

Abstract
Background:
Potential therapeutic targets and clinical trials for Huntington’s disease (HD) have grown immensely in the last decade. However, in order to improve clinical trial outcomes there is a need to better characterize sign and symptom profiles across different epochs of the disease to improve selection of participants.
Objective:
To best distinguish longitudinal trajectories across different HD progression groups.
Methods:
Clinical and morphometric imaging data from 1,082 participants across IMAGE-HD, TRACK-HD and PREDICT-HD studies were combined, with longitudinal timepoints ranging between 1–10 years. Participants were classified into four groups using CAG-Age Product (CAP). Using Multivariate Linear Mixed Modelling (MLMM), 63 different combinations of markers were tested for their sensitivity in differentiating CAP groups. Next, MLMM was applied to define the best combination of markers to track progression across individual CAP groups.
Results:
Putamen and caudate volumes, individually and/or combined, were identified as the best variables to both differentiate CAP groups and track progression within them. The model using only caudate volumes best described advanced disease progression in the combined dataset. Contrary to expectations, combining clinical markers and volumetric measures did not improve tracking longitudinal progression.
Conclusions:
Monitoring volumetric changes throughout a trial (alongside primary and secondary clinical endpoints) may provide a more comprehensive understanding of improvements in functional outcomes and help improve the design of clinical trials. Alternatively, our results suggest that imaging deserves consideration as an endpoint in clinical trials because of the prospect of greater sensitivity.
Keywords: Huntington’s Disease, symptom profiles, statistical modelling, tracking disease progression
Introduction
Huntington’s disease (HD) is a neurodegenerative condition caused by an autosomal dominant expansion of a cytosine-adenine-guanine (CAG) tract in the huntingtin (HTT) gene [1] and characterised by progressive decline in motor, cognitive, and psychiatric functions [2–4]. There is currently no disease-modifying treatment for HD, but pivotal trials are underway [5]. Over a decade of research has helped improve clinical trial design and identify group differences of trial outcome measures with high sensitivity to HD related changes [6, 7]. The earliest epoch of HD is marked by a protracted asymptomatic premanifest period spanning over decades followed by an early and then full manifestation of disease severity. The long premanifest period (and an early manifest stage) means there are rather diversified biophysical and physiological factors across the spectrum that may translate to a widely varied sign/symptom profiles [8], which might also play a role in responses to future treatments. Hence, one of the most critical impediments in conducting new clinical trials effectively is in how to assign heterogeneous individuals to homogenous subgroups. Identification of an effective stratification of HD will be significant to therapeutic development by selecting participants with the specific symptom profile and finding an optimal window for future pharmacological intervention.
The existence of a reliable test for the CAG expansion and the establishment of large natural history repositories allows the investigation of a range of biomarkers that may be more or less sensitive to progression depending on disease epochs. PREDICT-HD [6, 9], TRACK-HD/TRACK-ON [7, 10, 11] and IMAGE-HD [12, 13] datasets have been widely used, often in isolation, to identify markers of disease progression and early detection of disease. By investigating the longitudinal changes in a range of outcome measures from these studies, researchers have identified several markers of HD progression, amongst which the symbol digit modality test (SDMT) [10, 14, 15], Unified Huntington’s Disease Rating Scale - Total Motor Score (UHDRS-TMS) [10, 16], caudate and putamen volumes [6, 10, 17], and motor tapping [6, 7] are shown to be the most sensitive.
An opportunity to combine these datasets not only represents a larger sample of participants, but potentially a collection of participants with different genetic backgrounds from multiple geographic locations. This approach enables the study of a more representative sample from the HD population and cross-study validations to improve the robustness of results [18]. Additionally, combining these datasets captures wider segments of disease progression (e.g., while IMAGE and TRACK have a low number of participants in one group, PREDICT provides a larger sample of participants in the same) and hence can improve the range of characterization. Combining different datasets has proven fruitful, as illustrated with the ENIGMA consortium for studying brain disease. ENIGMA obtains small samples of data collected at different sites across the world, covering a wide range of brain diseases, aggregated to generate large datasets to improve the quality, generalizability and robustness of the findings [19].
In this study, we combined datasets from IMAGE, TRACK+TRACK-ON (referred to as TRACK hereafter), and PREDICT, and used specific measures with identical assessments across studies. We modelled disease related longitudinal changes through Multivariate Linear Mixed Models (MLMM) [20] simultaneously examining differences in patient groups stratified using the CAG and age product (CAP) score, as defined previously [9]. We used CAP groupings because it is based on both age and CAG repeats which are the two main factors to characterise disease progression and has been successfully used previously [9, 21]. We hypothesised that by combining motor, functional, cognitive and morphometric imaging markers we would be able to improve our ability to distinguish various epochs of disease. To identify potential sensitivity of marker effects, we also examined which combinations of markers will improve the longitudinal tracking of HD progression within each group.
Methodology
Ethics Statement
IMAGE was approved by the Monash University and Melbourne Health Human Research Ethics Committees as a single site study in Melbourne, Australia. TRACK was approved by the local ethics committees at each study site in the Netherlands, UK, France and Canada. PREDICT procedures were approved by institutional review boards at each site (32 sites across the United States, Canada, Australia and Europe). For all three studies, each participant provided written informed consent.
Participants
We assessed data collected from structural MRI scans and clinical assessments, from previously published cohorts’ studies of IMAGE [12, 15, 17], TRACK [7, 11] and PREDICT [6, 9, 14]. All were longitudinal observational studies and data collection covered repeated visits over 3-years, 7-years and 12-years, respectively. To produce comparable datasets, we adopted a set of exclusion criteria primarily based on MRI parameters, which included three main steps (see Appendix 1). To reduce the variability due to the magnetic field strength, we removed all data from 1.5T MRI scans and retained only 3T MRI scans; this step affected only the PREDICT dataset. A rigorous quality control (QC) procedure was then applied, as described below. After completing the QC procedure and removing participants with incomplete data, the number of time points per participant ranged between 1–3 for IMAGE, 1–7 for TRACK and 1–10 for PREDICT, and varied from one participant to another.
Overall, 1,082 participants were left in the combined dataset (360 healthy controls, 619 pre-HD and 254 symptomatic-HD participants), and each participant was assigned to one of four CAP groups. Participants with < 36 CAG repeats served as controls, labelled CAP group 1, and those with ≥ 36 were categorized into further 3 groups based on their baseline CAP score [1]. The CAP score was calculated using the formula CAP = Age * (CAG – 33.66) [1]. Cut-offs for the three HD groups were: 0 < CAP < 290 for CAP group 2 (far from onset), 290 ≤ CAP ≤ 368 for CAP group 3 (medium to onset), and CAP > 368 for CAP group 4 (close to onset) [9] (see a summary in Table 1).
