

Abstract
The 78 kilodalton glucose-regulated protein (GRP78) is a major endoplasmic reticulum (ER) molecular chaperone with antiapoptotic properties and a key regulator of the unfolded protein response (UPR). ER-stress induction of GRP78 in cancer cells represents a major pro-survival branch of the UPR. Pancreatic ductal adenocarcinoma (PDAC) remains a highly lethal disease and high level of GRP78 is associated with aggressive disease and poor survival. Recently, we reported that PDAC exhibited high level of ER stress and that GRP78 haploinsufficiency is sufficient to suppress pancreatic tumorigenesis in mice, suggesting the utility of inhibitors of GRP78 expression in combating pancreatic cancer. Screening of clinically relevant compound libraries revealed that cardiac glycosides (CGs) can inhibit ER-stress induction of GRP78 in pancreatic and other types of human cancers. Using the FDA-approved CG compound Lanatoside C (LanC) and human pancreatic cancer cell lines as model systems, we discovered that LanC preferably suppressed ER stress induction of GRP78 and to a lesser extent GRP94. The suppression is at the post-transcriptional level and dependent on the Na+/K+-ATPase ion pump. Overexpression of GRP78 mitigates apoptotic activities of LanC in ER stressed cells. Our study revealed a new function of CGs as inhibitor of stress induction of GRP78, and that this suppression at least in part contributes to the apoptotic activities of CGs in human pancreatic cancer cells in vitro. These findings support further investigation into CGs as potential antineoplastic agents for pancreatic and other cancers which depend on GRP78 for growth and survival.
Keywords: GRP78, BiP, ER-stress, Cardiac Glycosides, Lanatoside C, Pancreatic Cancer
Abbreviations: ER, Endoplasmic Reticulum; GRP78, 78-kilodalton glucose-regulated protein; UPR, Unfolded Protein Response; PERK, protein kinase R(PKR)-like endoplasmic reticulum kinase; IRE-1, inositol-requiring enzyme 1; ATF6, activating transcription factor 6; CG, cardiac glycoside; LanC, Lanatoside C; Na+/K+-ATPase, Sodium Potassium ATPase; ROS, reactive oxygen species; NF-κB, Nuclear factor-kappa B; Tg, thapsigargin; Tu, tunicamycin; 2-DG, 2-deoxy-D-glucose; 3-MA, 3-methyladenine; GRP94, Glucose-regulated protein 94; HSP70, 70-kilodalton heat shock protein; PDI, protein disulfide isomerase
Introduction
The endoplasmic reticulum (ER) is a major site of synthesis, folding, and maturation for membrane and secreted proteins as well as the main calcium ion storage inside the cell [1], [2], [3]. To maintain ER homeostasis, the ER houses a diverse repertoire of molecular chaperones to aid in the folding process and/or direct the degradation of misfolded proteins [4,5]. When the folding capacity of the ER is overwhelmed due to increased protein synthesis or excessive amount of misfolded proteins, the cell is subjected to ER-stress which activates an adaptive program called the Unfolded Protein Response (UPR) [1,2,6]. The UPR is a complex network of signaling pathways consisting of three branches (PERK, IRE1, and ATF6) aiming at restoring ER homeostasis or triggering apoptosis depending on context, duration, and intensity of the stress [6], [7]. Among the ER chaperones, the 78 kDa glucose-regulated protein (GRP78), also referred to as BiP/HSPA5, is one of the most abundant and well-studied [8]. It acts as the master regulator of the UPR by binding to and inhibiting the three main ER-stress sensors PERK, IRE1, and ATF6 [2,6]. Upon ER-stress, GRP78 is titrated away by unfolded proteins, thus relieving the inhibition of the transmembrane ER stress sensors (PERK, IRE1 and ATF6) allowing them to activate simultaneous signaling cascades to attenuate protein translation [9] and up-regulate the expression of more ER chaperones to help resolve the stress [6].
