

Abstract
The functionalization of C─H bonds is one of the most challenging transformations in synthetic chemistry. In biology, these processes are well known and are achieved with a variety of metalloenzymes, many of which contain a single metal center within their active sites. The most well studied are those with Fe centers and the emerging experimental data show that high valent Fe–oxido species are the intermediates responsible for cleaving the C─H bond. This Forum describes the state of this field with an emphasis on non-heme Fe enzymes and current experimental results that provide insights into the properties that make these species capable of C─H bond cleavage. These parameters are also briefly considered in regard to Mn-oxido complexes and Cu-containing metalloenzymes. Synthetic Fe–oxido complexes are discussed to highlight their utility as spectroscopic and mechanistic probes, and reagents for C─H bond functionalization. Avenues for future research are also examined.
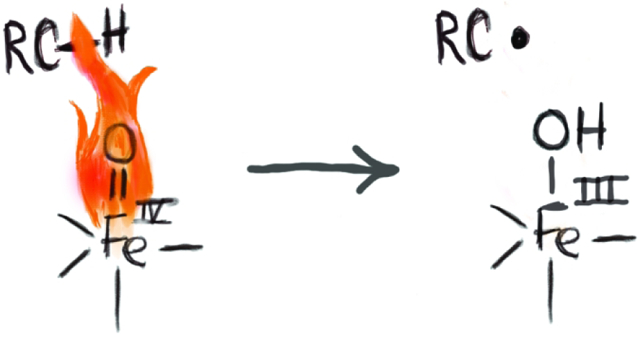
Synopsis Graphical Abstract
The cleavage of unactivated C─H bonds is a challenging transformation in synthetic chemistry yet is readily accomplished in biology by metalloenzymes. A sub-class of these enzymes contain active sites with single Fe centers that generate high valent Fe–oxido intermediates whose function is to cleave C─H bonds. This Forum describes the current state of this field from spectroscopic, mechanistic, and synthetic perspectives. Discussions on relevant Mn and Cu systems are also included.
Introduction
Functional group transformations are the heart of organic synthesis and many a student has been taught these concepts as a means of learning organic chemistry. For all the many types of reactions that have been developed, the most prevalent functional group, the C─H bond, is often overlooked. In fact, we usually neglect even showing these bonds and for the longest time, few practitioners even considered it a functional group. There are many good reasons for this neglect, the foremost being the large bond dissociation free energies associated with most C─H bonds – it is just too thermodynamically difficult to selectively cleave these bonds to afford targeted products.1 However, the situation is different in biology where C─H bonds can be selectively and efficiently functionalized. This difference rests on a cadre of enzymes that have evolved to perform these difficult reactions and their usefulness in biosynthetic processes is impressive.2 Although we recognize that there have been important advances in the development of synthetic reagents to accomplish these types of reactions, none compares to those that exist in nature. Understanding how enzymes accomplish C─H bond activation from both structure-function and mechanistic considerations remains an ongoing and challenging endeavor; yet these pursuits impact a wide area of science that encompass biochemistry, health, and synthetic chemistry. To the organic chemist, for instance, the ability to selectively convert a C─H bond at the late stage of a multi-step preparation would be a glad addition to any synthetic toolbox.
The aim of this Forum is to describe one of the main classes of enzymes that performs biological C─H bond functionalization, namely metalloenzymes that contain cofactors in which a non-heme, mononuclear Fe or Cu center has a central role. These metalloenzymes have been acknowledged to be potent at oxidizing organic substrates, yet we still are learning the details of how they manipulate C─H bonds. The focus will be on results over the last 5-10 years and covers both protein chemistry and synthetic complexes which have provided a wealth of information. The caveat is that this field is quite extensive with many outstanding contributions – this review is thus not meant to be comprehensive in content but rather, a selective Forum on some of the key accomplishments and controversies that still exist.
Cleavage of C─H Bonds by Metal Complexes: Comparisons.
Before going forward, it is instructive to briefly compare the two general mechanisms that transition metal complexes utilize to cleave C─H bonds. Within the organometallic field, it has been well documented that several 2nd and 3rd row transition metal complexes can cleave C─H bonds.3-6 This type of process is illustrated using Bergman's classic IrCp complexes in which an oxidative addition reaction occurs to produce an (alkyl)Ir-hydride complex (Scheme 1A).7,8 This two-electron process derives from the relatively low-valent Ir(I) complex that cleaves the C─H bond. Metal–oxido complexes function in a different manner, in which the metal center usually has a high formal oxidation state (greater than 3+).9 Gray has determined that the bonding between the metal center and oxido ligand contains substantial multiple bond character, resulting in an oxido ligand being electrophilic.10 The M–oxido unit is thus poised to cleave C─H bonds which typically occurs through a homolytic process to produce a hydrogen atom (H-atom) and a carbon-centered radical (Scheme 1B). The H-atom is transferred to the metal–oxido species with the oxido ligand being protonated to produce a hydroxido ligand and the metal center being reduced by one electron. Because the electron and proton end up on different atoms, this process is not actually an H-atom transfer but rather a proton-coupled electron transfer.11-13 More recently, it has been described as a concerted proton-electron transfer process to emphasize that both the electron and proton are transferred together at the transition state.1,14,15 We will return to this concept later in this Forum.
Scheme 1.
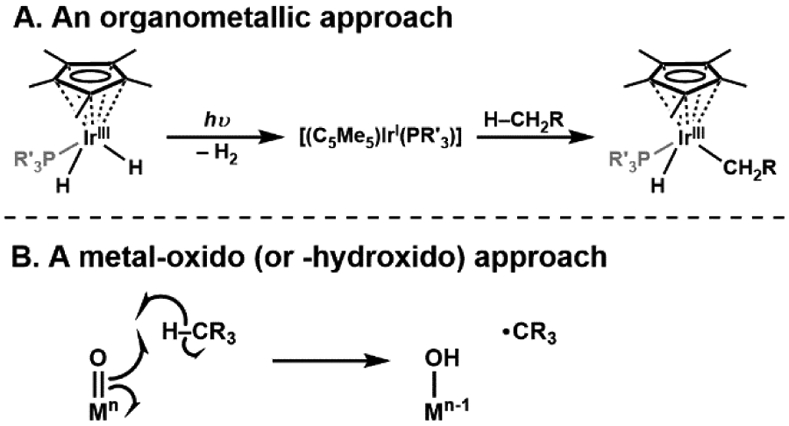
Cleavage of C─H bond by metal complexes using an organometallic approach (A) such as an IrCp-based precursor, or a metal-oxido (or hydroxido) approach (B).
The functionalization of C─H bonds comes after cleavage has occurred. To illustrate this point, we will use Fe=O and Fe–OH species because they are operative within proteins. High valent Fe=O intermediates are usually formed after the binding and activation of O2.16-22 For heme systems (e.g. cytochrome P450s), these intermediates are formally FeV=O (denoted, compound I) but spectroscopic studies conclusively show that it is an FeIV=O unit coupled to a ligand radical (Scheme 2).23 After C─H bond cleavage, an FeIV–OH species is produced (denoted, compound II) that recombines with the substrate carbon radical to form an FeIII–O(H)CR3 species in a process that is referred as a rebound step.24-26 It is the interplay between these two Fe species that drives biological C─H cleavage, which we will highlight in the ensuing sections.
Scheme 2.
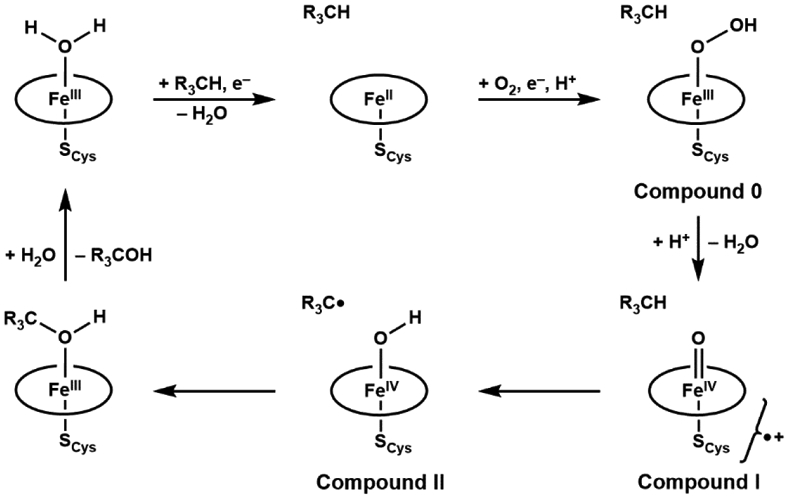
Catalytic mechanism of aliphatic hydroxylation by cytochrome P450 enzymes.
Non-Heme FeIV=O Species.
Metalloproteins.
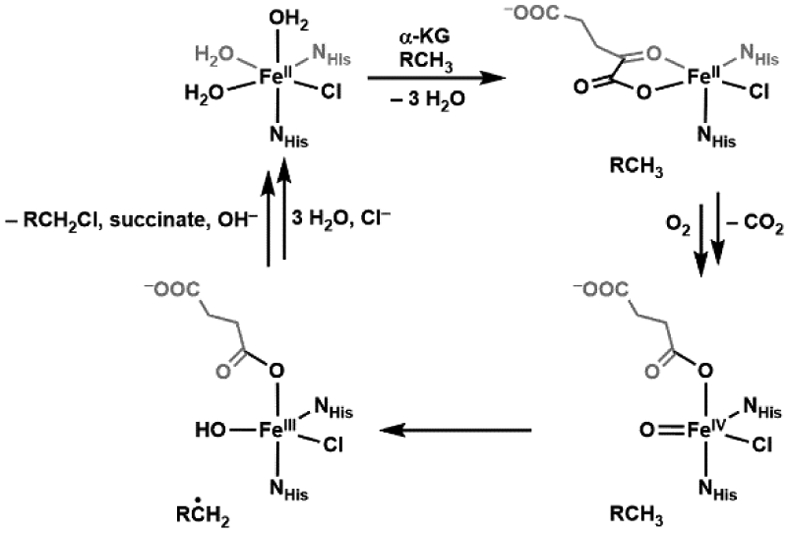
Bollinger and Krebs first reported the spectroscopic characterization of an enzymatic non-heme FeIV-oxido species in taurine dioxygenase (TauD), part of the superfamily of α-keto glutarate (α-KG) enzymes that has over a thousand members that catalyze a diverse set of reactions.27-29 The active site of these enzymes in its resting state consists of a ferrous ion bound by two histidine residues and a carboxylate (aspartate or glutamate) in a facial coordination, known as the “facial triad”, with the remaining coordination sites occupied by 3 water molecules.30-33 Water coordination is commonly found in resting states of non-heme proteins and serves important functions: 1) they stabilize the more reduced states of the metal centers and 2) they can be easily replaced by other ligands such as cofactors, small molecules that need to be activated, and substrates. All these roles are illustrated in α-KG enzymes and are represented in the consensus mechanism of action (Scheme 3).28 Upon binding of the cofactor α-KG to the Fe(II) center, two water molecules are displaced; docking of the taurine substrate occurs proximal to the active site which causes the last aqua ligand to dissociate. The vacant coordination site allows for dioxygen to bind to the iron center, but the exact coordination of this species has not been experimentally confirmed. A reasonable proposal is for dioxygen to coordinate end-on,34 leaving the distal O-atom to attack the α-keto carbon of the α-KG ligand, which leads to the dissociation of CO2 which is the driving force to generate the reactive FeIV=O species (denoted intermediate J).35 The FeIV=O species is then proposed to follow a similar pathway as proposed for P450s by hydroxylating taurine in a “rebound” mechanism, in which it first homolytically cleaves a C─H bond in taurine to form FeIII–OH and the carbon-radical.25,36 Recombination then occurs between the FeIII–OH species and the resultant carbon radical to yield an alcohol. Although this mechanistic proposal is similar to that for P450s, there are important differences in that intermediate J is formally at the 4+ oxidation level, whereas compound I is more oxidized with the additional oxidizing equivalent stored on the ligands.23 This difference also includes the rebound step which, for these non-heme oxygenases, is from an FeIII–OH species. These differences could impact function: intermediate J decays at a rate of 13 s−1 and has an observed kinetic isotope effect (KIE) of kH/kD ~ 50, both of which are different from those in heme monooxygenases For instance, a thermophilic P450 (CYP119) compound I decays at a rate of 220 s−1 in the presence of 20 μM dodecanoic acid and a KIE value can range from 1 – 18 for P450 enzyme-catalyzed C─H bond activation.23,37-39
Scheme 3.
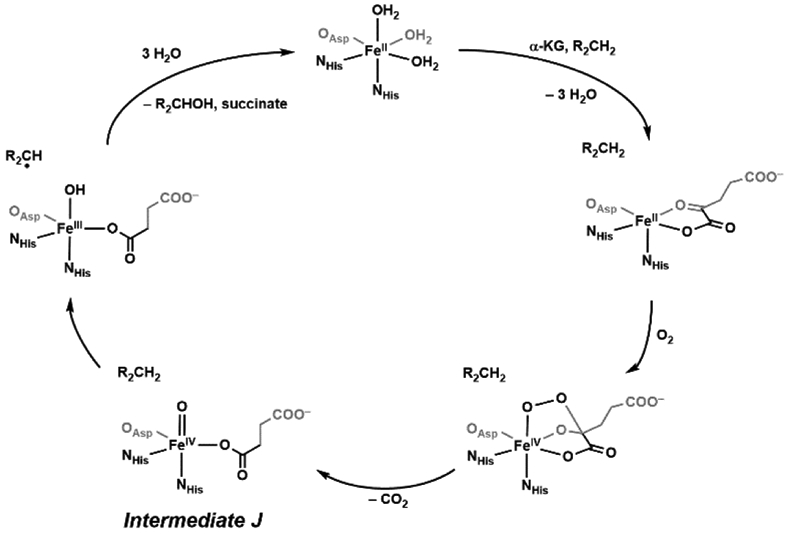
Catalytic mechanism of aliphatic hydroxylation by TauD.
The key discovery in this field was the detection of intermediate J by Bollinger and Krebs using rapid spectroscopic methods.35,38,40 They first employed stopped-flow UY-vis spectroscopy to observe the appearance of an absorbance feature at λmax = 318 nm. Knowing the time scale for detection of intermediate J, they then used freeze-quench methods coupled with 57Fe-Mössbauer spectroscopy to identify the oxidation level of the Fe center. Intermediate J has an experimentally determined isomer shift (δ) of 0.31 mm/s and quadrupole splitting (ΔEQ) of −0.88 mm/s;35 however, these commonly reported parameters are often not enough to conclusively determine the oxidation and spin states. For example, the parameters for intermediate J are not that different from the δ = 0.32 mm/s and ΔEQ = −1.54 mm/s obtained for a synthetic FeIII–oxido complex ([FeIIIH3buea(O)]2−) that will be discussed in a later section.41,42 However, for intermediate J there is additional evidence to support the assignment of an FeIV–oxido species: it lacks perpendicular-mode EPR signals and variable-field Mössbauer studies indicate it is an integer spin system. A subsequent report by Neese, Bollinger, and Krebs provided evidence to indicate it has an S = 2 spin ground state.43 In particular, they examined the A-tensor obtained from Mössbauer measurements (Ax and Ay) and computations (Az) to determine if there is an anisotropic component that arises from the spin-dipolar contributions, which cause one tensor component to be approximately twice the magnitude of the other two. For intermediate J these A-values are (ASD = (+5.4, +5.4, −11.9) T), indicative of an S = 2 ground spin state with an unoccupied dz2 orbital (Note: the ASD tensor for an S = 1 FeIV=O complex is similar but the signs on the A-values are reversed). These magnetic studies were followed by corroborative reports from other spectroscopic measurements on intermediate J. Extended X-ray absorption fine structure (EXAFS) spectroscopy revealed a short Fe–O distance of 1.62 Å which supports the assignment of an FeIV=O unit,44 and resonance Raman studies found an Fe–O vibrational feature at 821 cm−1 that shifts to 787 cm−1 upon substitution with 18O2.45 This shift of 34 cm−1 is consistent with that predicted using a harmonic Fe–O oscillator model.
Since the first report of intermediate J in TauD, many other α-KG/Fe-dependent enzymes have been investigated and found to form an FeIV–oxido species including intermediates that catalyze reactions other than substrate hydroxylation.27,46 One example is halogenation, as observed in SyrB2, which is a non-heme Fe halogenase.47,48 The active site of SyrB2 differs from that of TauD by replacing the aspartate amino residue with a halide anion in the primary coordination sphere of the Fe center. An FeIV–oxido species still forms and functions to abstract an H-atom from the substrate, but instead of “rebound” of the hydroxido ligand, the halide recombines with the carbon radical to form a C─X bond (Scheme 4). Bollinger and coworkers have demonstrated experimentally that substrate orientation is crucial in the chemoselectivity.49-51 By positioning the C─H bond of the substrate further away from the oxido moiety but closer to the chloro ligand, the rate of C─H cleavage decreases, but the hydroxylation pathway is suppressed, and the halogen-rebound is preferred. Other reactions catalyzed by these non-heme enzymes include substrate desaturation,52,53 epoxidation,54 and decarbonylation.55
Scheme 4.
Catalytic mechanism of aliphatic halogenation by SyrB2.
Synthetic Mononuclear Fe–Oxido Analogs: The Beginning.
The trapping of non-heme FeIV–oxido species within proteins inspired the preparation of synthetic analogs, yet stabilizing these reactive species in abiotic systems was challenging. There was one example of a stable Fe–oxido complex that preceded its detection in proteins, but it was an FeIII–oxido species. This complex, [FeIIIH3buea(O)]2− ([H3buea]3− = tris[(N'-tertbutylureaylato)-N-ethyl]aminato, Figure 1A) was generated from the activation of dioxygen by an FeII precursor, and spectroscopic and structural characterizations confirmed its formulation that includes a rather long Fe–O bond length of 1.816(3) Å.41 Prior to the discovery of [FeIIIH3buea(O)]2− all FeIII–oxido units were incorporated into bridging structures with other metal ions, though most are with other FeIII centers.56,57 These species arise because of the strong thermodynamic driving force to form FeIII–(μ-O)–FeIII units, which are the basic unit in rust. Formation of multinuclear products is prevented in [FeIIIH3buea(O)]2− because of the [H3buea]3− ligand which controls both the primary and secondary coordination spheres.58,59 Specifically, experimental and computational studies have directly linked the stability of this complex to the three intramolecular hydrogen bonds (H-bonds) involving the oxido ligand and the NH groups of [H3buea]3−.60 This complex is rare, and H-bonds appear to be needed for isolation as the only other two that are fully characterized in the condensed phase, which are reported by Fout (Figure 1B) and Agapie (Figure 1C), also have an intramolecular H-bonding network around the FeIII–oxido unit.61,62
Figure 1.