Table 1:
Baseline demographics, and summary of baseline clinical and volumetric data as a function of CAP group.
| CAP Category | IMAGE | TRACK | PREDICT | Combined data | |
|---|---|---|---|---|---|
| N | 1 | 36 | 155 | 139 | 330 |
| 2 | 5 | 13 | 143 | 161 | |
| 3 | 21 | 69 | 143 | 233 | |
| 4 | 46 | 188 | 124 | 358 | |
| Female (% within category) | 1 | 24 (67%) | 69 (44%) | 86 (62%) | 179 (54%) |
| 2 | 3 (60%) | 4 (31%) | 105 (73%) | 112 (69%) | |
| 3 | 9 (43%) | 32 (46%) | 102 (71%) | 143 (61%) | |
| 4 | 24 (52%) | 85 (45%) | 68 (55%) | 177 (49%) | |
| Age (years) | 1 | 42.67 (13.93) | 45.7 (10.29) | 44.81 (11.79) | 44.39 (12.00) |
| 2 | 38.05 (10.13) | 36.34 (9.96) | 33.19 (8.67) | 35.86 (9.58) | |
| 3 | 41.41 (10.19) | 41.12 (9.08) | 40.95 (10.02) | 41.16 (9.76) | |
| 4 | 50.25 (9.80) | 46.29 (10.16) | 45.24 (10.80) | 47.26 (10.25) | |
| Education (standardized levels) | 1 | 5.18 (0.43) a | 3.98 (1.28) | 4.35 (0.89) | 4.27 (1.13) |
| 2 | 5.58 (0.27) a | 4 (1.03) | 4.3 (0.89) | 4.31 (0.92) | |
| 3 | 5.02 (0.53) a | 4.07 (1.05) | 4.26 (0.87) | 4.27 (0.94) | |
| 4 | 5.04 (0.47) a | 3.68 (1.31) | 4.24 (0.98) | 4.04 (1.21) | |
| TMS | 1 | 0.63 (0.51) b | 1.42 (1.65) | 3.56 (4.18) | 2.48 (3.37) |
| 2 | 1.60 (0.86) | 4.38 (4.56) | 4.18 (6.13) | 4.11 (5.97) | |
| 3 | 1.71 (1.44) | 4.06 (4.71) | 8.11 (8.18) | 6.28 (7.36) | |
| 4 | 15.75 (13.42) | 16.42 (14.12) | 14.68 (13.11) | 15.73 (13.71) | |
| TFC | 1 | - | 12.98 (1.47) | 12.92 (0.51) | 12.95 (1.09) |
| 2 | - | 13.00 (0.00) | 12.74 (1.06) | 12.85 (0.25) | |
| 3 | - | 12.78 (1.46) | 12.37 (1.38) | 12.58 (1.23) | |
| 4 | - | 11.58 (1.46) | 12.16 (1.41) | 11.89 (1.36) | |
| SDMT | 1 | 56.28 (9.94) | 52.46 (9.46) | 53.52 (9.78) | 53.32 (9.72) |
| 2 | 56.00 (10.31) | 49.85 (12.12) | 56.68 (10.81) | 56.11 (11.07) | |
| 3 | 50.80 (8.90) | 52.54 (11.04) | 49.74 (10.79) | 50.66 (10.79) | |
| 4 | 38.88 (12.43) | 39.59 (12.84) | 43.04 (10.61) | 40.70 (12.19) | |
| Stroop (word) | 1 | 109.75 (16.34) | 105.27 (16.92) | 103.23 (17.27) | 104.90 (17.11) |
| 2 | 109.00 (8.15) | 97.00 (11.07) | 105.71 (15.46) | 103.43 (15.61) | |
| 3 | 101.20 (18.90) | 102.06 (17.03) | 95.32 (18.67) | 97.83 (18.49) | |
| 4 | 87.68 (23.16) | 85.84 (21.05) | 86.43 (18.42) | 86.28 (20.65) | |
| Putamen | 1 | 0.006 (0.001) | 0.006 (0.001) | 0.006 (0.001) | 0.006 (0.001) |
| 2 | 0.005 (0.001) | 0.006 (0.001) | 0.006 (0.001) | 0.006 (0.001) | |
| 3 | 0.005 (0.001) | 0.005 (0.001) | 0.005 (0.001) | 0.005 (0.001) | |
| 4 | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | |
| Caudate | 1 | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) |
| 2 | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | |
| 3 | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | 0.004 (0.001) | |
| 4 | 0.003 (0.001) | 0.003 (0.001) | 0.003 (0.001) | 0.003 (0.001) | |
| GM | 1 | 0.438 (0.0221) | 0.437 (0.048) | 0.426 (0.042) | 0.433 (0.044) |
| 2 | 0.430 (0.011) | 0.442 (0.051) | 0.445 (0.033) | 0.444 (0.035) | |
| 3 | 0.438 (0.018) | 0.436 (0.046) | 0.432 (0.037) | 0.434 (0.039) | |
| 4 | 0.408 (0.021) | 0.406 (0.043) | 0.411 (0.036) | 0.408 (0.038) | |
| WM | 1 | 0.310 (0.018) | 0.327 (0.035) | 0.320 (0.041) | 0.322 (0.037) |
| 2 | 0.305 (0.007) | 0.320 (0.039) | 0.316 (0.031) | 0.316 (0.031) | |
| 3 | 0.315 (0.015) | 0.325 (0.035) | 0.316 (0.032) | 0.319 (0.032) | |
| 4 | 0.291 (0.019) | 0.302 (0.030) | 0.314 (0.033) | 0.305 (0.031) |
Data represent mean of the number of participants and the standard deviation at baseline. Standard deviation is replaced with percentages for categorical variables. CAP: product of CAG repeat length and age calculated using CAP = Age × (CAG – 33.66); CAP category is 1 = controls, 2 = far from onset, 3 = medium to onset, 4 = close to onset.
UHDRS-TMS: Unified Huntington’s Disease Rating Scale-Total Motor Score; UHDRS-TFC: Unified Huntington’s Disease Rating Scale-Total Functional Capacity; SDMT: Symbol Digit Modality Test. Volumetric data are recorded as a percentage of Intracranial volume (ICV).
For IMAGE-HD cohort, education level was not recorded; this was estimated by re-scaling the National Adult Reading Test (NART) IQ scores that were acquired as part of the study following [23] where the NART score is found to be inter-related with the years of education.
For controls in IMAGE, UHDRS-TMS was not recorded; this was estimated using tapping task data (see Appendix 2 for details). Comparatively low UHDRS-TMS scores observed for CAP 3 in IMAGE is due to low CAG repeats in the IMAGE population compared to that of TRACK and PREDICT.