Cancer cells are subjected to intrinsic stress due to rapid and uncontrolled proliferation of mutant cells, which requires increased protein synthesis, and coupling with extrinsic stress such as hypoxia and nutrient deprivation due to insufficient vascularization, leaving those cells in a sustained state of ER stress [3,[10], [11], [12]. To gain survival advantage, cancer cells exploit the adaptive aspects of the UPR to enhance their survival such as the up-regulation of ER chaperones to augment folding capacity and homeostasis of the ER [1,2]. Indeed, GRP78 up-regulation has been reported in a wide variety of human cancers and well established to play critical roles in cancer growth in vitro and in vivo, as well as therapeutic resistance [2,[13], [14], [15]. For instance, genetic mouse models with heterozygous deletion or conditional knockout of GRP78 in specific tissues have demonstrated that GRP78 deficiency can arrest cancer progression or suppress tumorigenesis in leukemia, breast, prostate, pancreatic, and lung cancers [2,12,16,17]. Furthermore, a subfraction of GRP78 can translocate from the ER to the surface of stressed cancer cells and engage in many signaling pathways important for cancer and viral infection [18], [19], [20], [21], [22], [23], [24], [25], [26]. Targeting cell surface GRP78 with antibodies can suppress the growth in xenograft models of colon, lung, prostate, breast cancers and melanoma [2,[27], [28], [29]. Therefore, developing therapies against total and surface GRP78 represents a novel and promising approach to combat cancer.
Pancreatic ductal adenocarcinoma (PDAC) remains one of the deadliest cancers with limited therapeutic options and an overall 5-year survival rate of <10%. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer [30]. Utilizing the Pdx1-Cre; Kras G12D/+; p53f/+ (PKC) mouse model, we recently discovered that GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia and pancreatic tumorigenesis [12]. It is notable that pancreatic cancer cells could experience a high level of ER stress due to increased proliferation driven by oncogene activation and desmoplasia leading to tissue hypovascularity. Consistent with this notion, we observed that in PDAC of the PKC model, GRP78 is up-regulated, along with markers of ER stress. Thus, identification of agents that could suppress ER-stress induction of GRP78 in pancreatic cancer cells will offer a novel therapeutic option towards this highly lethal disease.
CGs have been used as herbal medicine for treatment of heart disorders for decades [31]. Among the CGs, Digoxin and Lanatoside C (LanC) are FDA-approved drugs to treat heart failure and atrial fibrillation [31,32]. The primary mechanism of action of CGs is to inhibit the Na+/K+-ATPase pump leading to an increase in intracellular Ca2+ which decreases the heart rate and increases cardiac contractility [31], [32], [33]. Subsequently, evidence has accumulated that CGs can suppress cancer growth in vitro and in vivo [31,32,34,35] and retrospective studies on cancer patients who received CGs along with conventional cancer therapies showed a significantly enhanced overall survival compared to those who received just the cancer therapies alone [36,37]. While diverse mechanisms for the antitumor effects of CGs have been reported including increase in ROS production, inhibition of NF-κB signaling, increase FasL expression and up-regulation of DR4/DR5 [32,38], additional aspects of their anticancer activity remain an active area of research. Here we report that a novel mechanism for anti-cancer activities of CGs, as exemplified by LanC, is suppression of stress induction of GRP78, which represents a critical pro-survival UPR branch in cancer. These findings support further investigation into CGs as potential antineoplastic agents for pancreatic and other deadly cancers which depend on GRP78 for growth and resistance.
Materials and Methods
Cell Culture
Human pancreatic cancer PANC-1, breast cancer MCF-7, cervical cancer HeLa, melanoma SK-MEL-28, squamous cell carcinoma SCC-25, mouse embryonic fibroblast, and mouse acinar pancreatic cancer 266-6 cell lines were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; GeminiBio, West Sacramento, CA) and 1% penicillin/streptomycin (pen/strep) (Corning Inc., Glendale, AZ). Human pancreatic adenocarcinoma cell line CFPAC-1 was cultured in Iscove's Modified Dulbecco's Medium (IMDM) containing 10% FBS and 1% pen/strep. Human lung adenocarcinoma cell line A549 was cultured in Kaighn's Modification of Ham's F-12 (F-12K) medium containing 10% FBS and 1% pen/strep. Human colon cancer cell line HCT116 was cultured in McCoy's 5A medium containing 10% FBS and 1% pen/strep. Human prostate cancer cell line C4-2B was cultured in RPMI-1640 medium containing 10% FBS and 1% pen/strep. Human endometrial adenocarcinoma cell line Ishikawa was cultured in Minimum Essential Media containing 2mM Glutamine, 1% Non-Essential Amino Acids, 10% FBS and 1% pen/strep. All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Mouse acinar pancreatic cancer 266-6 cell line was kindly provided by Dr. Hamid M. Said (University of California, Irvine).