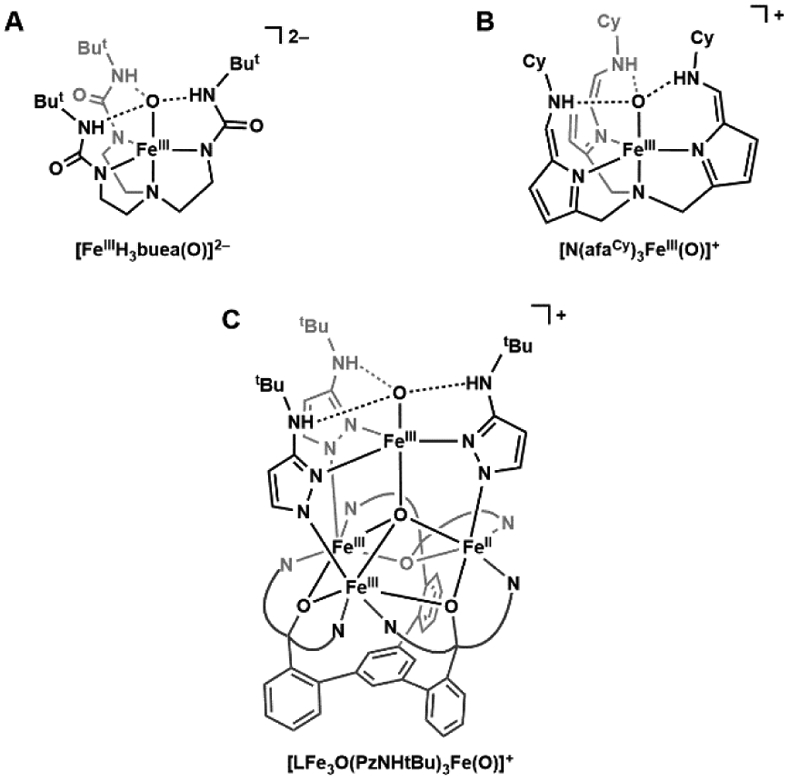
Terminal FeIII–oxido complexes: [FeIIIH3buea(O)]2− (A), [N(afaCy)3FeIII(O)]+ (B), and [LFe3O(PzNHtBu)3Fe(O)]+ (C).
S=1 FeIV–Oxido Complexes.
The detection of a synthetic FeIV–oxido complex was prompted by Wieghardt's observation of a new FeIV species produced from the ozonolysis of [(cyclam-acetato)FeIII(OTf)]+ (cyclam = 1,4,8, 11-tetraazacyclotetradecane).63 The magnetic properties obtained by Mössbauer spectroscopy, such as a low isomer shift and an anisotropic ASD tensor, were comparable to those of compound II in horseradish peroxidase and can best describe an S = 1 FeIV center. However, the low yielding generation of this species precluded further structural and spectroscopic characterizations. The breakthrough in preparation of synthetic FeIV–oxido complexes came from the laboratories of Nam and Que who reported the first crystallographically characterized FeIV-oxido complex in [FeIV(O)(TMC)(MeCN)]2+ (TMC = tetramethylcyclam, Figure 2A) and later followed that with the structure of [FeIV(O)(N4Py)]2+ (N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine, Figure 2B).64,65 The structural analyzes of these complexes showed relatively short Fe─O bond lengths of 1.646(3) and 1.636(3) Å that are indicative of FeIV–oxido species. There have been numerous spectroscopic and computational studies on these complexes that corroborate their assignments as FeIV species, but they both have spin ground states of S = 1 which differ from those found in proteins.20,28,66 Nevertheless, these complexes serve as important contributions to the field and there is now a library of over 100 examples of S = 1 FeIV–oxido complexes in similar ligand frameworks.67-76 They have been particularly useful in delineating properties of electronic structure and demonstrating how optical spectroscopy is a reliable spectroscopic handle for FeIV–oxido complexes. Signature features of synthetic mononuclear FeIV–oxido species are weak bands around 700 – 900 nm that are assigned to a d-d transition.66,67 The energy of the d-d transition is sensitive to the primary coordination sphere around the FeIV–oxido unit, both to the equatorial ligands within the N-ligand frameworks,67 or the exogenous ligand trans to the oxido ligand.77,78
Figure 2.
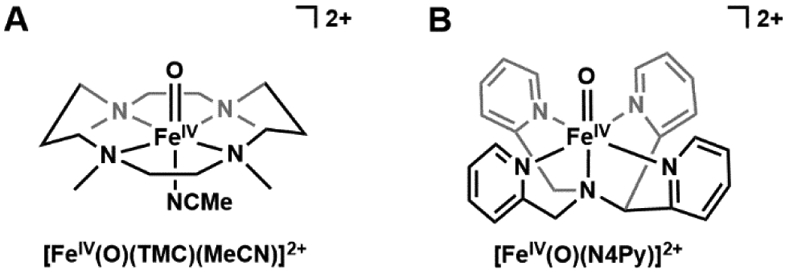
The first reported S = 1 FeIV-oxido complexes: [FeIV(O)(TMC)(MeCN2+ (A) and [FeIV(O)(N4Py)]2+ (B).
Biologically Relevant S = 2 FeIV–Oxido Complexes.
For all the success of FeIV–oxido complexes discussed above, their spin states differ from those found in proteins – this difference is important because electronic structure is often correlated with reactivity.79-81 Alternative design concepts were thus needed; one approach is to change the coordination geometry around the Fe(IV) center. The rationale for this approach is rooted in the fundamental concept of bonding within a M–oxido unit. Most FeIV–oxido complexes have tetragonal symmetry, which favors an S = 1 spin state because of the arrangement of the d orbitals (Figure 3). These insights have been known for over 50 years and come from the seminal work of Gray who first used molecular orbital theory to describe the bonding in M–oxido complexes.82,83 Gray's work was only for complexes in tetragonal symmetry and formed the basis for the Oxo-Wall concept.9,10 Although many have made claims of making complexes that "broke" this wall, most were incorrect or lack sufficient experimental evidence, making the topic still controversial.84-86 Within the context of FeIV–oxido complexes, a change in the coordination geometry to lower symmetry could afford high spin species. As first pointed out by Mayer and Thorn, C3-symmetric M–oxido complexes are good candidates to stabilize high spin species because this symmetry mandates that the manifold of d orbitals contains two doubly degenerate E states (Figure 3).87
Figure 3.
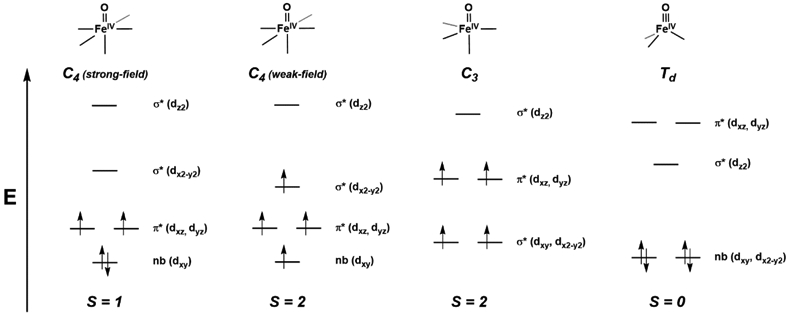
Qualitative d-orbital splitting diagrams for FeIV–oxido complexes with tetragonal (strong- and weak-field), trigonal, and tetrahedral symmetries.
This concept proved successful in preparing high spin FeIV–oxido species and there is now a small group of well-characterized trigonal bipyramidal FeIV–oxido complexes with S = 2 spin ground states. Members of this group use tripodal ligands to enforce the C3-symmetry and include the following complexes, [FeIV(O)(TMG3tren)]2+, [FeIVH3buea(O)]−, [FeIV(O)(tpaPh)]−, and [FeIVpoat(O)]− (Figure 4), where TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine, tpaPh = tris(5-phenylpyrrol-2-ylmethyl)amine), and [poat]3− = N,N’,N’-(nitrilotris(ethane-2,1-diyl))tris(P,P-diphenylphosphinic amido)).88-91 While Mössbauer spectroscopy confirmed the high-spin configuration for these systems by the anisotropic character of the ASD-tensor (see above), an S = 2 signal was also observed for some of these complexes using parallel-mode EPR spectroscopy to independently verify the spin state.42,89,90,92 Additionally, [FeIV(O)(TMG3tren)]2+ and [FeIVH3buea(O)]− were crystallographically characterized, with Fe─O bond lengths of 1.661(2) and 1.680(1) Å, respectively.89,93 From the works of Solomon who performed in-depth investigation into the electronic structure of [FeIV(O)(TMG3tren)]2+, we know the frontier molecular orbitals to be π*(xz, yz), which leads to the highly reactive nature of the complex in comparison to less reactive S = 1 FeIV–oxido complexes.94 In [FeIVH3buea(O)]−, the frontier molecular orbitals are dxy and dx2-y2 which are non-bonding with respect to the Fe–oxido unit, which is a consequence of the strong ligand field provided by the deprotonated urea donors within the trigonal plane.42
Figure 4.
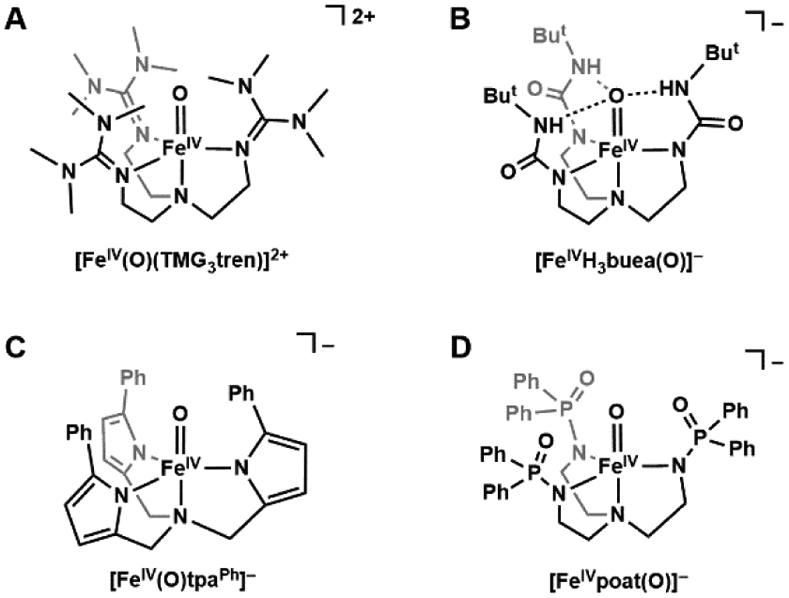
Trigonal FeIV–oxido complexes with S = 2 spin ground states: [FeIV(O)TMG3tren]2+ (A), [FeIVH3buea(O)]− (B), [FeIV(O)tpaPh]− (C), and [FeIVpoat(O)]− (D).
The generation of synthetic FeIV–oxido complexes allowed for further exploration into the bonding within the FeIV–oxido unit. Within a broader context, there has been much debate on whether Mn–oxido units, or its valence tautomer with Mn-1–O · (Mn-1–oxyl) units, are the active species during catalysis.95 Discussions on the relevance of Mn-1–O · species are driven by computational studies but there is little experimental support for these assignments, especially for complexes with Fe and Mn centers.96-98 The parallel-mode EPR signal of [FeIVH3buea(O)]− gave us the opportunity to experimentally address this question within an FeIV–oxido unit. The experiment was accomplished by measuring the broadening of the EPR signal at g = 8 of [FeIVH3buea(17O)]− (where I = 5/2 for an 17O nucleus).42 Substantial spin polarization was found on the oxido ligand as indicated by a ρp = 0.56 spins, where ρp is the spin population of the p orbitals on the oxido ligand. We have interpreted this large value of spin polarization on the oxido ligand to indicate significant covalency of the Fe–oxido bond, which is consistent with the assignment of an FeIV center from Mössbauer data (see above). This type of experiment has not yet been carried out on other non-heme FeIV–oxido systems, but DFT reported values of of 0.63 for [FeIV(O)TMG3tren]2+ and 0.6 for intermediate J agree with our experimental findings.43,88 Finally, we point out that if [FeIVH3buea(O)]− were to contain an oxyl radical, an oxygen-17 hyperfine interaction of nearly one full spin would have been observed on the oxido ligand (ρp ≈ 1).
The above examples of S = 2 FeIV–oxido species utilized tripodal ligands to promote C3 symmetry and high spin iron centers — an alternative approach to their preparation is to weaken the ligand fields in complexes with tetragonal geometry (Figure 3). The work of Que highlights this approach: he has studied the tuning of spin-state on Fe(II) and Fe(III) complexes in various TPA-derived ligands (TPA = tris(2-methylpyridine)amine) and discovered incorporation of bulky substituents at the 6-position of the pyridine donors lengthens the Fe–Npy bond and promotes high spin electron configuration.99 This observation led to a comparison of the tetragonal [FeIV(O)(L)(MeCN)]2+ complexes (L = TPA (tris(2-pyridylmethyl)amine), QBPA ((2-quinolylmethyl)bis(2-pyridylmethyl)amine), and TQA (tris(2-quinolylmethyl)amine), Figure 5), in which the pyridines in TPA were systematically replaced by the bulkier quinolines (QBPA contains 1 quinoline and 2 pyridines, TQA contains 3 quinolines).100-102 While [FeIV(O)(TPA)(MeCN)]2+ and [FeIV(O)(QBPA)(MeCN)]2+ adopt the standard low-spin configuration, [FeIV(O)(TQA)(MeCN)]2+ is S = 2. Importantly, this study showed that a high-spin FeIV-oxido unit can exist in a tetragonal geometry within an appropriate ligand field.
Figure 5.
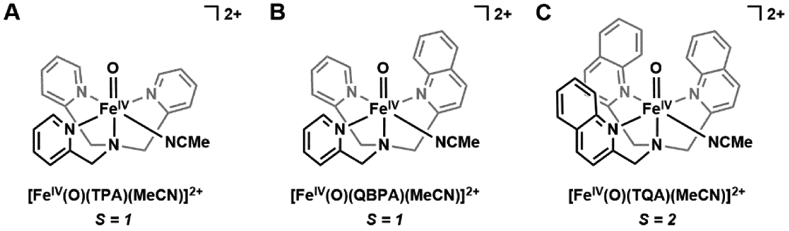
Systematic weakening of ligand field by incorporation of steric bulk in [FeIV(O)(TPA)(MeCN)]2+ (A), [FeIV(O)(QBPA)(MeCN)]2+ (B), and [FeIV(O)(TQA)(MeCN)]2+(C).
Role of Non-Covalent Interactions.
The [FeIVH3buea(O)]− (Figure 4B) complex also has a key structural difference when compared to most other FeIV–oxido complexes because of its intramolecular H-bonding network. Similar to the molecular structure of its FeIII–oxido analog, [FeIVH3buea(O)]− has the potential to have three H-bonds to the FeIV–oxido unit. While H-bonds are prevalent within the active sites of many metalloproteins, there is still limited experimental data on how they affect the physical and chemical properties of metallocofactors. Insights into their effects for FeIV–oxido complexes came from a comparative study of [FeIVH3buea(O)]− and [FeIVpoat(O)]− (Figure 4D), a complex with a tripodal ligand with phosphinic amido groups.91 Compared to the structure of [FeIVH3buea(O)]− the [poat]3− ligand produced a similar ligand field and coordination geometry to the Fe center, but [FeIVpoat(O)]− lacked the ability to form intramolecular H-bonds within the secondary coordination sphere. The experimental findings showed a significant weakening of the Fe=O bond in [FeIVH3buea(O)]− which was directly correlated to the presence of intramolecular H-bonds with the oxido ligand. Moreover, these studies showed how H-bonds affected the overall electronic structures in which the energies of the π*(xz, yz) orbitals are particularly sensitive. In the absence of H-bonding groups, [FeIVpoat(O)]− displays similar spectroscopic characteristics and instability as [FeIV(O)(TMG3tren)]2+.88,94
The effects of electrostatic interactions have also been explored with FeIV–oxido complexes. In many cases, the reactivity of the FeIV–oxido complexes was enhanced upon addition of a Lewis acid which usually is a redox-inactive metal ion (e.g. Ca2+ and Sc3+). One notable example is the work of Fukuzumi and Nam who showed that the electron transfer rate in [FeIV(O)N4Py]2+ is accelerated by a factor of 108 in the presence of redox-inactive metal ions (Figure 6A).103 We have found that the rate of O2 activation by [FeIIMST]−, a complex with a sulfonamido tripodal ligand ([MST]3− = N,N’,N”-[2,2’,2”-nitrilotris(ethane-2,1-diyl)]tris(2,4,6-trimethylbenzenesulfonamido)), is correlated with the Lewis acidity of group 2 metal ions used to produce heterobimetallic complexes (Figure 6B).104 These studies, and several others, demonstrate the influential and functional roles Lewis acids play in the chemistry of Fe complexes. However, an understanding of how Lewis acids interact with the Fe=O complexes to induce these functional changes is still lacking. In particular, there is little experimental evidence demonstrating that redox-inactive metal ions bond directly to the FeIV=O unit, which many have claimed is the principal reason for the changes in function.103,105,106 Two cautionary examples are worth a mention. Following their work on electron transfer, Fukuzumi and Nam reported the molecular structure from XRD methods of a related heterobimetallic complex with an FeIV–(μ-O)–ScIII core (Figure 6C), which was reasonably argued as the possible intermediate that gave rise to the increase in electron transfer rates.107 However, a subsequent computational study by Swart suggested that this structure was incorrectly assigned and should be formulated as an FeIII–(μ-O)–ScIII species.108 Que has repeated the experimental work and his findings agreed with those from the computational studies.109 Another possibility that is rarely discussed in the literature is that in the presence of (adventitious) water, the redox-inactive metal ions can function as simple Brønsted acids, [M(OH2)m]n+, that protonate the FeIV–oxido complexes rather than directly bind. This point was recently illustrated in a study reported by Jackson who examined the effects of Sc3+ and Al3+ ions on the reactivity of [MnIII(OH)(dpaq)]+ ([dpaq]− = 2-(bis(pyridine-2-ylmethyl))amino-N-quinolin-8-ylacetamidate, Scheme 5).110 While addition of trivalent metal ions to the MnIII–OH complex showed no significant spectroscopic perturbations, the resulting species demonstrated greatly enhanced reactivity towards hydrocarbons. These findings were comparable with the spectroscopic and reactivity properties of [MnIII(OH2)(dpaq)]2+,111 which suggest that addition of Sc3+/Al3+ ions results in protonation of the MnIII–OH complex, not metal binding.
Figure 6.
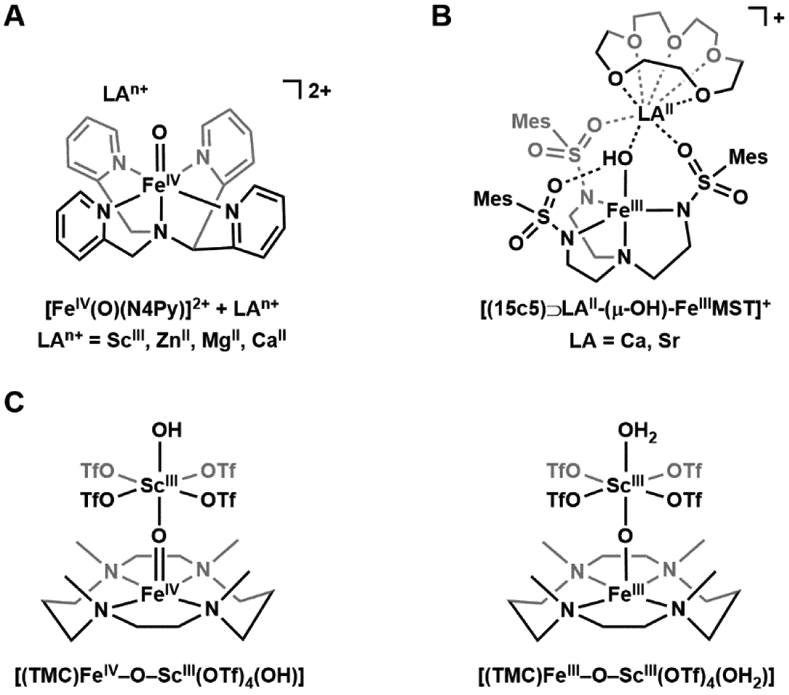
Effects of electrostatic interactions on metal-oxido and -hydroxido complexes. Addition of LAn+ accelerates the rate of electron transfer for [FeIV(O)(N4Py)]2+ by up to 108-fold (A). [(15c5)⊃LAII-(μ-OH)-FeIIIMST]+ (LA = Ca, Sr) (B). The previously characterized [(TMC)FeIV–O–ScIII(OTf)4(OH)] was reformulated as [(TMC)FeIII–O–ScIII(OTf)4(OH2)] (C).
Scheme 5.