Motor, functional and cognitive measures (clinical measures)
Since our main goal was to use MLMM to study the combined dataset, it was important to select identical measures across all three studies. After a thorough inspection of each dataset, we identified the following outcome measures as identical across studies and were thus used to study the complete dataset: SDMT, Stroop word test (Stroop(w)) and UHDRS-TMS. IMAGE did not include UHDRS-TMS assessment for controls. To overcome this limitation, we approximated UHDRS-TMS for IMAGE controls using a linear regression of the finger-tapping task (see Appendix 2 for details) as it has been shown that the number of taps and the inter-tap interval are significantly correlated with UHDRS-TMS [22]. We also applied MLMM for individual studies and in doing so UHDRS-TFC was included for TRACK and PREDICT cohorts in addition to the aforementioned markers but UHDRS-TFC was not available for IMAGE. Participant demographics, as well as a summary of clinical data for all study cohorts, are presented in Table 1.
Structural MRI (sMRI) data processing
sMRI data processing and segmentation was performed using FreeSurfer (version 5.3.0), for all data points per participant independently by following a consistent method and parameters starting from T1-weighted images. FreeSurfer analyses were performed using “recon-all” function on the Monash University computing cluster [24]. Following the segmentation procedure using Desikan-Killiany atlas, we selected the volumetric data of regions of interest that are widely established as regions with a higher sensitivity to HD-related longitudinal changes [25–28]. This included volumetric data for putamen, caudate, whole-brain grey matter (GM) and whole-brain white matter (WM). Even though the sMRI data were acquired at different sites, multi-site effects have shown to be relatively less pronounced in sMRI than in diffusion and functional MRI [29]. Therefore, we did not account for the multi-site effects of the sMRI data.
Based on our experience on FreeSurfer’s segmentation quality, occasional mis-segmentation may occur in key subcortical structures. For example, the putamen may be under-segmented due to the neighboring pallidus segmentation invading the true putamen structure. To help mitigate these segmentation issues, we created a semi-automatic QC routine. As the first step, putamen and caudate volume asymmetry was calculated for all the segmented images. The asymmetry formula is (left volume – right volume)/ (left volume + right volume), and the score range is −1 to 1. Then, the segmented images, at the top and bottom 5% of the asymmetry distribution, were selected and manually checked for erroneous segmentation. Approximately 5% of the segmented images were removed from each study due to erroneous segmentation. This number of identified failures is in line with a separate effort by one of the authors to manually check all FreeSurfer v6 segmentations from the three datasets used in this study (4.6% failure of putamen segmentation, unpublished data). Summary of baseline volumetric data are presented in Table 1. For more information, see Appendix 3.
Statistical analysis:
The main analysis focuses on finding the best combination of motor, functional, cognitive and imaging markers that would discriminate among CAP groups. A secondary analysis was conducted to find the combination of markers that could track disease progression within each CAP group. Our goal was to model the trend over time of several markers simultaneously while accounting for the correlations among variables and conditional dependencies due to repeated measures. Based on literature [9] we considered only a linear component of time in our analysis using MLMM [20] as the primary tool. Random intercept and slopes were specified in the model for all the biomarkers of each participant, while education and sex were used as fixed effects covariates. The random effects for a single variable and the random effects among different variables were allowed to correlate. Additionally, outcome variables were standardized using the mean and standard deviation of the entire stacked vector (subjects and visits). Model estimation for MLMM was performed using sampling methods of (Bayesian) Markov Chain Monte Carlo (MCMC) methods. The mvglmer() function of the JMBayes package [30] was used in R software (version 3.6.3) with uninformative (naive) priors for all parameters. Relative global model fit was indexed by the Deviance Information Criterion (DIC, smaller is better; [31]), which is a generalization of the Akaike information criterion (AIC). DIC calculates a posterior mean of the log likelihood and adds a penalty for model complexity allowing the comparison of complex hierarchical models. It identifies models that best explain the observed data, which are likely to minimize uncertainty [31]. It is similar to a Bayesian version of AIC. Within the Baysian framework, MLMM uses MCMC for parameter estimation and for MCMC, calculating the log likelihood is not straightforward for AIC as it is for DIC.
MLMM analysis was performed in two steps. First, MLMM was used to analyse the longitudinal trajectories of all the markers. This step involved running MLMM for single studies as well as for combined multi-study data using the most complex model that included all the markers. The purpose of this initial analysis was to identify the markers that best discriminated CAP groups. To assist in the comparison between all outcome measures, fitted slopes of each CAP group were compared with that of the controls.
The second step was to analyse models with different combinations of markers using MLMM for separate CAP groups, to identify the best combination of markers to track the longitudinal progression for each CAP group. There were 63 models (see Appendix 4) estimated from all possible combinations of the outcome variables (models with single variables, two variables, three variables, etc., up to six variables). For this analysis, out of the four volumetric measures only putamen and caudate volumes were used as they have been reported to be the most sensitive volumetric markers [21]. On the other hand, the longitudinal atrophy of GM and WM was significantly small compared to other markers (see Appendix 5).
Results
1. Combination of markers for distinguishing disease groups
1.1. MLMM results of the most complex model
Figure 1 illustrates the MLMM results in the combined dataset for the model with all seven markers (i.e., SDMT, Stroop(w), TMS, putamen, caudate, GM and WM). For ease of presentation, the predicted curves are for females between 1–6 standardized education levels as measured by the International Standard Classification of Education. Appendix 5 shows the fitted annual rate of change across CAP groups for female and male populations separately. The slope of CAP groups 2, 3 and 4 was significantly different from CAP group 1 (controls) with all (Bayesian) p-values < 0.001 for all predictors in both populations. Similarly, the slope of CAP groups 2, 3 and 4 was significantly different from each other with all (Bayesian) p-values < 0.001. Similar figures for individual study datasets can be found in Appendix 6.
