz to activate VIP neurons just prior to electrical stimulation, as expected for activation of VIP neurons. (IPSC with light = 80 ± 5% of control, n = 7; Wilcoxon signed rank test, p = 0.015; Fig. S2a).
Next, we examined whether optogenetic activation of VIP neurons expressing ChR2-YFP was sufficient to induce ITDP by recording the electrically-evoked SC PSP from CA2 PNs (in the absence of antagonists). Following a 5-min baseline recording period, we applied two 1-ms pulses of blue light separated by 20 ms for 90 s at 1 Hz to mimic the ITDP induction protocol using paired electrical stimulation. SC-elicited PSPs in slices injected with ChR2-YFP increased markedly to 217 ± 30% of baseline 30 min after paired activation (n = 8; Wilcoxon signed rank test, p = 0.02; Fig. 2d). To test whether the increase in the SC PSP following VIP neuron excitation depended on DOR activation (similar to CA2 ITDP) we performed the light stimulation in ChR2-YFP expressing slices in the presence of 5 μM naltrindole, a DOR antagonist that blocks CA2 ITDP [23]. Bath-application of naltrindole fully prevented the increase in the SC response following optogenetic activation of the VIP neurons, confirming the importance of DORs for this enhancement (108 ± 12% of baseline, n = 8; Wilcoxon signed rank test, p = 0.5; Mann-Whitney test compared to no naltrindole, p = 0.003; Fig. 2d).
To determine whether optogenetically induced ITDP results from the depression of feed-forward inhibition as does electrically induced ITDP, we repeated this experiment under voltage-clamp conditions, with the CA2 PN held at +10 mV to directly monitor the IPSC. Paired photostimulation of ChR2-YFP-expressing VIP neurons decreased the IPSC elicited by SC stimulation to 59 ± 18% of baseline 30 min after optogenetic stimulation (n = 9; Wilcoxon signed rank test, p = 0.008), similar to our findings with electrically induced ITDP [23]. Moreover, the addition of 0.1 μM naltrindole to the bath blocked the suppression (IPSC = 114 ± 12% of baseline, n = 5; Wilcoxon signed rank test, p = 0.4; Mann-Whiney test compared to absence of naltrindole, p = 0.02; Fig. 2e). Taken together, these data show that excitation of VIP neurons is both necessary and sufficient for the generation of DOR-dependent ITDP in CA2.
As VIP neurons form inhibitory synapses preferentially on other interneurons [14], including PV interneurons [13], we sought to determine whether GABA release during the induction of plasticity was required for CA2 ITDP. We therefore used paired electrical stimulation of the EC and SC inputs to induce ITDP with GABAergic transmission inhibited specifically during the induction protocol using 2 μM SR 95531 and 1 μM CGP 55845 to block GABAA and GABAB receptors, respectively. Application of the blockers prior to the induction of ITDP abolished the IPSC elicited in CA2 PNs by SC stimulation as expected. We then applied the ITDP electrical pairing protocol in the maintained presence of the GABA antagonists, and washed out the drugs 5 min after the pairing protocol to allow inhibitory transmission to recover, as previously described for CA1 ITDP [33]. GABA receptor blockade during the pairing protocol failed to suppress the ISPC reduction; 30 min after GABA blocker washout, the IPSC was reduced to 55 ± 19% of its initial level (− 20 ms pairing, n = 6 cells; Wilcoxon signed rank test, p = 0.03, Fig. S2b), similar to the reduction when the pairing protocol was applied in the absence of the GABA antagonists [23]. To ensure that there was sufficient time for full washout of the antagonists, we applied the antagonists during paired electrical stimulation but reversed the order of pairing (SC stimulus before PP stimulus, +20 ms pairing) since this procedure fails to induce ITDP [23]. In this case the SC-induced IPSC was maintained near its initial baseline level 30 min after the pairing protocol (106 ± 16%, n = 6 cells; Wilcoxon signed rank test, p = 0.6), arguing against a prolonged effect of the antagonists. Overall, our data suggest that, unlike enkephalin, GABAergic transmission is not required for the induction of ITDP.
Does the effect of optogenetic activation of VIP neuron result from release of enkephalin from these cells? To address this question, we synthesized an shRNA targeting Penk mRNA (shRNAPenk) [34], packaged it into an AAV also expressing GFP, and injected the virus into CA2. We also prepared a control AAV expressing a scrambled version of the shRNA (shRNACtrl, Fig. 3a) [35]. To assess the efficacy of the shRNA approach, we performed in situ hybridization to measure Penk, Gfp and Vip mRNA levels in slices expressing shRNAPenk or shRNACtrl (Fig. 3b–d). Widespread injection of AAV expressing shRNAPenk caused a marked reduction in Penk expression throughout the infected region (Fig. S3a). Importantly, Vip+ cells expressing Gfp (marker for shRNAPenk) showed a 3-fold decrease in the intensity of Penk labelling compared to nearby Vip+ cells that had not taken up the virus and did not express Gfp (Fig. 3b–d). Moreover, this effect was not seen in mice injected with the AAV expressing shRNACtrl (Fig. 3c–d). These results indicate that our strategy to reduce Penk mRNA levels in VIP interneurons is both specific and efficient.
Figure 3: Enkephalin is necessary for ITDP induction.
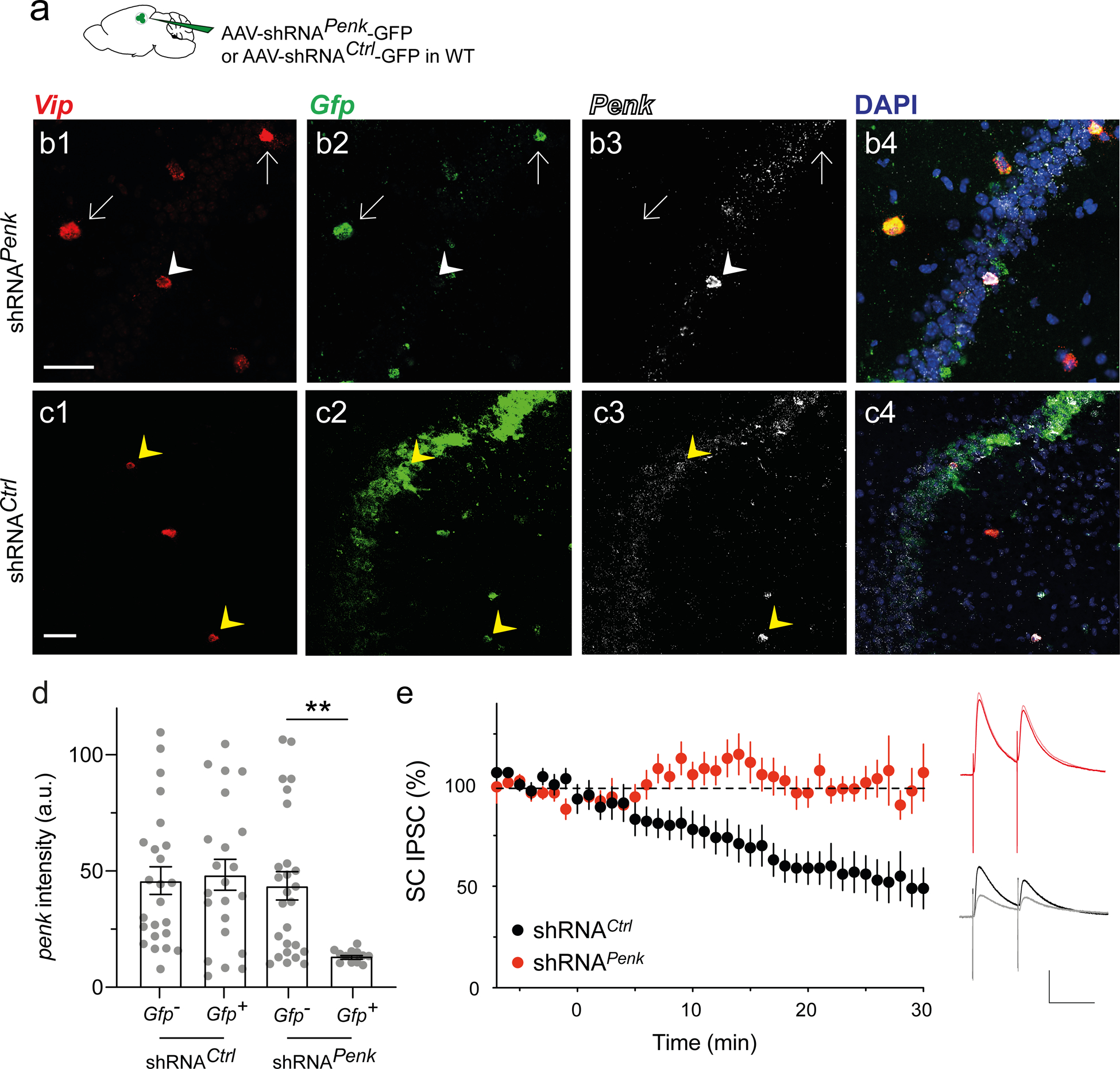
a. WT mice injected in dorsal CA2 with AAV2/8 U6.shRNAPenk.hSyn.GFP.WPRE to downregulate Penk or control AAV2/8 U6.shRNCtrl.hSyn.GFP.WPRE. b-c. In situ hybridization images of one hippocampal slice expressing shRNAPenk (b) and one expressing shRNCtrl (c) labelled for Vip, Gfp, and Penk mRNAs. White arrows denote Vip+ neurons that express shRNAPenk, showing reduced levels of Penk. White arrowheads denote a Vip+ neuron that does not express shRNAPenk and expresses Penk. Yellow arrowheads denote Vip+ neurons that express shRNACtrl and Penk. d. Quantification of Penk expression in Vip+ cells using in situ hybridization in slices from mice injected with AAV expressing control of Penk shRNAs. In each slice neurons were classified as to whether they were uninfected or infected with virus based on Gfp expression (3 mice per group; each point is from a different slice). Note reduction in Penk expression in neurons infected with Penk shRNA. e. Time course of SC IPSCs recorded in CA2 PNs before and after ITDP induction using paired electrical stimulation from slices expressing control or Penk shRNAs. Insets show sample IPSCs before and after ITDP induction in the two groups. Scale bars: b and c 50 μm, e 100 ms / 200 pA.
To examine the role of enkephalin release in ITDP, we prepared acute hippocampal slices from WT animals injected in the CA2 region with shRNAPenk or shRNACtrl expressing-viruses. We then induced ITDP using electrical pairing while monitoring the IPSC in CA2 PNs elicited by electrical stimulation of the SC. The IPSC in slices expressing shRNACtrl was decreased by the pairing protocol by an amount similar to what we previously reported (51 ± 11% of baseline, n = 6; Wilcoxon signed rank test, p = 0.03; Fig. 3e) [23]. In contrast, the pairing protocol caused no change in the SC-evoked IPSC in slices expressing shRNAPenk (102 ± 10% of baseline, n = 11; Wilcoxon signed rank test, p = 0.9; Mann-Whitney test compared to shRNACtrl, p = 0.003). We repeated this experiment in Vip-Cre animals using Cre-dependent viruses expressing the same shRNAs. As for WT mice, IPSCs recorded from CA2 PNs in slices expressing shRNACtrl were decreased by the ITDP pairing protocol (to 38 ± 4% of baseline, n = 7, Wilcoxon signed rank test, p = 0.02; Fig. S3b) while slices expressing shRNAPenk failed to exhibit ITDP (94 ± 31% of baseline, n = 4; Wilcoxon signed rank test, p = 0.8, Mann-Whitney test compared to shRNACtrl, p = 0.006). Taken together, these data suggest that enkephalin release from local VIP interneurons is necessary to trigger CA2 ITDP.