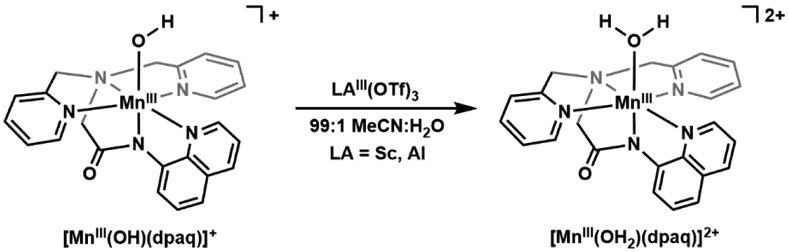
Addition of ScIII or AlIII ions to [MnIII(OH)(dpaq)]+ resulted in the protonation of the complex.
We have approached this problem differently by using an auxiliary binding site within the secondary coordination sphere of our [FeIVpoat(O)]− complex to coordinate a redox-inactive metal ion. The goal was to pinpoint where the redox-inactive metal ion was coordinated and to examine whether it had any effects on the properties of the FeIV–oxido unit. The [FeIVpoat(O)]− complex was designed for Lewis acids to bind in close proximity to the FeIV–oxido unit and our spectroscopic findings are consistent with a direct interaction between a redox-inactive metal ion and the oxido ligand (Figure 7).91 Thus, we were able to measure the changes in electronic and structural properties of the FeIV=O unit caused by the binding of Lewis acids, and our experimental and computational studies showed that the Lewis acid adducts modulate the strength of the π-bond within the FeIV=O unit. We could systematically tune the magnitude of the changes through the binding of Mg2+, Ca2+, Sr2+, Ba2+ ions to [FeIVpoat(O)]−. For instance, the electronic properties of [FeIVpoat(O)---MgII]+ varied significantly from those of [FeIVpoat(O)]−, but were identical to [FeIVH3buea(O)]− which used H-bonds to regulate the secondary coordination sphere. These studies allowed for the quantitative comparison of the effect of different types of non-covalent interactions to the FeIV–oxido moiety and illustrated that they can indeed have a strong impact on the properties of metal-oxido complexes.
Figure 7.
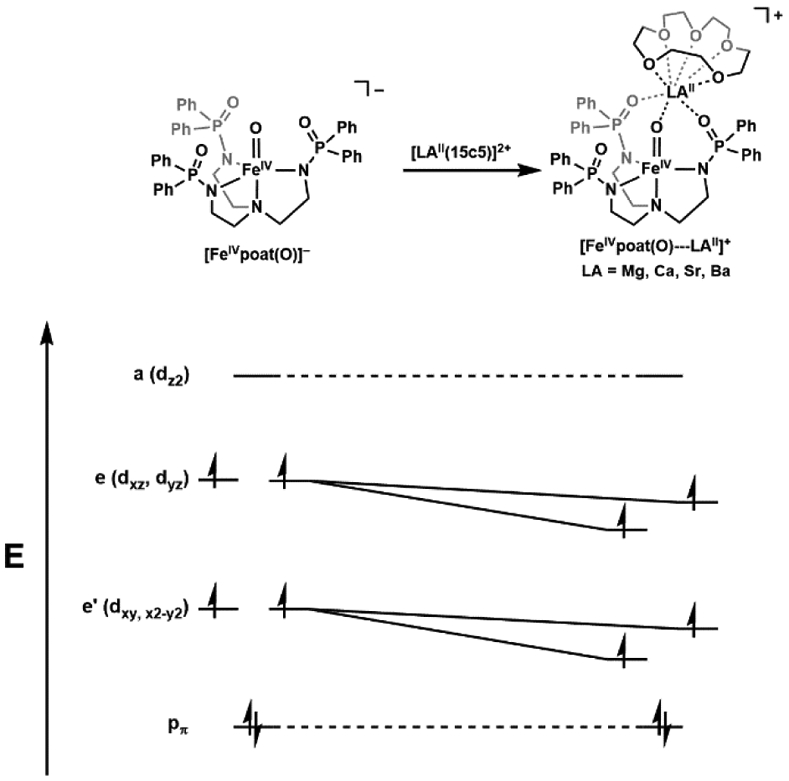
Binding and electrostatic effect of redox-inactive Group 2 metal ions on [FeIVpoat(O)]−.
Structural characterization of the series of [FeIVpoat(O)---LAII]+ complexes was unsuccessful due to their thermo-instability; however, we recently prepared and solved the molecular structure of the analogous FeIII-(μ-OH)–MgII species that provided useful structural insights. Addition of NMe4OAc to the starting synthon K[FeIIIpoat(OH)] in CH2Cl2 resulted in precipitation of the insoluble KOAc. The reaction mixture was then filtered and treated with [MgII(TMTACN)(OTf)2] (TMTACN = 1,4,7-trimethyl-1,4,7-triazacyclononane) to produce the desired [(TMTACN)MgII–(μ-OH)–FeIIIpoat]OTf complex (Figure 8A). Multiple recrystallizations provided single crystals suitable for X-ray diffraction that revealed a discrete bimetallic [FeIII–(μ-OH)–MgII] core (Figure 8B). The FeIII center adopts a trigonal bipyramidal geometry, comprising of the [poat]3− ligand and an hydroxido ligand; the MgII site retains the tridentate TMTACN capping ligand, and binds to the [FeIIIpoat(OH)]− complex via two O-atom donors from the phosphinic amido groups of [poat]3− and the bridging hydroxido ligand, as proposed in the series of [FeIVpoat(O)---LAII]+ complexes. Notably, binding of the MgII adduct is consistent with a Mg1–O1 distance of 1.983(3) Å. The remaining phosphinic amido arm that does not coordinate to the MgII ion participates in intramolecular H-bonding with the bridging hydroxido ligand, with an O1⋯O2 distance of 2.661(3) Å.
Figure 8.

Preparative route to [(TMTACN)MgII–(μ-OH)–FeIIIpoat]OTf from K[FeIIIpoat(OH)] (A). Thermal ellipsoid diagram depicting the molecular structure of [(TMTACN)MgII–(μ-OH)–FeIIIpoat]+ determined by X-ray diffraction (B). Ellipsoidsc are shown at 50% probability level, and only the hydroxido H atom is shown for clarity. Selected bond distances (Å) and angles (deg): Fe1–O1, 1.892(2); Fe1–N1, 2.236(3); Fe1–Neq,avg, 2.012(3); O1⋯O2, 2.661(3); Mg1–O1, 1.983(3); Mg1–O3, 2.015(3); Mg1–O4, 2.037(3); Mg1–NTMTACN,avg, 2.257(3); O1–Fe1–N1, 175.74(11); Fe1–O1–Mg1, 120.09(12).
Reactivity
C─H Bond Cleavage.
There have been many studies on the reactivity of synthetic FeIV–oxido complexes with organic substrates to model the C─H bond cleavage step found in non-heme monooxygenases. We have summarized some of those findings in Table 1 which also includes the rate of TauD. FeIV–oxido complexes with S = 2 spin ground states were calculated to be more reactive towards C─H bonds than their S = 1 counterparts.79-81 However, no significant trend can be found when comparing experimentally obtained reactivity data of complexes with different ground spin states. The S = 1 [FeIV(O)TMC(MeCN)]2+ displays sluggish reactivity towards 9,10-dihydroanthracene (DHA,112 BDEC─H = 78 kcal mol−1) and 1,4-cyclohexadiene (CHD,113 BDEC─H = 77 kcal mol−1),88,114 while the similarly thermostable S = 1 [FeIV(O)N4Py]2+ demonstrated comparable, if not better, substrate reactivity than the S = 2 [FeIV(O)TMG3tren]2+, [FeIVH3buea(O)]−, and [FeIV(O)tpaPh]−complexes.88,90,115 The diminished or absent substrate reactivity of the latter three complexes could be attributed to the steric bulk provided by the ligand frameworks. On the other hand, the S = 2 [FeIV(O)(TQA)(MeCN)]2+ displayed extraordinary reactivity towards DHA and cyclohexane (BDEC─H = 99.3 kcal mol−1),116 which is comparable with taurine oxidation by TauD-J after correction for the temperature difference.37 It is obvious that more mechanistic work is needed to understand the connections between spin states and C─H bond cleavage.
Table 1.
Reactivity of synthetic and enzymatic FeIV–oxido complexes with C-H substrates.
| Spin state | Complex | Substrate Reactivitya | References |
|---|---|---|---|
| 1 | [FeIV(O)TMC(MeCN)]2+ | DHA (0.016) CHD (0.018) |
88,114 |
| [FeIV(O)TPA*(MeCN)]2+b | DHA (0.042) | 117 | |
| [FeIV(O)N4Py]2+ | DHA (2) CHD (1.3) |
88 | |
| 2 | [FeIV(O)TMG3tren]2+ | DHA (0.090) CHD (1.2) |
88 |
| [FeIVH3buea(O)]− | No reactivity | 115 | |
| [FeIV(O)tpaPh]− | CHD (1.4) | 90 | |
| [FeIV(O)(TQA)(MeCN)]2+ | Cyclohexane (0.37, −40 °C) DHA (200, −80 °C) |
102 | |
| TauD-J | Taurine (kobs = 13 s−1, 5 °C, aqueous) | 37 |
Unless specified, rate constants (k2, M−1 s−1) were measured in MeCN at −30 °C.
TPA* = tris(4-methoxy-3,5-dimethylpyridyl-2-methyl)amine.
Rebound Step and FeIII–OH Species.
The proposed mechanisms for hydroxylation of substrates include a rebound step whereby a transient carbon radical combines with an Fe─OH species to produce a new C─OH bond (Schemes 2, 3).25,36 Synthetic FeIII–OR species that mimic the rebound step for the α-KG dependent, non-heme Fe proteins have been developed. Goldberg first reported the synthesis of an FeIII–OCH3 complex, [FeIII(N3PyO2Ph)(OCH3)]+, ((N3PyO2Ph)− = 2-(((bis(6-phenylpyridin-2-yl)methyl)(pyridin-2-ylmethyl)amino)methyl)phenolate) and its reactivity towards trityl radical (Ph3C•) to produce [FeII(N3PyO2Ph)(Solv)]+ and Ph3OCH3.118 Further mechanistic studies showed a concerted rebound process, with no initial charge transfer.119 Goldberg and Fout then independently demonstrated different FeIII–OH complexes that readily react with trityl radical to produce quantitative Ar3COH (Scheme 6A, B).120,121 Goldberg also investigated the selectivity of hydroxido vs. halogen rebound using a library of non-heme Fe complexes with the [BNPAph2O]− (2-(bis((6-(neopentylamino)pyridin-2-yl)methyl)amino)-1,1-diphenylethan-1-olate) ligand framework: FeIII(OH)(X) and FeIII(X)2 (X = Br, Cl).122 He showed that reactivity of FeIII(OH)(X) with trityl radical produced near-quantitative FeII(X) and Ar3COH, widi no Ar3CX detected (Scheme 6C). Moreover, the FeIII(X)2 complex does react with trityl radical to produce near-stoichiometric FeII(X) and Ar3CX (Scheme 6D). The preference of FeIII(OH)(X) towards hydroxylation is tentatively attributed to a thermodynamic factor, for which OH-rebound is calculated to be more favorable than halogenation by > 15 kcal/mol.123,124 Factors of selective halogenation by halogenase enzymes such as SyrB2 remain to be investigated.
Scheme 6.
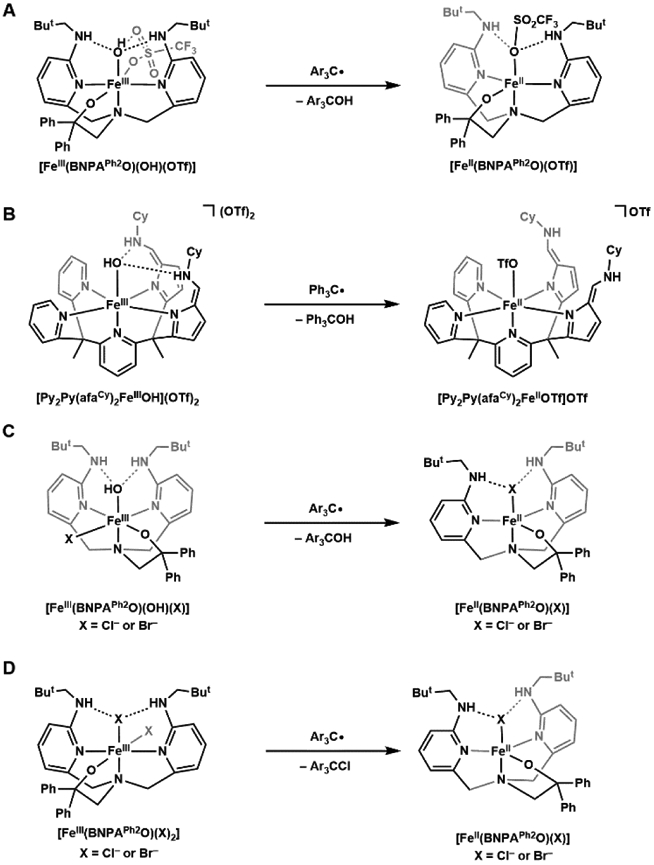
Synthetic non-heme Fe(III) complexes demonstrate rebound reactivity of hydroxido and halide ligands with trityl radical substrates.
The work of Goldberg and Fout illustrated that a rebound step is chemically possible; however, Shaik and Nam have questioned whether this step is mechanistically viable in all systems.125 They offered an alternate mechanistic possibility in which the substrate radical dissociates and reacts with another FeIV=O complex to produce an FeIII–OCR species. They support this premise with theoretical calculations that predicted dissociation of the substrate radical that is formed after C─H bond cleavage is more favorable than FeIII–OH rebound. Moreover, they offer experimental evidence using synthetic complexes. This idea is intriguing and offers a reasonable explanation for observations found with synthetic FeIV=O complexes. What is less clear is whether this proposal is relevant within the active site of non-heme metalloenzymes where the local environments around the Fe centers are more controlled than in synthetic systems and likely prevent dissociation of substrate radicals.
Rebound Step and FeIV–OH Species.
There is strong evidence for this step from studies using P450s with the key intermediate being compound II which is an FeIV–OH species — however, the role that this type of high valent metal-hydroxido species in other enzymatic systems remains controversial. Green has found that compound II has a pKa(OH) of 12, which is unusually high for comparable metal–hydroxido species which have estimated pKa(OH) values of less than 5.26,126 He has argued that the difference is because compound II has an axially coordinated cysteine thiolate which is trans to the hydroxido ligand and elevates the pKa(OH) value.
Other heme proteins with axial ligands containing N-atom donors produce intermediates that should not be protonated under normal conditions based on known thermodynamic information.127-131 The difficulties in producing FeIV–OH species can be understood from the simple equilibrium expression (eq 1) which favors the products. In general, terminal oxido ligands are electrophiles and are weakly basic, which makes protonation thermodynamically unattainable.10,126,132 For P450s, and other heme proteins with an axially coordinated thiolate, the strongly covalent Fe─S bond weakens the bonds within the FeIV=O unit, shifting the equilibrium to favor protonation. Other biologically relevant axial ligands do not appear to form Fe─X bonds that are sufficient to change the basicity of the oxido ligand; hence, to our knowledge there are no other biological examples, in both heme or non-heme proteins, where there is definitive experimental proof of an FeIV–OH species.
| (1) |
Our knowledge of non-heme FeIV–OH species is still incomplete, and we question whether they can be generated in a biologically relevant context. We make these points recognizing that there are a considerable number of mechanistic proposals that include FeIV–OH species as essential intermediates.133-135 Intuitively FeIV–OH species could be considered reasonable intermediates, but there is little experimental evidence to support their formation and, furthermore, there is no thermodynamic evidence (e.g., Ka values in eq 1) to validate their stability. Synthetic verification of FeIV–OH species has also been difficult to achieve. Efforts to model compound II led Goldberg to prepare and structurally characterize a formally FeIV–OH species in a corrole framework, [Fe(OH)(ttppc)] (H3ttppc = tris(2,4,6-triphenyl)-phenyl corrole, Figure 9A).136 Its ability to react with a carbon radical substrate to form a new C─OH bond makes it a suitable synthetic model for compound II. However, the oxidation assignments of the Fe center and corrole ligand are unclear, and the species can only be accurately interpreted as one oxidizing equivalent above FeIII–OH. Although the Mössbauer spectrum of [Fe(OH)(ttppc)] was reported, further variable-field experiments, as well as X-ray absorption spectroscopy, are required to elucidate the oxidation state of the Fe center.
Figure 9.
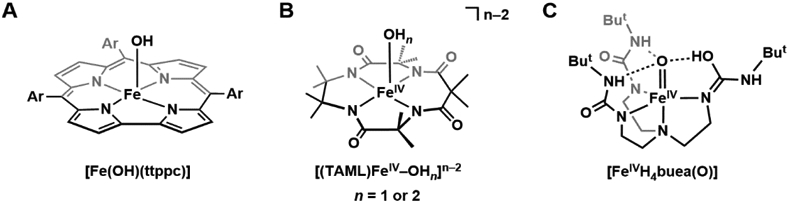
Synthetic attempts for FeIV-hydroxido complexes: [Fe(OH)(ttppc)] (A), [(TAML)FeIV-OHn]n-2 (B, n = 1 or 2), and [FeIVH4buea(O)] (C).
Hendrich has attempted to generate and study an FeIV–OH species by subjecting the complex [(TAML)FeIII–OH2]− ([TAML]4− = [Me4C2(NCOCMe2NCO)2CMe24−) to chemical oxidation or electrolysis.137 A new species was formed, and its EPR and Mössbauer parameters are consistent with a high-spin Fe(IV) center. However, the authors cannot definitively assign the FeIV species to be [(TAML)FeIV–OH]− or [(TAML)FeIV–OH2] (Figure 9B). No Fe–X vibrational features > 600 cm−1 were observed for this species, and the features observed between 447–475 cm−1 were proposed to be Fe–O vibrations, weakened due to protonation of the oxido ligand, and hydrogen-bonding with the bulk aqueous solvent. However, these features are comparable or weaker in energy than those of a series of FeIII–OH complexes we have developed (477–594 cm−1), which does not support the assignment for an FeIV–OH species.138 Additionally, electrochemical experiments on [(TAML)FeIII–OH2]− revealed a Nernstian one-electron redox event, which suggests oxidation does not result in geometric rearrangement or ligand modification, posing the possibility that an FeIV–OH2 species could be produced.
We have approached this problem with experiments to protonate [FeIVH3buea(O)]− (Figure 4B): a thermodynamic study provided an estimated pKa of ~11 for its conjugate acid, [FeIVH3buea(O)(H)], which is a similar value to that found for compound II in P450s.26 Treating [FeIVH3buea(O)]− with acids produced a new complex with distinct optical and magnetic properties.139 This complex reverted back to [FeIVH3buea(O)]− upon the addition of base. Moreover, this protonated species could be prepared from treatment of [FeIVH3buea(O)]− with an oxidant which is consistent with initial formation of a formally FeV=O species that further reacts through a similar mechanism as proposed for P450s to produce a species similar to compound II. However, the vibrational and structural properties do not support the formation of FeIV–OH species and computational studies suggest that the [H3buea]3− ligand is the site of the protonation (and not the oxido ligand, Figure 9C). These results demonstrate that care should be taken in assigning the protonation state of an oxido ligand in high valent Fe complexes, especially when other sites of protonation are present such as a basic site within a protein active site or synthetic ligand.
Use in Chemical Synthesis.