Figure 1:
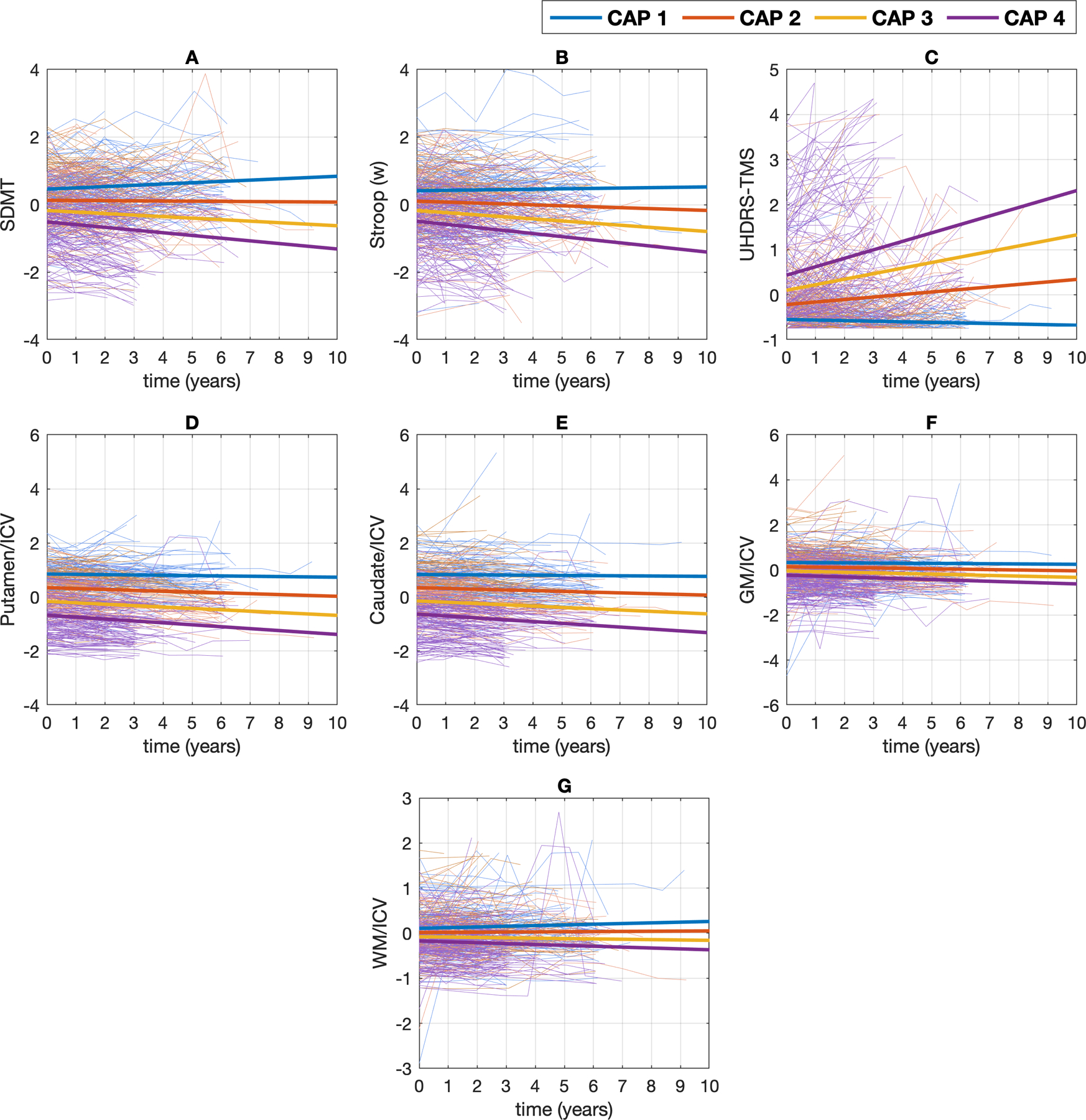
Standardized observed (thin lines) and predicted (thick lines) values for the female population as a function of time (0 = baseline), separated by CAP groups for (A). SDMT: Symbol Digit Modality Test, (B). Stroop(w), (C). UHDRS-TMS: Unified Huntington’s Disease Rating Scale-Total Motor Score, (D). Putamen volume and (E). Caudate volume, (F). GM volume and (G). WM volume. All the variables were standardized (see text). Each thin line represents the measurements for a single participant over time.
1.2. MLMM results of 63 models with different combinations of markers
Figure 2 presents the DIC for all the MLMMs (list of models - Appendix 4) for IMAGE, TRACK and PREDICT, as well as for the combined data. The top ranked model for IMAGE, as well as for combined data was the model with only putamen while for TRACK and PREDICT it was the model which included both putamen and caudate. Starting with this model as a base, the more variables added, the more complex the model gets, and the more the goodness of fit deteriorates. Interestingly, all models without any of the volumetric marker(s) had the worst model fits in PREDICT. This means that the volumetric markers are strongly associated with improved stratification of CAP groups coupled with different epochs of disease, compared to motor, functional and cognitive markers.
Figure 2:
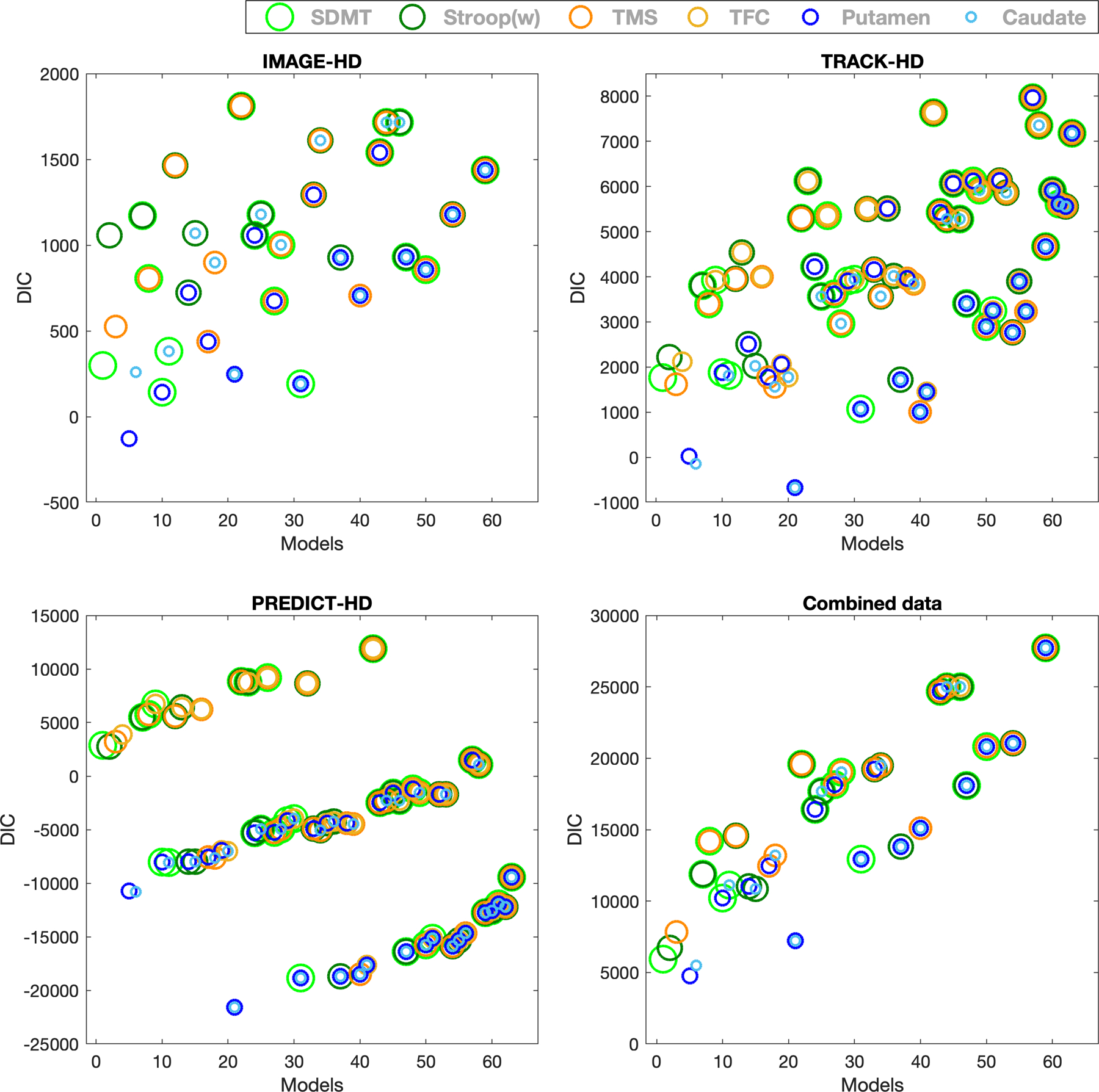
DIC is plotted for all the models based on the MLMM analysis (refer to Appendix 4 for the list of models) across IMAGE, TRACK, PREDICT and combined dataset. Circles of different colors represent six markers as follows (in order of decreasing radius): Light green: SDMT (Symbol Digit Modality Test), Green: Stroop(w), Dark orange: UHDRS-TMS (Unified Huntington’s Disease Rating Scale-Total Motor Score), Yellow: UHDRS-TFC (Unified Huntington’s Disease Rating Scale-Total Functional Capacity), Blue: Putamen volume and Light Blue: Caudate volume. Combined data = IMAGE, TRACK and PREDICT data combined. Two (or more) circles of different colors and sizes at a single point represents a MLMM with two (or more) variables with respective colors of the circles.
