

Abstract
Lung cancer is the second most common cancer in both men and women and is the leading cause of cancer death in the United States. The development of drug resistance to commonly used chemotherapeutics in non-small-cell lung cancer (NSCLC) poses significant health risks and there is a dire need to improve patient outcomes. In this study, we report the use of RNA nanotechnology to display ligand on exosome that was loaded with small interfering RNA (siRNA) for NSCLC regression in animal trials. Cholesterol was used to anchor the ligand targeting epidermal growth factor receptor on exosomes that were loaded with siRNA to silence the antiapoptotic factor survivin. The cytosolic delivery of siRNA overcame the problem of endosome trapping, leading to potent gene knockdown, chemotherapy sensitization, and tumor regression, thus achieving a favorable IC50 of 20 nmol/kg siRNA encapsulated by exosome particles in the in vivo gene knockdown assessment.
Keywords: RNA nanotechnology, exosomes, non-small-cell lung cancer, siRNA, EGFR, aptamer
Introduction
Lung cancer is the most common cancer worldwide, resulting in 2.09 million new patient cases per year, and causing 1.76 million deaths per year in 2018 according to World Health Organization (WHO) reports [1]. Non-small-cell lung cancer (NSCLC) accounts for more than 80% of all lung cancer cases [2]. First-line treatment for metastatic NSCLC includes chemotherapy, targeted therapy, and immunotherapy. Treatments targeting epidermal growth factor receptor (EGFR), including tyrosine kinase inhibitors (eg, erlotinib, gefitinib, osimertinib, etc.) [3–5] and monoclonal antibodies (e.g., bevacizumab, cetuximab, necitumumab, etc.) [6–8] have been widely used for various molecular forms of NSCLC. An RNA aptamer selectively targeting EGFR was discovered by Systematic Evolution of Ligands by Exponential enrichment (SELEX) and had been applied with RNA nanotechnology for targeting breast cancer [9–11]. Studies have shown that BIRC5, which codes for the survivin protein, is overexpressed in NSCLC and correlates with the development of resistance [12,13]. Moreover, downregulation of BIRC5 can induce apoptosis, which has been proven to be a potent strategy for treatment in multiple cancer types [14–17].
RNA interference (RNAi), discovered by Andrew Fire and Craig Mello in 1998 and recognized by the Noble Prize in 2006, was one of the most important breakthroughs in the history of gene therapy [18]. Instead of introducing the deficient gene, RNAi enables post-transcriptional gene silencing (PTGS) by suppressing mRNA translation or inducing mRNA cleavage. Small interfering RNA (siRNA) is double-strand artificially synthesized RNA ranging from 19 to 27 bp that can utilize the Dicer-RISC pathway to silence target mRNA [19]. This novel strategy created a new direction for gene therapy by allowing the specific design of siRNA to achieve precise manipulation of the target and minimize off-target effects.
However, challenges still exist and the development of RNAi-based gene therapy was more difficult than expected. The major challenge is drug delivery, the same problem facing the field of whole gene therapy [20,21]. First, naked RNA is vulnerable to RNases that widely exist in the circulatory system. Second, as a negatively charged macromolecule, the cellular uptake of RNA is low due in part to the lack of specific transporters that many small molecule drugs utilize for cellular entry. Third, due to these challenges, higher doses are needed, which increases the risk of side effects due to unwanted gene manipulation in healthy cells. Even though a small portion of RNA enters the cell, most of them will be trapped in the endosome that prevents the RNA from reaching the RNAi machinery, as well as its mRNA target located in the cytoplasm. Therefore, the development of a drug delivery vehicle that can protect RNA and improve target cell uptake, especially cytosolic distribution, is urgently needed. Despite the challenges aforementioned, three siRNA drugs (Patisiran, Gibosiran, and Lumasiran) have been approved by the U.S. Food and Drug Administration (FDA) since 2018, leveraging the advantage of nucleic acid chemistry, GalNac conjugation, and nanoparticle-mediated delivery. However, the current success of systemic siRNA administration is still limited to liver applications, and the targeting strategy for other organ-associated disease is needed.
RNA nanotechnology was first proven by Peixuan Guo in 1998, a decade after his discovery of packaging RNA (pRNA) in bacteriophage phi29 DNA packaging motor [22,23]. The single-strand pRNA contains a 3-way-junction (3WJ) core that retains high thermostability in the physiological condition when truncated to three 16 ∼ 20 nt single strands [24]. This unique feature provides feasibility to design self-assembling nanoparticles in a bottom-up strategy using single-strand RNA as a building block with precise control of size, shape, stoichiometry, and homogeneity [25–29]. Meanwhile, both the thermostability and the enzymatic stability can be significantly improved with the chemical modification of 2′-OH on ribose sugar [30]. Moreover, the cytotoxicity and immunogenicity can be controlled to a negligible level by rational design of sequence, size, shape, and chemical modifications [31–34]. Thereby, the development of RNA nanotechnology rebuilds the concept of RNA as a stable and biocompatible nanomaterial that can be used for drug delivery. The fully controllable feature of RNA nanotechnology enables the programmable design for tuning the pharmacokinetic (PK) and distribution profiles. In general, desired drug delivery should maximize the accumulation in the disease site and minimize healthy tissue accumulations. The nanometer scale allows RNA nanoparticles by themselves to nonspecifically accumulate in tumor tissue through enhanced permeability and retention effect due to the leaky texture in overgrowing tumor tissue [35,36]. The controllable size, along with the negatively charged and hydrophilic property, of RNA nanoparticle also eliminate the liver accumulation that reduces potential toxicity and immunogenicity [32,37,38]. Besides the passive targeting effect, RNA nanoparticles can also be actively internalized by cancer cells by the conjugation of ligands that can target to cancer cells, including RNA aptamers [10,39,40] and small molecule ligands [41–43]. However, challenges such as relatively low drug payload, endosome entrapment after receptor-mediated endocytosis, and rapid clearance are problems that remain to be addressed for RNA nanoparticle as a delivery vehicle for gene therapy. Nevertheless, RNA nanotechnology as an emerging solution holds great promise for RNAi therapeutics delivery.