Because of the utility of Fe-oxido as the active oxidant in nature, much attention has turned to the environmentally green Fe/O2 (or H2O2, bleach) system for chemical processes. While the identity of the catalytically competent Fe species in many instances is not fully elucidated, this has led to the discovery of many new chemical transformations. Costas and White have independently made important contributions using molecular iron precursors in tetradentate N-based ligand frameworks, but most work is based on the N2Py2 scaffold, in which two pyridines are attached via a diamine linker.140-142 They have developed various catalytic processes that include those for hydroxylation, dihydroxylation, and epoxidation and have modified these catalysts to improve site- and chemo-selectivities. They have shown that increasing the rigidity of the diamine backbone,143-145 as well as protecting sites that are prone to oxidation on the ligand (e.g. by deuteration of the benzylic hydrogens),146,147 can improve the robustness of the catalysts. Costas has further found that modulating the electronic and steric properties of the pyridine arms can have significant effect on chemoselectivity. For instance, installation of more electron donating groups at the γ-pyridine position increases the preference and yield for olefin epoxidation (Figure 10A),148 while alkyl substitution at the α-position promotes the selectivity for olefin syn-dihydroxylation (Figure 10B).149 Using a more supramolecular approach, it was found that installation of directing groups in the catalyst/substrate through covalent or non-covalent interactions can improve regioselectivity. The work of Costas illustrated this concept when he incorporated benzocrown-6-ethers onto [FeII(PDP)]2+ complexes that served as receptors for alkyl substrates with protonated primary amines (Figure 10C).150 Upon treatment with H2O2, the complex can selectively perform C─H hydroxylations at the C8 and C9 positions of the alkyl chain; this regioselectivity is attributed to controlling the orientation of the substates via docking the R-NH4+ within the internal crown ether. The docking of the alkyl substrates to promote specificity is reminiscent of how proteins increase selectivity through positioning of a C─H bond near reactive Fe-oxido centers by orienting substrates within an active site using H-bonds.151,152 White has demonstrated the utility of late-stage site- and stereo-selective C-H bond functionalization of a variety of natural products using the catalyst [FeII(PDP)]2+ and [FeII(CF3-PDP)]2+; the oxidized derivatives are compounds of pharmacological interests.142 For instance, selective hydroxylation of artemisinin can be achieved by [FeII(PDP)]2+ with comparable yields to the native P450 enzyme (Figure 10D).153 White has also explored functionalization of unnatural amino acids using the same catalysts to provide new building blocks for oligopeptides and proteins.154 To showcase a different type of reactivity of Fe–oxido complexes we turn to the work of Collins who has demonstrated the viability of using Fe catalysts in waste treatment industry (Figure 10E).155,156 Using Fe-TAML (tetraamido macrocyclic ligands) complexes as catalysts and O2/H2O2/NaOCl as the oxidant, he has been able to degrade explosives (e.g. trinitrotoluene, trinitrobenzene157), endocrine disruptors (e.g. bisphenol A158, nitrophenols159), and other pollutants (e.g. commercial dyes160,161, metaldehyde162) and demonstrated that these systems can be adapted to an industrial setting. The identity of the competent oxidants for all these processes has often been proposed to be a high-valent Fe-oxido species; however, there is still little experimental evidence to verify these claims, leaving much to investigate.
Figure 10.

Representative work in Fe molecular catalysis in chemical synthesis. Electronic and steric modifications in the N4-based ligands modulate chemoselectivity (A & B). Incorporation of supramolecular directing groups improve regioselectivity of C─H bond activation (C). Late-stage transformations of various natural products and their derivatives (D). Use of Fe-TAML catalysts in environmentally beneficial catalysis, such as waste treatment (E).
Mechanistic Considerations
Cleavage of C─H bonds by Metal-Oxido Species.
Within this Forum we have highlighted the utility of metal-oxido species for the cleavage of C─H bonds and we now address ideas on why they are able perform this function. There are two mechanistic camps that have guided our thinking on this process, one that emphasizes ground state thermodynamics,1,12,163 while the other suggests that the radical character of the oxido ligand drives the reactivity. The attributes of both mechanistic proposals of C─H bond cleavage have been reviewed extensively and we will not duplicate those discussions here. We highlight the thermodynamic approach and how it can be applied to biological and synthetic C─H bond cleavage. This approach is rooted in physical organic chemistry112,164,165 and adapted by Mayer for the cleavage of X─H bonds (X = C, N, O).1,12 The central tenet is that the reactivity of a Mn=O center can be evaluated by comparing the bond dissociation free energy (BDFE) of the C─H bond to be cleaved to that of the O─H bond formed in the resulting Mn-1–OH species. We note that the literature interchanges BDFE and BDE values with the older literature often using BDEs which is purely enthalpic.1 The merits of using BDFEs values have been discussed by Mayer and the field has generally adopted its usage. In this Forums article, the terminology used by the authors of the articles to which we refer is used to most accurately report the data. Thermodynamic square schemes (Figure 11) are useful to illustrate the various connections between proton transfer (pKa electron transfer (E1/2), and BDFE. Square schemes also provide a convenient way to represent the three limiting mechanistic pathways for C─H bond cleavage: 1) stepwise proton transfer-electron transfer (PT-ET); 2) stepwise electron transfer-proton transfer (ET-PT); and 3) concerted proton electron transfer (CPET).
Figure 11.
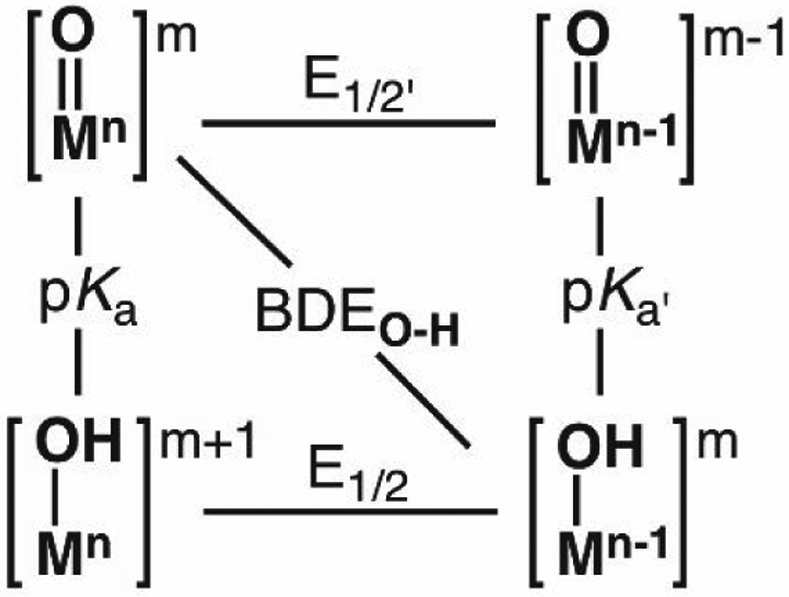
Thermodynamic scheme for a metal-oxido complex.
The literature indicates that CPET pathways are the most common for reactions involving FeIV=O complexes and substrates with a C─H bond. This conclusion comes from numerous studies with synthetic FeIV=O species that show strong correlations between the log (k2) and the BDFEC─H of the substrates (where k2 is the second-order rate constant of a reaction).1,15,76,90,115,166-171 Linear free energy relationships of this type follow from the Bell-Evans-Poyanyi (BEP) principle172,173 which predicts that experimentally obtained rate constants increase with either a decrease in the BDFEC─H of the substrate or the increase in energy released from forming the MO─H bond (that is, BDFEO─H). These BDFEO─H values, however, are missing from the literature – although there are reports for some systems, there are still too few examples that are derived from experiments. However, BDFEO─H values can be determined from the E1/2 value of the M=O species and the pKa value of its conjugate acid, that is M–OH (eq 2), where the coefficients are derived from fundamental thermodynamic relationships and CG is a constant that is dependent on the reference electrode and solvent.1 The experimental difficulties are usually in determination of the pKa value because these metal-hydroxido complexes are difficult to prepare (see above for FeIV–OH). Nevertheless, eq 2 illustrates the close relationship with reduction potential and pKa values in determining the BDFEO─H, which influences the overall driving force for the reactivity of a M=O species with C─H bonds.
| (2) |
The cleavage of C─H bonds is an oxidative process and chemical intuition would suggest that a stronger oxidant (i.e., M=O species) would increase the efficiency of a given reaction. However, highly positive potentials have the disadvantage of poor selectivity and/or irreversible destruction of proteins or ligands. Eq 2 shows that this condition can be circumvented – in fact, a less positive potential and a large pKa value (more basic M=O species) can produce large BDFEO─H values and reactive M=O species. This relationship can be shown graphically for the cleavage of the C─H bond in methane (Figure 12).174 For a M=O species with a relatively low pKa value of 5, the corresponding positive potential of 1.75 V vs NHE is large enough to oxidize the nearby aromatic side chains within the active site of a protein. When the pKa value is raised to 17.5, the potential is lowered to ~1 V vs NHE which will inhibit off-path and destructive oxidations. Therefore, the basicity of the M=O unit is an important parameter that regulates reactivity toward C─H bonds.
Figure 12.

Relationship between reduction potential and pKa for generic metal-oxido species in the homolytic cleavage of a C─H bond in methane with BDFEC─H = 102 kcal/mol from eq 2. The vertical lines indicate regions of relatively high (blue) and low (red) potentials.
Basic M–Oxido Species.
This role of the basicity of the M=O unit has been studied in both proteins and synthetic complexes. Green was the first to show the relevance of this concept in the high valent Fe–oxido intermediate of cytochrome P450s (compound I). He demonstrated experimentally and computationally that the reduced intermediate of compound I is sufficiently basic to promote the abstraction of H-atoms from strong C─H bonds at relatively low one-electron reduction potentials. We have studied how the basicity of the metal-oxido unit in synthetic complexes can be used to regulate the cleavage of C─H bonds. In one study, we examined the reactivity of [MnIIIH3buea(O)]2− with the common test substrate 9,10-dihydroanthracene (DHA), and found clean conversion to anthracene and [MnIIH3buea(OH)]2−, indicating C─H bond cleavage had occurred (Scheme 7).175 However, the one-electron reduction potential for [MnIIIH3buea(O)]2− is quite negative and we were only able to estimate its value at less than −2.0 V versus [FeIII/IICp2]+/0.13 This potential is in the range found for strong reductants such as cobaltocene and just based on its electrochemical potential should not be able to cleave C─H bonds. However, we measured the pKa(OH) value of 28 in DMSO for [MnIIIH3buea(OH)]−, the conjugate acid of the MnIII–oxido complex.176 This value indicates that the MnIII–oxido unit in [MnIIIH3buea(O)]2− (its conjugate base) is sufficiently basic to compensate for the negative potential to promote reactivity.
Scheme 7.
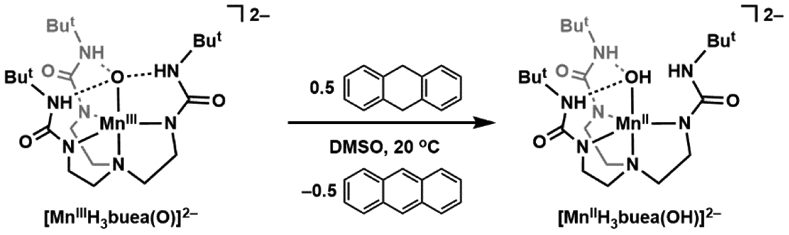
Reactivity of [MnIIIH3buea(O)]2− towards 9,10-dihydroanthracene.
The idea that the basicity of a M–oxido unit is an important parameter suggests that it can be tuned to regulate reactivity toward C─H bonds. We tested this premise by developing a system that permitted us to systematically modulate the basicity of the MnIII–oxido unit and determine whether these changes correlated with the rate of C─H bond cleavage. We redesigned [H3buea]3− into a series of new hybrid ligands [H3bpuea-R]3− (R = OMe, H, F, Cl, CF3) in which one of the tripodal arms contains a para-substituted phenylurea unit, and we prepared the corresponding series of [MnIIIH3bpuea-R(O)]2− complexes (Figure 13).177 These MnIII–oxido complexes differed only in their H-bond involving the phenylurea groups – our experimental data showed that the changes in H-bond donor strength modulated by the R-group regulated the basicity of each complex. Moreover, we found a correlation between the pKa(MnIIIOH) values and the second order rate constants for the reaction of the [MnIIIH3bpuea-R(O)]2− complex with DHA. Additional studies on these MnIII–oxido complexes did not show any correlation between log(k2) and the BDFEC─H of DHA which would be characteristic of a synchronous CPET. However, our data also did not support a two-step process for rate-limiting PT followed by ET.
Figure 13.
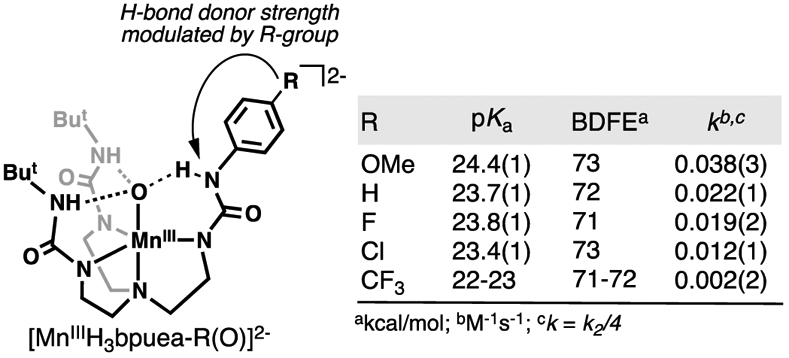
The intramolecular H-bond donor strength in a series of [MnIIIH3bpuea-R(O)]2− complexes is modulated remotely by the R-group, which affects the basicity of the complex and the reaction rate towards 9,10-dihydroanthracene. The experimentally determined second order rate constant k2 was corrected to k to account for 4 equivalents of C─H bonds that can possibly be cleaved in DHA.
A detailed description of the mechanism for this reaction is still not known, but it appears that it does not follow the BEP principle and a conventional CPET in which the proton and electron move together at the transition states (a synchronous process). We hypothesize that this process is still CPET but involves an asynchronous transition state in which PT precedes ET.15,178 Similar asynchronous CPET (Figure 14A) has been suggested to occur in other systems and appears to be a new mechanistic description of C─H bond cleavage by metal–oxido species. The experimental work of Anderson on a unique CoIII–oxido complex provides strong evidence for a PT-dominated asynchronous CPET mechanism (Figure 14B),179,180 as does work by Tolman on a CuIII–O2CAr complex (Figure 14C).181 The link between these complexes and [MnIIIH3buea(O)]2− is the high basicity of the metal–X unit (X = oxido, hydroxido, carboxylato) which seems to be characteristic of this type of mechanism. Moreover, computational studies by Srnec on FeIV=O complexes provide a theoretical framework to evaluate asynchronous processes and introduced an asynchronicity parameter to assess the contributions of PT and ET within transition states.182 However, additional experimental evidence is needed to evaluate whether this parameter will be useful in designing new reagents and understanding their mechanisms. It is obvious that the field would benefit from additional methods to analyze data to indicate which of the possible mechanisms is operative for a given system. This type of analysis has implications beyond garnering fundamental insights as it could establish a practical advantage for developing complexes toward chemical synthesis. Rather than using complexes that rely on BDFEC─H to induce selectivity, M–oxido complexes that are sufficiently basic could lead to reagents with enhanced selectivity that favor targets based on acidity of the C─H bonds.
Figure 14.

Square scheme for asynchronous CPET (red arrows, (A)), Anderson’s CoIII-oxido (B), Tolman’s CuIII-O2CAr (C). Scheme in A is adapted from Anderson.177
Some Remaining Challenges
Throughout this Forum article we presented topics on M–oxido and –hydroxido chemistry with an eye to highlight important accomplishments and point out where additional experiments might be needed. We end with two short discussions on areas that are not as complete, are somewhat controversial, have importance in chemistry and biology, and from our perspective are ripe for additional studies.
The Relevance of FeV=O in Biology and Beyond.
We have addressed many important aspects of FeIV-oxido and - hydroxido cofactors in metalloenzymes and synthetic analogues, but at this point one may wonder if higher valent analogous are known and useful. In heme systems there is no evidence that higher valent Fe systems are operative. The formation of the key oxidized intermediate occurs from the heterolytic cleavage of an FeIII–OOH species to produce water and compound I, which is not an FeV=O species, but rather an FeIV=O unit coupled to a ligand-based radical (Scheme 2).23 There is consensus supported from both computational and experimental studies for this assignment. These finding underline an important feature of active sites in metalloenzymes: instead of incorporating weakly-donating amino acid-based ligands or other scaffolds to support a thermodynamically unfavorable, high-valent metal ion (e.g. FeV), these enzymes are equipped to store additional oxidative equivalents on a redox-active organic moiety.23,183
For non-heme systems the role of an FeV-oxido in both proteins and synthetic systems is controversial and the subject of continued debate.184,185 Mechanistic proposals for the Rieske oxygenases provide an illustrative example of where there are concerns. Early mechanistic thoughts centered on the possible similarities between the O2 activation pathways of these non-heme Fe monooxygenases and cytochrome P450s: O2 binding was thought to result in heterolytic cleavage of an O─OH bond to generate a new Fe intermediate that is commonly proposed as an FeV(O)(OH) species (Scheme 8A).133,186,187 However, there is still no experimental data to support the formation of an FeV-oxido species within Rieske proteins or for that matter, any metalloprotein. For Rieske proteins, recent investigations have supported alternative mechanisms that avoid the formation of high valent Fe intermediates. Solomon and Lipscomb investigated the native O2 reaction in the Rieske enzyme benzoate 1,2-dioxygenase (BZDO), and determined in the native O2 reaction an FeIII-superoxo species reacts directly with the benzoate substrate without accessing higher valence states (Scheme 8B).188 Additional experiments using a peroxide shunt route found that an FeIII–OOH species is produced which also leads to product formation. This study again emphasizes nature’s ability to circumvent the need to access the high oxidation states by employing other mechanistic pathways: not only is it thermodynamically challenging to achieve an FeV species, but a highly reactive FeV cofactor could also lead to nonproductive pathways that can cause irreversible damage to the protein.
Scheme 8.
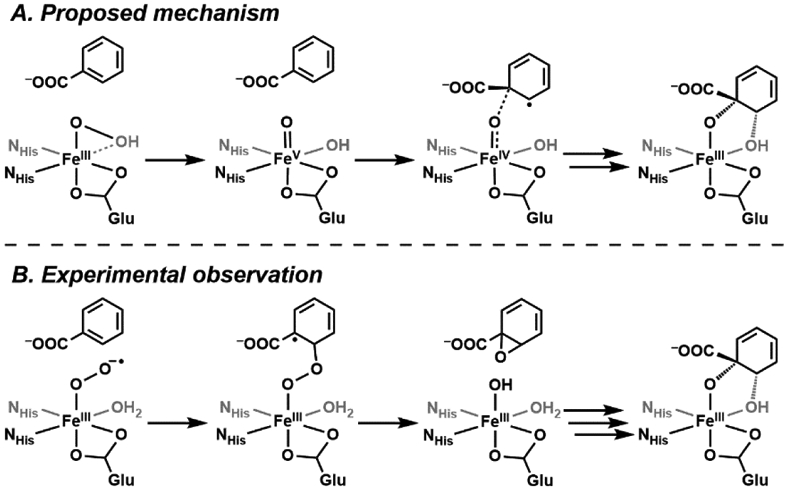
The proposed (A) and experimentally verified (B) mechanisms of benzoate 1,2-dioxygenase, a Rieske enzyme. An FeV intermediate was not observed.