2. Combination of markers for tracking the longitudinal trajectory of progression within each disease group
Figure 3 shows the DIC for fitted MLMMs across each CAP group for the combined dataset (for individual studies see Appendix 7). Table 2 provides a high-level summary of the model comparison across CAP groups across IMAGE, TRACK, PREDICT and combined datasets. In all groups, and across datasets, the model with putamen and/or caudate ranked as the model with the best fit for the longitudinal changes.
Figure 3:
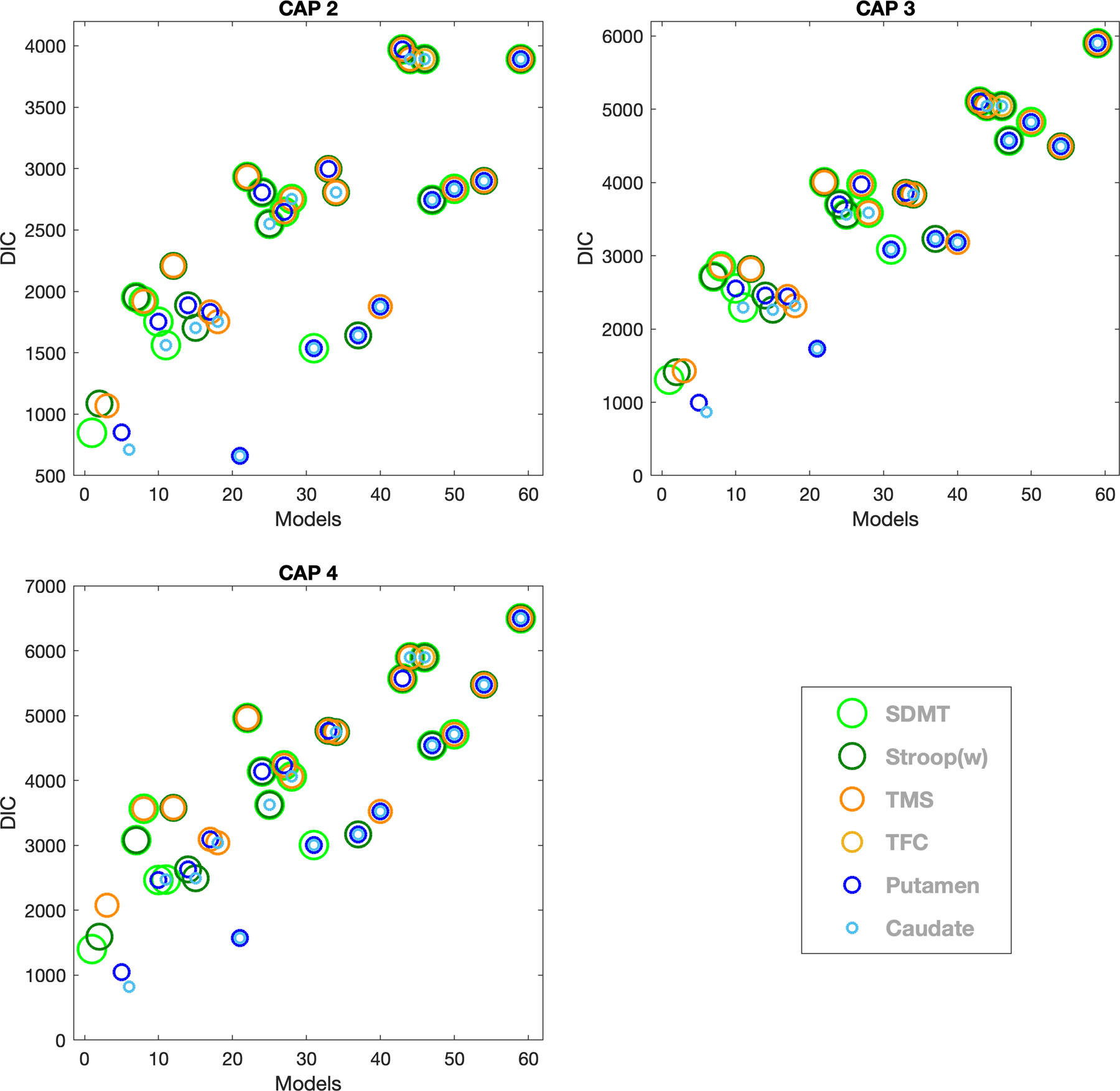
DIC is plotted for CAP groups 2, 3 and 4 for all models based on the MLMM analysis (refer to Appendix 4 for the list of models) using the combined dataset. Circles of different colors represent six markers as follow (in order of decreasing radius): Light green: SDMT (Symbol Digit Modality Test), Green: Stroop (w), Dark orange: UHDRS-TMS (Unified Huntington’s Disease Rating Scale-Total Motor Score), Yellow: UHDRS-TFC (Unified Huntington’s Disease Rating Scale-Total Functional Capacity), Blue: Putamen volume and Light Blue: Caudate volume. Two (or more) circles of different colors and sizes at a single point represents a MLMM with two (or more) variables with respective colors of the circles. CAP 1 is not shown as all participants in this group are healthy controls and the purpose of this analysis is to find the model that best fits the disease progression.
Table 2:
Summary of the best model fit per CAP group across each study separately and combined.
| Study | CAP Group | ||
|---|---|---|---|
| CAP 2 (Low) | CAP 3 (Medium) | CAP 4 (High) | |
| IMAGE | aTMS | Putamen | Putamen |
| TRACK | aTMS + TFC + Putamen + Caudate | Caudate | Putamen + Caudate |
| PREDICT | Putamen + Caudate | Putamen + Caudate | Putamen + Caudate |
| Combined data | Putamen + Caudate | Caudate | Caudate |
In IMAGE and TRACK cohorts CAP 2 included only 5 and 13 participants, respectively. Therefore, we were not able to draw any conclusions regarding the best model in this scenario.