Integration of inputs by VIP interneurons in CA2
Since we found that activation of VIP neurons in CA2 is necessary for the ITDP induction, we next asked whether these neurons could be a site of integration of the SC and PP inputs. We first used a monosynaptic rabies virus tracing approach to determine which neurons form synapses onto CA2 VIP neurons. We injected an AAV helper virus expressing the TVA receptor, G protein and GFP in CA2 of Vip-Cre mice, followed 15 days later by injection of G-deleted pseudotyped rabies virus expressing mCherry (EnvA-ΔG-mCherry; Fig. 4a). We observed mCherry+ cells in CA3 and in layer II of the medial and lateral entorhinal cortex (MEC and LEC; Fig. 4b–c, Fig. S4a). Because CA3 is physically close to CA2, we confirmed that the CA3 GFP+ cells were labelled retrogradely based on the fact that they did not express the helper virus marker GFP (Fig. 4b2).
Figure 4: VIP neurons receive excitatory inputs from both CA3 and EC layer II stellate cells.
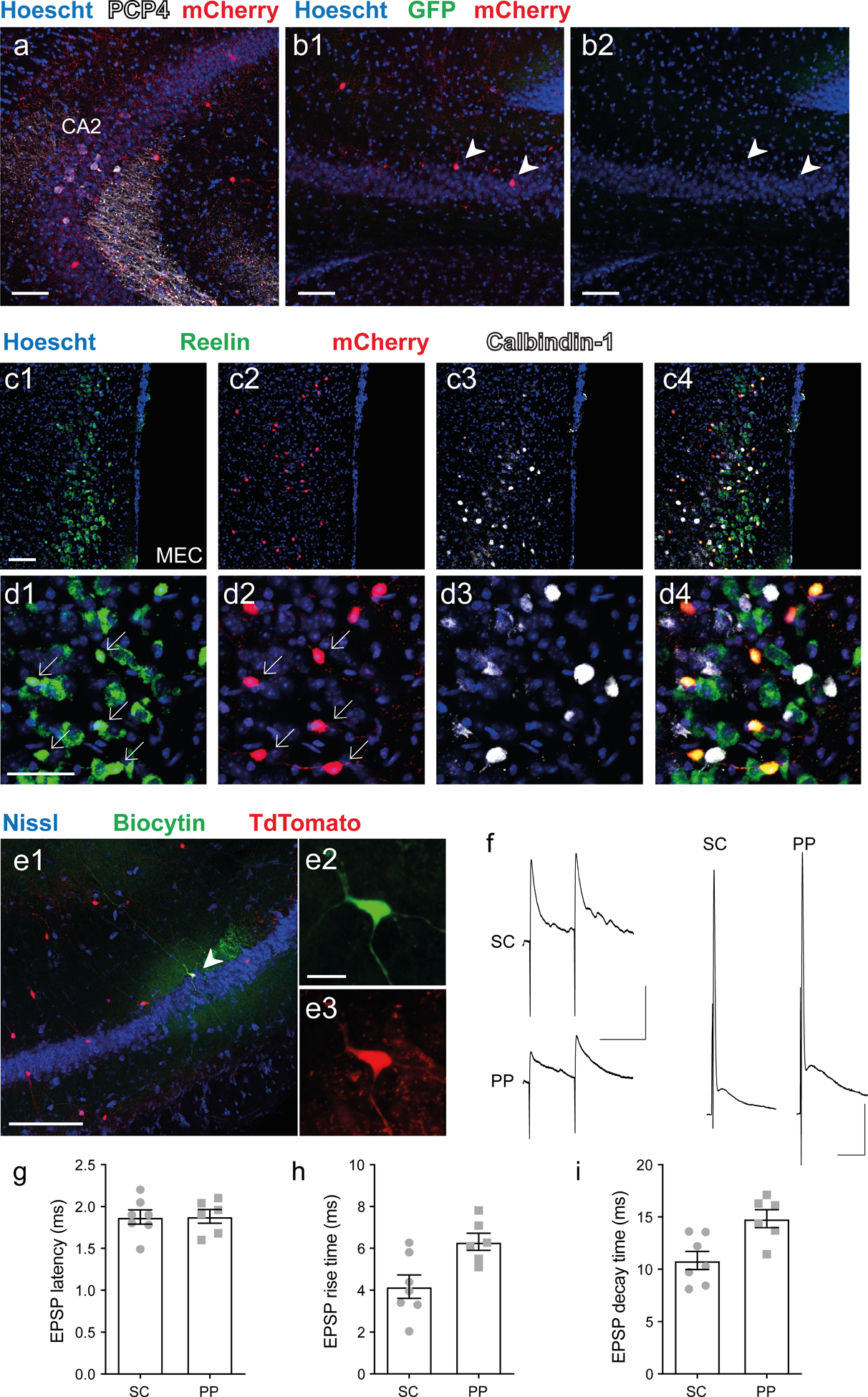
a-d. Vip-Cre mouse injected in dorsal CA2 with AAV2/8 syn.DIO.TVA.2A.GFP.2A.B19G and rabies virus SAD.B19.EnvA.ΔG.mCherry to label monosynaptic inputs to VIP neurons. Immunohistochemistry of hippocampal and MEC slices showing: (a) CA2 PN marker PCP4 and rabies-based mCherry-labeled VIP neurons in CA2 region; (b) mCherry-labeled pyramidal neurons in CA3 (arrowheads); (c) Low- and (d) high-magnification images of MEC showing mCherry labeling of reelin-expressing LII stellate cells but not of calbindin-expressing LII pyramidal cells. e. Low (e1) and high (e2–3) magnification immunohistochemistry images of an acute hippocampal slice from a Vip-Cre × Ai14 mouse showing biocytin (e2) and TdTomato labeling (e3) of a patch-clamped VIP neuron. f. Representative PSPs recorded from VIP neuron following electrical stimulation of PP and SC inputs using low intensity stimulation that evoked subthreshold PSPs (left) or high intensity stimulation that evoked a spike (right). g-i, PSP parameters elicited by PP or SC stimulation. g. PSP latency. h. PSP rise time. i. PSP decay time. Scale bars: a, b1, b3, and c 100 μm; d 50 μm; e1 200 μm; e2 20 μm; f left 100 ms / 5 mV; f right 10 ms / 10 mV.
To identify the rabies labeled cells in LII of entorhinal cortex, we co-labeled neurons with either an anti-reelin antibody or an anti-calbindin-1 antibody, markers for stellate/fan cells and pyramidal cells, respectively [36]. All rabies-labeled mCherry+ cells in LEC and MEC co-expressed reelin but not calbindin-1 (Fig. 4c–d and Fig. S4), thus confirming that CA2 VIP neurons receive direct inputs from the LII stellate and fan cells of the medial and lateral entorhinal cortices, respectively. Taken together, the retrograde rabies tracing experiment demonstrate that CA2 VIP neurons receive most of their direct inputs from CA3 and layer II stellate/fan cells of the entorhinal cortices, which is in accordance with a previous characterization of CA2 inputs required for ITDP [23].
We then crossed the VIP-Cre mouse line with the Ai14 TdTomato reporter line to obtain patch-clamp recordings from identified CA2 VIP neurons so as to determine whether they receive functional synaptic inputs from EC LII and CA3 neurons (Fig. 4e). We obtained whole-cell current-clamp recordings from TdTomato+ neurons and first characterized their intrinsic properties (Fig. S5). Due to their high input resistance (391 ± 24 MΩ, n = 15), CA2 VIP neurons readily fired spikes in response to direct current injection. We next asked whether a VIP neuron could integrate PP and SC inputs, using a stimulating electrode in the stratum lacunosum moleculare (SLM) to activate the PP inputs and a second stimulating electrode in SR to activate the SC inputs (Fig. 4F–I). Stimulation of either the SC or PP inputs elicited large PSPs with short latencies in all VIP neurons examined (SC = 1.49 ± 0.08 ms, n = 7 and PP = 1.60 ± 0.08 ms, n = 6, Fig. 4g), suggesting monosynaptic connections. In addition, CA2 VIP neurons fired action potentials in response to electrical stimulation of either the SC or PP inputs, with 10 out of 10 neurons firing to SC stimulation and 9 out of 10 cells firing to PP stimulation. In conclusion, CA2 VIP neuron intrinsic and synaptic properties enable them to readily fire action potentials following activation of their SC and PP inputs.
VIP neuron activity is necessary for social memory storage
Our previous results indicate that ITDP and DOR activation are necessary for social memory storage [23]. Here, we asked whether the activity of VIP neurons in CA2 and their release of enkephalin during social interactions are also critical for social memory. To investigate these questions, we examined the effect of silencing the VIP neurons during an open arena test for social memory (Fig. 5a) [37]. In this test a subject mouse is exposed to two novel stimulus mice inside wire cup cages in opposite corners of a square open arena. After 5 min of social exploration in a social learning trial, the subject mouse is removed from the arena and one of the two stimulus mice is replaced by a third novel mouse. After a 30 min inter-trial interval the subject mouse is reintroduced into the arena in a social memory recall trial. Social memory normally manifest as a greater time spent by the subject mouse exploring the third novel stimulus mouse (tN) compared to the now familiar stimulus mouse that was present in the learning trial (tF). We quantified the preference for exploring the novel mouse by calculating a discrimination index: DI = (tN – tF) / (tN + tF).
Figure 5: Silencing VIP neurons impairs social memory formation.