Exosomes have been reported as a novel drug delivery vehicle with great potential [16,44–47]. Exosomes are derived from late endosome/multivesicular body with a diameter between 50 and 150 nm [48]. Exosomes have an endomembrane-like membrane property (structure, lipid, peptides, protein, etc.), which offers an innate ability to fuse with recipient plasma membrane or the membrane of the cellular organelles [47,49–52]. Exosomes were considered to be a relative “garbage disposal bucket” [53] until 2007, when three research groups discovered that genetic materials, especially RNA, can be transferred and communicated in long distance among cells through exosomes [54–56]. In the following decade, exosomes have been demonstrated to serve as carriers for direct delivery of their RNA payloads into the cytosol, which enables the full functionality [47,57–59]. The advantages of exosomes include that they are the natural carriers of proteins and RNAs [55,60,61], and the high payloads they can carry while remaining a favorable size and are well tolerated in vivo [51]. Therapeutic payloads, such as siRNA, can remain fully functional after delivery to cells by exosomes [45,49,52,55,62]. However, the selectivity of naive exosomes is relatively low and vary from sources, which can cause random uptake in healthy cells. To generate cell-specific targeting exosomes, we used RNA nanotechnology for ligand displaying on naive exosomes to promote efficient cell targeting, cytosolic siRNA delivery, and cancer regression [16].
In this study, to leverage the targeting effect and therapeutic potential of EGFR RNA aptamer for NSCLC treatment, we designed an arrowtail RNA nanoparticle to display EGFR RNA aptamer on the surface of exosomes to deliver survivin siRNA (EGFRApt/Exo/siSurvivin) for NSCLC treatment as the formulation schematic shows in Fig. 1A.
FIG. 1.

Assembly of exosome vesicle loaded with siRNA and RNA nanotechnology decoration. (A) Formulation scheme of EGFRApt displaying exosomes loaded with siRNA. (B) Size distribution of exosomes. Purified exosomes were analyzed by NTA with peak size at 109 nm and average size of 177 ± 6 nm. (C) Surface marker characterization. Purified exosomes from four independent purification were assayed by immunoblotting to confirm exosome surface marker CD63 and TSG 101. (D) Design of EGFRApt arrowtail pRNA nanoparticles for ligand displaying. (E) Assembly of EGFRApt arrowtail pRNA nanoparticles assay by 10% Native PAGE in TBE buffer stained by EtBr. (F) Loading efficiency of siRNA into exosomes assayed by comparing fluorescent intensity in unloaded supernatant of siRNA loading with Exo and without Exo. EGFR, epidermal growth factor receptor; EtBr, ethidium bromide; NTA, nanoparticle tracking analysis; PAGE, polyacrylamide gel electrophoresis; pRNA, packaging RNA; siRNA, small interference RNA; TBE, tris/borate/EDTA buffer.
Materials and Methods
Cell culture, chemicals, and antibodies
Human lung cancer cell line H596 and H1568 were obtained from American Type Culture Collection and maintained in Rosewell Park Memorial Institute (RPMI) 1640 medium (ATCC; Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS) (GE Healthcare, Chicago, IL) and 1% penicillin/streptomycin (Life Technologies, Carlsbad, CA). Cells were cultured in 37°C with 5% CO2. Cisplatin (Sigma, St. Louis, MO) was dissolved in dimethyl formamide (Sigma). Anti-CD63, TSG101, Survivin, EGFR, cleaved-poly (ADP-ribose) polymerase (PARP), cleaved-Caspase3, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-rabbit, and anti-mouse secondary antibodies were purchased from LI-COR Bioscience (Lincoln, NE).
Purification and characterization of exosomes
Exosomes were purified using a modified differential ultracentrifugation method as previously described [16,17,63]. HEK293T cells obtained from ATCC were cultured in FiberCell Hollow Fiber Bioreactor (C2011, 20 kDa MWCO) using Dulbecco's modified Eagle's medium (DMEM) with 10% exosome-free FBS, and the exosome-enriched medium was collected each week. Exosome-enriched medium was spun down at 300 rcf for 10 min to remove dead cells, followed by spinning at 10,000 rcf for 30 min at 4°C followed by 22 nm filtration to remove cell debris and microvesicles and store at −80°C until 500 mL accumulated.
Preprocessed medium was thawed slowly at 4°C overnight, then loaded into preconditioned Pall Minimate™ tangential flow filtration (TFF) system with 100 kDa MWCO capsule (OA100C12), precondition follows the standard operation by the manufacturer. Five hundred milliliters of exosome-enriched medium was processed at 6 mL/min by setting pump speed at 40 mL/min. When the volume reduced to ∼5 mL, 200 mL sterile Dulbecco's phosphate buffered saline (DPBS) was added as washing step and continued to run until the volume reduced to ∼5 mL again, then collected. Twice the 15 mL DPBS wash was performed and total 30 mL wash was collected and combined with the 5 mL sample from the last step.