Treatment of drug compounds and small molecule inhibitors
The compound libraries used in the drug screen, their sources and drug screen are described in Supplemental information. Lanatoside C (LanC), Digoxin, Bufalin, Chloroquine, 3- Methyladenine (3-MA) were purchased from Millipore Sigma (St. Louis, MO). The proteasome inhibitors MG101, MG115, and MG132 were purchased from Selleck Chemicals (Houston, TX). The ER-stress inducers thapsigargin (Tg), tunicamycin (Tu), and 2-deoxy-D-glucose (2-DG), the autophagy inhibitor Bafilomycin A1, and the calcium chelator BAPTA-AM were purchased from Cayman Chemical (Ann Arbor, MI). All drugs were dissolved in DMSO except for 3-MA and 2-DG which were dissolved in sterile double distilled water. For ER-stress induction, the cells were treated with Tg at 300nM, Tu at 1.78μM, or 2-DG at 10mM for 24 hr. For combination treatment with cardiac glycosides, the cells were treated with the indicated ER-stress inducers and the indicated cardiac glycosides at the indicated dose at the same time and incubated for 24 hr. For combination treatment with the proteasome inhibitors, the cells were treated with MG101 (10μM), MG115 (10μM), and MG132 (10μM) concurrently with Tg and LanC for 24 hr. For combination treatment with the autophagy inhibitors, the cells were treated with 3-MA (10mM), Chloroquine (20μM), and Bafilomycin A1 (100nM) with Tg and LanC for 24 hr. For calcium sequestration, the cells were treated with BAPTA-AM (5μM, 10μM, 20μM) with Tg and LanC for 24 hr. DMSO was used as vehicle control for all drug treatment experiments.
Hypoxia Induction and Glucose Starvation
For hypoxia induction, we utilized the Galaxy 48r Incubator CO2/O2/N2 (Eppendorf, Hamburg Germany). The chamber was set at 0.1% O2 and 5% CO2. Regular culture medium (DMEM 10% FBS 1% pen/strep) was incubated at 0.1% O2 and 5% CO2 for 48 hr before the start of the experiment to decrease the oxygen concentration in the medium. This is the “equilibrated medium”. PANC-1 cells were seed at 500,000 cells per 6cm dish and allowed to attached overnight. Next day, the cells were switched to the “equilibrated medium” prepared above and treated with DMSO vehicle control or 1μM of LanC and incubated at 0.1% O2 and 5% CO2 for 24 hr. An identical set of cells were incubated at regular culture condition (20% O2 and 5% CO2). After 24 hr incubation period, cells were washed once with ice cold PBS and immediately lysed with RIPA buffer. Cell lysates were subjected to western blot analysis to detect the proteins of interest.
For Glucose starvation, PANC-1 were seeded at 500,000 cells per 6 cm dish and allowed to attach overnight. Next day, cells were wash twice with PBS to remove any trace of glucose and incubated in Glucose-free DMEM media supplemental with 1% pen/strep (Thermo Fisher Scientific, Cat# 11966025, Waltham, MA) and treated with DMSO vehicle control or LanC (0.5-1μM) for 24 hr. Cells were then washed with PBS and lysed with RIPA buffer. Cell lysates were subjected to western blot analysis to detect the proteins of interest.
Immunoblot Analysis
Cell lysates preparation and immunoblot analysis has been described in detail previously [39]. Proteins were electrophoresed in 8%, 10%, or 12% SDS-PAGE gels and probed with the following antibodies. The primary antibodies used in this study and their conditions are as followed: mouse anti-GRP78 (1:1000, BD Biosciences, San Jose, CA, 610979), rat anti-GRP94 (1:1000, Enzo Life Sciences, Farmingdale, NY, SPA-851), mouse anti-HSP70 (1:1000, Santa Cruz Biotechnology, Inc., Dallas, TX, sc-66048), rabbit anti-calnexin (1:2000, Enzo Life Sciences, ADI-SPA-860), rabbit anti-PDI (1:2000, Enzo Life Sciences, ADI-SPA-890), mouse anti-β-actin (1:5000, ProteinTech, Rosemont, IL, 66009-1-Ig), mouse anti-FLAG (1:2000, Millipore-Sigma, F1804), rabbit anti-cleaved PARP (Asp214) (1:1000, Cell Signaling, Danvers, MA, #5625), rabbit anti-cleaved Caspase-3 (Asp175) (1:1000, Cell Signaling, #9661), rabbit anti-cleaved Caspase-7 (Asp198) (1:1000, Cell Signaling, #8438). The secondary antibodies used in this study and their conditions are as followed: horseradish peroxidase (HRP) conjugated goat anti-mouse (sc-2005), goat anti-rabbit (sc-2004), and goat anti-rat (sc-2006) antibodies (1:1000, Santa Cruz Biotechnology, Inc.), mouse IgG1 binding protein conjugated to HRP (1:1000, Santa Cruz Biotechnology, Inc., sc-525408), mouse anti-rabbit IgG conjugated to HRP (1:1000, Santa Cruz Biotechnology, Inc., sc-2357), goat anti-mouse IRDye® 800CW (1:1000, LI-COR Biosciences), goat anti-rabbit IRDye® 680RD (1:1000, LI-COR Biosciences).