The implication of FeV intermediates in non-heme enzymes has inspired attempts to prepare similar species within synthetic systems.189-193 However, unlike the FeIV=O analogs, preparative difficulties in accessing FeV state, increased instability, and lack of structural data of proposed FeV complexes make the assignment of iron oxidation state challenging. Moreover, there is a general lack of identifiable spectroscopic features for non-heme FeV–oxido species that makes unambiguous assignments difficult. Two spectroscopic handles for FeV valence states have been proposed: 1) the Griffith-Taylor model is used to predict the g factors of S = 1/2 FeIII centers, in which a large g-anisotropy is expected (gmax – gmin > 0.8), while an S = 1/2 EPR feature with a small g-anisotropy has been observed and proposed for low-spin FeV complexes;192,194-196 and 2) anisotropy in the 57Fe A-tensor is proposed to be a spectroscopic marker to identify 3d electron configuration and to assign FeV centers.189 Recently, Ye and Neese performed detailed computational studies into several proposed FeV=O complexes, and concluded the x/y anisotropy of the A-matrix to be an unreliable spectroscopic handle for the assignment of the FeV state.197 In this study, most of these reported FeV complexes are reformulated to have dominant FeIV character coupled with a ligand radical which is not unlike what occurs in cytochrome P450s. They only assigned [FeV(O)(TAML)]− as a genuine FeV=O species, and stabilization of the high-valent Fe center is attributed to the tetraanionic ligand field.155 The shortening of the Fe–O bond (1.58 Å) as well as a significantly more negative isomer shift (−0.42 mm/s) in [FeV(O)(TAML)]− distinguish it from FeIV=O and other proposed FeV complexes.190 Generation of an FeV-oxido species has also been proposed in the gas phase using helium tagging infrared photodissociation (IRPD) spectroscopy, a powerful technique used to measure IR spectra of mass-selected ions.198 However, magnetic and redox properties of these species, which have proven essential for determining the oxidation level of Fe=O complexes, are not currently available in the gas phase to corroborate these claims.
For the above description it is apparent that the quest to prepare and characterize FeV–oxido complexes is still ongoing. Regardless if FeV=O species turn out to be relevant in biology, developing a complete understanding of their properties and reactivities is important in determining their use in chemical synthesis.184,185 In many ways, the progress in FeV=O chemistry is reminiscent of where the field was 20 years ago in the pursuit of FeIV=O complexes – breakthroughs were made only after detailed investigations into their molecular/electronic properties which were coupled to vibrational data. The key lesson learned from these studies is that one technique alone cannot be used to make definitive assignments of oxidation state and structure, and computational findings need to be supported by the appropriate experimental results. We still have much to learn before consensus on FeV=O systems match that achieved for heme and non-heme FeIV=O complexes.
Single Site Cu Centers for C─H Bond Functionalization.
Research into the structure and function of metalloenzymes with single Cu centers has increased in the last 10 years, being spurred on by studies of particulate monooxygenases (pMMOs) and lytic polysaccharide monooxygenases (LPMOs). pMMOs have been known for some time199-204 and have been reviewed previously205-208 – our focus will be on LPMOs in which the involvement of Cu and O2 has only been confirmed within the last dozen years.209 These metalloenzymes are commonly isolated from bacteria and fungi within compost heaps210 and promote the degradation of biopolymers such as cellulose and chitin via oxidative cleavage of polysaccharide chains at either the C1 or C4 positions of the glucose unit (Scheme 9).211,212
Scheme 9.
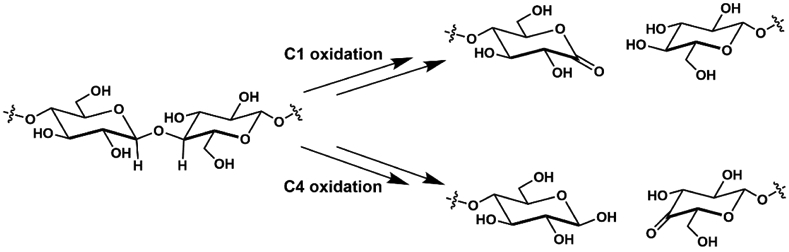
Substrate transformation performed by LPMOs on cellulose.
From an applied perspective, LPMOs have attracted much attention because of their role in enhancing the conversion of biomass to biofuels.211,213-215 From a fundamental perspective, this transformation has intriguing mechanistic implications: it can be performed via activation of dioxygen in conjunction with exogenous electron and proton sources or with hydrogen peroxide. Dioxygen was reported to be involved in catalysis in 2010.209 Later in 2017, it was proposed that hydrogen peroxide could form in situ from dioxygen and then lose water resulting in a CuII-oxyl intermediate which abstracts a hydrogen atom from substrate and finally hydroxylates the polysaccharide.216 Now evidence suggests that H2O2 can be used to initiate catalysis anaerobically.217,218
The active site, which lies along the surface of the protein (Figure 15A), consists of a single Cu-center coordinated to three nitrogen atoms from two histidine side chains and the N-terminus amine (denoted the histidine brace, Figures 15B and C).211,212,215,219,220 While the histidine brace is highly conserved across LPMOs with some exception,221 there are significant structural variations in the proteins from different types of organisms.222 For example, the N-terminal histidine in fungal LPMOs, with exception in one organism,223 undergoes post-translational modification to insert a methyl group (Figure 15B).212,223 This alteration has been shown to provide protection for the enzyme in environments required for function and reactive oxygen species produced in catalysis.223 Fungal LPMOs also have a tyrosine residue in the axial position ~2.7 Å away from the Cu ion (Figure 15B).220 This tyrosine is present in some bacterial LPMOs; however, many have a phenylalanine (Phe) occupying this position (Figure 15C).224,225 Although it is generally agreed upon that the resting form of the enzyme has a CuII center, photoreduction often plagues crystallographic data which has made it difficult to interpret the metrical data. For example, photoreduction caused lengthening of the distances between water molecules and the metal in the structure of the bacterial LMPO (denoted AA10) from 2.6 and 3.2 Å, which is more consistent with a CuI center.226,227 However, insight into substrate interaction with the protein has also been provided by X-ray diffraction studies. Crystallographic data has shown a cellotriose (G3) substrate docked over the face of the active site via H-bonding to water molecules and nearby amino acid residues (Figure 15D). Upon substrate docking, the axial water molecule is displaced and the Cu⋯O (Tyr164) distance decreases from 2.7 to 2.5 Å.220
Figure 15.

Crystallographic data for LPMOs. Surface representation of AA9 showing the protein in purple and the active site in pink (A), and the molecular structures of a fungal LPMO (AA9, PDB: 5ACG) (B), a bacterial LPMO (AA10, PDB: 6RW7) (C), and an AA9 in the presence of substrate analog (G3, pink, PDB: 5ACJ) (D). Secondary coordination sphere interactions are represented by dashed lines. Cu(II) is shown as a cyan sphere and Cu(I) is shown as a brass sphere.
The catalytic transformation performed by LPMOs requires dioxygen, two electrons, and two protons; however, the mechanism(s) that govern this reactivity is still debated.228 We will not discuss the merits of the various proposals in this Forum other than to mention that several possible reactive intermediates are proposed from computational studies that include CuII—O2•−, CuII—OOH, CuII—O•, or a CuIII—OH species.229-236 At this juncture, there is little experimental evidence to support that these species are formed during turnover and it is obvious that more work is needed to validate which are relevant. It has also been found that the enzyme can function with H2O2 under anaerobic conditions.216 The direct biological relevance of this discovery has sparked numerous reports. In one study, Marletta compared O2 and H2O2 reactivity with the CuI intermediate in fungal LMPOs (denoted AA9s) and found that the reaction with O2 gave regioselective substrate oxidation while the H2O2 reaction gave a faster rate with nonspecific reactivity consistent with formation of hydroxyl radicals.237 In a different investigation, Solomon found the catalytic rate was greatly enhanced (1000-fold) when H2O2 was used instead of O2 in the reaction with a CuI AA9 LPMO which agrees with the faster rate observed for H2O2 by Marletta. However, in contrast to the report by Marletta, Solomon found the product profile of reactions for an AA9 LPMO with O2 and H2O2 to be indistinguishable.217 Väljamäe also observed an increased rate for oxidation of chitin by a bacterial LPMO when the transformation was driven by H2O2 rather than O2.225
There have been several reported synthetic complexes that have attempted to model various species associated with the proposed mechanisms of LPMOs that include a variety of Cu-hydroxido, Cu-superoxido, and Cu-hydroperoxido complexes.238,239 The possibility of CuIII–OH being the active species of the LPMO catalytic cycle is reinforced by the synthetic analogue developed by Tolman which, when generated at −80 °C, can activate C─H bonds upwards of 90 kcal/mol (Figure 16A).238 Itoh has developed a CuII complex, CuII(LH3)(tfa)2, that incorporates the tridentate ligand (N-(2-(1H-imidazol-4-yl)-benzyl)histamine) that accurately models the histidine brace (Figure 16B).239 This complex has twist angles close to those found in active sites of LPMOs giving spectroscopic parameters similar to those of the enzyme and a quasi-reversible CuII/I redox couple within the range reported for the enzymes. Moreover, this model complex was able to catalytically cleave the glycosidic bond in 4-nitrophenyl β-D-glucopyranoside (PNPG) with a turnover number (TON) of 58 after 24 hours.
Figure 16.
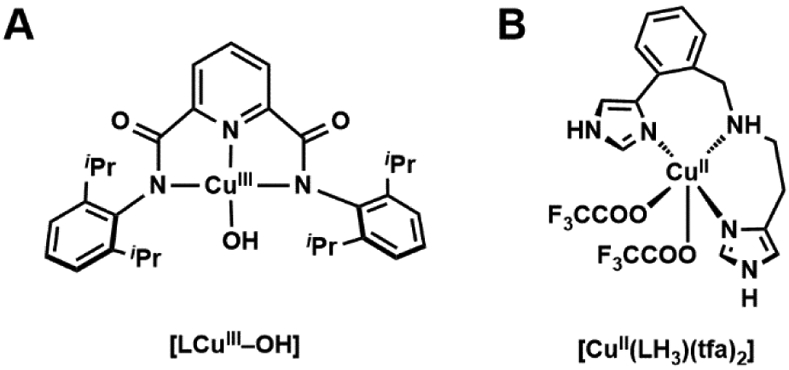
Synthetic CuIII–OH complex by Tolman (A), and synthetic CuII complex by Itoh with a histidine brace-type ligand (B).
Most of the above mechanistic and synthetic studies have focused on what occurs directly at the Cu center, yet it is plausible that effects within the secondary coordination sphere could contribute to catalysis.228 Marletta has examined the role of proximal amino acid residues on stability of a possible CuII–OOH intermediate.228 Using a combination of activity assays, site directed mutagenesis, and EPR spectroscopy, he proposed that residues H161 and Q167 within the secondary coordination sphere form H-bonds with the distal O-atom of putative Cu–OO and Cu–OOH intermediates (Figure 17) – these H-bonds assist in the subsequent O–O cleavage from the Cu-hydroxoperoxido species. The proximity of a redox-active tyrosine residue within 3 Å of the Cu-center in fungal LPMOs has also been proposed as possibly being involved catalysis. Walton has suggested that a CuII-tyrosyl radical species forms as a product when H2O2 reacts with the CuI intermediate.220 While the role of this intermediate is currently unknown, Walton proposes it could be forming in lieu of a CuIII–OH or CuII–oxyl to act as the competent oxidant. However, work from Solomon states that this tyrosyl species is a minor product that is unlikely to be involved in catalysis because the tyrosine residue is not conserved across all LPMOs. His studies indicate formation of tryptophanyl and tyrosyl radicals, however, further study suggested they were generated only in small quantities during catalysis.217 The small amounts of these radical species generated was suggested to indicated that oxidation of these amino acid residues is a minor, off-path route.
Figure 17.

Proposed CuII-OOH showing H-bonds to the distal O-atom involving H161 and Q167.
Vistas
This Forum provided an overview on FeIV=O, related Fe–OH, and Cu complexes as a means to understand C─H bond functionalization. The field has made great strides in recent years but there is much space to explore until we have a full appreciation for how these complexes can be applied to chemical problems. The presented topics were described with an eye to critique issues and point out where additional experiments might be beneficial. We have learned that regardless of the systems or method, few synthetic Fe=O complexes can compete with the selectivities and efficiencies of their biological counterparts. We have outlined selective key achievements by Costas and White, who explored increasing chemo- and regioselectivity of C─H bond functionalization, as well as by Collins, whose Fe(TAML) systems have been demonstrated to react efficiently towards the degradation of pollutants reminiscent of P450 enzymes, and have been adapted for green chemical and waste treatment industries (Figure 10). One obvious difference between the synthetic and biological systems is the extent to which the microenvironments around metal center(s) are controlled.19 Microenvironments are defined as the volume of space that surrounds a metal complex which includes the secondary coordination sphere. In metalloproteins, microenvironments around metallocofactors are highly regulated by the protein host and there is sufficient data to show that this regulation has a substantial impact on function. Our description of the possible role of secondary coordination sphere amino acid residues in the catalytic function of LPMOs offers one example of these effects. Another relevant example is to compare the non-heme Fe monooxygenase discussed above to their related halogenase counterparts. In halogenases such as SyrB2, the organic substrate is positioned in a designated orientation with respect to the metal cofactor in the active site such that, in spite of kinetic deficiencies (slower C─H activation) and thermodynamic considerations (OH-rebound is computed to be more favorable than halide-rebound by > 15 kcal/mol),49,123,124 the halide-rebound pathway is preferred. Replicating this control of the microenvironment in synthetic systems is challenging: to-date only several Fe(O(H))(halide) complexes have been reported, and they demonstrate little selectivity towards halogenation without regulation in the secondary coordination sphere in bulk organic solvent.122,240
Several advances in ligand design have produced synthetic systems that model some aspects found within active sites – these studies have increased our understanding of the parallels in the structures of synthetic constructs to architectural aspects found in metalloprotein active sites. However, microenvironments within synthetic systems have not yet achieved the regulatory control found within metalloproteins. Therefore, one of the next challenges is to increase the complexity of the microenvironments surrounding metal centers and to more accurately simulate the intricacies found within protein active sites; yet an increase in complexity comes with significant synthetic difficulties. Furthermore, the added complexity needs to accommodate interactions with water molecules that have been shown to be essential in regulating function within metalloproteins, a feature that is incompatible with most synthetic systems. In order to achieve these goals, scientists have turned to artificial metalloproteins (ArMs) in which the benefits of synthetic inorganic chemistry are leveraged with the power of protein hosts.241-244 The utility of this approach can be found in the pioneering work of Arnold on directed evolution of cytochrome P450s that has spawned a wave of highly functional ArMs.245-247 Other notable methods include de novo-designed proteins,248,249 metal templated interface redesign assembly of proteins,250,251 and inserting artificial metallocofactors into reconstituted/repurposed metalloproteins.252-255
One additional method for generating ArMs is to selectively insert a synthetic metallocofactor into a non-metalloprotein host. Examples of ArMs that use this method are those prepared with biotin-streptavidin (Sav) technology.256,257 First introduced by Whitesides258 and refined by Ward,259-261 this method has been used to generate ArMs for a variety of different transformations. Within the context of this Forum, we describe two recent reports of non-heme Fe artificial proteins. Ward has engineered new ArMs made from biotinylated Fe(TAML) complexes and Sav that show enhanced reactivity compared to the free Fe complexes.262 By systematically modulating the positioning of the Fe(TAML) complex within the protein pocket, they showed increased enantioselectivity in benzylic C─H bond activation, demonstrating the microenvironment of the active site plays an important role in dictating the transition state of the reaction. Additionally, mutations at key amino acid positions served to modulate both the primary and secondary coordination spheres of the metal complex, which allowed for structural characterization of the metalloproteins, and enhanced enantioselectivity and turnover capability towards substrate oxidation were also observed. We have used this approach to model the active site of non-heme Fe monooxygenase by engineering an Fe artificial protein with a facial triad active site.263 We biotinylated the common tridentate ligand dpa (di-(2-pyridylmethyl)amine) and prepared the corresponding FeII complex. Sav was modified by placing a glutamate at position 112 (S112E) and the resulting ArM had an Fe site with a facial triad composed of two N-atom donors from the dpa ligand and one O-atom donor from the carboxylate side chain from the glutamate with remaining ligands being water molecules. X-ray diffraction studies confirmed the structure and show a κ1-coordination of the carboxylate that is identical to that found in the native monooxygenases. Moreover, the carboxylate forms an intramolecular H-bond with one of the aqua ligands which also matches what is found in natural proteins. These examples illustrate that biologically relevant Fe sites can be designed within protein hosts and thus offer another way to model both primary and secondary coordination sphere effects to produce functional systems.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health USA (GM120349 and GM050781) for financial support.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publication website at DOI: 10.1021/acs.inorgchem.1c01754 The following file is available free of charge.
Synthetic details (PDF) and X-ray diffraction information (CIF) for [(TMTACN)MgII–(μ-OH)–FeIIIpoat]OTf.
Accession Codes
CDCC 2088881 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk.
The authors declare no competing financial interest.