The 35 mL post TFF medium was then further purified by 100,000 rcf ultracentrifugation using a SW28 rotor (Beckman Coulter) for 90 min at 4°C. Two-hundred microliters of 60% iodixanol (Sigma) was added to the bottom of each tube to serve as iso-osmotic cushion as we previously reported [16,17,63]. The supernatant was carefully removed from the top, and around 2 mL of the fraction close to the interface and cushion were collected. Nanoparticle tracking analysis (NTA) was performed using the Malvern NanoSight NS300 system following the standard protocol developed and reported before [16,17,63].
Assembly of exosomes loaded with siRNA and RNA nanotechnology decoration
Exosomes (0.05 pmole) and Alexa647 or Alexa750-conjugated siRNA (0.15 nmole) were mixed in 100 μL of 1 × PBS with 2.5 μL of ExoFect exosome transfection (System Bioscience) following the manufacturer's instruction. Unloaded siRNA from the supernatant was collected and Alexa647 intensity on supernatant was measured to evaluate loading efficiency compared with the control group. EGFRApt/3WJ arrowtail RNA nanoparticles were self-assembled in a one-pot manner by mixing individual RNA strands at equimolar concentrations (25 μM) in PBS buffer. Assembled RNA nanoparticles were confirmed by electrophoresis. Then RNA nanoparticles were incubated with exosomes at 37°C for 45 min, then left on ice for 1 h to prepare the RNA-decorated Exosome.
In vitro exosomes cellular binding assay
The ability of exosomes binding to human lung cancer cells was assessed in vitro by flow cytometry. In brief, 1 × 105 H596 and H1568 cells were resuspended in 100 μL PBS buffer, and incubated with 50 nM Alexa647-labeled exosomes loaded with siRNA at 37°C for 1 h. After washing three times with PBS, the cells were subjected to flow cytometry analysis using the BD LSRFortessa flow cytometer. Data were analyzed using the FlowJo 7.6.1 software.
Real-time quantitative polymerase chain reaction analysis
Survivin gene silencing by siRNA delivered by exosomes was detected by real-time quantitative polymerase chain reaction (PCR) assay. Total cellular RNA was isolated using TRIzol (Invitrogen), and one microgram of total RNA was reverse transcribed using Supercript reverse transcriptase (Bio-Rad, Hercules, CA). PCR was performed on iCYCLER real-time PCR machine (Bio-Rad) using SYBR-Green chemistry (Bio-Rad). The gene expression levels were normalized to housekeeping gene GAPDH. The sequences of used primers are shown in Supplementary Table S1.
Immunoblotting
Cell lysates were prepared using RIPA buffer (Thermo Fisher, Waltham MA) supplemented with 1 × protease inhibitors (Complete; Roche, Indianapolis, IN) and phosphatase inhibitors (PhosSTOP; Roche) followed by protein quantification with the Dc Protein Assay Kit (Bio-Rad). Equal amounts of protein were loaded and resolved by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were incubated in 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween-20 (TBST) blocking buffer for 1 h at room temperature. Primary antibodies with dilution of 1:200–1,000 were allowed to bind overnight at 4°C, or for 2 h at room temperature. After washing in TBST, the membranes were incubated with immunofluorescent secondary antibodies at a 1:5,000 dilution for 1 h at room temperature. Membranes were washed with TBST and allowed to air dry before imaging through LI-COR Odyssey® CLx Imaging System (Thermo Fisher). Original data refer to Supplementary Figure S3.
IncuCyte cell proliferation assay
Cells were treated with various exosomes accordingly for 48 h, and then seeded at 1,000–2,000 cells per well in 96-well plates. Cell confluence as a measure of cell growth over time was monitored every 4 h for up to 6 days using the IncuCyte ZOOM Live-Cell Imaging System (Essen Biosciences). Cell proliferation curve was plotted using GraphPad Prism.
Cell apoptosis analysis by flow cytometry
Cells were seeded into 6-well plates at a density of 200,000 cells per well in 2 mL medium for 16 h, then treated with exosomes for 48 h, followed by 5 μM cisplatin treatment for another 24 h. Cell apoptosis was assessed by Annexin V-FITC (Invitrogen) and Propidium Iodide (PI; Sigma-Aldrich) staining coupled with flow cytometry analysis using the BD LSRFortessa flow cytometer. Data were analyzed using the FlowJo 7.6.1 software.
Biodistribution and antitumor activity of exosomes in vivo
Animal studies were conducted in accordance with an approved protocol adhering to the Institutional Animal Care and Use Committee (IACUC) policies and procedures at The Ohio State University. Six-to eight-week-old male athymic nude mice (Taconic Farms, Inc., NY) were caged in groups of five or less, and fed with a diet of animal chow and water ad libitum. H596 cells were injected subcutaneously into the flanks of each mouse at 5 × 106 per injection. When the tumor sizes reached 150–200 mm3, the mice were randomly divided into four groups and tail vein injected with exosomes three times a week for 2 weeks. The tumor volume was monitored three times a week, and tumor size was calculated using the formula: 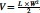 . For in vivo exosomes targeting and tumor imaging, mice were tail vein injected with 100 μL of 20 μM of RNA Alexa750-labeled siSurvivin loaded with exosomes, and animal or tissue was imaged using the in vivo imaging system (IVIS) Lumina imaging system with Living Images 3.0 software (Caliper Life Sciences).
. For in vivo exosomes targeting and tumor imaging, mice were tail vein injected with 100 μL of 20 μM of RNA Alexa750-labeled siSurvivin loaded with exosomes, and animal or tissue was imaged using the in vivo imaging system (IVIS) Lumina imaging system with Living Images 3.0 software (Caliper Life Sciences).