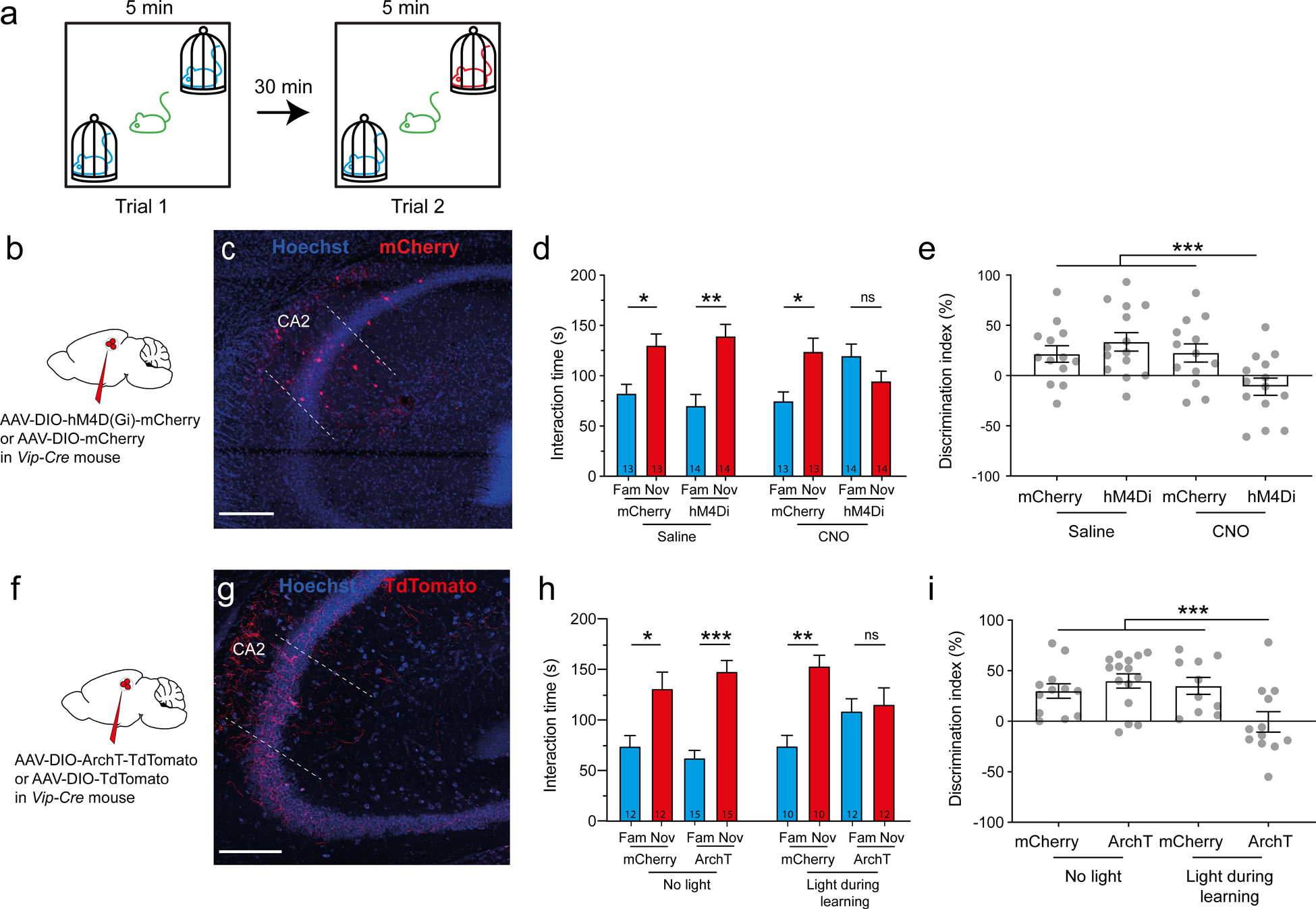
a. Schematic of the social memory test. In trial 1 a subject mouse explored two novel mice for 5 min. The subject was then removed from the arena. In trial 2 (memory recall trial), after a 30 min intertrial interval the subject mouse was reintroduced into the arena for 5 min, during which time it was allowed to explore one of the two mice encountered in trial 1 and a new novel mouse. b. Vip-Cre mouse injected in dorsal CA2 with AAV2/8 hSyn.DIO.hM4D(Gi)-mCherry to express the inhibitory DREADD (hM4Di) or AAV2/8 hSyn.DIO.mCherry to express mCherry in the control group. c. Immunohistochemistry showing mCherry expression in hippocampal slice. d. Interaction time with novel or familiar mouse during trial 2 in mice expressing mCherry or hM4Di; both groups were injected with saline (left groups) or 5 mg/kg of the DREADD agonist CNO (right group). Number of mice indicated in each bar. Paired t-tests: p = 0.02, p = 0.0001, p = 0.004 and p = 0.8. e. Social memory assessed by discrimination index of four groups during recall trial. ANOVA F(3,50) = 4.991, p = 0.004; Dunnett’s multiple comparisons tests: hM4Di + CNO vs. mCherry + saline, p = 0.03; hM4Di + CNO vs. mCherry + CNO, p = 0.002; hM4Di + CNO vs. mCherry + CNO, p = 0.02. f. Vip-Cre mouse injected in dorsal CA2 with AAV2/2 CAG.DIO.ArchT-TdTomato or AAV2/2 CAG.DIO.TdTomato. g. Immunohistochemistry of hippocampal slice. h. Interaction time with novel or familiar mouse during recall trial in mice expressing mCherry or ArchT in absence (left groups) or present (right groups) of yellow light. Paired t-tests: p = 0.01, p = 0.005, p = 0.03 and p = 0.2. i. Discrimination index during recall. ANOVA F(3,45) = 5.007, p = 0.004; Dunnett’s multiple comparisons tests: ArchT + light vs. mCherry + no-light, p = 0.03; ArchT + light vs. ArchT + no-light, p = 0.002; ArchT + light vs. mCherry + light, p = 0.02. Scale bars: a and b 200 μm.
We first confirmed the role of CA2 PNs in this task using a chemogenetic approach in which an inhibitory DREADD (hM4Di) [38] was expressed in CA2 PNs. We injected a Cre-dependent AAV expressing hM4Di-mCherry in the CA2 region of the Amigo2-Cre mouse line, which expresses Cre selectively in CA2 PNs (Fig. S6a–b) [39]. Three weeks later, mice were injected intraperitoneally with either saline or 5 mg/kg of the DREADD agonist CNO 20 min prior to conducting the social memory test. We compared social memory performance in three control groups (WT littermate mice given saline, Cre+ mice given saline, WT littermate mice given CNO) with the test group (Cre+ mice given CNO). Importantly, during learning or recall, the total exploration time of the mice was similar across groups (Fig. S6c–d), suggesting that CA2 PN silencing did not affect social exploration. During recall, all 3 control groups spent more time exploring the novel animals (DI of the 3 control groups pulled together: 40 ± 3%, N = 48; Fig. S6e), which is a hallmark of social memory. However, in the test group in which CA2 was silenced, the subject mice explored the novel and familiar mice to the same extent (Fig. S6e), so that the discrimination index was not different from zero (DI = −5 ± 10%, N = 12; t-test compared to control groups, p < 0.0001; Fig. S6f). This is consistent with previous studies showing that social memory is abolished when CA2 is silenced [39–41]. Taken together, these data confirm that this new task of social memory is indeed dependent on CA2.
Next, we examined the effect of silencing VIP neurons in CA2 on social memory. Since VIP-Cre mice are homozygous for Cre, we injected mice in the CA2 region with Cre-dependent AAVs expressing either hM4Di-mCherry or mCherry (Fig. 5b), which resulted in viral expression in VIP neurons in CA2 and neighboring CA3a (Fig. 5c). We then administered saline or 5 mg/kg CNO 20 min prior to testing for anxiety and social behavior. First, we examined the mice in an open field test by measuring the total distance travelled (Fig. S7a) and the time spent in the center and surround of the open space (Fig. S7b–c). We saw no difference in open field measures upon VIP neuron silencing, which indicates that locomotion and anxiety were not affected. Next, we ran the mice in the social memory task described above. All groups showed similar duration of social investigation during learning and recall trials (Fig. S7d–e), demonstrating that their motivation to explore was not affected by the manipulation. However, during recall, whereas control groups showed a marked preference for the novel animal, the VIP-silenced test group showed no social novelty preference (Fig. 5d), indicating a deficit in social memory (DI of the 3 control groups pulled together: 26 ± 5%, N = 40 versus DI of test group: −11 ± 8%, N = 14; t-test, p = 0.0005; Fig. 5e). Taken together, these results indicate that the activity of VIP neurons in the CA2/CA3a region is necessary for social memory.
In the above test, the VIP neurons were silenced during both the learning and recall trials so we could not distinguish whether these neurons were important for the memory acquisition and consolidation (learning), recall, or both. To explore whether VIP neurons are necessary for social memory learning, we used an optogenetic approach in which the social memory test was performed on mice expressing ArchT in VIP neurons in CA2/CA3a (Fig. 5f–g). We selectively illuminated the CA2 regions with yellow light during the learning phase of the trial. All groups showed a similar extent of social investigation during learning and recall trials (Fig. S7f–g). However, silencing VIP neurons during the learning trial significantly impaired social memory in ArchT-expressing mice compared to mCherry-expressing mice and compared to the same groups that were not illuminated (Average DI for the 3 control groups: 35 ± 5%, N = 37; DI for test group: −1 ± 10%, N = 12; t-test, p = 0.0004; Fig. 5h–i). Overall, this optogenetic silencing experiment allows us to conclude that the activity of VIP neurons in the CA2 region is necessary for the formation of social memory.
Enkephalin release from VIP neurons is required for social memory
The above experiments indicate that VIP interneurons are necessary for social memory storage. However, these experiments do not allow us to conclude whether it is enkephalin release that is important for social memory, and if so, whether the enkephalin is released from VIP neurons. We first determined the general importance of enkephalin using the shRNA strategy to knock down levels of Penk mRNA in the CA2 region as described above (Fig. 3). WT littermates were injected in CA2 with AAVs expressing shRNAPenk or shRNACtrl, and after 3 weeks were tested for social behavior (Fig. 6a). All groups showed a similar extent of social investigation during both learning and recall trials (Fig. S8a–c). However, whereas mice expressing shRNACtrl showed a normal preference for the novel mouse in the social memory recall trial (DI = 33 ± 5%, N = 16), the mice expressing shRNAPenk showed a reduced preference (DI = 10 ± 6%, N = 14; t-test for shRNAPenk versus shRNACtrl, p = 0.006; Fig. 6c–d). Altogether, these findings confirm that enkephalin expression in the CA2/CA3a region is necessary for social memory.
Figure 6: Enkephalin expression in CA2 region is necessary for social memory.
a-d. WT mice injected in dCA2 with AAV2/8 U6.shRNAPenk.hSyn.GFP or AA2/8 U6.shRNACtrl.hSyn.GFP. b. Immunohistochemistry of hippocampal slice. c. Interaction time with novel or familiar mouse during recall trial in mice expressing shRNAPenk or control shRNA. Number of mice indicated in each bar. Paired t-test, p < 0.0001 and p = 0.2. d. Discrimination index during recall trial. e-k. Vip-Cre mice injected in dCA2 with AAV2/8 U6.FLEX.shRNAPenk.hSyn.GFP or AA2/8 U6.FLEX.shRNACtrl.hSyn.GFP. f. Immunohistochemistry of hippocampal slice. g. Interaction time with novel or familiar mouse during recall trial. Paired t-test, p = 0.03 and p = 0.4. h. Discrimination index during recall trial. i. Interaction times during repetitive presentation of the same ovariectomized female (trials 1–4) and a novel ovariectomized female (trial 5), a test of social memory. j. Normalized interaction times during presentations. 5 mice per group. Two-way ANOVA; trial × virus F(3,52) = 1.9, p = 0.1; trial F(3,52) = 3.6, p = 0.02; virus F(1,52) = 9.7, p = 0.002. Scale bars: b and f 200 μm.
Although enkephalin is predominantly expressed by VIP neurons in CA2 (Fig. 1) a small fraction of enkephalin-expressing cells did not express VIP. Thus, to determine whether it is enkephalin expressed by VIP neurons that is important for social memory, we used Cre-dependent viruses expressing shRNAPenk or shRNACtrl in VIP-Cre mice to downregulate enkephalin selectively in VIP neurons. We recombined the shRNAs in a DIO construct that was packaged into AAV8 capsids (AAV2/8 FLEX.shRNA; see Methods). We then injected the Cre-dependent viruses into the CA2 region of Vip-Cre mice (Fig. 6e–f) and repeated the social memory test. Mice expressing shRNAPenk in VIP neurons showed no preference for the novel animal (DI = 2 ± 9%, N = 14) while control mice expressing shRNACtrl showed a marked preference (DI = 30 ± 5%, N = 14; t-test, p = 0.01; Fig. 6g–h). Total interaction times for learning and recall were similar across groups (Fig. S8d–f). Next, we verified that the loss of social memory was not secondary to a deficit in sociability. Mice were free to explore for 5 min a wire cup cage containing a mouse or an empty cup. Both groups showed the same preference for the cup containing the mouse (Fig. S8g–h). This is consistent previous reports that CA2 silencing has no effect on sociability [39]. Finally, we tested the effects of downregulating enkephalin in VIP neurons on a second social memory task: the repetitive presentation of an ovariectomized female (Fig. 6i) [39]. Mice expressing control shRNA showed a progressive decrease in interaction time with repeated presentations of the same female. Importantly, when a novel female was presented in the final trial, the interaction time increased to its initial level, demonstrating that the decreased interaction was not due to fatigue or loss of engagement in the task. In contrast, mice expressing shRNAPenk showed no decrease in interaction with a female during the repeated presentations (Fig. 6j). Taken together these experiments provide strong support for the view that enkephalin expression in CA2 VIP neurons is necessary for social memory.