Discussion
We comprehensively analysed motor, functional, cognitive and imaging markers across IMAGE, TRACK and PREDICT cohorts to determine the best combination of markers to distinguish longitudinal trajectories of pre-defined HD progression (CAP) groups. The results showed that the model with caudate volume only, or with caudate and putamen volumes, performed best in distinguishing among CAP groups. Considering additional motor, functional and cognitive outcomes did not improve the model fit. Our results confirm the importance of tracking striatal deterioration when monitoring HD progression.
Caudate and putamen volume combined and/or individually, ranked as the best model(s) to fit the longitudinal changes in each CAP group in most of the individual datasets (except CAP group 2 in IMAGE and TRACK, due to the low number of participants). This finding is in agreement with previous studies, which have shown that structural brain imaging measures in general are the most powerful at detecting disease progression in premanifest and early HD participants [9, 10, 15, 32]. Interestingly though, and we demonstrated for the first time, when investigating different combinations of motor, functional, cognitive and volumetric markers, the model that included only the caudate volume best described more advanced disease progression as noted in CAP groups 3 and 4 for the combined dataset. In the absence of volumetric markers, SDMT ranked as the best marker to describe longitudinal changes across all CAP groups (Figure 3). This finding is in accord with the original TRACK analysis, suggesting early changes on SDMT [33].
The longitudinal trajectories of the brain volume markers were found to have the strongest differences among the CAP groups. The intercept and slope of the linear trajectory increased as the CAP group increased (Figure 1), which is consistent with previous findings where imaging markers overall have been found to be the most sensitive in detecting longitudinal differences in disease epochs [10, 32]. Overall, our findings confirm an acceleration effect in the reduction of striatal volume over time in HD.
The significance of volume atrophy over other clinical measures is linked to its relationship with the functional outcome measures. A strong association between the observed changes in volumetric markers and the underlying genetic disease burden—as well as functional outcome measures like tapping task and TFC—has been reported previously [10, 32, 34], suggesting a significant correlation between brain structural measures and function. Volumetric markers are also categorized as objective markers, which have shown longitudinal consistency across different studies [35].
Our findings also suggest that, in the absence of volumetric data, the most powerful model to distinguish CAP groups is the univariate model with SDMT, followed by the other univariate models with Stroop(w) and TMS (Figure 2 - panel 4: combined data). SDMT, Stroop(w) and TMS are also among the most sensitive clinical markers in previous studies [9, 10, 35], which showed that cognitive decline, as measured by SDMT and Stroop (w), to be strongly associated with changes in TMS.
Contrary to our expectations, when motor, functional and cognitive markers were included in the model with volumetric markers there was no improvement in the model fitting over volumetric markers alone. Thus, comparatively more objective markers, such as caudate and putamen volumes, seem to be better differentiators of disease epochs compared to more subjective clinical markers/functional outcome measures like SDMT, TMS etc. [35]. According to previous work, not only the effect sizes of the volumetric markers are larger compared to that of the clinical measures [7, 10], they also have a steady atrophy within disease epochs. In addition, in a recent phase II clinical trial cohort of at-risk HD participants, treatment-related slowing of volume atrophy was strikingly sensitive to the drug (high-dose creatine) compared to controls but had no significant effect on clinical markers [36]. Thus, a trial selection criterion that includes sMRI could help better stratify participants resulting in improved outcomes and better evaluation of the candidate therapeutic. Additionally, monitoring volumetric changes throughout a trial alongside the primary and secondary endpoints (which are predominantly clinical functional markers, such as TMS) [14, 18, 35, 37, 38] could improve clinical trial design with an overall improvement in functional outcomes [39, 40]. Moreover, since there could be drugs that directly affect volume atrophy in the trial but only leads to improvements in clinical measures later, and vice versa [36], it would be beneficial and cost effective to perform longitudinal sMRI first in a phase II trial with small sample sizes to identify clinical endpoints. sMRI could also be used in combination with sample enrichment recruitment methods (described in [41]) to increase the likelihood of developing cost-effective efficient therapeutic strategies by enrolling an optimal sample of participants.
In PREDICT, the study cohort with the longest time span, there was a clear separation of models that discriminated the CAP groups, based on the inclusion/exclusion of volumetric data in the models. Interestingly, the stratification pattern among the models (Figure 2) was most pronounced for PREDICT, slightly visible for TRACK but not for the IMAGE cohort. We speculate that this might be due to the longer time span of data (10 years, even after data filtering), as well as a higher number of participants per CAP group in PREDICT. Even though TRACK included a large number of participants in CAP groups 3 and 4 (Table 1), data collection spanned only 7 years, resulting in comparatively fewer time points per participant compared to PREDICT. This may suggest that to identify subtle deviations between different disease epochs above and beyond already established knowledge, collecting continuous longitudinal data for longer periods and with more time points per participant (e.g., collecting annual data for >10 years) is required. This, together with also including follow up of younger, more diverse samples [42, 43], could be argued as being more important than having a bigger sample with follow ups of 4–5 years. This further supports previous discussions on the importance of investing in long-term longitudinal studies that span five years or more with annual follow-ups, which has been posited for HD [44, 45] as well as other neurodegenerative diseases like Alzheimer’s disease [46, 47]. This approach will improve the clinical heterogeneity and help accelerate the development of new therapeutics by gaining a deeper and more robust understanding of disease progression. An example is Enroll-HD, a worldwide observational study that annually collects clinical data and biospecimens for biobanking [44]. The Enroll-HD database has been used in many studies to date, not only to broaden understanding of disease progression [48– 49] but also to enable improved clinical assessment techniques to improve trial design [50].
One limitation of our study is that we inspected only five markers for the combined dataset that were common across studies, including one motor (TMS), one functional (TFC), two cognitive (SDMT and Stroop(w)), and two volumetric (putamen and caudate volume) markers. By including additional markers, such as finger tapping that has already been shown to be associated with disease progression during different epochs [34, 50], could result in a more robust and complete model to track longitudinal progression and to identify different sign/symptom profiles. Future research could also investigate whether combining white matter diffusion measures in the models with volumetric markers could capture additional subtle differences between different CAP groups, as well as different sign/symptom profiles within the CAP groups, to improve stratification and clinical trial selection criteria.