References
- (1).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Que L; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- (3).Arndtsen BA; Bergman RG; Mobley TA; Peterson TH Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res 1995, 28, 154–162. [Google Scholar]
- (4).Neufeldt SR; Sanford MS Controlling Site Selectivity in Palladium-Catalyzed C─H Bond Functionalization. Acc. Chem. Res 2012, 45, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thirunavukkarasu VS; Kozhushkov SI; Ackermann L C─H Nitrogenation and Oxygenation by Ruthenium Catalysis. Chem. Commun 2014, 50, 29–39. [DOI] [PubMed] [Google Scholar]
- (6).Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C─C Bond Formation via Heteroatom-Directed C─H Bond Activation. Chem. Rev 2010, 110, 624–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bergman RG Activation of Alkanes with Organotransition Metal Complexes. Science 1984, 223, 902–908. [DOI] [PubMed] [Google Scholar]
- (8).Bergman RG Activation of Carbon-Hydrogen Bonds in Alkanes and Other Organic Molecules Using Organotransition Metal Complexes. In Homogeneous Transition Metal Catalyzed Reactions; Moser WR, Slocum DW, Eds.; American Chemical Society: Washington D.C., 1992; Vol. 230, pp 211–220. [Google Scholar]
- (9).Winkler JR; Gray HB Electronic Structures of Oxo-Metal Ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos DMP, Day P, Dahl JP, Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 17–28. [Google Scholar]
- (10).Gray HB; Winkler JR Living with Oxygen. Acc. Chem. Res 2018, 51, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mayer JM Thermodynamic Influences on C-H Bond Oxidation. In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier B, Ed.; Imperial College Press, 2000; pp 1–43. [Google Scholar]
- (12).Mayer JM Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]
- (13).Borovik AS Role of Metal–Oxo Complexes in the Cleavage of C─H Bonds. Chem. Soc. Rev 2011, 40, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Costentin C; Robert M; Savéant J-M Concerted Proton-Electron Transfers: Electrochemical and Related Approaches. Acc. Chem. Res 2010, 43, 1019–1029. [DOI] [PubMed] [Google Scholar]
- (15).Darcy JW; Koronkiewicz B; Parada GA; Mayer JM A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res 2018, 51, 2391–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Costas M; Mehn MP; Jensen MP; Que L Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (19).Shook RL; Borovik AS Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Guo M; Corona T; Ray K; Nam W Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci 2019, 5, 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kovaleva EG; Lipscomb JD Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (24).Groves JT; McClusky GA Aliphatic Hydroxylation via Oxygen Rebound. Oxygen Transfer Catalyzed by Iron. J. Am. Chem. Soc 1976, 98, 859–861. [Google Scholar]
- (25).Huang X; Groves JT Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. J. Biol. Inorg. Chem 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)Hydroxide pKa and the Role of Thiolate Ligation in C─H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).2-Oxoglutarate-Dependent Oxygenases; Schofield CJ, Hausinger RP, Eds.; Royal Society of Chemistry: Cambridge, 2015. [Google Scholar]
- (28).Krebs C; Fujimori DG; Walsh CT; Bollinger JM Non-Heme Fe(IV)–Oxo Intermediates. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hausinger RP Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- (30).Kal S; Que L Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. J. Biol. Inorg. Chem 2017, 22, 339–365. [DOI] [PubMed] [Google Scholar]
- (31).Valegard K; Terwisscha van Scheltinga AC; Lloyd MD; Hara T; Ramaswamy S; Perrakis A; Thompson A; Lee H-J; Baldwin JE; Schofield CJ; Andersson JH; Anderrson I Structure of a Cephalosporin Synthase. Nature 1998, 394, 805–809. [DOI] [PubMed] [Google Scholar]
- (32).Neidig ML; Brown CD; Light KM; Fujimori DG; Nolan EM; Price JC; Barr EW; Bollinger JM; Krebs C; Walsh CT; Solomon EI CD and MCD of CytC3 and Taurine Dioxygenase: Role of the Facial Triad in α-KG-Dependent Oxygenases. J. Am. Chem. Soc 2007, 129, 14224–14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Koehntop KD; Emerson JP; Que L The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-Heme Iron(II) Enzymes. J. Biol. Inorg. Chem 2005, 10, 87–93. [DOI] [PubMed] [Google Scholar]
- (34).Diebold AR; Brown-Marshall CD; Neidig ML; Brownlee JM; Moran GR; Solomon EI Activation of α-Keto Acid-Dependent Dioxygenases: Application of an {FeNO}7/{FeO2}8 Methodology for Characterizing the Initial Steps of O2 Activation. J. Am. Chem. Soc 2011, 133, 18148–18160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Price JC; Barr EW; Tirupati B; Bollinger JM; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia Coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (36).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JM Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc 2003, 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- (38).Bollinger JM; Krebs C Stalking Intermediates in Oxygen Activation by Iron Enzymes: Motivation and Method. J. Inorg. Biochem 2006, 100, 586–605. [DOI] [PubMed] [Google Scholar]
- (39).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Krebs C; Price JC; Baldwin J; Saleh L; Green MT; Bollinger JMJ Rapid Freeze-Quench 57Fe Mössbauer Spectroscopy: Monitoring Changes of an Iron-Containing Active Site during a Biochemical Reaction. Inorg. Chem 2005, 44, 742–757. [DOI] [PubMed] [Google Scholar]
- (41).MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)–Oxo Complex Derived From O2. Science 2000, 289, 938–941. [DOI] [PubMed] [Google Scholar]
- (42).Gupta R; Lacy DC; Bominaar EL; Borovik AS; Hendrich MP Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV-Oxo Complex. J. Am. Chem. Soc 2012, 134, 9775–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sinnecker S; Svensen N; Barr EW; Ye S; Bollinger JMJ; Neese F; Krebs C Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates In Taurine: α-Ketoglutarate Dioxygenase from Escherichia Coli and Its His99Ala Ligand Variant. J. Am. Chem. Soc 2007, 129, 6168–6179. [DOI] [PubMed] [Google Scholar]
- (44).Riggs-Gelasco PJ; Price JC; Guyer RB; Brehm JH; Barr EW; Bollinger JM; Krebs C EXAFS Spectroscopic Evidence for an Fe=O Unit in the Fe(IV) Intermediate Observed during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc 2004, 126, 8108–8109. [DOI] [PubMed] [Google Scholar]
- (45).Proshlyakov DA; Henshaw TF; Monterosso GR; Ryle MJ; Hausinger RP Direct Detection of Oxygen Intermediates in the Non-Heme Fe Enzyme Taurine/α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc 2004, 126, 1022–1023. [DOI] [PubMed] [Google Scholar]
- (46).Chang W. chen; Liu P; Guo Y Mechanistic Elucidation of Two Catalytically Versatile Iron(II)- and α-Ketoglutarate-Dependent Enzymes: Cases Beyond Hydroxylation. Comments Inorg. Chem 2018, 38, 127–165. [Google Scholar]
- (47).Matthews ML; Krest CM; Barr EW; Vaillancourt FH; Walsh CT; Green MT; Krebs C; Bollinger JM Substrate-Triggered Formation and Remarkable Stability of the C─H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Srnec M; Wong SD; Matthews ML; Krebs C; Bollinger JM; Solomon EI Electronic Structure of the Ferryl Intermediate in the α-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. J. Am. Chem. Soc 2016, 138, 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Substrate Positioning Controls the Partition between Halogenation and Hydroxylation in the Aliphatic Halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM; Solomon EI Elucidation of the Fe(IV)=O Intermediate in the Catalytic Cycle of the Halogenase SyrB2. Nature 2013, 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Martinie RJ; Livada J; Chang WC; Green MT; Krebs C; Bollinger JM; Silakov A Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc 2015, 137, 6912–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Dunham NP; Chang WC; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Two Distinct Mechanisms for C─C Desaturation by Iron(II)- and 2-(Oxo)Glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance. J. Am. Chem. Soc 2018, 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Liao HJ; Li J; Huang JL; Davidson M; Kurnikov I; Lin TS; Lee JL; Kurnikova M; Guo Y; Chan NL; Chang WC Insights into the Desaturation of Cyclopeptin and Its C3 Epimer Catalyzed by a Non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chemie Int. Ed 2018, 57, 1831–1835. [DOI] [PubMed] [Google Scholar]
- (54).Li J; Liao HJ; Tang Y; Huang JL; Cha L; Lin TS; Lee JL; Kurnikov IV; Kurnikova MG; Chang WC; Chan NL; Guo Y Epoxidation Catalyzed by the Nonheme Iron(II)- and 2-Oxoglutarate-Dependent Oxygenase, AsqJ: Mechanistic Elucidation of Oxygen Atom Transfer by a Ferryl Intermediate. J. Am. Chem. Soc 2020, 142, 6268–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yu CP; Tang Y; Cha L; Milikisiyants S; Smirnova TI; Smirnov AI; Guo Y; Chang WC Elucidating the Reaction Pathway of Decarboxylation-Assisted Olefination Catalyzed by a Mononuclear Non-Heme Iron Enzyme. J. Am. Chem. Soc 2018, 140, 15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kurtz DM Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem. Rev 1990, 90, 585–606. [Google Scholar]
- (57).Tshuva EY; Lippard SJ Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev 2004, 104, 987–1012. [DOI] [PubMed] [Google Scholar]
- (58).MacBeth CE; Gupta R; Mitchell-Koch KR; Young VG; Lushington GH; Thompson WH; Hendrich MP; Borovik AS To Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J. Am. Chem. Soc 2004, 126, 2556–2567. [DOI] [PubMed] [Google Scholar]
- (59).Mukherjee J; Lucas RL; Zart MK; Powell DR; Day VW; Borovik AS Synthesis, Structure, and Physical Properties for a Series of Monomeric Iron(III) Hydroxo Complexes with Varying Hydrogen-Bond Networks. Inorg. Chem 2008, 47, 5780–5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Dey A; Hocking RK; Larsen P; Borovik AS; Hodgson KO; Hedman B; Solomon EI X-Ray Absorption Spectroscopy and Density Functional Theory Studies of [(H3Buea)FeIII–X]n− (X = S2−, O2−, OH−): Comparison of Bonding and Hydrogen Bonding in Oxo and Sulfido Complexes. J. Am. Chem. Soc 2006, 128, 9825–9833. [DOI] [PubMed] [Google Scholar]
- (61).Matson EM; Park YJ; Fout AR Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc 2014, 136, 17398–17401. [DOI] [PubMed] [Google Scholar]
- (62).Reed CJ; Agapie T A Terminal FeIII-Oxo in a Tetranuclear Cluster: Effects of Distal Metal Centers on Structure and Reactivity. J. Am. Chem. Soc 2019, 141, 9479–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Grapperhaus CA; Mienert B; Bill E; Weyhermüller T; Wieghardt K Mononuclear (Nitrido)Iron(V) and (Oxo)Iron(IV) Complexes via Photolysis of [(Cyclam-Acetato)FeIII(N3)]+ and Ozonolysis of [(Cyclam-Acetato)FeIII(O3SCF3)]+ in Water/Acetone Mixtur. Inorg. Chem 2000, 39, 5306–5317. [DOI] [PubMed] [Google Scholar]
- (64).Rohde J-U; In J-H; Lim MH; Brennessel WW; Bukowski MR; Stubna A; Münck E; Nam W; Que L Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science 2003, 299, 1037–1039. [DOI] [PubMed] [Google Scholar]
- (65).Klinker EJ; Kaizer J; Brennessel WW; Woodrum NL; Cramer CJ; Que L Structures of Nonheme Oxoiron(IV) Complexes from X-Ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem. Int. Ed 2005, 44, 3690–3694. [DOI] [PubMed] [Google Scholar]
- (66).Puri M; Que L Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res 2015, 48, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Que L The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res 2007, 40, 493–500. [DOI] [PubMed] [Google Scholar]
- (68).Nam W High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res 2007, 40, 522–531. [DOI] [PubMed] [Google Scholar]
- (69).Meyer S; Klawitter I; Demeshko S; Bill E; Meyer F A Tetracarbene-Oxoiron(IV) Complex. Angew. Chemie Int. Ed 2013, 52, 901–905. [DOI] [PubMed] [Google Scholar]
- (70).Sahu S; Zhang B; Pollock CJ; Dürr M; Davies CG; Confer AM; Ivanović-Burmazović I; Siegler MA; Jameson GNL; Krebs C; Goldberg DP Aromatic C─F Hydroxylation by Nonheme Iron(IV)–Oxo Complexes: Structural, Spectroscopic, and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12791–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Chanda A; Shan X; Chakrabarti M; Ellis WC; Popescu DL; De Oliveira FT; Wang D; Que L; Collins TJ; Münck E; Bominaar EL (TAML)FeIV=O Complex in Aqueous Solution: Synthesis and Spectroscopic and Computational Characterization. Inorg. Chem 2008, 47, 3669–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chantarojsiri T; Sun Y; Long JR; Chang CJ Water-Soluble Iron(IV)-Oxo Complexes Supported by Pentapyridine Ligands: Axial Ligand Effects on Hydrogen Atom and Oxygen Atom Transfer Reactivity. Inorg. Chem 2015, 54, 5879–5887. [DOI] [PubMed] [Google Scholar]
- (73).Rana S; Biswas JP; Sen A; Clémancey M; Blondin G; Latour JM; Rajaraman G; Maiti D Selective C─H Halogenation over Hydroxylation by Non-Heme Iron(IV)-Oxo. Chem. Sci 2018, 9, 7843–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Wang D; Ray K; Collins MJ; Farquhar ER; Frisch Jonathan R; Gomez L; Jackson TA; Kerscher M; Waleska A; Comba P; Costas M; Que L Nonheme Oxoiron(IV) Complexes of Pentadentate N5 Ligands: Spectroscopy, Electrochemistry, and Oxidative Reactivity. Chem. Sci 2013, 4, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wegeberg C; Skavenborg ML; Liberato A; McPherson JN; Browne WR; Hedegård ED; McKenzie CJ Engineering the Oxidative Potency of Non-Heme Iron(IV) Oxo Complexes in Water for C─H Oxidation by a Cis Donor and Variation of the Second Coordination Sphere. Inorg. Chem 2021, 60, 1975–1984. [DOI] [PubMed] [Google Scholar]
- (76).Monte Pérez I; Engelmann X; Lee Y-M; Yoo M; Kumaran E; Farquhar ER; Bill E; England J; Nam W; Swart M; Ray K A Highly Reactive Oxoiron(IV) Complex Supported by a Bioinspired N3O Macrocyclic Ligand. Angew. Chemie Int. Ed 2017, 129, 14576–14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Sastri CV; Park MJ; Ohta T; Jackson TA; Stubna A; Seo MS; Lee J; Kim J; Kitagawa T; Münck E; Que L; Nam W Axial Ligand Substituted Nonheme FeIV=O Complexes: Observation of Near-UV LMCT Bands and Fe=O Raman Vibrations. J. Am. Chem. Soc 2005, 127, 12494–12495. [DOI] [PubMed] [Google Scholar]
- (78).Jackson TA; Rohde J-U; Seo MS; Sastri CV; DeHont R; Stubna A; Ohta T; Kitagawa T; Münck E; Nam W; Que L Axial Ligand Effects on the Geometric and Electronic Structures of Nonheme Oxoiron(IV) Complexes. J. Am. Chem. Soc 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Shaik S; Hirao H; Kumar D Reactivity of High-Valent Iron–Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Acc. Chem. Res 2007, 40, 532–542. [DOI] [PubMed] [Google Scholar]
- (80).Decker A; Rohde J-U; Klinker EJ; Wong SD; Que L; Solomon EI Spectroscopic and Quantum Chemical Studies on Low-Spin FeIV=O Complexes: Fe─O Bonding and Its Contributions to Reactivity. J. Am. Chem. Soc 2007, 129, 15983–15996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Geng C; Ye S; Neese F Analysis of Reaction Channels for Alkane Hydroxylation by Nonheme Iron(IV)–Oxo Complexes. Angew. Chemie Int. Ed 2010, 49, 5717–5720. [DOI] [PubMed] [Google Scholar]
- (82).Ballhausen CJ; Gray HB The Electronic Structure of the Vanadyl Ion. Inorg. Chem 1962, 1, 111–122. [Google Scholar]
- (83).Gray HB; Hare CR The Electronic Structures and Spectra of Chromyl and Molybdenyl Ions. Inorg. Chem 1962, 1, 363–368. [Google Scholar]
- (84).O’Halloran KP; Zhao C; Ando NS; Schultz AJ; Koetzle TF; Piccoli PMB; Hedman B; Hodgson KO; Bobyr E; Kirk ML; Knottenbelt S; Depperman EC; Stein B; Anderson TM; Cao R; Geletii YV; Hardcastle KI; Musaev DG; Neiwert WA; Fang X; Morokuma K; Wu S; Kogerler P; Hill CL Revisiting the Polyoxometalate-Based Late-Transition-Metal-Oxo Complexes: The “Oxo Wall” Stands. Inorg. Chem 2012, 51, 7025–7031. [DOI] [PubMed] [Google Scholar]
- (85).Ray K; Heims F; Pfaff FF Terminal Oxo and Imido Transition-Metal Complexes of Groups 9-11. Eur. J. Inorg. Chem 2013, 2013, 3784–3807. [Google Scholar]
- (86).Wang B; Lee Y-M; Tcho W-Y; Tussupbayev S; Kim S-T; Kim Y; Seo MS; Cho K-B; Dede Y; Keegan BC; Ogura T; Kim SH; Ohta T; Baik M-H; Ray K; Shearer J; Nam W Synthesis and Reactivity of a Mononuclear Non-Haem Cobalt(IV)-Oxo Complex. Nat. Commun 2017, 8, 14839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Mayer JM; Thorn DL; Tulip TH Synthesis, Reactions, and Electronic Structure of Low-Valent Rhenium–oxo Compounds. Crystal and Molecular Structure of Re(O)I(MeCCMe)2. J. Am. Chem. Soc 1985, 107, 7454–7462. [Google Scholar]
- (88).England J; Martinho MM; Farquhar ER; Frisch JR; Bominaar EL; Münck E; Que L A Synthetic High-Spin Oxoiron(IV) Complex: Generation, Spectroscopic Characterization, and Reactivity. Angew. Chem. Int. Ed 2009, 48, 3622–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Lacy DC; Gupta R; Stone KL; Greaves J; Ziller JW; Hendrich MP; Borovik AS Formation, Structure, and EPR Detection of a High Spin FeIV–Oxo Species Derived from Either an FeIII–Oxo or FeIII–OH Complex. J. Am. Chem. Soc 2010, 132, 12188–12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Bigi JP; Harman WH; Lassalle-Kaiser B; Robles DM; Stich TA; Yano J; Britt RD; Chang CJ A High-Spin Iron(IV)–Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J. Am. Chem. Soc 2012, 134, 1536–1542. [DOI] [PubMed] [Google Scholar]
- (91).Oswald VF; Lee JL; Biswas S; Weitz AC; Mittra K; Fan R; Li J; Zhao J; Hu MY; Alp EE; Bominaar EL; Guo Y; Green MT; Hendrich MP; Borovik AS Effects of Noncovalent Interactions on High-Spin Fe(IV)–Oxido Complexes. J. Am. Chem. Soc 2020, 142, 11804–11817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).England J; Guo Y; Van Heuvelen KM; Cranswick MA; Rohde GT; Bominaar EL; Münck E; Que L A More Reactive Trigonal-Bipyramidal High-Spin Oxoiron(IV) Complex with a Cis-Labile Site. J. Am. Chem. Soc 2011, 133, 11880–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).England J; Guo Y; Farquhar ER; Young VG; Münck E; Que L The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J. Am. Chem. Soc 2010, 132, 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Srnec M; Wong SD; England J; Que L; Solomon EI π-Frontier Molecular Orbitals in S = 2 Ferryl Species and Elucidation of Their Contributions to Reactivity. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Shimoyama Y; Kojima T Metal–Oxyl Species and Their Possible Roles in Chemical Oxidations. Inorg. Chem 2019, 58, 9517–9542. [DOI] [PubMed] [Google Scholar]