Activity of CA2 VIP neurons increases during interaction with a novel mouse
To explore further how the activity of VIP neurons in the CA2 region may contribute to social memory, we imaged calcium signals in these neurons in freely-moving mice during social and non-social interactions. We injected a Cre-dependent AAV expressing GCaMP6f in the CA2 region of Vip-Cre mice and then implanted a GRIN lens and baseplate to attach a micro-endoscope (Fig. 7a). Test mice were habituated to an open area before a wire cup cage was introduced in the middle of the arena. The mice were free to interact with this novel object before a novel mouse was introduced under the cup. After 5 min, the novel mouse was exchanged for a familiar littermate (familiar mouse). The familiar mouse was then removed and the test mouse was free to interact for another 5 min with the now familiar wire cup (familiar object). Each session lasted 5 min. Mean calcium event rate across an entire session was greatest in the session with the novel mouse, but remained at baseline levels during the familiar mouse and familiar object sessions (Fig. 7b). Since the test mice spent more time interacting with the novel mouse than with the familiar mouse, the novel object or the familiar object (107 ± 12 s; 55 ± 19 s; 27 ± 11 s and 28 ± 7 s respectively, N = 6), it is possible that the VIP neurons responded equally during the different interactions, with the higher calcium event rate in the novel mouse session reflecting the greater fraction of time engaged in an interaction. To test this possibility, we restricted the measure of the calcium event rate to the periods of interaction with the wire cup (Fig. 7c). The calcium event rate was still greater during interactions with the novel mouse compared to interactions with the familiar mouse or novel or familiar object. We also compared the calcium event rates within a given session measured during interactions to baseline periods without interactions (Fig. 7d–g). The calcium event rate increased compared to baseline only during interactions with a novel mouse (Fig. 7e); no significant increase was observed during interactions with a familiar mouse Fig. 7f), novel object (Fig. 7d), or familiar object (Fig. 7g). Taken together, these data suggest that VIP neurons in CA2 encode social novelty, similar to findings for CA2 pyramidal neurons based on extracellular electrophysiological recordings [42].
Figure 7: Activity of VIP neurons in CA2 increases specifically during interaction with a novel mouse.
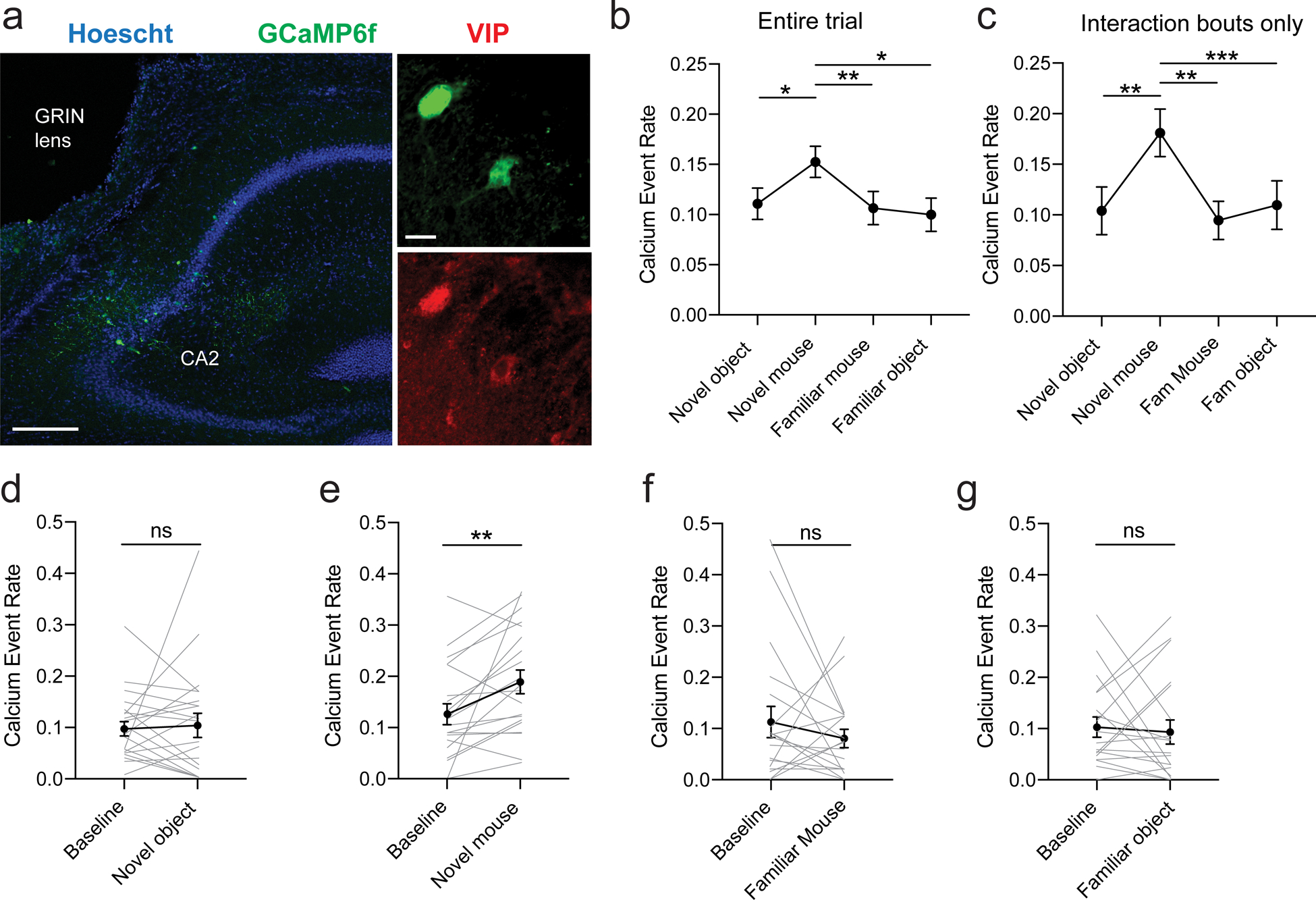
a. Immunohistochemistry of hippocampal slices in Vip-Cre mouse injected with AAV2/1.syn.FLEX.GCaMP6f.WPRE.SV40 in dorsal CA2. Scale bars: left 200 μm, right 20 μm. b. Calcium event rate (events/s) during each session (novel object, novel mouse, familiar mouse and familiar object, 21 cells, 4 mice). ANOVA: F(2.358, 41.16) = 7.054, p = 0.001. Dunnett’s multiple comparisons tests: novel mouse vs. novel object, p = 0.02; novel mouse vs. familiar mouse, p = 0.006; novel mouse vs. familiar object, p = 0.01. c. Calcium event rate during each interaction. ANOVA F(2.519, 50.39) = 6.961, p = 0.001. Dunnett’s multiple comparisons tests: novel mouse vs. novel object, p = 0.007; novel mouse vs. familiar mouse, p = 0.002; novel mouse vs. familiar object, p = 0.0002. d. Calcium event rate during novel object exploration compared to exploration of the arena within the same session (baseline). Individual lines represent cells. Paired t-test, p = 0.8. e. Calcium event rate during novel mouse exploration compared to baseline. Individual lines represent cells. Paired t-test, p = 0.007. f. Calcium event rate during familiar mouse exploration compared to baseline. Individual lines represent cells. Paired t-test, p = 0.3. g. Calcium event rate during familiar mouse exploration compared to baseline. Individual lines represent cells. Paired t-test, p = 0.8.
Discussion
In this study, we have expanded our understanding as to how inhibitory neurons may regulate information flow through defined neural circuits and behavior by defining the role of enkephalin release from VIP-positive interneurons within the CA2 region of the hippocampus in both inhibitory plasticity (ITDP) and social memory. VIP interneurons have been long known to selectively form GABAergic synapses on other inhibitory neurons [13–15], including fast-spiking parvalbumin-expressing basket cells that strongly inhibit excitatory neuron activity, thereby producing disinhibition of excitatory neuron output [16–18]. As a result, recent studies implicating VIP neurons in regulating circuit function and learning behavior, including amygdala-dependent fear memory [5], hippocampal-dependent goal learning[4] and avoidance behavior[6], and sensory perceptual learning [7], have focused on this rapid disinhibitory action, either explicitly or implicitly. However, VIP-expressing neurons, like many other classes of inhibitory neurons, express a number of neuropeptides [8] known to exert powerful modulatory actions on the intrinsic excitability and pre- and postsynaptic properties of various neurons [9–12]. Our study is one of the first to elucidate how release of neuropeptides contributes to the physiological and behavioral effects of a given class of interneurons.
Peptide release in the hippocampus supports memory formation through heterosynaptic plasticity
Research into the neurobiological basis of memory has primarily focused on LTP and LTD in excitatory neurons as a key mechanism of synaptic plasticity [43]. However, inhibitory interneurons and plasticity of inhibitory synapses are also critical for episodic memory [1–7]. Hippocampal interneurons typically contact thousands of pyramidal neurons which makes them ideally positioned to regulate their activity [44]. In addition, interneurons that specifically inhibit other interneurons, and thereby disinhibit principal excitatory cells, provide a common mechanism to enhance excitability throughout the cortex [3, 17, 45].
Interneurons have been categorized based on specific protein expression[8] and classes of interneurons secrete various neuropeptides (VIP, enkephalin, galanin, somatostatin, BDNF) whose role in synaptic plasticity and in learning and memory are little understood. Enkephalin is known to play a major role in pain perception, cognitive functions and affective behavior [46–48] and DORs, their main target, have been implicated in emotional control [49]. However, due to the extensive distribution of the ENK / DOR signaling throughout the brain and spinal cord [50], knock-out studies [47] failed to provide a detailed description of its role in each brain region. On the other hand, pharmacological studies that have characterized how enkephalin in CA1 modulates interneuron activity and CA1 excitability [51, 52] have not identified the source of enkephalin or a link between modulation of hippocampal activity and memory formation.
Our present work has expanded on these previous studies by showing that enkephalin release from local VIP interneurons in the CA2/3a region of the hippocampus is responsible for mediating the actions of these interneurons in both the induction of ITDP and in social memory storage. Thus, viral-mediated shRNA depletion of enkephalin from the CA2 region blocked ITDP. In addition, silencing CA2 enkephalin-expressing cells or depleting enkephalin altogether entirely impaired the ability of mice to form social memory. Finally, in vivo imaging of calcium in CA2 VIP neurons revealed a selective increase in neural activity during exploration of a novel mouse that was not observed during exploration of an object or familiar animal. Taken together, our results show that local VIP interneurons and their release of enkephalin in the CA2 region are critical for both CA2 input-timing-dependent plasticity and the formation of social memories.