In summary, we comprehensively analysed the composite dataset by combining five motor, functional, cognitive and volumetric markers via MLMM and found that putamen and caudate volumes individually and/or in combination (without any other variable) provide the best model to differentiate CAP groups, as well as being more sensitive in tracking progression within each CAP group. Of all five markers, the model using only caudate volume was best in describing more advanced disease progression (CAP groups 3 and 4). Contrary to expectations, clinical markers in combination with volumetric markers did not substantially improve disease progression prediction. Based on our findings we raise three implications that could have application in HD clinical trials: i) volumetric markers could be considered as a primary endpoint along with other clinical endpoints since they are more sensitive than clinical/functional markers; ii) volumetric markers could be combined with other sample enrichment recruitment methods to optimize cost-effective participant enrollments and increase trial efficiency; and iii) monitoring volumetric changes alongside primary and secondary clinical endpoints may provide more insight into functional outcome improvements and inform clinical trial design.
Supplementary Material
Acknowledgements
This study was supported by CHDI Foundation research agreement A–3433 (N.G.‐K.) and the National Health and Medical Research Council Australia grant 606650 (N.G.‐K.). AR is funded by the Australian Research Council (Refs: DE170100128 and DP200100757) and National Health and Medical Research Council (Ref: APP1194910). IMAGE-HD data used in this work was generously provided by the participants in the IMAGE-HD study. TRACK-HD data used in this work was generously provided by the participants in the TRACK-HD study and made available by Dr Sarah Tabrizi, Principal Investigator, University College London. PREDICT-HD data used in this work was generously provided by the participants in PREDICT-HD study and made available by the PREDICT-HD investigators and coordinators of the Huntington study group, Jane S Paulsen, Principal Investigator (NS040068).
Funding sources: This study was funded by research grants from NHMRC Australia and NIH NS040068.
Financial Disclosure of all authors (for the preceding 12 months)
Dr. Pubu M. Abeyasinghe reports no disclosure.
Dr. Jeffrey D. Long is a paid Advisory Board member for F. Hoffmann-La Roche Ltd and uniQure biopharma B.V., and has been a paid consultant for Vaccinex Inc, Wave Life Sciences USA Inc, Genentech Inc, Triplet Inc, and PTC Therapeutics Inc.
Dr. Adeel Razi is funded by the Australian Research Council (Refs: DE170100128 and DP200100757) and National Health and Medical Research Council (Ref:1194910).
Dr. Dorian Pustina reports no disclosure.
Dr. Jane S. Paulsen is funded by NIH, the University of Wisconsin, Madison, and consultant work for Acadia Pharmaceuticals, Wave Life Sciences, and CHDI.
Dr. Sarah J. Tabrizi receives grant funding for her research from the Medical Research Council UK, the Wellcome Trust, the Rosetrees Trust, Takeda Pharmaceuticals Ltd, Vertex Pharmaceuticals, Cantervale Limited, NIHR North Thames Local Clinical Research Network, UK Dementia Research Institute, and the CHDI Foundation. In the past 12 months, S.J.T. has undertaken consultancy services, including advisory boards, with F. Hoffmann-La Roche Ltd, Novartis Pharma, Spark Therapeutics, Takeda Pharmaceuticals Ltd, Triplet Therapeutics, and Vertex Pharmaceuticals Incorporated. All honoraria for these consultancies were paid through the offices of UCL Consultants Ltd., a wholly owned subsidiary of University College London.
Dr. Govinda Poudale reports no disclosure
Dr. Nellie Georgiou-Karistianis is funded by the National Health and Medical Research Council (606650).
Footnotes
Conflict of Interest: None
References
- 1.Walker FO (2007). Huntington’s disease. The Lancet, 369(9557), 218–228. [DOI] [PubMed] [Google Scholar]
- 2.Soloveva MV, Jamadar SD, Velakoulis D, Poudel G, & Georgiou-Karistianis N (2020). Brain compensation during visuospatial working memory in premanifest Huntington’s disease. Neuropsychologia, 136, 107262. [DOI] [PubMed] [Google Scholar]
- 3.Lo J, Reyes A, Pulverenti TS et al. (2020). Dual tasking impairments are associated with striatal pathology in Huntington’s disease. Annals of Clinical and Translational Neurology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poudel G, Stout JC, Gray M et al. (2017). Longitudinal changes in the fronto-striatal network are associated with executive dysfunction and behavioral dysregulation in Huntington’s disease: 30 months IMAGE-HD data. Cortex, 92, 139–149. [DOI] [PubMed] [Google Scholar]
- 5.Tabrizi SJ, Flower MD, Ross CA, & Wild EJ (2020). Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nature Reviews Neurology, 1–18. [DOI] [PubMed] [Google Scholar]
- 6.Paulsen JS, Langbehn DR, Stout JC et al. (2008). Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. Journal of Neurology, Neurosurgery & Psychiatry, 79(8), 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabrizi SJ, Scahill RI, Owen G et al. (2013). Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. The Lancet Neurology, 12(7), 637–649. [DOI] [PubMed] [Google Scholar]
- 8.Nana AL, Kim EH, Thu DC et al. (2014). Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. Journal of Huntington’s disease, 3(1), 45–64. [DOI] [PubMed] [Google Scholar]
- 9.Paulsen JS, Long JD, Johnson HJ et al. (2014). Clinical and biomarker changes in premanifest Huntington disease show trial feasibility: a decade of the PREDICT-HD study. Frontiers in aging neuroscience, 6, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabrizi SJ, Reilmann R, Roos RA et al. (2012). Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. The Lancet Neurology, 11(1), 42–53. [DOI] [PubMed] [Google Scholar]
- 11.Klöppel S, Gregory S, Scheller E et al. (2015). Compensation in preclinical Huntington’s disease: evidence from the track-on HD study. EBioMedicine, 2(10), 1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poudel GR, Stout JC, Churchyard A, Chua P, Egan GF, & Georgiou-Karistianis N (2015). Longitudinal change in white matter microstructure in Huntington’s disease: the IMAGE-HD study. Neurobiology of disease, 74, 406–412. [DOI] [PubMed] [Google Scholar]
- 13.Dominguez JF, Stout JC, Poudel G et al. (2016). Multimodal imaging biomarkers in premanifest and early Huntington’s disease: 30-month IMAGE-HD data. The British Journal of Psychiatry, 208(6), 571–578. [DOI] [PubMed] [Google Scholar]
- 14.Paulsen JS, Smith MM, Long JD, PREDICT HD investigators, & Coordinators of the Huntington Study Group. (2013). Cognitive decline in prodromal Huntington Disease: implications for clinical trials. Journal of Neurology, Neurosurgery & Psychiatry, 84(11), 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan GF, Gray MA, Poudel GR et al. (2013). Multi-modal neuroimaging in premanifest and early Huntington’s disease: 18-month longitudinal data from the IMAGE-HD study. PloS one, 8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biglan KM, Ross CA, Langbehn DR et al. (2009). Motor abnormalities in premanifest persons with Huntington’s disease: the PREDICT‐HD study. Movement Disorders, 24(12), 1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgiou-Karistianis N, Gray MA, Dymowski AR et al. (2013). Automated differentiation of pre-diagnosis Huntington’s disease from healthy control individuals based on quadratic discriminant analysis of the basal ganglia: the IMAGE-HD study. Neurobiology of disease, 51, 82–92. [DOI] [PubMed] [Google Scholar]