RNA Extraction and Reverse Transcription Quantitative Real-Time PCR
Cells were scraped from 60mm cell culture dishes and lysed with TRI reagent (Millipore-Sigma) and total RNA was extracted according to manufacturer's recommendation. RNA concentration and purity were measured by the NanoDrop 1000 UV Visible Spectrophotometers Machine (ThermoFisher, Waltham, MA). 1μg of total RNA was used to synthesize complementary DNA (cDNA) using the qScript cDNA SuperMix (QuantaBio, Beverly, MA) according to manufacturer's instructions. Quantitative real-time PCR analysis of GRP78 and β-actin cDNA was performed using KAPA SYBR® FAST qPCR Master Mix (Roche Sequencing and Life Science, Wilmington, MA) and detected by the Stratagene MX3000P Real-Time QPCR System (Agilent, Santa Clara, CA) with the following PCR conditions (40 cycles, 15s at 95°C, 15s at 55°C, 30s at 72°C). Melting curve analysis was performed to ascertain the specificity of the primers. One single peak was observed for each PCR product. The primers for human GRP78 are 5’-GGTGAAAGACCCCTGACAAA-3’ and 5’-GTCAGGCGATTCTGGTCATT-3’, for human β-actin are 5’-TCCCTGGAGAAGAGCTACGA-3’ and 5’-AGCACTGTGTTGGCGTACAG-3’.
WST-1 Cell Viability Assay
PANC-1 cells were seeded at a density of 10,000 cells per well in a 96-well plate and allowed to attach overnight. Next day, the cells were treated with Tg and LanC at the indicated concentrations and incubated for 24 hr. Cell viability was measured using the (4-[3-(4-Iodophenyl)-2-(4-nitro-phenyl)-2H-5-tetrazolio]-1,3-benzene sulfonate) WST-1 Roche Cell Proliferation Reagent (Millipore Sigma, Burlington, MA) following manufacturer's recommendations. Colorimetric readout was detected at 450nm wavelength and quantified using a Model 680 Microplate Reader (Bio-Rad Laboratories, Hercules, CA).
Flow Cytometry Analysis
PANC-1 cells were seeded at a density of 200,000 cells per well in a 6-well plate and allowed to attach overnight. Next day, the cells were treated with 1μM of LanC alone or in combination with Tg (300nM) and incubated for 24 hr. Next day, the cells were washed twice with PBS and incubated with PerCP-Cyanine5.5-conjugated Propidium Iodide staining solution for 15 min according to manufacturer's recommendation (BD Biosciences, Cat# 556547, San Jose, CA). Cells were then washed twice with PBS and incubated with PBS+10mM EDTA solution for 10 min at 37°C to detach. Detached cells were spun down at 1000g for 5 min and washed 5 times with DPBS supplemented with Calcium and Magnesium (Millipore Sigma, D8662, Burlington, MA) to completely remove EDTA and strained with BD Falcon 100μm Nylon Cell Strainer. Cells were then resuspended in Annexin V Binding Buffer and stain with FITC-conjugated Annexin V for 15 min according to manufacturer's protocol (BD Biosciences, Cat# 556547, San Jose, CA). Stained cells were analyzed by BD FACSVerseTM Cell Analyzer and BD FACSuiteTM Software (BD Biosciences, San Jose, CA)
Transfection of plasmid DNA expression vector
The construction of the expression plasmid for FLAG-GRP78 (F-GRP78) has been described previously [40]. The empty vector pcDNA3 or the expression vector for F-GRP78 were transfected into PANC-1 cells using the BioT Transfection Reagent (Bioland Scientific, Paramount, CA) following the manufacturer's recommendation. The cells were incubated with the transfection mix for 24 hr to allow for protein expression before the addition of Tg and LanC which were incubated for a further 24 hr before harvesting cells for immunoblot analysis of apoptotic markers.
TCGA Analysis
GRP78 gene expression analysis in human normal pancreas and pancreatic adenocarcinoma tissues is obtained from the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) [41]. The “normal tissues” include the matched adjacent normal pancreatic tissues from the same patients in the TCGA database in addition to normal tissue samples from unrelated donors from the Genotype-Tissue Expression (GTEx) database [42]
Statistical Analysis
All pair-wise comparisons were made using the one-tailed Student's t-test in Microsoft Excel. Data are presented as the mean ± Standard Error of the mean (S.E.M.). A p-value of ≤ 0.05 is considered statistically significant.