Although the Vip-Cre mouse line has been reported to have a reduced expression of Vip in the suprachiasmatic nucleus [29] and we also found a 22% reduction in Vip mRNA intensity in CA2 of these mice (Fig. S1d), we do not think that this decrease in Vip level is likely to contribute to our results in this mouse line. Thus, when injected with control Cre-dependent viral vectors, these mice show robust social memory (Fig. 5e,i and Fig. 6h) and CA2 ITDP (Fig. S3b), similar to that seen in our previous study [23]. Moreover, all of our experiments in the Vip-Cre mouse line using Cre-dependent viral vectors to express optogenetic tools or anti-Penk shRNA selectively in VIP neurons always compared results of such manipulations to those in the same Vip-Cre line injected with a control virus (e.g., expressing GFP). Thus, any results cannot be explained by differences in background levels of Vip expression alone. Although it is possible that the strong behavioral and electrophysiological phenotypes that we see using shRNA to downregulate Penk may depend to some extent on the background effects of reduced levels of Vip, our finding that non-Cre-dependent expression of Penk shRNA in CA2/CA3a of wild-type mice is also highly effective in suppressing ITDP (Fig. 3e) and social memory (Fig. 6a–d), supports the view that it is indeed the enkephalin release that is the critical factor.
Our finding that the VIP neurons receive strong excitatory inputs from both the direct performant path from entorhinal cortex and from hippocampal CA3 neurons also provides an explanation for how weak paired activation of the entorhinal cortical inputs and the Schaffer collateral inputs to CA2, or strong activation of either input alone, is likely to lead to activation of delta opioid receptors on PV-expressing basket cells in the CA2 region [22, 23, 53]. How enkephalin depresses release of GABA from PV interneurons is not fully understood, although it may depend on opposing PKA-mediated inactivation of K+ channels [54]. Ultimately, long-term depression of PV neurons decreases the amount of feed-forward inhibition recruited by the SC inputs and allows CA2 pyramidal neurons (PNs) to be more readily excited by the CA3 region for an extended period of time after VIP neurons were active. While it is of interest that VIP neuron activation also provides for a transient inhibition of PV neurons through fast inhibitory GABAergic synapses onto these cells [17], our data do not indicate that this short-lasting inhibition is necessary for ITDP or sufficient for social memory.
How does CA2 ITDP support the acquisition and consolidation of social memory?
Our study provides new mechanistic insights into inhibitory neuron plasticity in CA2 and its role in social memory, while also raising a number of important questions. For example, is feedforward inhibition on CA2 PNs uniformly modulated during social learning or are there selective effects on a subset of CA2 PNs that participate in a unique social memory – perhaps creating a CA2 engram? As mentioned above, it is still unclear whether enkephalin release operates in a global or cell-specific manner. This is an important consideration given that a number of in vivo electrophysiological studies have shown that CA2 PNs have mixed selectivity and heterogenous responses, showing differential activation in response to spatial, contextual and social stimuli [37, 42, 55–58]. Consistent with this idea, our laboratory found that a fraction of CA2 PNs respond to social novelty, enabling CA2 population activity to decode a novel from a familiar mouse [42]. Moreover, about half of CA2 neurons encode the location of a conspecific whereas a smaller fraction encode allocentric location [37]. We suggest that CA2 ITDP may provide a mechanism for the activity dependent enhancement of CA3 input to specific CA2 PNs to encode different aspects of a social experience.
It is still unclear how changes in activity in key limbic areas such as the hippocampus translate into the acquisition and consolidation of episodic memories. We find that VIP neurons respond to social novelty similarly to what has been described for CA2 PNs [42]. At present it is not known whether this reflects the fact that the two classes of neurons have shared inputs, or whether VIP neuron activity enables CA2 PNs to respond to social novelty through disinhibition. Given the rather slow time course of ITDP, which requires several minutes to develop, it is unlikely that the social novelty response, which is manifest within 1–2 minutes of social exposure, requires ITDP. Rather, ITDP may be important for social memory consolidation during sharp-wave ripples, population events reflecting the concerted firing of hippocampal neurons during slow wave sleep or quiet rest [59], in the period immediately following social learning. Thus, Oliva et al. (2020) found that the fraction of sharp-wave ripples originating in CA2 [60] are necessary for social memory consolidation. In addition, CA2 regulates hippocampal and prefrontal cortical low-gamma oscillations [61]. Thus, the consolidation of social memories likely involves the formation of a distributed memory engram encompassing CA1 and CA3 – as these hippocampal subfields are critical for various forms of declarative memory [62].
However, one difficulty in reconciling results from different laboratories concerning the hippocampal social memory circuit is that the hippocampus is an elongated structure, extending along a longitudinal dorsal to ventral axis. Our studies have focused on the role of dorsal CA2, as dorsal hippocampus is implicated in more cognitive functions whereas ventral hippocampus is thought to be important for emotional responses. However, other studies have shown that ventral, but not dorsal, regions of CA1 [63] and CA3 [64] are involved in social memory. In a previous study [40], we provided a unitary circuit model for social memory that reconciles these results by showing that dorsal CA2 sends strong excitatory longitudinal projections to ventral CA1. We further showed that these CA2 projections are necessary for social memory [40]. As sharp-wave ripples propagate along the longitudinal axis from dorsal to ventral hippocampus [65], we propose that social memory formation and consolidation involve a distributed EC-hippocampal network, in which social information propagates from dorsal CA2 to ventral CA1, where it may contribute to a social memory engram [63].
Opioid antagonists and schizophrenia
Postmortem studies have reported a specific loss of PV neurons in CA2 of schizophrenic patients [25, 26]. A role for CA2 in schizophrenia is consistent with studies of a mouse model of the 22q11.2 microdeletion, which confers a 30-fold increase in the risk of developing schizophrenia in humans [66]. The mouse model line exhibits reduced PV neuron density in CA2, resulting in a loss of feed-forward inhibition from CA3 to CA2, an increase in resting K+ conductance, which leads to CA2 hyperpolarization, and a profound loss of social memory [27]. Furthermore, the representation of social novelty in CA2 neuron firing is disrupted in the same mouse model and social memory deficits can be rescued by genetic downregulation of CA2 resting K+ conductance [28]. Finally, a recent meta-study confirmed the beneficial effect of opioid antagonists in treating both positive and negative symptoms (such as social withdrawal) of schizophrenia [67]. Our results suggest, but do not definitively demonstrate, that some of the beneficial effects of opioid antagonists on the negative symptoms of schizophrenia may be due to their action on DOR expressed by CA2 PV neurons. Additional studies investigating the dysregulation of CA2 DOR-mediated plasticity in mouse models of neuropsychiatric disorders will be of great future interest.
In conclusion, our study provides a novel mechanism for memory storage in which the action of a local inhibitory neuron produces a long-lasting synaptic plasticity through the release of the neuropeptide enkephalin. In a previous study, we provided evidence that an episode of social learning was able to recruit ITDP in vivo [23]. It will be of interest in future studies to examine how coordinated activity of entorhinal and CA3 inputs is recruited to induce ITDP and to understand how the subsequent disinhibition of CA2 PNs allows the emergence of a distributed engram that captures the various aspects of social interactions to form the unique memories that are critical to social interactions.
MATERIAL & METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to Steven A. Siegelbaum (sas8@columbia.edu) or Felix Leroy (felxfel@aol.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal procedures were performed in accordance with the regulations of the Columbia University IACUC. We used 6- to 12-week-old C57BL6/J (Jackson Laboratories #000664) male mice. When required, we also used males of the same age range from the following transgenic mouse lines: Vip-Cre mice (Jackson Laboratories #010908), Ai14 mice (Jackson Laboratories #007914) and Amigo2-Cre (Jackson Laboratories #030215). We also used C57BL6/J ovariectomized females (Jackson Laboratories #000664) as stimulus mice for the repetitive presentation test.
METHOD DETAILS
Plasmids and Virus Production
The shRNA sequence for pre-pro-enkephalin (GGATAACATCGACATGTAC; shRNAPenk) has been previously shown to reduce Penk mRNA transcript levels in rodents [34] and has 100% homology to the C57B6/J genome. The control shRNA (shRNACtrl) is a commonly used shRNA sequence that does not align to any mammalian RNA transcript (GCGCGATAGCGCTAATAAT; shRNACtrl) [35]. To continuously express shRNA constructs, both shRNA sequences were synthesized to contain an identical stem loop sequence (TTCAAGAGA) and cloned into an AAV plasmid (PX552; Addgene #60958) containing a U6 promoter to yield AAV2/8 U6.shRNA.hSyn.GFP.WPRE. For FLEX constructs, we created a plasmid to make both the shRNA and GFP expression dependent on recombination. The shRNA (shRNAPenk or shRNACtrl) and hSyn promoter sequences were synthesized to harbor a loxp and lox2272 on both end of the fragment. This DNA was then cloned into a pUC vector after a U6 promoter sequence and followed by a GFP sequence to yield AAV2/8 U6.FLEX.shRNA.hSyn.GFP.WPRE (Epoch life science). Similar constructs have been used in the past to attain Cre-inducible shRNA knockdown [70]. The transgenes were then packaged (HHMI-Janelia, Virus Services) into AAV2/8 capsids and purified with an iodixanol gradient to obtain a high titer (>1013 pp/mL).
Virus injections
For all injections, animals were anesthetized using isoflurane and given analgesics. A craniotomy was performed above the target region and a glass pipette was stereotaxically lowered down the desired depth. Injections were performed using a nano-inject II (Drummond Scientific). 23 nL were delivered 10 s apart until total amount was reached. The pipette was retracted after 5 min. With homozygous animals (WT, Vip-Cre), injection of the virus injection expressing DREADD, ArchT, shRNAPenk and their control virus (fluorophore only) was randomized within each cage.
AAV injections in dCA2
We injected AAV2/5 EF1a.DIO.eYFP (Fig. 1, UNC vector core, Addgene Cat# 27056-AAV5), AAV2/9 CBA.FLEX.Arch3.0-GFP.WPRE.SV40 (Fig. 2, UPenn vector core, Cat# AV-5-PV2432), AAV2/9 CAG.FLEX.eGFP.WPRE.bGH (Fig. 2, Addgene Cat# 51502-AAV9), AAV2/5 EF1a.DIO.hChR2(E123T/T159C)-eYFP (Fig. 2, Addgene, Cat# 35509-AAV5), AAV2/8 U6.shRNAPenk.hSyn.GFP.WPRE or AAV2/8 U6.shRNCtrl.hSyn.GFP.WPRE (Fig. 3 and 6, HHMI-Janelia, Virus Services, see above), AAV2/5 hSyn.DIO.hM4D(Gi)-mCherry (Fig. S5, Addgene, Cat# 44362-AAV5), AAV2/8 hSyn.DIO.hM4D(Gi)-mCherry (Fig. 5, Addgene, Cat# 44362-AAV8) AAV2/8 hSyn.DIO.mCherry (Fig. 5, Addgene, Cat# 50459-AAV8), AAV2/2 CAG.DIO.ArchT-TdTomato (Fig. 5, UNC vector core) or AAV2/2 CAG.DIO.TdTomato (Fig. 5, UNC vector core), AA2/8 U6.FLEX.shRNAPenk.hSyn.GFP.WPRE or AA2/8 U6.FLEX.shRNACtrl.hSyn.GFP.WPRE (Fig. 6, HHMI-Janelia, Virus Services, see above) into the hippocampi of WT C57BL6/J, Vip-Cre or Amigo2-Cre mice. Injections were done bilaterally at 2 sites with 100 nl injected per site. Injection coordinates were the following (in mm from Bregma): AP: −1.6, ML: ±1.8, DV: −1.5 and AP: −2.5, ML: ±2.5, DV: −2. Animals were processed starting 3 weeks after injection.