- (96).Ye S; Neese F Nonheme Oxo-Iron(IV) Intermediates Form an Oxyl Radical upon Approaching the C─H Bond Activation Transition State. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Siegbahn PEM Structures and Energetics for O2 Formation in Photosystem II. Acc. Chem. Res 2009, 42, 1871–1880. [DOI] [PubMed] [Google Scholar]
- (98).Cox N; Pantazis DA; Neese F; Lubitz W Biological Water Oxidation. Acc. Chem. Res 2013, 46, 1588–1596. [DOI] [PubMed] [Google Scholar]
- (99).Zang Y; Kim J; Dong Y; Wilkinson EC; Appelman EH; Que L Models for Nonheme Iron Intermediates: Structural Basis for Tuning the Spin States of Fe(TPA) Complexes. J. Am. Chem. Soc 1997, 119, 4197–4205. [Google Scholar]
- (100).Lim MH; Rohde J-U; Stubna A; Bukowski MR; Costas M; Ho RYN; Münck E; Nam W; Que LJ An FeIV=O Complex of a Tetradentate Tripodal Nonheme Ligand. Proc. Natl. Acad. Sci 2003, 100, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Paine TK; Costas M; Kaizer J; Que LJ Oxoiron(IV) Complexes of the Tris(2-Pyridylmethyl)Amine Ligand Family: Effect of Pyridine a-Substituents. J. Biol. Inorg. Chem 2006, 11, 1098–1099. [DOI] [PubMed] [Google Scholar]
- (102).Biswas AN; Puri M; Meier KK; Oloo WN; Rohde GT; Bominaar EL; Münck E; Que L Modeling TauD-J: A High-Spin Nonheme Oxoiron(IV) Complex with High Reactivity toward C─H Bonds. J. Am. Chem. Soc 2015, 137, 2428–2431. [DOI] [PubMed] [Google Scholar]
- (103).Morimoto Y; Kotani H; Park J; Lee Y-M; Nam W; Fukuzumi S Metal Ion-Coupled Electron Transfer of a Nonheme Oxoiron(IV) Complex: Remarkable Enhancement of Electron-Transfer Rates by Sc3+. J. Am. Chem. Soc 2011, 133, 403–405. [DOI] [PubMed] [Google Scholar]
- (104).Park YJ; Cook SA; Sickerman NS; Sano Y; Ziller JW; Borovik AS Heterobimetallic Complexes with MIII-(μ-OH)-MII Cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): Structural, Kinetic, and Redox Properties. Chem. Sci 2013, 4, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Park J; Morimoto Y; Lee Y-M; Nam W; Fukuzumi S Metal Ion Effect on the Switch of Mechanism from Direct Oxygen Transfer to Metal Ion-Coupled Electron Transfer in the Sulfoxidation of Thioanisoles by a Non-Heme Iron(IV)–Oxo Complex. J. Am. Chem. Soc 2011, 133, 5236–5239. [DOI] [PubMed] [Google Scholar]
- (106).Park J; Morimoto Y; Lee Y-M; You Y; Nam W; Fukuzumi S Scandium Ion-Enhanced Oxidative Dimerization and N-Demethylation of N,N-Dimethylanilines by a Non-Heme Iron(IV)-Oxo Complex. Inorg. Chem 2011, 50, 11612–11622. [DOI] [PubMed] [Google Scholar]
- (107).Fukuzumi S; Morimoto Y; Kotani H; Naumov PP; Lee YMM; Nam W Crystal Structure of a Metal Ion-Bound Oxoiron(IV) Complex and Implications for Biological Electron Transfer. Nat. Chem 2010, 2, 756–759. [DOI] [PubMed] [Google Scholar]
- (108).Swart M A Change in the Oxidation State of Iron: Scandium Is Not Innocent. Chem. Commun 2013, 49, 6650–6652. [DOI] [PubMed] [Google Scholar]
- (109).Prakash J; Rohde GT; Meier KK; Jasniewski AJ; Van Heuvelen KM; Münck E; Que L Spectroscopic Identification of an FeIII Center, Not FeIV, in the Crystalline Sc-O-Fe Adduct Derived from [FeIV(O)(TMC)]2+. J. Am. Chem. Soc 2015, 137, 3478–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Rice DB; Grotemeyer EN; Donovan AM; Jackson TA Effect of Lewis Acids on the Structure and Reactivity of a Mononuclear Hydroxomanganese(III) Complex. Inorg. Chem 2020, 59, 2689–2700. [DOI] [PubMed] [Google Scholar]
- (111).Sankaralingam M; Lee YM; Karmalkar DG; Nam W; Fukuzumi SA Mononuclear Non-Heme Manganese(III)-Aqua Complex as a New Active Oxidant in Hydrogen Atom Transfer Reactions. J. Am. Chem. Soc 2018, 140, 12695–12699. [DOI] [PubMed] [Google Scholar]
- (112).Bordwell FG; Cheng JP; Ji GZ; Satish AV; Zhang X Bond Dissociation Energies in DMSO Related to the Gas Phase Values. J. Am. Chem. Soc 1991, 113, 9790–9795. [Google Scholar]
- (113).Roth JP; Mayer JM Hydrogen Transfer Reactivity of a Ferric Bi-Imidazoline Complex That Models the Activity of Lipoxygenase Enzymes. Inorg. Chem 1999, 38, 2760–2761. [DOI] [PubMed] [Google Scholar]
- (114).Sastri CV; Lee J; Oh K; Lee YJ; Lee J; Jackson TA; Ray K; Hirao H; Shin W; Halfen JA; Kim J; Que L; Shaik S; Nam W Axial Ligand Tuning of a Nonheme Iron(IV)–oxo Unit for Hydrogen Atom Abstraction. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19181–19186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Usharani D; Lacy DC; Borovik AS; Shaik S Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X─H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity. J. Am. Chem. Soc 2013, 135, 17090–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Kaizer J; Klinker EJ; Oh NY; Rohde J-U; Song WJ; Stubna A; Kim J; Münck E; Nam W; Que L Nonheme FeIVO Complexes That Can Oxidize the C─H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc 2004, 126, 472–473. [DOI] [PubMed] [Google Scholar]
- (117).Xue G; Wang D; De Hont R; Fiedler AT; Shan X; Münck E; Que L A Synthetic Precedent for the [FeIV2(μ-O)2] Diamond Core Proposed for Methane Monooxygenase Intermediate Q. Proc. NatI. Acad. Sci. U. S. A 2007, 104, 20713–20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP Observation of Radical Rebound in a Mononuclear Nonheme Iron Model Complex. J. Am. Chem. Soc 2018, 140, 4191–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Pangia TM; Yadav V; Gérard EF; Lin YT; De Visser SP; Jameson GNL; Goldberg DP Mechanistic Investigation of Oxygen Rebound in a Mononuclear Nonheme Iron Complex. Inorg. Chem 2019, 58, 9557–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Yadav V; Gordon JB; Siegler MA; Goldberg DP Dioxygen-Derived Nonheme Mononuclear FeIII(OH) Complex and Its Reactivity with Carbon Radicals. J. Am. Chem. Soc 2019, 141, 10148–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Drummond MJ; Ford CL; Gray DL; Popescu CV; Fout AR Radical Rebound Hydroxylation Versus H-Atom Transfer in Non-Heme Iron(III)-Hydroxo Complexes: Reactivity and Structural Differentiation. J. Am. Chem. Soc 2019, 141, 6639–6650. [DOI] [PubMed] [Google Scholar]
- (122).Yadav V; Rodriguez RJ; Siegler MA; Goldberg DP Determining the Inherent Selectivity for Carbon Radical Hydroxylation versus Halogenation with FeIII(OH)(X) Complexes: Relevance to the Rebound Step in Non-Heme Iron Halogenases. J. Am. Chem. Soc 2020, 142, 7259–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Srnec M; Solomon EI Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. J. Am. Chem. Soc 2017, 139, 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Quesne MG; De Visser SP Regioselectivity of Substrate Hydroxylation versus Halogenation by a Nonheme Iron(IV)–oxo Complex: Possibility of Rearrangement Pathways. J. Biol. Inorg. Chem 2012, 17, 841–852. [DOI] [PubMed] [Google Scholar]
- (125).Cho K-B; Wu X; Lee Y-M; Kwon YH; Shaik S; Nam W Evidence for an Alternative to the Oxygen Rebound Mechanism in C─H Bond Activation by Non-Heme FeIVO Complexes. J. Am. Chem. Soc 2012, 134, 20222–20225. [DOI] [PubMed] [Google Scholar]
- (126).Behan RK; Green MT On the Status of Ferryl Protonation. J. Inorg. Biochem 2006, 100, 448–459. [DOI] [PubMed] [Google Scholar]
- (127).Yosca TH; Behan RK; Krest CM; Onderko EL; Langston MC; Green MT Setting an Upper Limit on the Myoglobin Iron(IV)Hydroxide pKa: Insight into Axial Ligand Tuning in Heme Protein Catalysis. J. Am. Chem. Soc 2014, 136, 9124–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Ledray AP; Krest CM; Yosca TH; Mittra K; Green MT Ascorbate Peroxidase Compound II Is an Iron(IV) Oxo Species. J. Am. Chem. Soc 2020, 142, 20419–20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Casadei CM; Gumiero A; Metcalfe CL; Murphy EJ; Basran J; Concilio MG; Teixeira SCM; Schrader TE; Fielding AJ; Ostermann A; Blakeley MP; Raven EL; Moody PCE Neutron Cryo-Crystallography Captures the Protonation State of Ferryl Heme in a Peroxidase. Science 2014, 345, 193–197. [DOI] [PubMed] [Google Scholar]
- (130).Kwon H; Basran J; Pathak C; Hussain M; Freeman SL; Fielding AJ; Bailey AJ; Stefanou N; Sparkes HA; Tosha T; Yamashita K; Hirata K; Murakami H; Ueno G; Ago H; Tono K; Yamamoto M; Sawai H; Shiro Y; Sugimoto H; Raven EL; Moody PCE XFEL Crystal Structures of Peroxidase Compound II. Angew. Chemie Int. Ed 2021, 60, 14578–14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Chreifi G; Baxter EL; Doukov T; Cohen AE; McPhillips SE; Song J; Meharenna YT; Soltis SM; Poulos TL Crystal Structure of the Pristine Peroxidase Ferryl Center and Its Relevance to Proton-Coupled Electron Transfer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Green MT Application of Badger’s Rule to Heme and Non-Heme Iron-Oxygen Bonds: An Examination of Ferryl Protonation States. J. Am. Chem. Soc 2006, 128, 1902–1906. [DOI] [PubMed] [Google Scholar]
- (133).Barry SM; Challis GL Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013, 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Wang Y; Li J; Liu A Oxygen Activation by Mononuclear Nonheme Iron Dioxygenases Involved in the Degradation of Aromatics. J. Biol. Inorg. Chem 2017, 22, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Schulz CE; Castillo RG; Pantazis DA; DeBeer S; Neese F Structure–Spectroscopy Correlations for Intermediate Q of Soluble Methane Monooxygenase: Insights from QM/MM Calculations. J. Am. Chem. Soc 2021, 143, 6560–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Weitz AC; Mills MR; Ryabov AD; Collins TJ; Guo Y; Bominaar EL; Hendrich MP A Synthetically Generated LFeIVOHn Complex. Inorg. Chem 2019, 58, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Weitz AC; Hill EA; Oswald VF; Bominaar EL; Borovik AS; Hendrich MP; Guo Y Probing Hydrogen Bonding Interactions to Iron-Oxido/Hydroxido Units by 57Fe Nuclear Resonance Vibrational Spectroscopy. Angew. Chemie Int. Ed 2018, 57, 16010–16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Hill EA; Weitz AC; Onderko E; Romero-Rivera A; Guo Y; Swart M; Bominaar EL; Green MT; Hendrich MP; Lacy DC; Borovik AS Reactivity of an Fe(IV)–Oxo Complex with Protons and Oxidants. J. Am. Chem. Soc 2016, 138, 13143–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Milan M; Salamone M; Costas M; Bietti M The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C─H Bond Oxygenation. Acc. Chem. Res 2018, 51, 1984–1995. [DOI] [PubMed] [Google Scholar]
- (141).Vicens L; Olivo G; Costas M Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar]
- (142).White MC; Zhao J Aliphatic C─H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Grau M; Kyriacou A; Cabedo Martinez F; De Wispelaere IM; White AJP; Britovsek GJP Unraveling the Origins of Catalyst Degradation in Non-Heme Iron-Based Alkane Oxidation. Dalt. Trans 2014, 43, 17108–17119. [DOI] [PubMed] [Google Scholar]
- (144).Canta M; Font D; Gõmez L; Ribas X; Costas M The Iron(II) Complex [Fe(CF3SO3)2(Mcp)] as a Convenient, Readily Available Catalyst for the Selective Oxidation of Methylenic Sites in Alkanes. Adv. Synth. Catal 2014, 356, 818–830. [Google Scholar]
- (145).Chen MS; White MC A Predictably Selective Aliphatic C─H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]
- (146).Codolà Z; Gamba I; Acuna-Parés F; Casadevall C; Clémancey M; Latour JM; Luis JM; Lloret-Fillol J; Costas M Design of Iron Coordination Complexes as Highly Active Homogenous Water Oxidation Catalysts by Deuteration of Oxidation-Sensitive Sites. J. Am. Chem. Soc 2019, 141, 323–333. [DOI] [PubMed] [Google Scholar]
- (147).Chen J; Klein Gebbink RJM Deuterated N2Py2 Ligands: Building More Robust Non-Heme Iron Oxidation Catalysts. ACS Catal. 2019, 9, 3564–3575. [Google Scholar]
- (148).Cussó O; Garcia-Bosch I; Ribas X; Lloret-Fillol J; Costas M Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-Heme Iron Catalysts. J. Am. Chem. Soc 2013, 135, 14871–14878. [DOI] [PubMed] [Google Scholar]
- (149).Prat I; Font D; Company A; Junge K; Ribas X; Beller M; Costas M Fe(PyTACN)-Catalyzed Cis-Dihydroxylation of Olefins with Hydrogen Peroxide. Adv. Synth. Catal 2013, 355, 947–956. [Google Scholar]
- (150).Olivo G; Farinelli G; Barbieri A; Lanzalunga O; Di Stefano S; Costas M Supramolecular Recognition Allows Remote, Site-Selective C─H Oxidation of Methylenic Sites in Linear Amines. Angew. Chemie Int. Ed 2017, 56, 16347–16351. [DOI] [PubMed] [Google Scholar]
- (151).O’Brien JR; Schuller DJ; Yang VS; Dillard BD; Lanzilotta WN Substrate-Induced Conformational Changes in Escherichia Coli Taurine/α-Ketoglutarate Dioxygenase and Insight into the Oligomeric Structure. Biochemistry 2003, 42, 5547–5554. [DOI] [PubMed] [Google Scholar]
- (152).Saban E; Chen YH; Hangasky JA; Taabazuing CY; Holmes BE; Knapp MJ The Second Coordination Sphere of FIH Controls Hydroxylation. Biochemistry 2011, 50, 4733–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Gormisky PE; White MC Catalyst-Controlled Aliphatic C─H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc 2013, 135, 14052–14055. [DOI] [PubMed] [Google Scholar]
- (154).Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Oxidative Diversification of Amino Acids and Peptides by Small-Molecule Iron Catalysis. Nature 2016, 537, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Collins TJ; Ryabov AD Targeting of High-Valent Iron-TAML Activators at Hydrocarbons and Beyond. Chem. Rev 2017, 117, 9140–9162. [DOI] [PubMed] [Google Scholar]
- (156).Ellis WC; Tran CT; Roy R; Rusten M; Fischer A; Ryabov AD; Blumberg B; Collins TJ Designing Green Oxidation Catalysts for Purifying Environmental Waters. J. Am. Chem. Soc 2010, 132, 9774–9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Kundu S; Chanda A; Khetan SK; Ryabov AD; Collins TJ TAML Activator/Peroxide-Catalyzed Facile Oxidative Degradation of the Persistent Explosives Trinitrotoluene and Trinitrobenzene in Micellar Solutions. Environ. Sci. Technol 2013, 47, 5319–5326. [DOI] [PubMed] [Google Scholar]
- (158).Onundi Y; Drake BA; Malecky RT; DeNardo MA; Mills MR; Kundu S; Ryabov AD; Beach ES; Horwitz CP; Simonich MT; Truong L; Tanguay RL; Wright LJ; Singhal N; Collins TJ A Multidisciplinary Investigation of the Technical and Environmental Performances of TAML/Peroxide Elimination of Bisphenol A Compounds from Water. Green Chem. 2017, 19, 4234–4262. [Google Scholar]
- (159).Kundu S; Chanda A; Thompson JVK; Diabes G; Khetan SK; Ryabov AD; Collins TJ Rapid Degradation of Oxidation Resistant Nitrophenols by TAML Activator and H2O2. Catal. Sci. Technol 2015, 5, 1775–1782. [Google Scholar]
- (160).Beach ES; Malecky RT; Gil RR; Horwitz CP; Collins TJ Fe-TAML/Hydrogen Peroxide Degradation of Concentrated Solutions of the Commercial Azo Dye Tartrazine. Catal. Sci. Technol 2011, 1, 437–443. [Google Scholar]
- (161).Warner GR; Mills MR; Enslin C; Pattanayak S; Panda C; Panda TK; Gupta S. Sen; Ryabov AD; Collins TJ Reactivity and Operational Stability of N-Tailed TAMLs through Kinetic Studies of the Catalyzed Oxidation of Orange II by H2O2: Synthesis and X-Ray Structure of an N-Phenyl TAML. Chem. - A Eur. J 2015, 21, 6226–6233. [DOI] [PubMed] [Google Scholar]
- (162).Tang LL; DeNardo MA; Gayathri C; Gil RR; Kanda R; Collins TJ TAML/H2O2 Oxidative Degradation of Metaldehyde: Pursuing Better Water Treatment for the Most Persistent Pollutants. Environ. Sci. Technol 2016, 50, 5261–5268. [DOI] [PubMed] [Google Scholar]
- (163).Saouma CT; Mayer JM Do Spin State and Spin Density Affect Hydrogen Atom Transfer Reactivity? Chem. Sci 2014, 5, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Bordwell FG Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res 1988, 21, 456–463. [Google Scholar]
- (165).Parker VD; Handoo KL; Roness F; Tilset M Electrode Potentials and the Thermodynamics of Isodesmic Reactions. J. Am. Chem. Soc 1991, 113, 7493–7498. [Google Scholar]
- (166).Hong S; Lee Y-M; Cho K-B; Sundaravel K; Cho J; Kim MJ; Shin W; Nam W Ligand Topology Effect on the Reactivity of a Mononuclear Nonheme Iron(IV)-Oxo Complex in Oxygenation Reactions. J. Am. Chem. Soc 2011, 133, 11876–11879. [DOI] [PubMed] [Google Scholar]
- (167).Sacramento JJD; Goldberg DP Factors Affecting Hydrogen Atom Transfer Reactivity of Metal-Oxo Porphyrinoid Complexes. Acc. Chem. Res 2018, 51, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Ehudin MA; Quist DA; Karlin KD Enhanced Rates of C─H Bond Cleavage by a Hydrogen-Bonded Synthetic Heme High-Valent Iron(IV) Oxo Complex. J. Am. Chem. Soc 2019, 141, 12558–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Dantignana V; Serrano-Plana J; Draksharapu A; Magallon C; Banerjee S; Fan R; Gamba I; Guo Y; Que L; Costas M; Company A Spectroscopic and Reactivity Comparisons between Nonheme Oxoiron(IV) and Oxoiron(V) Species Bearing the Same Ancillary Ligand. J. Am. Chem. Soc 2019, 141, 15078–15091. [DOI] [PubMed] [Google Scholar]
- (170).Kupper C; Mondal B; Serrano-Plana J; Klawitter I; Neese F; Costas M; Ye S; Meyer F Nonclassical Single-State Reactivity of an Oxo-Iron(IV) Complex Confined to Triplet Pathways. J. Am. Chem. Soc 2017, 139, 8939–8949. [DOI] [PubMed] [Google Scholar]
- (171).Seo MS; Kim NH; Cho K-B; So JE; Park SK; Clénmancey M; Garcia-Serres R; Latour J-M; Shaik S; Nam W A Mononuclear Nonheme Iron(IV)-Oxo Complex Which Is More Reactive than Cytochrome P450 Model Compound I. Chem. Sci 2011, 2, 1039. [Google Scholar]
- (172).Bell RP The Theory of Reactions Involving Proton Transfers. Proc. R. Soc. London. Ser. A - 1936, 154, 414–429. [Google Scholar]
- (173).Evans MG; Polanyi M Inertia and Driving Force Of Chemical Reactions. Trans. Faraday Soc 1938, 34, 11–23. [Google Scholar]
- (174).Stone KL; Borovik AS Lessons from Nature: Unraveling Biological C─H Bond Activation. Curr. Opin. Chem. Biol 2009, 13, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Parsell TH; Yang M-Y; Borovik AS C─H Bond Cleavage with Reductants: Re-Investigating the Reactivity of Monomeric MnIII/IV–Oxo Complexes and the Role of Oxo Ligand Basicity. J. Am. Chem. Soc 2009, 131, 2762–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Gupta R; Borovik AS Monomeric MnIII/II and FeIII/II Complexes with Terminal Hydroxo and Oxo Ligands: Probing Reactivity via O─H Bond Dissociation Energies. J. Am. Chem. Soc 2003, 125, 13234–13242. [DOI] [PubMed] [Google Scholar]
- (177).Barman SK; Jones JR; Sun C; Hill EA; Ziller JW; Borovik AS Regulating the Basicity of Metal–Oxido Complexes with a Single Hydrogen Bond and Its Effect on C─H Bond Cleavage. J. Am. Chem. Soc 2019, 141, 11142–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Darcy JW; Kolmar SS; Mayer JM Transition State Asymmetry in C─H Bond Cleavage by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 10777–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Goetz MK; Anderson JS Experimental Evidence for pKa-Driven Asynchronicity in C─H Activation by a Terminal Co(III)–Oxo Complex. J. Am. Chem. Soc 2019, 141, 4051–4062. [DOI] [PubMed] [Google Scholar]