- 18.Wijeratne PA, Johnson EB, Eshaghi A et al. (2020). Robust Markers and Sample Sizes for Multicenter Trials of Huntington Disease. Annals of Neurology, 87(5), 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson PM, Stein JL, Medland SE et al. (2014). The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain imaging and behavior, 8(2), 153–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sammel M, Lin X, & Ryan L (1999). Multivariate linear mixed models for multiple outcomes. Statistics in medicine, 18(17‐18), 2479–2492. [DOI] [PubMed] [Google Scholar]
- 21.Paulsen JS, Long JD, Ross CA et al. (2014). Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. The Lancet Neurology, 13(12), 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michell AW, Goodman AOG, Silva AHD, Lazic SE, Morton AJ, & Barker RA (2008). Hand tapping: a simple, reproducible, objective marker of motor dysfunction in Huntington’s disease. Journal of neurology, 255(8), 1145–1152. [DOI] [PubMed] [Google Scholar]
- 23.Kiely KM, Luszcz MA, Piguet O, Christensen H, Bennett H, & Anstey KJ (2011). Functional equivalence of the National Adult Reading Test (NART) and Schonell reading tests and NART norms in the Dynamic Analyses to Optimise Ageing (DYNOPTA) project. Journal of Clinical and Experimental Neuropsychology, 33(4), 410–421. [DOI] [PubMed] [Google Scholar]
- 24.Goscinski WJ, McIntosh P, Felzmann UC et al. (2014). The multi-modal Australian ScienceS Imaging and Visualization Environment (MASSIVE) high performance computing infrastructure: applications in neuroscience and neuroinformatics research. Frontiers in Neuroinformatics, 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aylward EH, Sparks BF, Field KM et al. (2004). Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology, 63(1), 66–72. [DOI] [PubMed] [Google Scholar]
- 26.Egan GF, Gray MA, Poudel GR et al. (2013). Multi-modal neuroimaging in premanifest and early Huntington’s disease: 18 month longitudinal data from the IMAGE-HD study. PloS one, 8(9), e74131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulsen JS, Nopoulos PC, Aylward E et al. (2010). Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain research bulletin, 82(3–4), 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabrizi SJ, Reilmann R, Roos RA et al. (2012). Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. The Lancet Neurology, 11(1), 42–53 [DOI] [PubMed] [Google Scholar]
- 29.Gregory S, Lohse KR, Johnson EB et al. (2020). Longitudinal Structural MRI in Neurologically Healthy Adults. Journal of Magnetic Resonance Imaging [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rizopoulos D, Rizopoulos MD, Imports MASS, SystemRequirements JAGS, & Rcpp L (2020). Package ‘JMbayes’ [Google Scholar]
- 31.Spiegelhalter DJ, Best NG, Carlin BP, & Van Der Linde A (2002). Bayesian measures of model complexity and fit. Journal of the royal statistical society: Series b (statistical methodology), 64(4), 583–639. [Google Scholar]
- 32.Tabrizi SJ, Scahill RI, Durr A et al. (2011). Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. The Lancet Neurology, 10(1), 31–42. [DOI] [PubMed] [Google Scholar]
- 33.Stout JC, Jones R, Labuschagne I et al. (2012). Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington’s disease. Journal of Neurology, Neurosurgery & Psychiatry, 83(7), 687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bechtel N, Scahill RI, Rosas HD et al. (2010). Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology, 75(24), 2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross CA, Aylward EH, Wild EJ et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature Reviews Neurology, 10(4), 204–216. [DOI] [PubMed] [Google Scholar]
- 36.Rosas HD, Doros G, Gevorkian S et al. (2014). PRECREST: a phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology, 82(10), 850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulsen JS, Hayden M, Stout JC et al. (2006). Preparing for preventive clinical trials: the Predict-HD study. Archives of neurology, 63(6), 883–890. [DOI] [PubMed] [Google Scholar]
- 38.Bates GP, Dorsey R, Gusella JF et al. (2015). Huntington disease. Nature reviews Disease primers, 1(1), 1–21. [DOI] [PubMed] [Google Scholar]
- 39.Dash D, & Mestre TA (2020). Therapeutic update on Huntington’s disease: symptomatic treatments and emerging disease-modifying therapies. Neurotherapeutics, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troncoso-Escudero P, Sepulveda D, Pérez-Arancibia R et al. (2020). On the right track to treat movement disorders: Promising therapeutic approaches for Parkinson’s and Huntington’s disease. Frontiers in Aging Neuroscience, 12, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulsen JS, Lourens S, Kieburtz K, & Zhang Y (2019). Sample enrichment for clinical trials to show delay of onset in huntington disease. Movement Disorders, 34(2), 274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langbehn KE, Cochran AM, van der Plas E et al. (2020). Behavioral Deficits in Juvenile Onset Huntington’s Disease. Brain Sciences, 10(8), 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moser AD, Epping E, Espe-Pfeifer P et al. (2017). A survey-based study identifies common but unrecognized symptoms in a large series of juvenile Huntington’s disease. Neurodegenerative disease management, 7(5), 307–315. [DOI] [PubMed] [Google Scholar]
- 44.Landwehrmeyer GB, Fitzer‐Attas CJ, Giuliano JD et al. (2017). Data analytics from Enroll‐HD, a global clinical research platform for Huntington’s disease. Movement disorders clinical practice, 4(2), 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bashir H (2019). Emerging therapies in Huntington’s disease. Expert review of neurotherapeutics, 19(10), 983–995. [DOI] [PubMed] [Google Scholar]
- 46.Bhagwat N, Viviano JD, Voineskos AN, Chakravarty MM, & Alzheimer’s Disease Neuroimaging Initiative. (2018). Modeling and prediction of clinical symptom trajectories in Alzheimer’s disease using longitudinal data. PLoS computational biology, 14(9), e1006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eskildsen SF, Coupé P, García-Lorenzo D et al. (2013). Prediction of Alzheimer’s disease in subjects with mild cognitive impairment from the ADNI cohort using patterns of cortical thinning. Neuroimage, 65, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghazaleh N, Houghton R, Palermo G, Schobel SA, Wijeratne P, & Long J (2020). Discovering Dominant Features Associated with Disease Progression in Huntington’s Disease (HD): A Data-driven Approach Using the Enroll-HD Database (1603) [Google Scholar]
- 49.Schultz JL, Kamholz JA, Nopoulos PC, & Killoran A (2019). Comparing Risperidone and Olanzapine to Tetrabenazine for the Management of Chorea in Huntington Disease: An Analysis from the Enroll‐HD Database. Movement disorders clinical practice, 6(2), 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman JS, Cody FWJ, O’Boyle DJ, Craufurd D, Neary D, & Snowden JS (1996). Abnormalities of motor timing in Huntington’s disease. Parkinsonism & related disorders, 2(2), 81–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.