Rabies and AAV helper virus injection in CA2
We delivered 50 nl of a the helper virus AAV2/8 syn.DIO.TVA.2A.GFP.2A.B19G (UNC vector core, Addgene Cat# 52473) into the dorsal hippocampus of Vip-Cre mouse at the following coordinates AP −1.8, ML +2.5, DV −1.7 (in mm from Bregma). Following 2 weeks of recovery and rAAV expression, a secondary surgery was performed by the same technique and 300 nl of rabies SAD.B19.EnvA.ΔG.mCherry (SAD-B19 strain, Addgene Cat# 32636 prepared by the Salk institute vector core) was injected at the same coordinates. Mice were killed 7 d later and the brains cut sagittaly for entorhinal cortex imaging or coronally for hippocampus imaging.
Colchicine injection in CA2
We delivered 1 μL of 6 mg/mL colchicine (Tocris, Cat# 13–641-G) into the dorsal hippocampus at the following coordinates AP −1.8, ML +2.5, DV −1.7 (in mm from Bregma) in order to block axonal transport and increase the peptide amount in cell bodies. Animals were perfused two days later and processed for immunohistochemistry.
Immunohistochemistry (IHC)
Mice were anesthetized using isoflurane then perfused in the heart with 10 mL saline and their brains were quickly extracted and incubated in 4% PFA overnight. After 1 h washing in PBS, 60 μm slices were prepared using a Leica VT1000S vibratome (Leica Biosystems). Unless indicated otherwise, slices were permeabilized for 2h in PBS with 0.5% Triton-X100 (T9284, Sigma) in PBS before being incubated overnight at 4°C with primary antibodies diluted in PBS with 0.5% Triton-X in PBS. The slices were washed in PBS for 1 h then incubated overnight at 4°C with secondary antibodies from Thermo Fisher Scientific at a concentration of 1:500 diluted in PBS with 0.1% Triton-X. Hoechst counterstain was applied (Hoechst 33342 at 1:1000 for 30 min in PBS at RT) prior to mounting the slice using fluoromount (Sigma-Aldricht). Images were acquired using an inverted confocal microscope (LSM 700, Zeiss). For post-hoc immunocytochemistry after patch-clamp recordings, slices were fixed for 1 h in PBS with 4% PFA and streptavidin was applied during secondary incubation. For ENK labelling, colchicine injections and antigen retrieval procedure[39] were used.
For ENK and VIP labelling (Fig. 1d–e), primary incubation was performed for 3 days at 4°C with mouse anti-ENK (1:500, Santa Cruz Biotechnologies, Cat# sc-47705) and rabbit anti-VIP (1:100, Immunostar, Cat# 20077). Secondary incubation was performed with anti-mouse isotype 1 conjugated to Alexa 488 (Cat# A21121) and anti-rabbit conjugated to Alexa 568 (Cat# A11036).
For GFP, VIP and ENK labelling (Fig. 1g), primary incubation was performed for 3 days at 4°C with chicken anti-GFP (1:1000, Aves, Cat# GFP-1020), rabbit anti-VIP (1:100, Immunostar, cat# 20077, lot#) and mouse anti-ENK (1:100, Fitzgerald, Cat# 10-L20A). Secondary incubation was performed with anti-chicken conjugated to 488 (Cat# A11039), anti-rabbit conjugated to Alexa 568 (Cat# A11036) and anti-mouse conjugated to Alexa 633 (Cat# A21052).
For GFP/YFP and biocytin post-hoc labelling (Fig. 2), incubation was performed overnight at 4°C with anti-GFP conjugated to Alexa 488 (1:500, Thermo Fisher Scientific, Cat# A21311) and streptavidin conjugated to Alexa 555 (1:500, Thermo Fisher Scientific, Cat# s21381).
For mCherry and PCP4 labelling (Fig. 4), no IHC was performed against mCherry and incubation was performed overnight at 4°C with rabbit anti-PCP4 (1:400, Sigma-Aldrich, Cat# HPA005792). Secondary incubation was performed with anti-rabbit conjugated to Alexa 633 (Cat# A21070).
For GFP and mCherry labelling (Fig. 4), no IHC was performed against mCherry and incubation was performed overnight at 4°C with anti-GFP conjugated to Alexa 488 (1:500, Thermo Fisher Scientific, Cat# A21311).
For reelin, mCherry and calbindin-1 labelling (Fig. 4), primary incubation was performed for 2 days at 4°C with mouse anti-reelin (1:250, MBL international, Cat# D223–3) and rabbit anti-calbindin-1 (1:1000, Abcam, Cat# ab11426). Secondary incubation was performed with anti-mouse isotype 1 conjugated to Alexa 488 (Cat# A21121) and anti-rabbit conjugated to Alexa 633 (Cat# A21070).
For Biocytin and TdTomato post-hoc labelling (Fig. 4), incubation was performed overnight with streptavidin conjugated to Alexa 488 (1:500, Thermo Fisher Scientific, Cat# S32354).
For mCherry or TdTomato labelling (Fig. 5&6), primary incubation was performed overnight at 4°C with anti-RFP antibody (1:500, Rockland Antibody, Cat# 600-401-379). Secondary incubation was performed with anti-rabbit conjugated to Alexa 568 (Cat# A11036).
For GFP labelling (Fig. 7), incubation was performed overnight at 4°C with anti-GFP conjugated to Alexa 488 (1:500, Thermo Fisher Scientific, Cat# A21311).
In situ hybridization (ISH)
Mice were anesthetized using isoflurane then decapitated and their brain quickly extracted. Brains were then immersed in dry-ice cold Butan X for 6 s before being stored at −80°C. 16 μm-thick slices were prepared using a Leica cryostat (CM3050 S, Leica Biosystems) and mounted on Superfrost Plus microscope slides (12-550-15, FisherBrand). We processed the slices following the RNAscope Fluorescent Multiplex protocol (ACD Bio) with the probes for Vip in C1 (Cat# 415961), Gfp in C2 (Cat# 409011-C2) and Penk in C3 (Cat# 318761-C3). For Penk and Vip labelling (Fig. 1), we used the Amp4 Alt-C color module. For Gfp, Vip and Penk labelling (Fig. 3&7), we used the Amp4 Alt-B color module. DAPI was apply for 1 min prior to mounting using fluoromount. Images were acquired using an inverted confocal microscope (LSM 700, Zeiss). To analyze the overlap (Fig. 1c and Fig. S1c), we set a threshold in FIDJI to remove background noise and then manually counted cells positive for Penk, Vip or both. The oval aspect of the cells was used as a criterion to reject any bright noise speck. The CA1 pyramidal layer sometimes showed a faint signal for Penk mRNA that fell below threshold and was typically 5-fold less intense than the interneuron signal. We did not see any corresponding ENK protein signal using immunohistochemistry. When analyzing Penk expression following shRNA expression (Fig. 3d) or Vip expression (Fig. S1d), the observer was blinded to the identity of the virus.
In vitro electrophysiological recordings
We prepared transverse hippocampal slices from 8- to 12-week-old C57BL6/J male mice. Animals were killed under isoflurane anesthesia by perfusion into the right ventricle of ice-cold solution containing the following (in mM): 10 NaCl, 195 sucrose, 2.5 KCl, 10 glucose, 25 NaHCO3, 1.25 NaH2PO4, 3 Na Pyruvate, 0.5 CaCl2, and 7 MgCl2. Hippocampi were dissected from the brain in the same dissecting solution, placed upright into a 4% agar mold, and cut into 400 μm slices with a vibratome (VT1200S, Leica) in the same ice-cold dissection solution. Slices were then transferred to a chamber containing 50% dissecting solution and 50% ACSF (in mM: 125 NaCl, 2.5 KCl, 22.5 glucose, 25 NaHCO3, 1.25 NaH2PO4, 3 Na Pyruvate, 1 Ascorbic acid, 2 CaCl2 and 1 MgCl2). The chamber was kept at 34°C for 30 min and then at room temperature for at least 1 h before recording. All experiments were performed at 33°C. Dissecting and recording solutions were both saturated with 95% O2 and 5% CO2, pH 7.4.
Slices were mounted in the recording chamber under a microscope. Recordings were acquired using the Multiclamp 700A amplifier (Molecular Device), data acquisition interface ITC-18 (Instrutech) and the Axograph software. We targeted CA2 PNs based somatic location and size in both deep and superficial layer. Whole-cell recordings were obtained from CA2 PNs in current-clamp mode held at −73 mV with a patch pipette (3–5 MΩ) containing the following (in mM): 135 K-gluconate, 5 KCl, 0.2 EGTA-Na, 10 HEPES, 2 NaCl, 5 ATP, 0.4 GTP, 10 phosphocreatine, and 5 μM biocytin, pH 7.2 (280–290 mOsm). The liquid junction potential was 1.2 mV and was left uncorrected. Inhibitory currents were recorded in voltage-clamp mode at +10 mV with a pipette solution containing 135 Cs-gluconate instead of K-gluconate. Series resistance (15–25 MΩ) was monitored throughout each experiment; cells with a >20% change in series resistance were discarded. Once whole-cell recording was achieved we confirmed the cell-type based on the following electrophysiological properties: input resistance (60 to 120 MΩ), resting membrane potential (below −70 mV), sag amplitude (smaller than 5 mV) and presence of a slow depolarization prior discharge upon the injection of a long current pulse.
For electrical stimulation, synaptic potentials from the Schaffer collateral (SC) and direct entorhinal cortex fibers (EC) were evoked by monopolar stimulation with pipettes filled with 1-M NaCl and positioned in the stratum radiatum (SR) of CA3 (SC stimulation) as well as in the stratum lacunosum moleculare (SLM) of CA3, near the hippocampal fissure (EC stimulation). The ITDP induction protocol (90 paired pulses at 1 Hz with EC stimulation 20 ms prior to the SC stimulation was applied after a stable baseline of 5 min, at least 10 min following breaking into the cell. For light stimulation, pulses of blue or yellow light (pE-100, Cool LED) were delivered through a 40x immersion objective and illuminated an area of 0.2 mm2. The illumination field was centered over the CA2 pyramidal cell layer. In a subset of experiments, the following drugs were used at the following concentrations via bath application (unless otherwise noted): SR95531 (2 mM, Tocris #1262), CGP55845 (1 mM, Tocris #1248), CNQX (2 mM, Cayman Chemical #14618), D-AP5 (50 mM, Cayman Chemical #14539) and naltrindole (0.1 μM, Sigma-Aldrich #N115). Naltrindole application (Fig. 2d–e) or electrical stimulation interval (Fig. S2b) were randomized within each animal.
The amplitudes of the PSPs were normalized to the baseline. The magnitude of plasticity was estimated by comparing averaged responses at 25–30 min after the induction protocol with baseline-averaged responses 0–5 min before the induction protocol. All drugs were bath-applied following dilution into the external solution from stock solutions. We used the Axograph software for data acquisition, and Excel (Microsoft) and PRISM (Graphpad) for data analysis.
Behavioral tests
Based on our experience conducting similar social behavior experiments, we used group size of 10–15 animals. Animals that had viral expression outside of CA2/CA3a were excluded from analysis. This criterion was pre-established since we wanted to investigate the role of local interneurons. The observer was blinded to the identity of the mice while performing the behavioral experiments and the subsequent analyses.