Results
ER-stress induction of GRP78 is suppressed by the cardiac glycoside Lanatoside C
Analysis of the Cancer Genome Atlas (TCGA) database by the GEPIA2 tool showed that GRP78 mRNA expression in human pancreatic adenocarcinoma is significantly higher than in normal pancreatic tissues (Fig. 1). In performing a high-throughput drug screening of clinically relevant compounds, we discovered that a class of compound called cardiac glycosides (CGs) can suppress ER-stress induced GRP78 expression. To study the effect of CG, as exemplified by LanC, in pancreatic cancer, we utilized two human pancreatic cancer cell lines (PANC-1 and CFPAC-1) treated with well-established pharmacological inducers of ER stress as model systems. First, we examined the effects of LanC on these human pancreatic cancer cell lines under non-stressed and ER stress conditions. To rule out potential bias by any one ER stress inducer and to establish the generality of our observations, we utilized three different ER stress inducers that can trigger ER stress by three distinct mechanisms. Thapsigargin (Tg), a classical ER stress inducer well-established in many studies, induces ER stress by blocking the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump which leads to depletion of the calcium storage in the ER resulting in ER stress. Another ER stress inducer is Tunicamycin (Tu) which blocks the N-linked glycosylation of proteins in the ER leading to misfolded proteins, ER stress, and activation of the UPR. The third ER stress inducer used in our study is 2-deoxy-D-glucose (2-DG) which is a glucose analog that acts as an inhibitor of glycolysis and mimics glucose starvation. Similar to Tu, 2-DG blocks protein glycosylation leading to accumulation of underglycosylated proteins and ER stress. Human pancreatic cancer cell lines PANC-1 and CFPAC-1 treated with Tg (300nM), Tu (1.78μM), or 2-DG (10mM) showed 2.5-to-3.5-fold increase of GRP78 expression after 24 hr (Fig. 2 A and B). In cells treated with Tg, Tu, or 2-DG in combination with LanC, the GRP78 protein level in the combination treatment was similar to that of untreated cells suggesting a nearly complete inhibition of ER-stress induced GRP78 expression (Fig. 2 A and B). For the basal expression of GRP78 under non-stressed condition, within 24 hr of LanC treatment, due to long half-life of GRP78 (>24 hr), the GRP78 levels were minimally affected, however, upon treatment for 48 hr, LanC is able to decrease GRP78 expression in a dosage-dependent manner (Supp. Fig. S1). To mimic the natural tumor microenvironment in which rapidly proliferating cancer cells are frequently under oxygen deprivation and glucose starvation conditions due to poor vascularization [2], we subjected PANC-1 cells to hypoxia (0.1% O2) or cultured the cells in glucose-free media and treated them with LanC concurrently for 24 hr. Under hypoxic culture condition, PANC-1 cells induced GRP78 protein expression by about 3-fold which was blunted in combination treatment with LanC (Fig. 2 C). Similarly, glucose starvation resulted in 4.5-fold increase in GRP78 protein level, and this increase was effectively inhibited in a dosage-dependent manner with LanC treatment (Fig. 2 D). Collectively, these results indicate that LanC is a suppressor of GRP78 expression in pancreatic cancer cells and capable in curbing induction of GRP78 under a variety of ER stress conditions induced by pharmacological agents or natural physiological settings commonly found in the tumor microenvironment.
Fig. 1.