- (180).Goetz MK; Anderson JS Erratum: Experimental Evidence for pKa-Driven Asynchronicity in C─H Activation by a Terminal Co(III)–Oxo Complex. J. Am. Chem. Soc 2020, 142, 5439–5441. [DOI] [PubMed] [Google Scholar]
- (181).Mandal M; Elwell CE; Bouchey CJ; Zerk TJ; Tolman WB; Cramer CJ Mechanisms for Hydrogen-Atom Abstraction by Mononuclear Copper(III) Cores: Hydrogen-Atom Transfer or Concerted Proton-Coupled Electron Transfer? J. Am. Chem. Soc 2019, 141, 17236–17244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Bím D; Maldonado-Domínguez M; Rulísek L; Srnec M Beyond the Classical Thermodynamic Contributions to Hydrogen Atom Abstraction Reactivity. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E10287–E10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Greene BL; Kang G; Cui C; Bennati M; Nocera DG; Drennan CL; Stubbe J Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem 2020, 89, 45–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Kal S; Xu S; Que L Bio-Inspired Nonheme Iron Oxidation Catalysis: Involvement of Oxoiron(V) Oxidants in Cleaving Strong C─H Bonds. Angew. Chemie Int. Ed 2020, 59, 7332–7349. [DOI] [PubMed] [Google Scholar]
- (185).Dantignana V; Company A; Costas M Oxoiron(V) Complexes of Relevance in Oxidation Catalysis of Organic Substrates. Isr. J. Chem 2020, 60, 1004–1018. [Google Scholar]
- (186).Chakrabarty S; Austin RN; Deng D; Groves JT; Lipscomb JD Radical Intermediates in Monooxygenase Reactions of Rieske Dioxygenases. J. Am. Chem. Soc 2007, 129, 3514–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Wolfe MD; Parales JV; Gibson DT; Lipscomb JD Single Turnover Chemistry and Regulation of O2 Activation by the Oxygenase Component of Naphthalene 1,2-Dioxygenase. J. Biol. Chem 2001, 276, 1945–1953. [DOI] [PubMed] [Google Scholar]
- (188).Sutherlin KD; Rivard BS; Böttger LH; Liu LV; Rogers MS; Srnec M; Park K; Yoda Y; Kitao S; Kobayashi Y; Saito M; Seto M; Hu M; Zhao J; Lipscomb JD; Solomon EI NRVS Studies of the Peroxide Shunt Intermediate in a Rieske Dioxygenase and Its Relation to the Native FeII O2 Reaction. J. Am. Chem. Soc 2018, 140, 5544–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Fan R; Serrano-Plana J; Oloo WN; Draksharapu A; Delgado-Pinar E; Company A; Martin-Diaconescu V; Borrell M; Lloret-Fillol J; García-España E; Guo Y; Bominaar EL; Que L; Costas M; Münck E Spectroscopic and DFT Characterization of a Highly Reactive Nonheme FeV–Oxo Intermethate. J. Am. Chem. Soc 2018, 140, 3916–3928. [DOI] [PubMed] [Google Scholar]
- (190).Tiago de Oliveira F; Chanda A; Banerjee D; Shan X; Mondal S; Que L; Bominaar EL; Münck E; Collins TJ Chemical and Spectroscopic Evidence for an FeV-Oxo Complex. Science 2007, 315, 835–838. [DOI] [PubMed] [Google Scholar]
- (191).Van Heuvelen KM; Fiedler AT; Shan X; De Hont RF; Meier KK; Bominaar EL; Münck E; Que L One-Electron Oxidation of an Oxoiron(IV) Complex to Form an [O=FeV=NR]+ Center. Proc. Natl Acad. Sci. U. S. A 2012, 109, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Lyakin OY; Bryliakov KP; Britovsek GJP; Talsi EP EPR Spectroscopic Trapping of the Active Species of Nonheme Iron-Catalyzed Oxidation. J. Am. Chem. Soc 2009, 131, 10798–10799. [DOI] [PubMed] [Google Scholar]
- (193).Ezhov R; Ravari AK; Pushkar Y Characterization of the FeV=O Complex in the Pathway of Water Oxidation. Angew. Chemie Int. Ed 2020, 59, 13502–13505. [DOI] [PubMed] [Google Scholar]
- (194).Taylor CPS The EPR of Low Spin Heme Complexes. Relation of the t2g Hole Model to the Directional Properties of the g Tensor, and a New Method for Calculating the Ligand Field Parameters. Biochim. Biophys. Acta - Protein Struct 1977, 491, 137–148. [DOI] [PubMed] [Google Scholar]
- (195).Lyakin OY; Bryliakov KP; Talsi EP EPR, 1H and 2H NMR, and Reactivity Studies of the Iron–Oxygen Intermediates in Bioinspired Catalyst Systems. Inorg. Chem 2011, 50, 5526–5538. [DOI] [PubMed] [Google Scholar]
- (196).Oloo WN; Meier KK; Wang Y; Shaik S; Münck E; Que L Identification of a Low-Spin Acylperoxoiron(III) Intermediate in Bio-Inspired Non-Heme Iron-Catalysed Oxidations. Nat. Commun 2014, 5, 1–9. [DOI] [PubMed] [Google Scholar]
- (197).Mondal B; Neese F; Bill E; Ye S Electronic Structure Contributions of Non-Heme Oxo-Iron(V) Complexes to the Reactivity. J. Am. Chem. Soc 2018, 140, 9531–9544. [DOI] [PubMed] [Google Scholar]
- (198).Borrell M; Andris E; Navrátil R; Roithová J; Costas M Characterized Cis-FeV(O)(OH) Intermethate Mimics Enzymatic Oxidations in the Gas Phase. Nat. Commun 2019, 10, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Nguyen HHT; Shiemke AK; Jacobs SJ; Hales BJ; Lidstrom ME; Chan SI The Nature of the Copper Ions in the Membranes Containing the Particulate Methane Monooxygenase from Methylococcus Capsulatus (Bath). J. Biol. Chem 1994, 269, 14995–15005. [PubMed] [Google Scholar]
- (200).Semrau JD; Chistoserdov A; Lebron J; Costello A; Davagnino J; Kenna E; Holmes AJ; Finch R; Murrell JC; Lidstrom ME Particulate Methane Monooxygenase Genes in Methanotrophs. J. Bacteriol 1995, 177, 3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Nguyen H-HT; Elliott SJ; Yip JH-K; Chan SI The Particulate Methane Monooxygenase from Methylococcus Capsulatus (Bath) Is a Novel Copper-Containing Three-Subunit Enzyme: Isolation and Characterization. J. Biol. Chem 1998, 273, 7957–7968. [DOI] [PubMed] [Google Scholar]
- (202).Lieberman RL; Rosenzweig AC Crystal Structure of a Membrane-Bound Metalloenzyme That Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. [DOI] [PubMed] [Google Scholar]
- (203).Sirajuddin S; Barupala D; Helling S; Marcus K; Stemmler TL; Rosenzweig AC Effects of Zinc on Particulate Methane Monooxygenase Activity and Structure. J. Biol. Chem 2014, 289, 21782–21794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Ross MO; MacMillan F; Wang J; Nisthal A; Lawton TJ; Olafson BD; Mayo SL; Rosenzweig AC; Hoffman BM Particulate Methane Monooxygenase Contains Only Mononuclear Copper Centers. Science 2019, 364, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Chan SI; Chen KHC; Yu SSF; Chen CL; Kuo SSJ Toward Delineating the Structure and Function of the Particulate Methane Monooxygenase from Methanotrophic Bacteria. Biochemistry 2004, 43, 4421–4430. [DOI] [PubMed] [Google Scholar]
- (206).Lieberman RL; Rosenzweig AC Biological Methane Oxidation: Regulation, Biochemistry, and Active Site Structure of Particulate Methane Monooxygenase. Crit. Rev. Biochem. Mol. Biol 2004, 39, 147–164. [DOI] [PubMed] [Google Scholar]
- (207).Culpepper MA; Rosenzweig AC Architecture and Active Site of Particulate Methane Monooxygenase. Crit. Rev. Biochem. Mol. Biol 2012, 47, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Koo CW; Rosenzweig AC Biochemistry of Aerobic Biological Methane Oxidation. Chem. Soc. Rev 2021, 50, 3424–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Vaaje-Kolstad G; Westereng B; Horn SJ; Liu Z; Zhai H; Sørlie M; Eijsink VGH An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [DOI] [PubMed] [Google Scholar]
- (210).Johansen KS Lytic Polysaccharide Monooxygenases: The Microbial Power Tool for Lignocellulose Degradation. Trends Plant Sci. 2016, 21, 926–936. [DOI] [PubMed] [Google Scholar]
- (211).Phillips CM; Beeson WT; Cate JH; Marletta MA Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora Crassa. ACS Chem. Biol 2011, 6, 1399–1406. [DOI] [PubMed] [Google Scholar]
- (212).Quinlan RJ; Sweeney MD; Lo Leggio L; Otten H; Poulsen J-CNJ-CN; Johansen KS; Krogh KBRM; Jørgensen CI; Tovborg M; Anthonsen A; Tryfona T; Walter CP; Dupree P; Xu F; Davies GJ; Walton PH Insights into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme That Exploits Biomass Components. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Gardner JG; Crouch L; Labourel A; Forsberg Z; Bukhman YV; Vaaje-Kolstad G; Gilbert HJ; Keating DH Systems Biology Defines the Biological Significance of Redox-Active Proteins during Cellulose Degradation in an Aerobic Bacterium. Mol. Microbiol 2014, 94, 1121–1133. [DOI] [PubMed] [Google Scholar]
- (214).Simmons TJ; Frandsen KEH; Ciano L; Tryfona T; Lenfant N; Poulsen JC; Wilson LFL; Tandrup T; Tovborg M; Schnorr K; Johansen KS; Henrissat B; Walton PH; Lo Leggio L; Dupree P Structural and Electronic Determinants of Lytic Polysaccharide Monooxygenase Reactivity on Polysaccharide Substrates. Nat. Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Walton PH Enzymes Knuckle down to the Job. Nat. Chem. Biol 2020, 16, 812–816. [DOI] [PubMed] [Google Scholar]
- (216).Bissaro B; Røhr ÅK; Müller G; Chylenski P; Skaugen M; Forsberg Z; Horn SJ; Vaaje-Kolstad G; Eijsink VGH Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol 2017, 13, 1123–1128. [DOI] [PubMed] [Google Scholar]
- (217).Jones SM; Transue WJ; Meier KK; Kelemen B; Solomon EI Kinetic Analysis of Amino Acid Radicals Formed in H2O2-Driven CuI LPMO Reoxidation Implicates Dominant Homolytic Reactivity. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 11916–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Singh RK; Blossom BM; Russo DA; Singh R; Weihe H; Andersen NH; Tiwari MK; Jensen PE; Felby C; Bjerrum MJ Detection and Characterization of a Novel Copper-Dependent Intermethate in a Lytic Polysaccharide Monooxygenase. Chem. - A Eur. J 2020, 26, 454–463. [DOI] [PubMed] [Google Scholar]
- (219).Vu VV; Hangasky JA; Detomasi TC; Henry SJW; Ngo ST; Span EA; Marletta MA Substrate Selectivity in Starch Polysaccharide Monooxygenases. J. Biol. Chem 2019, 294, 12157–12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Frandsen KEH; Simmons TJ; Dupree P; Poulsen JCN; Hemsworth GR; Ciano L; Johnston EM; Tovborg M; Johansen KS; Von Freiesleben P; Marmuse L; Fort S; Cottaz S; Driguez H; Henrissat B; Lenfant N; Tuna F; Baldansuren A; Davies GJ; Lo Leggio L; Walton PH The Molecular Basis of Polysaccharide Cleavage by Lytic Polysaccharide Monooxygenases. Nat. Chem. Biol 2016, 12, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Frandsen KEH; Tovborg M; Jorgensen CI; Spodsberg N; Rosso MN; Hemsworth GR; Garman EF; Grime GW; Poulsen JCN; Batth TS; Miyauchi S; Lipzen A; Daum C; Grigoriev IV; Johansen KS; Henrissat B; Berrin JG; Leggio L. Lo. Insights into an Unusual Auxiliary Activity 9 Family Member Lacking the Histidine Brace Motif of Lytic Polysaccharide Monooxygenases. J. Biol. Chem 2019, 294, 17117–17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Vaaje-Kolstad G; Forsberg Z; Loose JS; Bissaro B; Eijsink VG Structural Diversity of Lytic Polysaccharide Monooxygenases. Curr. Opin. Struct. Biol 2017, 44, 67–76. [DOI] [PubMed] [Google Scholar]
- (223).Petrović DM; Bissaro B; Chylenski P; Skaugen M; Sørlie M; Jensen MS; Aachmann FL; Courtade G; Várnai A; Eijsink VGH Methylation of the N-Terminal Histidine Protects a Lytic Polysaccharide Monooxygenase from Auto-Oxidative Inactivation. Protein Sci. 2018, 27, 1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Gregory RC; Hemsworth GR; Turkenburg JP; Hart SJ; Walton PH; Davies GJ Activity, Stability and 3-D Structure of the Cu(II) Form of a Chitin-Active Lytic Polysaccharide Monooxygenase from Bacillus Amyloliquefaciens. Dalt. Trans 2016, 45, 16904–16912. [DOI] [PubMed] [Google Scholar]
- (225).Kuusk S; Bissaro B; Kuusk P; Forsberg Z; Eijsink VGH; Sørlie M; Valjamae P Kinetics of H2O2-Driven Degradation of Chitin by a Bacterial Lytic Polysaccharide Monooxygenase. J. Biol. Chem 2018, 293, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Fowler CA; Sabbadin F; Ciano L; Hemsworth GR; Elias L; Bruce N; McQueen-Mason S; Davies GJ; Walton PH Discovery, Activity and Characterisation of an AA10 Lytic Polysaccharide Oxygenase from the Shipworm Symbiont Teredinibacter Turnerae. Biotechnol. Biofuels 2019, 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Gudmundsson M; Kim S; Wu M; Ishida T; Momeni MH; Vaaje-Kolstad G; Lundberg D; Royant A; Stahlberg J; Eijsink VGH; Beckham GT; Sandgren M Structural and Electronic Snapshots during the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-Ray Photoreduction. J. Biol. Chem 2014, 289, 18782–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Span EA; Suess DLM; Deller MC; Britt RD; Marletta MA The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem. Biol 2017, 12, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Walton PH; Davies GJ On the Catalytic Mechanisms of Lytic Polysaccharide Monooxygenases. Curr. Opin. Chem. Biol 2016, 31, 195–207. [DOI] [PubMed] [Google Scholar]
- (230).Kracher D; Scheiblbrandner S; Felice AKG; Breslmayr E; Preims M; Ludwicka K; Haltrich D; Eijsink VGH; Ludwig R Extracellular Electron Transfer Systems Fuel Cellulose Oxidative Degradation. Science 2016, 352, 1098–1101. [DOI] [PubMed] [Google Scholar]
- (231).O’Dell WB; Agarwal PK; Meilleur F Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angew. Chemie Int. Ed 2017, 56, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Kracher D; Andlar M; Furtmüller PG; Ludwig R Active-Site Copper Reduction Promotes Substrate Binding of Fungal Lytic Polysaccharide Monooxygenase and Reduces Stability. J. Biol. Chem 2018, 293, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Wang B; Johnston EM; Li P; Shaik S; Davies GJ; Walton PH; Rovira C QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal. 2018, 8, 1346–1351. [Google Scholar]
- (234).Bertini L; Breglia R; Lambrughi M; Fantucci P; De Gioia L; Borsari M; Sola M; Bortolotti CA; Bruschi M Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations. Inorg. Chem 2018, 57, 86–97. [DOI] [PubMed] [Google Scholar]
- (235).Wang B; Walton PH; Rovira C Molecular Mechanisms of Oxygen Activation and Hydrogen Peroxide Formation in Lytic Polysaccharide Monooxygenases. ACS Catal. 2019, 4958–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Hangasky JA; Detomasi TC; Marletta MA Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases. Trends Chem. 2019, 1, 198–209. [Google Scholar]
- (237).Hangasky JA; Iavarone AT; Marletta MA Reactivity of O2 versus H2O2 with Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C─H Bond Activation by a Monocopper(III)–hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Fukatsu A; Morimoto Y; Sugimoto H; Itoh S Modelling a ‘Histidine Brace’ Motif in Mononuclear Copper Monooxygenases. Chem. Commun 2020, 56, 5123–5126. [DOI] [PubMed] [Google Scholar]
- (240).Puri M; Biswas AN; Fan R; Guo Y; Que L Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)-Halide Complexes That Halogenate C─H Bonds. J. Am. Chem. Soc 2016, 138, 2484–2487. [DOI] [PubMed] [Google Scholar]
- (241).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (242).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Simmons TR; Berggren G; Bacchi M; Fontecave M; Artero V Mimicking Hydrogenases: From Biomimetics to Artificial Enzymes. Coord. Chem. Rev 2014, 270–271, 127–150. [Google Scholar]
- (244).Yu F; Cangelosi VM; Zastrow ML; Tegoni M; Plegaria JS; Tebo AG; Mocny CS; Ruckthong L; Qayyum H; Pecoraro VL Protein Design: Toward Functional Metalloenzymes. Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Yang Y; Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Coelho PS; Brustad EM; Kannan A; Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- (247).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic Assembly of Carbon–Carbon Bonds via Iron-Catalysed Sp3 C─H Functionalization. Nature 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Lombardi A; Pirro F; Maglio O; Chino M; DeGrado WF De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res 2019, 52, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Korendovych IV; DeGrado WF De Novo Protein Design, a Retrospective. Q. Rev. Biophys 2020, 53, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Churchfield LA; Tezcan FA Design and Construction of Functional Supramolecular Metalloprotein Assemblies. Acc. Chem. Res 2019, 52, 345–355. [DOI] [PubMed] [Google Scholar]
- (251).Song WJ; Tezcan FA A Designed Supramolecular Protein Assembly with in Vivo Enzymatic Activity. Science 2014, 346, 1525–1528. [DOI] [PubMed] [Google Scholar]
- (252).Mirts EN; Bhagi-Damodaran A; Lu Y Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Ueno T; Ohashi M; Kono M; Kondo K; Suzuki A; Yamane T; Watanabe Y Crystal Structures of Artificial Metalloproteins: Tight Binding of FeIII(Schiff-Base) by Mutation of Ala71 to Gly in Apo-Myoglobin. Inorg. Chem 2004, 43, 2852–2858. [DOI] [PubMed] [Google Scholar]
- (254).Berggren G; Adamska A; Lambertz C; Simmons TR; Esselborn J; Atta M; Gambarelli S; Mouesca JM; Reijerse E; Lubitz W; Happe T; Artero V; Fontecave M Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499, 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Cavazza C; Bochot C; Rousselot-Pailley P; Carpentier P; Cherrier MV; Martin L; Marchi-Delapierre C; Fontecilla-Camps JC; Ménage S Crystallographic Snapshots of the Reaction of Aromatic C─H with O2 Catalysed by a Protein-Bound Iron Complex. Nat. Chem 2010, 2, 1069–1076. [DOI] [PubMed] [Google Scholar]
- (256).Ward TR Artificial Metalloenzymes Based on the Biotin–Avidin Technology: Enantioselective Catalysis and Beyond. Acc. Chem. Res 2011, 44, 47–57. [DOI] [PubMed] [Google Scholar]
- (257).Liang AD; Serrano-Plana J; Peterson RL; Ward TR Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res 2019, 52, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Wilson ME; Whitesides GM Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc 1978, 100, 306–307. [Google Scholar]
- (259).Skander M; Humbert N; Collot J; Gradinaru J; Klein G; Loosli A; Sauser J; Zocchi A; Gilardoni F; Ward TR Artificial Metalloenzymes: (Strept)Avidin as Host for Enantioselective Hydrogenation by Achiral Biotinylated Rhodium-Diphosphine Complexes. J. Am. Chem. Soc 2004, 126, 14411–14418. [DOI] [PubMed] [Google Scholar]
- (260).Hyster TK; Knörr L; Ward TR; Rovis T Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C─H Activation. Science 2012, 338, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed Evolution of Artificial Metalloenzymes for in Vivo Metathesis. Nature 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- (262).Serrano-Plana J; Rumo C; Rebelein JG; Peterson RL; Barnet M; Ward TR Enantioselective Hydroxylation of Benzylic C(Sp3)–H Bonds by an Artificial Iron Hydroxylase Based on the Biotin–Streptavidin Technology. J. Am. Chem. Soc 2020, 142, 10617–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Miller KR; Paretsky JD; Follmer AH; Heinisch T; Mittra K; Gul S; Kim IS; Fuller FD; Batyuk A; Sutherlin KD; Brewster AS; Bhowmick A; Sauter NK; Kern J; Yano J; Green MT; Ward TR; Borovik AS Artificial Iron Proteins: Modeling the Active Sites in Non-Heme Dioxygenases. Inorg. Chem 2020, 59, 6000–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.