Data analysis
Data are presented as the mean ± standard deviation or standard error of the mean of at least three independent experiments. The difference among groups were calculated using Student's t test or one-way analysis of variance (ANOVA) analysis followed by Tukey's post-hoc test (GraphPad Prism).
Result
Isolation and characterization of exosomes derived from HEK293T cells
We first produced and isolated bulk exosomes from HEK293T cells using a hollow-fiber bioreactor, TFF, and cushion ultracentrifugation (Supplementary Fig. S1). With this workflow, the volume of exosomes can be concentrated from liter to microliter scale in a relatively straightforward process. Postexosome production, size distribution was analyzed by the NTA method, showing the size peak at 109 nm, and average size of exosomes as 177 ± 6 nm (Fig. 1B). We further characterized exosome marker expression by immunoblotting, which showed high expression of CD63 and TSG101 in various batches of the exosome solution (Fig. 1C).
Assembly, loading, and ligand displaying of exosomes by RNA nanotechnology
Following our rationale design reported previously to increase thermodynamic and enzymatic stability [16,17,24,63], we harbored survivin siRNA onto the pRNA 3WJ core with a 2’F modification on the sense strand. To determine siRNA loading efficiency into exosomes, the remaining siRNA was fused to an Alexa647-labeled 3WJ core (Fig. 2) and then was loaded into exosomes using ExoFect method as we reported previously [16,17,63]. In short, 3WJ-siSurvivin-Alexa647 mixed with equal volume of PBS and treated with Exofect was used as background to evaluate loading efficacy. As shown in Fig. 1F, 88.8% of siRNA was loaded into exosomes. EGFR RNA aptamer (EGFRApt) was adapted to generate arrowtail RNA nanoparticles (Fig. 1D), which we had developed for ligand displaying on exosome surface [16,17,63]. As shown in Fig. 1E, EGFRApt arrowtail RNA nanoparticles were assembled with very high efficiency as indicated by gel shift assays showing stepwise assembly of the complex.
FIG. 2.

Design and assembly of si-survivin harboring 3WJ RNA nanoparticle. Design of RNA nanoparticle shown on the left panel, 2′ F-modified sequence in lower case. Self-assembly of RNA nanoparticles was verified by 12% TBE PAGE, EtBr staining channel in green, Alexa647 channel in red (right panel). 3WJ, 3-way-junction.
Specific binding and siRNA delivery to cells in vitro using EGFRApt displaying exosome
To prove the ligand-displaying strategy can improve NSCLC cell targeting, we next examined the cellular uptake of EGFRApt/Exo/siSurvivin on two EGFR-expressing lung cancer cell lines, H596, and H1568 (Fig. 3). As shown in Fig. 4A and Supplementary Fig. S2, flow cytometry revealed significantly higher binding efficiency (78.8% and 85.6%) for EGFRApt-decorated exosomes, compared with no EGFRApt controls (71.2% and 48%) in two cell lines. The results suggest that EGFRApt is successfully displayed onto exosome surface by arrowtail RNA nanoparticles and enhance the uptake by cancer cell.
FIG. 3.

Protein expression level in different NSCLC cell lines and a human bronchial epithelial cell line. Western blot was conducted to screen the protein expression level of EGFR receptor (upper panel) and survivin (lower panel). NSCLC, non-small-cell lung cancer.
FIG. 4.

EGFRApt/Exo/siSurvivin suppresses survivin expression in human lung cancer. (A) Cellular uptake of siSurvivin delivered by EGFRApt/Exo and Exo only compared by flow cytometry on both H596 and H1568 NSCLC cell lines, **P < 0.01, ***P < 0.001 by Student's t-test, n = 3. Raw data and gate setting in Supplementary Fig. S4. (B) mRNA level of survivin gene quantified by qRT-PCR assay post 48 h treatment of EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr, Exo/siSurvivin, and TES buffer in 50 nM of siRNA concentration on both H596 and H1568 NSCLC cell lines. **P < 0.01, ****P < 0.0001 by Student's t-test, n = 3. (C) Protein expression level of Survivin evaluated by western blot post 48 h treatment of EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr, Exo/siSurvivin and TES buffer in 50 nM of siRNA concentration on both H596 and H1568 NSCLC cell line. qRT-PCR, real-time quantitative reverse transcription polymerase chain reaction; TES, tris/EDTA/saline buffer.
To further demonstrate the therapeutic potential of EGFRApt/Exo/siSurvivin, we then evaluated the target gene expression and protein expression in vitro. We treated cancer cells with EGFRApt/Exo/siSurvivin along with control groups at 50 nM of siRNA concentration for 48 h. The expression of survivin was assayed by real-time PCR analysis and western blot. We found that Exo/siSurvivin can suppress survivin mRNA expression in H596 and H1568 cells, and EGFRApt/Exo/siSurvivin can further suppress mRNA levels of survivin (Fig. 4B), which was consistent with Fig. 4A and Supplementary Fig. S2 that EGFR aptamer enhanced the ability of exosomes binding to human lung cancer cells. Western blotting further confirmed that EGFRApt/Exo/siSurvivin suppressed expression of survivin at the protein level (Fig. 4C).