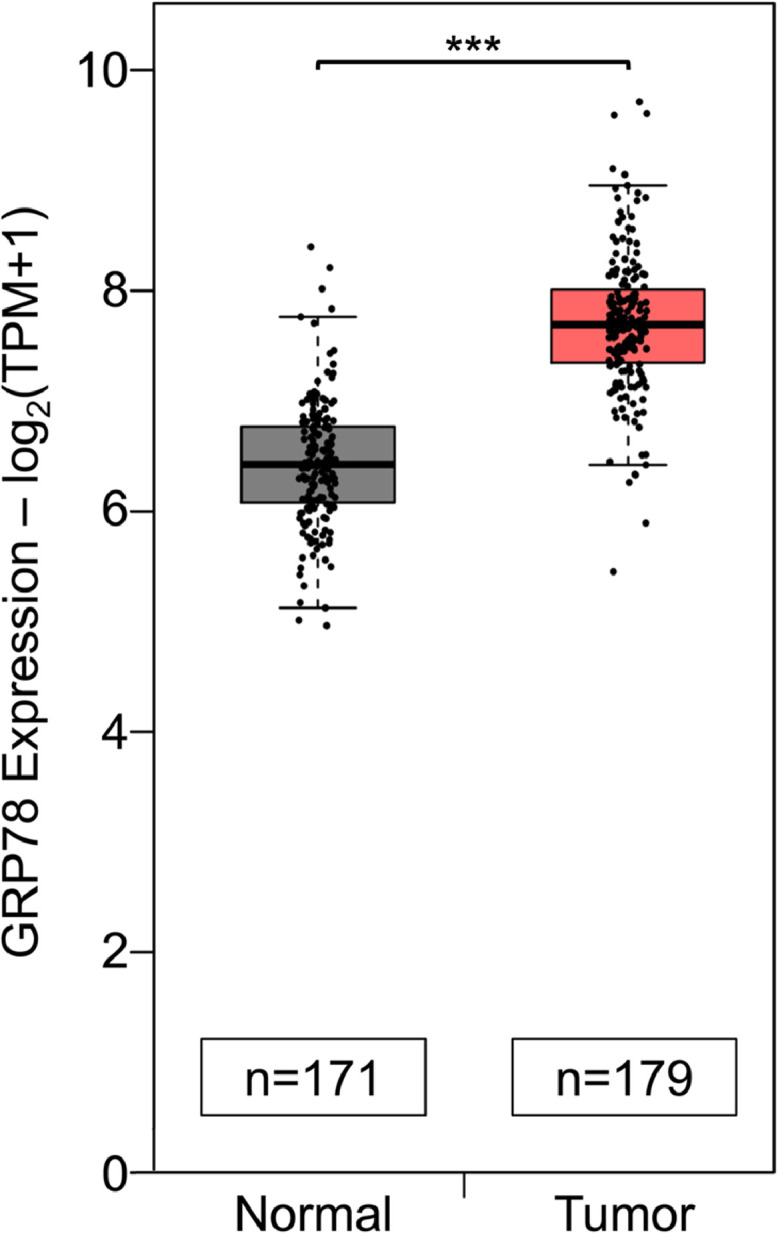
Comparative analysis of GRP78 mRNA expression in human pancreatic tissues. GRP78 mRNA expression in human normal pancreatic (n=171) and pancreatic adenocarcinoma tumor tissues (n=179) from TCGA and GTEx databases. Data analysis was performed by the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) tool. *** P ≤ 0.001 (Student's t test).
Fig. 2.

LanC inhibits stress induction of GRP78 protein expression by three distinct ER stress inducers in two different human pancreatic cancer cell lines. (A) PANC-1 cells were treated with 1μM of LanC alone or in combination with three different ER stress inducers Tg (300nM), Tu (1.78μM), or 2-DG (10mM) for 24 hr. Whole cell lysates (WCL) were subjected to western blot analysis for GRP78 protein level with β-actin serving as loading control. Quantitation of the relative protein level of GRP78 was shown in the graphs below. (B) Same as in (A) except human pancreatic adenocarcinoma cell line CFPAC-1 was used. (C) PANC-1 cells were cultured in normoxia (20% O2) or hypoxia (0.1% O2) conditions and treated with 1μM of LanC for 24 hr. WCL were subjected to western blot analysis for GRP78 protein level with β-actin as loading control. (D) Similar to (C) but PANC-1 cells were cultured in high glucose (4.5 g/l) or glucose-free (0 g/l) conditions and treated with 0.5-1μM of LanC for 24 hr. All drugs were added to the cells at the same time. Data are presented as mean ± S.E.M. * P ≤ 0.05, ** P ≤ 0.01 (Student's t test).
General inhibition of GRP78 stress induction by cardiac glycosides in human cancers
Next, we investigate whether there is a class effect of cardiac glycosides inhibition of ER-stress induced GRP78 expression by testing two other cardiac glycosides: Digoxin (Dig) and Bufalin (Buf). LanC and Digoxin belong to the sub-family of cardiac glycosides called cardenolides and Bufalin belongs to the other sub-family called bufadienolides which contains a six-membered lactone ring at the C-17 position. In contrast, cardenolides have a five-membered lactone ring at the same position [32]. As shown in Fig. 3 A, treatment of PANC-1 cells with Digoxin and Bufalin for 24 hr robustly suppressed ER stress induction of GRP78 expression similar to LanC. These results imply that cardiac glycosides, as a family of diverse molecules with very different structures, are effective in suppressing GRP78 up-regulation under ER stress.