Open arena test:
Test mouse was introduced into a new arena (60 cm × 60 cm). It was allowed to roam freely for 10 min. Using automatic tracking of the test mouse (Any-Maze 7, Stoelting), we quantified the total distance travelled as well as time spent in the surround (20% of the surface) or center (remaining 80% of the surface).
Sociability test:
Test mouse was introduced into a same open arena and allowed to roam freely for 10 min. Then wire cup cages were introduced at opposite corners. One cup had a novel mouse from the same sex, same strain background and similar age underneath it. The test mouse was allowed to explore each wire cups for 5 min. Using automatic tracking of the test mice (Any-Maze 7, Stoelting), we quantified the time spent in the area surrounding each cup. The preference exhibited by the test mouse for the novel animal compared to the empty cup was used as an index for social memory. We also calculated the total time spent interacting with stimulus mice during each trial in order to verify that the sociability drive was not affected.
Social memory test:
This test was performed in the same arena as the open-arena and sociability test. Test mouse was allowed to explore the arena for 10 min. Then, the test mouse explored for 5 min two wire cups on opposite corners (learning phase). Each cup contained a novel mouse. Stimulus mice were removed from the cups and the test mouse were placed in an empty cage, the size of its home cage for 30 min. Then, one of the now familiar mice was returned under its cup while a novel mouse was introduced under the other cup (recall phase). The test mouse was free to explore each cup again for 5 min. Importantly, all 3 stimulus mice were littermates housed in the same cage (thus preventing the test mice to use any cage-specific odor clue). We believe this new task allows for better exploration of each cup than the classical 3-chamber test while still giving the test mouse freedom to explore the novel or familiar mice unlike the direct interaction test. Using automatic tracking of the test mice (ANY-Maze, Stoelting), we quantified the time the test mice spent in the area surrounding each cup - and therefore, interacting with each stimulus mice - during learning and recall. The preference exhibited by the test mouse for the novel compared to the familiar animal was used as an index for social memory. We also calculated the total time spent interacting with stimulus mice during each trial in order to verify that the sociability drive was not affected.
Repetitive presentation of a gonadectomized female:
Test mice were introduced in a clean cage the same size than their home cage and left to explore for 5 min. Then, a gonadectomized C57Bl6/J female mouse was introduced for 2 min 4 times with 10 min inter-trial. For the 5th trial, a novel gonadectomized C57Bl6/J female mouse housed in the same cage than the first one was presented. Films were scored offline for social interaction by a trained observer blind to the experimental condition. Sniffing of any part of the body, allo-grooming and close following of the female counted as social. Interaction times were normalized with respect to the first interaction.
Viral injections and GRIN lens implantations for in vivo calcium imaging
We injected 200 nl unilaterally (right hemisphere) of AAV2/1.syn.FLEX.GCaMP6f.WPRE.SV40 virus (titer: 6.5 × 1011 GC per mL, Addgene, Cat# 100833-AAV1) at 150 nl/min into the right hemisphere above the hippocampal area CA2, using stereotactic coordinates: AP −2.0 mm, ML +1.8 mm, DV −1.7 mm from Bregma in Vip-Cre mice. Three weeks following injection, a 1.2 mm diameter circular craniotomy was centered at the following coordinates: AP −2.0 mm, ML +2.5 mm. To position the lens above CA2 such that the bottom of the lens was parallel to the pyramidal layer, we inserted the lens at a 10° angle so that the lens was angled towards the midline. The GRIN lens (1.0 mm diameter, 4.0 mm height, Inscopix) was inserted into the site of the craniotomy at a depth of −1.4 mm relative to Bregma. We used the Proview system (Inscopix), which allows site imaging during the lens implantation process. We adjusted the lens position such that the greatest region of fluorescence was visible underneath the lens. KWIK-SIL was placed around the craniotomy site to prevent cement from contacting the brain surface. The lens was secured in place using dental cement (C&B Metabond® Quick Adhesive Cement System), and the top of the Proview cuff around the lens was filled with KWIK-CAST to protect the lens. Mice were housed with littermates for one week before the plastic baseplate was placed over the lens and secured with dental cement. The baseplate and microscope were placed over the lens and the position was adjusted until cells were maximally in focus. Mice were handled and habituated for three days before the experiments, on the following schedule: handling (day 1), handling and exposure to oval arena for 15 minutes (day 2), handling, exposure to the oval arena for 15 minutes, and insertion of the microscope (day 3). No changes in subject mouse behavior, including during social interaction, were observed.
Data acquisition
On the day of the experiment, mice were moved to the behavior room and littermates were separated into different holding cages. Mice were allowed to acclimate to the environment for 30 min. A nVista 3.0 miniaturized microscope (Inscopix) was inserted into the baseplate and used to record calcium fluorescence videos at 20 Hz with a LED power of 0.2 mW from VIP neurons during social and non-social behavior using the nVista acquisition software (Inscopix). The working distance between the microscope objective and the lens was adjusted using electronic focusing through the Inscopix Data Acquisition software to maximize cell focus, and this distance was maintained between separate trials. To align behavior and calcium videos, a TTL pulse was triggered through ANY-maze and the AMi-2 Optogenetic interface at the start of each trial while the behavior video was simultaneously triggered and recorded. Behavior recordings were also collected at a rate of 20 Hz to allow ease of alignment with calcium recordings.
We imaged Vip-Cre mice over a course of five consecutive trials, with the subject mouse removed to its holding cage between trials, to determine how the presence of social conspecifics and non-social objects alters calcium activity in VIP neurons. First, subject mice were placed into an oval arena, which consisted of two half-circles with radius 15cm connected to a central square area with length of 30 cm (total dimensions: length 60 cm, width 30 cm, height 45 cm). During this empty trial, no other objects were in the arena. Next, a novel object consisting of an empty wire pencil cup (2 mice) or a similarly sized glass media storage container (2 mice) was placed on one side of the arena. In the third trial, a novel age- and sex-matched conspecific was placed underneath the pencil cup. In the fourth trial, a familiar age- and sex-matched conspecific was placed underneath the pencil cup. A littermate was used as the familiar mouse, except for one subject mouse that did not have a littermate in which case the subject mouse was co-housed with an age- and sex-matched conspecific for two hours with barrier separation to prevent fighting. The conspecific and subject mouse were separated for 30 minutes prior to testing. Finally, the mouse was allowed to interact with a familiar object, consisting of the empty wire pencil cup.
In vivo imaging analysis
In each trial, the subject mouse was free to explore the arena and its interaction with other objects in the arena were manually scored and its position tracked using ANY-maze tracking software (Stoelting). To improve tracking, the behavior videos were later run through a deep neural network trained using DeepLabCut [69] to recognize the position of the mouse head and body, as well as location of the objects placed in the arena. The raw calcium imaging videos from separate sessions were concatenated and then run through Inscopix Data Analysis software to correct defective pixels, spatially down-sample the videos by a factor of 4 to increase processing speed, de-noise with a spatial band-pass filter, and perform motion correction. The preprocessed and motion corrected videos were then exported for ROI identification and signal deconvolution.
We extracted ROIs using the Python CaImAn package for large-scale calcium imaging data [68]. The spatial footprints and deconvolved signal for the active sources (ROIs) were extracted using the extended version of constrained non-negative matrix factorization (CNMF-E) on spatial patches. The minimum peak noise ratio and minimum correlation were set to 12 and 0.9 respectively. The z-scored extracted noisy traces and spatial footprints were exported into MATLAB. We used a custom graphical user interface to evaluate individual ROIs and spatial footprints, and those with non-spherical or non-oval shapes caused by motion artifacts were excluded from analysis. The computed traces, separated by session, were deconvolved using the OASIS algorithm for nonnegative signal deconvolution [71]. The spike thresholds were set to twice the noise level, the baseline was calculated as the median of the signal and the noise levels were calculated by the median absolute deviation from baseline. The event rate was calculated across the entire trial by dividing the number of deconvolved and binarized spike events by the trial duration (5 min). We quantified the duration of interaction using a tracking software and setting up a perimeter around the object or wire cage. The event rate restricted to the of interactions was calculated by dividing the number of spike events occurring in the perimeter by the total duration of interaction.
Statistics
Statistical tests were performed with PRISM. Results presented in the text, figures and figure legends are reported as the mean ± SEM. * is for p < 0.05, ** is for p < 0.01 and *** is for p < 0.001.
Supplementary Material
Acknowledgments
This work was supported by: a 2019 NARSAD young investigator grant to F.L. from the Brain and Behavior research foundation, funded by the Osterhaus Family; a F32 MH122147-01A1 to C.d.S.; Howard Hughes Medical Institute support to E.R.K and R01-MH104602 and R01-MH106629 to S.A.S.
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest.