Suppressing of tumor cell growth, and sensitizing tumor cell to chemotherapy in vitro by EGFRApt/Exo/siSurvivin
To assess the effects of depletion of survivin by siRNA delivered by exosomes on cell growth, IncuCyte Cell Count proliferation assay was carried out. As shown in Fig. 5A, Exo/siSurvivin suppressed tumor cell growth, and the presence of EGFR aptamer further suppressed cell growth. Impaired apoptosis is one of the mechanisms for chemotherapy resistance in cancer cells. We also investigated the potential treatment option in combination with cisplatin, a common first-line chemotherapy drug for NSCLC. Research had been shown that inhibition of survivin can promote cisplatin potency [64,65]. Cells were pretreated with EGFRApt/Exo/siSurvivin for 48 h, followed by 24 h cisplatin treatment. Apoptotic cells were stained with Annexin V and PI, then the population of cells were detected by flow cytometry. As shown in Fig. 5B and Supplementary Fig. S4, Exo/siSurvivin treatment sensitized cells to cisplatin-induced apoptosis, and the EGFRApt/Exo/siSurvivin had further increased efficacy compared with the non-target control, which consisted of both binding and gene knockdown experiments (Fig. 4). Moreover, we validated the sensitization of cisplatin-induced apoptosis by evaluating the cleavage of PARP (c-PARP) and Caspase3 (c-Cas3), which are widely used as indicators of apoptosis [66,67]. Cotreatment of cisplatin and siSurvivin induced more cleavage of both indicator proteins with the same trends observed between EGFR-targeted and nontargeted formulation (Fig. 5C). The results above validated the siSurvivin delivery by EGFRApt/Exo nanoparticle and showed the potential application for combination therapy with cisplatin.
FIG. 5.

Silencing of survivin suppressed tumor cell growth and sensitized cisplatin cytotoxicity in NSCLC Cells. (A) IncuCyte cell proliferation assay on both H596 and H1568 NSCLC cell lines posttreating with EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr, Exo/siSurvivin, and TES buffer in 50 nM of siRNA concentration. n = 3, error bar ± SEM. (B) Cell apoptosis evaluation by Annexin V-positive cells using flow cytometry on both H596 and H1568 NSCLC cell lines posttreating with EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr, Exo/siSurvivin, and TES buffer in 50 nM of siRNA concentration for 48 h followed by 24 h treatment with 5 μM cisplatin, n = 3, *P < 0.05, **P < 0.01, error bar ± SD. Original data and gating refer to Supplementary Fig. S4. (C) Cisplatin-induced apoptosis marker evaluation by western blot on both H596 and H1568 NSCLC cell lines posttreating with EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr,/Exo/siSurvivin, and TES buffer in 50 nM of siRNA concentration for 48 h followed by 24 h treatment with or without 5 μM cisplatin. SD, standard deviation; SEM, standard error of the mean.
In vivo lung cancer cell targeting, and antitumor efficacy of EGFRApt/Exo/siSurvivin using tumor xenograft mouse model
To determine the in vivo siRNA delivery effects of EGFRApt/Exo/siSurvivin, we established nude mouse tumor xenografts with H596 cells. Live animal imaging demonstrated that Alexa750-conjugated EGFRApt/Exo/siSurvivin nanoparticles strongly accumulated in the xenograft tumor 24–48 h after tail vein injection of exosomes (Fig. 6A, Left). Further ex vivo biodistribution analysis confirmed that EGFR aptamer facilitated exosome accumulation in the xenograft tumors (Fig. 6A, Right). To optimize the best dose of exosomes for in vivo treatment, serial doses of EGFRApt/Exo/siSurvivin were intravenous (I.V.) administrated every 2 days four times over 8 days, and tumors were isolated for extraction of total RNA and protein (Fig. 6B). As shown in Fig. 6C and D, survivin RNA and protein expression was suppressed in a concentration-dependent manner. Forty nanomole per kilogram dose has been shown to be the most effective dose for in vivo silencing of survivin expression. We chose this dose to compare the antitumor efficacy along with all different control groups. Finally, we found that treatment of tumor-bearing mice with EGFRApt/Exo/siSurvivin led to marked tumor growth regression, compared with the other treatment groups (Fig. 6E).
FIG. 6.

In vivo evaluation of EGFRApt/Exo/siSurvivin on NSCLC tumor xerograph mice. (A) Biodistribution study by tracing Alexa750-labeled siSurvivin delivered by exosomes. H596 NSCLC cell-developed tumor xenograft mice were I.V. administrated with 100 μL of 20 μM of RNA Alexa750-labeled siSurvivin loaded in exosomes or EGFRApt/Exo and imaging by IVIS post 1, 4, 8, 24, 32, and 48 h shown on the left. Organ was dissected at 48 h and imaged by IVIS shown on the right. (B) Scheme of exosome injection and tumor isolation time frame for dose optimization study. (C) mRNA level of survivin gene in tumor quantified by qRT-PCR assay post three repeated I.V. administration of EGFRApt/Exo/siSurvivin in 4, 10, 20, and 40 nmol/kg of siRNA concentration and TES buffer on H596 NSCLC cell-developed tumor xenograft mice, n = 3, error bar ± SD. Protein level assayed by western blot shown in (D). (E) Tumor growth curve traced during six repeated I.V. administration of EGFRApt/Exo/siSurvivin, EGFRApt/Exo/siCtr, Exo/siSurvivin, and TES buffer in 40 nmol/kg of siRNA concentration on H596 NSCLC cell-developed tumor xenograft mice. ***P < 0.001, ****P < 0.0001, two-way ANOVA (mix model repeated measurement), n = 5, error bar ± SEM. ANOVA, analysis of variance; I.V., intravenus; IVIS, in vivo imaging system; ns, no significance.