Fig. 3.

General inhibition of GRP78 stress induction by cardiac glycosides in human cancers. (A) Human pancreatic cancer cell line PANC-1 was treated with 1μM of LanC, Digoxin (Dig), or Bufalin (Buf) alone or in combination with the ER stress inducer Tg (300nM) for 24 hours. Whole cell lysates were subjected to western blot analysis for GRP78 protein level with β-actin serving as loading control. Quantitation of the relative protein level of GRP78 was shown in the graphs below. Data are presented as mean ± S.E.M. * P ≤ 0.05, ** P ≤ 0.01 (Student's t test). (B) Human lung adenocarcinoma A549, squamous cell carcinoma SCC-25, pancreatic adenocarcinoma CFPAC-1, cervical cancer HeLa, melanoma SK-MEL-28, and breast cancer MCF-7 cell lines were treated with 1μM or 5μM of LanC alone or in combination with the ER stress inducer Tg (300nM) for 24 hr. Whole cell lysates were subjected to western blot analysis for GRP78 protein level with β-actin serving as loading control. All drugs were added to the cells at the same time.
Beyond pancreatic cancer cells, we next determined whether LanC has similar effects in other human cancers. We tested 5 additional human cancer cell lines representing different types of cancers including: A549 (Lung), SCC-25 (Head and neck), HeLa (Cervical), SK-MEL-28 (Skin), and MCF-7 (Breast) along with CFPAC-1 (Pancreas). Remarkably, LanC can suppress ER-stress induced GRP78 expression in all cancer cell lines being tested (Fig. 3 B). Collectively, these results indicate a class effect of cardiac glycosides in inhibiting GRP78 expression under ER stress and this effect is generally applicable among different cancer types.
The endoplasmic reticulum is a major site of protein synthesis and folding. As such, it contains a diverse repertoire of molecular chaperones to facilitate these protein folding activities with GRP78 being one of the major chaperones. Thus, it is important to examine the effect of LanC on other chaperones besides GRP78. To answer this question, PANC-1 cells were treated with increasing concentration of LanC from 0.1μM to 1μM alone or in combination with the ER stress inducer Tg (300nM) for 24 hr and whole cell lysates were analyzed by western blot. Notably, we observed that LanC can inhibit ER stress induction of GRP78 in a dosage-dependent manner, with an estimated IC50 value around 0.5μM (Fig. 4 A). GRP94 is an ER chaperone that is coordinately up-regulated with GRP78 under ER stress [2,43]. Treatment with increasing concentration of LanC under ER stress showed a trend of moderate suppression of GRP94 expression level and higher doses of LanC (0.75μM and 1μM) significantly inhibited GRP94 protein level (Fig. 4 B). HSP70 is closely related to GRP78 and is the cytosolic counterpart of GRP78 [2]. Treatment with increasing concentrations of LanC had no effect on the protein level of HSP70 either in non-stressed or ER stress conditions (Fig. 4 C). Similarly, LanC treatment under non-stressed or stressed conditions did not result in any changes in protein levels of two other ER chaperones calnexin and protein disulfide isomerase (PDI) (Fig. 4 C). Similar results were observed in another pancreatic cancer cell line CFPAC-1 (Supp. Fig. S2) and three other cancer cell lines from different types of cancer (colon, prostate and endometrial) (Supp. Fig. S3). Taken together, these results suggest that LanC treatment of cancer cells under ER stress mostly affects GRP78 and to a lesser extent GRP94 while having no significant impact on other chaperones.
Fig. 4.

LanC inhibits ER stress induction of GRP78 protein expression in a dose-dependent manner without significant effects on other chaperones in human pancreatic cancer cell line. PANC-1 cells were treated with increasing concentrations of LanC from 0.1μM to 1μM alone or in combination with the ER stress inducer Tg (300nM) for 24 hr. Whole cell lysates were subjected to western blot analysis for the (A) GRP78, (B) GRP94, (C) HSP70, Calnexin, and PDI protein levels with β-actin serving as loading control. Quantitation of the relative levels of the indicated proteins are shown in the graphs below. All drugs were added to the cells at the same time. Data are presented as mean ± S.E.M. * P ≤ 0.05 (Student's t test).