References
- 1.Basu J, Zaremba JD, Cheung SK, Hitti FL, Zemelman BV, Losonczy A, et al. Gating of hippocampal activity, plasticity, and memory by entorhinal cortex long-range inhibition. Science (80-). 2016;351:aaa5694–aaa5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Letzkus JJ, Wolff SBE, Meyer EMM, Tovote P, Courtin J, Herry C, et al. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature. 2011;480:331–335. [DOI] [PubMed] [Google Scholar]
- 3.Letzkus JJ, Wolff SBE, Lüthi A. Disinhibition, a Circuit Mechanism for Associative Learning and Memory. Neuron. 2015;88:264–276. [DOI] [PubMed] [Google Scholar]
- 4.Turi GF, Li W-K, Chavlis S, Pandi I, O’Hare J, Priestley JB, et al. Vasoactive Intestinal Polypeptide-Expressing Interneurons in the Hippocampus Support Goal-Oriented Spatial Learning. Neuron. 2019;101:1150–1165.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krabbe S, Paradiso E, D’Aquin S, Bitterman Y, Courtin J, Xu C, et al. Adaptive disinhibitory gating by VIP interneurons permits associative learning. Nat Neurosci. 2019;22:1834–1843. [DOI] [PubMed] [Google Scholar]
- 6.Lee AT, Cunniff MM, See JZ, Wilke SA, Luongo FJ, Ellwood IT, et al. VIP Interneurons Contribute to Avoidance Behavior by Regulating Information Flow across Hippocampal-Prefrontal Networks. Neuron. 2019;102:1223–1234.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams LE, Holtmaat A. Higher-Order Thalamocortical Inputs Gate Synaptic Long-Term Potentiation via Disinhibition. Neuron. 2019;101:91–102.e4. [DOI] [PubMed] [Google Scholar]
- 8.DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14:202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ögren SO, Kuteeva E, Elvander-Tottie E, Hökfelt T. Neuropeptides in learning and memory processes with focus on galanin. Eur J Pharmacol. 2010;626:9–17. [DOI] [PubMed] [Google Scholar]
- 10.Marder E Neuromodulation of Neuronal Circuits: Back to the Future. Neuron. 2012;76:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borbély É, Scheich B, Helyes Z. Neuropeptides in learning and memory. Neuropeptides. 2013;47:439–450. [DOI] [PubMed] [Google Scholar]
- 12.Gøtzsche CR, Woldbye DPD. The role of NPY in learning and memory. Neuropeptides. 2016;55:79–89. [DOI] [PubMed] [Google Scholar]
- 13.Dávid C, Schleicher A, Zuschratter W, Staiger JF. The innervation of parvalbumin-containing interneurons by VIP-immunopositive interneurons in the primary somatosensory cortex of the adult rat. Eur J Neurosci. 2007;25:2329–2340. [DOI] [PubMed] [Google Scholar]
- 14.Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acsády L, Görcs TJ, Freund TF. Different populations of vasoactive intestinal polypeptide-immunoreactive interneurons are specialized to control pyramidal cells or interneurons in the hippocampus. Neuroscience. 1996;73:317–334. [DOI] [PubMed] [Google Scholar]
- 16.Tyan L, Chamberland S, Magnin E, Camire O, Francavilla R, David LS, et al. Dendritic Inhibition Provided by Interneuron-Specific Cells Controls the Firing Rate and Timing of the Hippocampal Feedback Inhibitory Circuitry. J Neurosci. 2014;34:4534–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberland S, Topolnik L. Inhibitory control of hippocampal inhibitory neurons. Front Neurosci. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pi H-J, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennett JE, Poletini MO, Freeman ME. Vasoactive intestinal polypeptide modulates the estradiol-induced prolactin surge by entraining oxytocin neuronal activity. Brain Res. 2008;1196:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blasco-Ibáñez JM, Martínez-Guijarro FJ, Freund TF. Enkephalin-containing interneurons are specialized to innervate other interneurons in the hippocampal CA1 region of the rat and guinea-pig. Eur J Neurosci. 1998;10:1784–1795. [DOI] [PubMed] [Google Scholar]
- 21.Botcher NA, Falck JE, Thomson AM, Mercer A. Distribution of interneurons in the CA2 region of the rat hippocampus. Front Neuroanat. 2014;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piskorowski RA, Chevaleyre V. Delta-Opioid Receptors Mediate Unique Plasticity onto Parvalbumin-Expressing Interneurons in Area CA2 of the Hippocampus. J Neurosci. 2013;33:14567–14578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leroy F, Brann DH, Meira T, Siegelbaum SA. Input-Timing-Dependent Plasticity in the Hippocampal CA2 Region and Its Potential Role in Social Memory. Neuron. 2017;95:1089–1102.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasrallah K, Piskorowski RA, Chevaleyre V. Inhibitory Plasticity Permits the Recruitment of CA2 Pyramidal Neurons by CA3. Eneuro. 2015;2:ENEURO.0049–15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benes FM, Kwok EW, Vincent SL, Todtenkopf MS. A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry. 1998;44:88–97. [DOI] [PubMed] [Google Scholar]
- 26.Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF. Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry. 2004;9:609–620. [DOI] [PubMed] [Google Scholar]
- 27.Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, et al. Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome. Neuron. 2016;89:163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donegan ML, Stefanini F, Meira T, Gordon JA, Fusi S, Siegelbaum SA. Coding of social novelty in the hippocampal CA2 region and its disruption and rescue in a 22q11.2 microdeletion mouse model. Nat Neurosci. 2020;23:1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng AH, Fung SW, Cheng H-YM. Limitations of the Avp-IRES2-Cre (JAX #023530) and Vip-IRES-Cre (JAX #010908) Models for Chronobiological Investigations. J Biol Rhythms. 2019;34:634–644. [DOI] [PubMed] [Google Scholar]
- 30.Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O’Shea DJ, Prakash R, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2012;9:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudman JT, Tsay D, Siegelbaum SA. A Role for Synaptic Inputs at Distal Dendrites: Instructive Signals for Hippocampal Long-Term Plasticity. Neuron. 2007;56:866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevaleyre V, Siegelbaum SA. Strong CA2 Pyramidal Neuron Synapses Define a Powerful Disynaptic Cortico-Hippocampal Loop. Neuron. 2010;66:560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basu J, Srinivas KV., Cheung SK, Taniguchi H, Huang ZJ, Siegelbaum SA. A Cortico-Hippocampal Learning Rule Shapes Inhibitory Microcircuit Activity to Enhance Hippocampal Information Flow. Neuron. 2013;79:1208–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bérubé P, Poulin J-F, Laforest S, Drolet G. Enkephalin Knockdown in the Basolateral Amygdala Reproduces Vulnerable Anxiety-Like Responses to Chronic Unpredictable Stress. Neuropsychopharmacology. 2014;39:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Marshall PR, Leighton LJ, Zajaczkowski EL, Wang Z, Madugalle SU, et al. The DNA Repair-Associated Protein Gadd45γ Regulates the Temporal Coding of Immediate Early Gene Expression within the Prelimbic Prefrontal Cortex and Is Required for the Consolidation of Associative Fear Memory. J Neurosci. 2019;39:970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura T, Pignatelli M, Suh J, Kohara K, Yoshiki A, Abe K, et al. Island Cells Control Temporal Association Memory. Science (80-). 2014;343:896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliva A, Fernández-Ruiz A, Leroy F, Siegelbaum SA. Hippocampal CA2 sharp-wave ripples reactivate and promote social memory. Nature. 2020;587:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urban DJ, Roth BL. DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu Rev Pharmacol Toxicol. 2015;55:399–417. [DOI] [PubMed] [Google Scholar]
- 39.Hitti FL, Siegelbaum SA. The hippocampal CA2 region is essential for social memory. Nature. 2014;508:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meira T, Leroy F, Buss EW, Oliva A, Park J, Siegelbaum SA. A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nat Commun. 2018;9:4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevenson EL, Caldwell HK. Lesions to the CA2 region of the hippocampus impair social memory in mice. Eur J Neurosci. 2014;40:3294–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donegan ML, Stefanini F, Meira T, Gordon JA, Fusi S, Siegelbaum SA. Coding of social novelty in the hippocampal CA2 region and its disruption and rescue in a mouse model of schizophrenia. BioRxiv. 2019. 2019. 10.1101/833723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Citri A, Malenka RC. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology. 2008;33:18–41. [DOI] [PubMed] [Google Scholar]
- 44.Andersen P, Morris R, Amaral D, Bliss T, O’ Keefe J. The Hippocampus Book. Oxford University Press; 2006. [Google Scholar]
- 45.Wilmes KA, Clopath C. Inhibitory microcircuits for top-down plasticity of sensory representations. Nat Commun. 2019;10:5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Díaz J-L, Asai M. Dominant mice show much lower concentrations of methionine-enkephalin in brain tissue than subordinates: Cause or effect? Behav Brain Res. 1990;39:275–280. [DOI] [PubMed] [Google Scholar]
- 47.König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, et al. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. [DOI] [PubMed] [Google Scholar]
- 48.Roques BP. Novel approaches to targeting neuropeptide systems. Trends Pharmacol Sci. 2000;21:475–483. [DOI] [PubMed] [Google Scholar]
- 49.Calenco-Choukroun G, Daugé V, Gacel G, Féger J, Roques BP. Opioid δ agonists and endogenous enkephalins induce different emotional reactivity than μ agonists after injection in the rat ventral tegmental area. Psychopharmacology (Berl). 1991;103:493–502. [DOI] [PubMed] [Google Scholar]
- 50.François A, Low SA, Sypek EI, Christensen AJ, Sotoudeh C, Beier KT, et al. A Brainstem-Spinal Cord Inhibitory Circuit for Mechanical Pain Modulation by GABA and Enkephalins. Neuron. 2017;93:822–839.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicoll RA, Alger BE, Jahr CE. Enkephalin blocks inhibitory pathways in the vertebrate CNS. Nature. 1980;287:22–25. [DOI] [PubMed] [Google Scholar]
- 52.Madison DV, Nicoll RA. Enkephalin hyperpolarizes interneurones in the rat hippocampus. J Physiol. 1988;398:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nasrallah K, Therreau L, Robert V, Huang AJY, McHugh TJ, Piskorowski RA, et al. Routing Hippocampal Information Flow through Parvalbumin Interneuron Plasticity in Area CA2. Cell Rep. 2019;27:86–98.e3. [DOI] [PubMed] [Google Scholar]
- 54.Domínguez S, Rey CC, Therreau L, Fanton A, Massotte D, Verret L, et al. Maturation of PNN and ErbB4 Signaling in Area CA2 during Adolescence Underlies the Emergence of PV Interneuron Plasticity and Social Memory. Cell Rep. 2019;29:1099–1112.e4. [DOI] [PubMed] [Google Scholar]
- 55.Mankin EA, Diehl GW, Sparks FT, Leutgeb S, Leutgeb JK. Hippocampal CA2 Activity Patterns Change over Time to a Larger Extent than between Spatial Contexts. Neuron. 2015;85:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander GM, Farris S, Pirone JR, Zheng C, Colgin LL, Dudek SM. Social and novel contexts modify hippocampal CA2 representations of space. Nat Commun. 2016;7:10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kay K, Sosa M, Chung JE, Karlsson MP, Larkin MC, Frank LM. A hippocampal network for spatial coding during immobility and sleep. Nature. 2016;531:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wintzer ME, Boehringer R, Polygalov D, McHugh TJ. The Hippocampal CA2 Ensemble Is Sensitive to Contextual Change. J Neurosci. 2014;34:3056–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buzsáki G Hippocampal sharp wave‐ripple: A cognitive biomarker for episodic memory and planning. Hippocampus. 2015;25:1073–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oliva A, Fernández-Ruiz A, Buzsáki G, Berényi A. Role of Hippocampal CA2 Region in Triggering Sharp-Wave Ripples. Neuron. 2016;91:1342–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexander GM, Brown LY, Farris S, Lustberg D, Pantazis C, Gloss B, et al. CA2 neuronal activity controls hippocampal low gamma and ripple oscillations. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Josselyn SA, Tonegawa S. Memory engrams: Recalling the past and imagining the future. Science (80-). 2020;367:eaaw4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okuyama T, Kitamura T, Roy DS, Itohara S, Tonegawa S. Ventral CA1 neurons store social memory. Science (80-). 2016;353:1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiang M-C, Huang AJY, Wintzer ME, Ohshima T, McHugh TJ. A role for CA3 in social recognition memory. Behav Brain Res. 2018;354:22–30. [DOI] [PubMed] [Google Scholar]
- 65.Patel J, Schomburg EW, Berényi A, Fujisawa S, Buzsáki G. Local Generation and Propagation of Ripples along the Septotemporal Axis of the Hippocampus. J Neurosci. 2013;33:17029–17041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drew LJ, Crabtree GW, Markx S, Stark KL, Chaverneff F, Xu B, et al. The 22q11.2 microdeletion: Fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int J Dev Neurosci. 2011;29:259–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clark SD, Van Snellenberg JX, Lawson JM, Abi-Dargham A. Opioid antagonists are associated with a reduction in the symptoms of schizophrenia: a meta-analysis of controlled trials. Neuropsychopharmacology. 2020;45:1860–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giovannucci A, Friedrich J, Gunn P, Kalfon J, Brown BL, Koay SA, et al. CaImAn an open source tool for scalable calcium imaging data analysis. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mathis A, Mamidanna P, Cury KM, Abe T, Murthy VN, Mathis MW, et al. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat Neurosci. 2018;21:1281–1289. [DOI] [PubMed] [Google Scholar]
- 70.Favuzzi E, Deogracias R, Marques-Smith A, Maeso P, Jezequel J, Exposito-Alonso D, et al. Distinct molecular programs regulate synapse specificity in cortical inhibitory circuits. Science (80-). 2019;363:413–417. [DOI] [PubMed] [Google Scholar]
- 71.Chechik G, Sharma V, Shalit U, Bengio S. Large scale online learning of image similarity through ranking. J Mach Learn Res. 2010;11:1109–1135. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.