Discussion
Given the success in treating liver-associated disease by siRNA-based therapies, the exploration of nonliver therapeutic areas is of high interest. In this study, we report a novel strategy of delivering siRNA specific to NSCLC tumor, leveraging the advantage of exosomes and RNA nanotechnology, and demonstrating the antitumor effect on a xenograft mice model. With the engineerable arrow-tail RNA nanoparticle platform we developed [16,17,63], we expand the pipeline of ligand-displaying exosomes as vehicles for delivering anticancer siRNA on lung cancer treatment, meanwhile, demonstrating again the flexibility of this platform technology. Notably, with the combination of hollow-fiber bioreactor, TFF system, and cushion ultracentrifugation, we increased the scale of exosome production over 80 times, demonstrating the efficiency of this technique (data not shown). This new exosome production workflow not only expands our bandwidth to pursue comprehensive studies in animals, but introducing these processes that are widely used in industrial manufacturing also support the potential for further scaling up for additional animal or human studies.
The display of EGFRApt ligands on exosomes shows obvious enhancement on cellular uptake, and gene knockdown in our in vitro experiments. It is also noticed that the HEK293T cell-derived exosomes had passive delivery capacity in both in vitro and in vivo settings, which is consistent with both of research previously published by this team, and exosome uptake published by peers [47,48,57,59,63,68–72]. Nevertheless, it is observed that more tumor accumulation and higher potency of EGFRApt/Exo/siSurvivin compared with nontargeting Exo/siSurvivin in the mouse model (Fig. 6A–E). The enhancement caused by EGFR targeting is favorable for consideration of dose reduction. Moreover, the in vivo gene knockdown assessment among different doses shown in Fig. 6C and D indicates that EGFRApt/Exo/siSurvivin at 20 nmol/kg already reaches the <50% knockdown efficacy, while no significant improvement was observed by increasing the dose to 40 nmol/kg. These results might explain the potency of nontargeting exosome groups and indicate the possibility for reducing dosing for future study.
We also reported the proof-of-concept study of combination therapy by cotreating NSCLC cells with EGFRApt/Exo/siSurvivin and first-line chemotherapy reagent cisplatin (Fig. 5). The results clearly showed the potential of this combination strategy for either reducing the doses of the toxic chemotherapy or enhancing the treatment of patients that have developed resistance to chemotherapy, both highly clinically relevant and translatable options. Furthermore, exosomes had been reported to improve the delivery of chemotherapy reagents like Taxol [73,74], which raises the possibility of coencapsulating the siRNA and cisplatin into our EGFRApt/Exo system. Taken together, these results demonstrate continued advances in combining RNA nanotechnology with the exosome delivery platform for enhanced targeting of cancer therapy.
Supplementary Material
Data and Materials Availability
All data generated or analyzed during this study can be found in the article or supplementary materials.
Author Disclosure Statement
P.G. is the consultant of Oxford Nanopore Technologies; the cofounder of Shenzhen P&Z Biomedical Co. Ltd., as well as the cofounder of ExonanoRNA, LLC and its subsidiary Weina Biomedical (Guangdong), Ltd.
Funding Information
The research was supported by the NIH grants U01CA207946 and R01EB019036 to P.G.; U01CA151648 to P.G.; and ACS RSG-17-221-01-TBG to T.M.W. and P.G. P.G.'s Sylvan G. Frank Endowed Chair position in Pharmaceutics and Drug Delivery is funded by the CM Chen Foundation. The work involving flow cytometry and confocal microscopy was supported by OSU's Comprehensive Cancer Center (CCC) Shared Resources. This facility is supported in part by grant P30 CA016058, National Cancer Institute.
Supplementary Material
References
- 1. Molina JR, Yang P, Cassivi SD, Schild SE and Adjei AA. (2008). Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 83:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duma N, Santana-Davila R and Molina JR. (2019). Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc 94:1623–1640. [DOI] [PubMed] [Google Scholar]
- 3. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, et al. (2011). Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12:735–742. [DOI] [PubMed] [Google Scholar]
- 4. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362:2380–2388. [DOI] [PubMed] [Google Scholar]
- 5. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, et al. (2018). Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 378:113–125. [DOI] [PubMed] [Google Scholar]
- 6. Di Costanzo F, Mazzoni F, Micol Mela M, Antonuzzo L, Checcacci D, Saggese M and Di Costanzo F. (2008). Bevacizumab in non-small cell lung cancer. Drugs 68:737–746. [DOI] [PubMed] [Google Scholar]
- 7. Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT, et al. (2009). Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 373:1525–1531. [DOI] [PubMed] [Google Scholar]
- 8. Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, Vynnychenko I, Park K, Yu CT, et al. (2015). Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol 16:763–774. [DOI] [PubMed] [Google Scholar]
- 9. Esposito CL, Passaro D, Longobardo I, Condorelli G, Marotta P, Affuso A, de Franciscis V and Cerchia. L (2011). A neutralizing RNA aptamer against EGFR causes selective apoptotic cell death. PLoS One 6:e24071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shu D, Li H, Shu Y, Xiong G, Carson WE, 3rd, Haque F, Xu R and Guo P. (2015). Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano 9:9731–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim MW, Jeong HY, Kang SJ, Jeong IH, Choi MJ, You YM, Im CS, Song IH, Lee TS, et al. (2019). Anti-EGF receptor aptamer-guided co-delivery of anti-cancer siRNAs and quantum dots for theranostics of triple-negative breast cancer. Theranostics 9:837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okamoto K, Okamoto I, Hatashita E, Kuwata K, Yamaguchi H, Kita A, Yamanaka K, Ono M and Nakagawa K. (2012). Overcoming erlotinib resistance in EGFR mutation-positive non-small cell lung cancer cells by targeting survivin. Mol Cancer Ther 11:204–213. [DOI] [PubMed] [Google Scholar]
- 13. Okamoto K, Okamoto I, Okamoto W, Tanaka K, Takezawa K, Kuwata K, Yamaguchi H, Nishio K and Nakagawa K. (2010). Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res 70:10402–10410. [DOI] [PubMed] [Google Scholar]
- 14. Wheatley SP and Altieri DC. (2019). Survivin at a glance. J Cell Sci 132:jcs223826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li F. (2013). Discovery of survivin inhibitors and beyond: FL118 as a proof of concept. Int Rev Cell Mol Biol 305:217–252. [DOI] [PubMed] [Google Scholar]
- 16. Pi F, Binzel DW, Lee TJ, Li Z, Sun M, Rychahou P, Li H, Haque F, Wang S, et al. (2018). Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat Nanotechnol 13:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z, Wang H, Yin H, Bennett C, Zhang HG and Guo. P (2018). Arrowtail RNA for ligand display on ginger exosome-like nanovesicles to systemic deliver siRNA for cancer suppression. Sci Rep 8:14644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE and Mello CC. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. [DOI] [PubMed] [Google Scholar]
- 19. Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ and Carthew RW. (2004). Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117:69–81. [DOI] [PubMed] [Google Scholar]
- 20. Tiemann K and Rossi JJ. (2009). RNAi-based therapeutics-current status, challenges and prospects. EMBO Mol Med 1:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krieg AM. (2011). Is RNAi dead? Mol Ther 19:1001–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo PX, Erickson S and Anderson D. (1987). A small viral RNA is required for in vitro packaging of bacteriophage phi 29 DNA. Science 236:690–694. [DOI] [PubMed] [Google Scholar]
- 23. Guo P, Zhang C, Chen C, Trottier M and Garver K. (1998). Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol Cell 2:149–155. [DOI] [PubMed] [Google Scholar]
- 24. Shu D, Shu Y, Haque F, Abdelmawla S and Guo P. (2011). Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat Nanotechnol 6:658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shu Y, Pi F, Sharma A, Rajabi M, Haque F, Shu D, Leggas M, Evers BM and Guo P. (2014). Stable RNA nanoparticles as potential new generation drugs for cancer therapy. Adv Drug Deliv Rev 66C:74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shu D, Khisamutdinov EF, Zhang L and Guo. P (2014). Programmable folding of fusion RNA in vivo and in vitro driven by pRNA 3WJ motif of phi29 DNA packaging motor. Nucleic Acids Res 42:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shu Y, Shu D, Haque F and Guo P. (2013). Fabrication of pRNA nanoparticles to deliver therapeutic RNAs and bioactive compounds into tumor cells. Nat Protoc 8:1635–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khisamutdinov EF, Jasinski DL and Guo P. (2014). RNA as a boiling-resistant anionic polymer material to build robust structures with defined shape and stoichiometry. ACS Nano 8:4771–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khisamutdinov EF, Li H, Jasinski DL, Chen J, Fu J and Guo P. (2014). Enhancing immunomodulation on innate immunity by shape transition among RNA triangle, square and pentagon nanovehicles. Nucleic Acids Res 42:9996–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piao X, Wang H, Binzel DW and Guo P. (2018). Assessment and comparison of thermal stability of phosphorothioate-DNA, DNA, RNA, 2'-F RNA, and LNA in the context of Phi29 pRNA 3WJ. RNA 24:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo S, Li H, Ma M, Fu J, Dong Y and Guo P. (2017). Size, shape, and sequence-dependent immunogenicity of RNA nanoparticles. Mol Ther Nucleic Acids 9:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo S, Xu C, Yin H, Hill J, Pi F and Guo. P (2020). Tuning the size, shape and structure of RNA nanoparticles for favorable cancer targeting and immunostimulation. Wiley Interdiscip Rev Nanomed Nanobiotechnol 12:e1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ke W, Hong E, Saito RF, Rangel MC, Wang J, Viard M, Richardson M, Khisamutdinov EF, Panigaj M, et al. (2019). RNA-DNA fibers and polygons with controlled immunorecognition activate RNAi, FRET and transcriptional regulation of NF-kappaB in human cells. Nucleic Acids Res 47:1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong E, Halman JR, Shah AB, Khisamutdinov EF, Dobrovolskaia MA and Afonin KA. (2018). Structure and composition define immunorecognition of nucleic acid nanoparticles. Nano Lett 18:4309–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maeda H, Nakamura H and Fang J. (2013). The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev 65:71–79. [DOI] [PubMed] [Google Scholar]
- 36. Yin H, Wang H, Li Z, Shu D and Guo P. (2019). RNA micelles for systemic delivery of anti-miRNA for cancer targeting and inhibition without ligand. ACS Nano 13:706–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jasinski DL, Yin H, Li Z and Guo P. (2018). Hydrophobic effect from conjugated chemicals or drugs on in vivo biodistribution of RNA nanoparticles. Hum Gene Ther 29:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jasinski DL, Li H and Guo P. (2018). The effect of size and shape of RNA nanoparticles on biodistribution. Mol Ther 26:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Binzel DW, Shu Y, Li H, Sun M, Zhang Q, Shu D, Guo B and Guo P. (2016). Specific delivery of MiRNA for high efficient inhibition of prostate cancer by RNA nanotechnology. Mol Ther 24:1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin H, Xiong G, Guo S, Xu C, Xu R, Guo P and Shu D. (2019). Delivery of anti-miRNA for triple-negative breast cancer therapy using RNA nanoparticles targeting stem cell marker CD133. Mol Ther 27:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haque F, Shu D, Shu Y, Shlyakhtenko LS, Rychahou PG, Evers BM and Guo P. (2012). Ultrastable synergistic tetravalent RNA nanoparticles for targeting to cancers. Nano Today 7:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shu Y, Haque F, Shu D, Li W, Zhu Z, Kotb M, Lyubchenko Y and Guo P. (2013). Fabrication of 14 different RNA nanoparticles for specific tumor targeting without accumulation in normal organs. RNA 19:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abdelmawla S, Guo S, Zhang L, Pulukuri SM, Patankar P, Conley P, Trebley J, Guo P and Li QX. (2011). Pharmacological characterization of chemically synthesized monomeric phi29 pRNA nanoparticles for systemic delivery. Mol Ther 19:1312–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Batrakova EV and Kim MS. (2015). Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release 219:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. El-Andaloussi S, Lakhal S, Mager I and Wood MJ. (2013). Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev 65:391–397. [DOI] [PubMed] [Google Scholar]
- 46. Sun D, Zhuang X, Zhang S, Deng ZB, Grizzle W, Miller D and Zhang HG. (2013). Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv Drug Deliv Rev 65:342–347. [DOI] [PubMed] [Google Scholar]
- 47. varez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S and Wood MJ. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 29:341–345. [DOI] [PubMed] [Google Scholar]
- 48. Mathieu M, Martin-Jaular L, Lavieu G and Thery C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 21:9–17. [DOI] [PubMed] [Google Scholar]
- 49. EL Andaloussi S, I Mager, XO Breakefield and MJ Wood. (2013). Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12:347–357. [DOI] [PubMed] [Google Scholar]
- 50. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, et al. (2013). Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 21:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV and Filatov. MV (2013). Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ and Schiffelers RM. (2012). Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release 161:635–644. [DOI] [PubMed] [Google Scholar]
- 53. Johnstone RM, Mathew A, Mason AB and Teng K. (1991). Exosome formation during maturation of mammalian and avian reticulocytes: evidence that exosome release is a major route for externalization of obsolete membrane proteins. J Cell Physiol 147:27–36. [DOI] [PubMed] [Google Scholar]
- 54. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A and Rak J. (2008). Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10:619–624. [DOI] [PubMed] [Google Scholar]
- 55. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ and Lotvall JO. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. [DOI] [PubMed] [Google Scholar]
- 56. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM and Breakefield XO. (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Didiot MC, Hall LM, Coles AH, Haraszti RA, Godinho BM, Chase K, Sapp E, Ly S, Alterman JF, et al. (2016). Exosome-mediated delivery of hydrophobically modified siRNA for Huntingtin mRNA silencing. Mol Ther 24:1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL and Wood MJ. (2012). Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc 7:2112–2126. [DOI] [PubMed] [Google Scholar]
- 59. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ and Kalluri R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dreyer F and Baur A. (2016). Biogenesis and functions of exosomes and extracellular vesicles. Methods Mol Biol 1448:201–216. [DOI] [PubMed] [Google Scholar]
- 61. Zhang D, Lee H, Zhu Z, Minhas JK and Jin Y. (2017). Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 312:L110–L121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nordmeier S, Ke W, Afonin KA and Portnoy. V (2020). Exosome mediated delivery of functional nucleic acid nanoparticles (NANPs). Nanomedicine 30:102285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zheng Z, Li Z, Xu C, Guo B and Guo P. (2019). Folate-displaying exosome mediated cytosolic delivery of siRNA avoiding endosome trapping. J Control Release 311–312:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ueno T, Uehara S, Nakahata K and Okuyama H. (2016). Survivin selective inhibitor YM155 promotes cisplatin induced apoptosis in embryonal rhabdomyosarcoma. Int J Oncol 48:1847–1854. [DOI] [PubMed] [Google Scholar]
- 65. Hu W, Jin P and Liu W. (2016). Periostin contributes to cisplatin resistance in human non-small cell lung cancer A549 cells via activation of Stat3 and Akt and upregulation of survivin. Cell Physiol Biochem 38:1199–1208. [DOI] [PubMed] [Google Scholar]
- 66. Mullen P. (2004). PARP cleavage as a means of assessing apoptosis. Methods Mol Med 88:171–181. [DOI] [PubMed] [Google Scholar]
- 67. Henkels KM and Turchi JJ. (1999). Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res 59:3077–3083. [PubMed] [Google Scholar]
- 68. O'Brien K, Breyne K, Ughetto S, Laurent LC and Breakefield XO. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 21:585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Batrakova EV and Kim MS. (2015). Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release 219:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ha D, Yang N and Nadithe V. (2016). Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B 6:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lakhal S and Wood MJ. (2011). Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays 33:737–741. [DOI] [PubMed] [Google Scholar]
- 72. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV and Filatov. MV (2013). Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Guo S, Vieweger M, Zhang K, Yin H, Wang H, Li X, Li S, Hu S, Sparreboom A, et al. (2020). Ultra-thermostable RNA nanoparticles for solubilizing and high-yield loading of paclitaxel for breast cancer therapy. Nat Commun 11:972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Piao X, Yin H, Guo S, Wang H and Guo. P (2019). RNA nanotechnology to solubilize hydrophobic antitumor drug for targeted delivery. Adv Sci (Weinh) 6:1900951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.