Inhibition of ER-stress induced GRP78 expression by LanC leads to enhanced apoptosis and reduced cell viability in human pancreatic cancer cells
As the master regular of the UPR and a major ER chaperone, GRP78 has potent anti-apoptotic effects [1,2,4] and suppression of GRP78 under ER stress could lead to cell death. Brightfield microscopy images of PANC-1 cells treated with LanC alone or in combination of ER stress inducer Tg for 24 hr revealed altered cell morphology especially in the combination treatment with more rounded up cells and presence of large vacuoles inside the cells as well as floating dead cells (Fig. 5 A). Beyond cell morphology, we examined the apoptotic effect of LanC in pancreatic cancer cells by probing for the biochemical markers of apoptosis activation such as the cleavage of PARP, Caspase-3, and Caspase-7, with the activation of Caspase-7 directly regulated by GRP78 upon ER stress [53]. Treatment with increasing concentration of LanC from 0.1μM to 1μM alone led to a dose-dependent increase in the cleavage of PARP, Caspase-3 and Caspase-7 (Fig. 5 B-D) indicating more apoptosis activation. Interestingly, combination treatment of LanC and the ER stress inducer Tg resulted in even more cleaved PARP, cleaved Caspase-3, and cleaved Caspase-7 expression levels compared to the same dose of LanC treatment alone (Fig. 5 B-D). To further verify the apoptotic effects of LanC treatment, we treated PANC-1 cells with 1μM of LanC alone or in combination with Tg (300nM) for 24 hr and performed flow cytometry analysis of these cells after staining with Propidium Iodide (PI) and Annexin V to measure apoptotic activation. We observed that 1μM of LanC treatment alone (25.21%) resulted in a marked increase in the numbers of cells stained positive for both PI and Annexin V which is indicative of late-stage apoptosis, as compared to DMSO treated cells (2.57%) (Fig. 5 E). Treatment with the ER-stress inducer Tg alone led to a moderate increase in the percentage of PI/Annexin V positive cells (8.37%). Importantly, combination treatment of LanC and Tg triggered a notable increase in the percentage of cells stained positive for both PI and Annexin V (39.59%) (Fig. 5 E). These results suggest that LanC treatment aggravates cell death under ER stress condition in human pancreatic cancer cells. Combination treatment of LanC and Tg also led to greatly increased apoptosis in another pancreatic cancer cell line CFPAC-1 (Fig. 5 F). In addition to biochemical markers of apoptosis, we also measured cell viability under LanC treatment by the WST-1 assay. In agreement with the biochemical markers, treatment with increasing concentrations of LanC alone led to a dose-dependent decrease in cell viability (Fig. 5 G). Combination treatment of LanC and Tg resulted in even more reduction in cell viability with the effect appeared to be additive rather than synergistic (Fig. 5 G). Taken together, these results indicate that inhibition of ER-stress induced GRP78 expression by LanC leads to enhanced apoptosis and reduced cell viability in human pancreatic cancer cells.
Fig. 5.

Treatment with LanC under ER stress condition exacerbated apoptosis and further reduced cell viability in human pancreatic cancer cells. (A) Brightfield microscopy images of PANC-1 cells treated with 1μM of LanC alone or in combination with the ER stress inducer Tg (300nM) for 24 hours. Pictures were taken at 10x magnification. Scale bar represents 20μm. (B) PANC-1 cells were treated with increasing concentrations of LanC from 0.1μM to 1μM alone or in combination with the ER stress inducer Tg (300nM) for 24 hr. Whole cell lysates were subjected to western blot analysis for cleaved PARP (C-PARP), cleaved Caspase-3 (C-Cas 3) and cleaved Caspase-7 (C-Cas 7) protein levels with β-actin serving as loading control. (C) Quantitation of the relative levels of cleaved PARP are shown. (D) Quantitation of the relative levels of cleaved Caspase-3 and cleaved Caspase-7 are shown. (E) PANC-1 cells were treated with 1μM of LanC alone or in combination with Tg (300nM) for 24 hr and subjected to flow cytometry analysis for Annexin V and Propidium Iodide staining. (F) Same as in (B) but CFPAC-1 cells were used. (G) Same as in (B) but cell viability was measured by WST-1 assay and the percentage of non-viable cells was quantified in the graph. All drugs were added to the cells at the same time. Data are presented as mean ± S.E.M. * P ≤ 0.05, ** P ≤ 0.01 (Student's t test).
LanC inhibition of ER-stress induction of GRP78 is at the post-transcriptional level and dependent on the Na+/K+-ATPase ion pump
The major mechanism to upregulate GRP78 expression under ER stress is through transcriptional activation to produce more GRP78 mRNA leading to increased protein level [44,45]. To examine whether LanC suppresses the ER-stress induced increase in GRP78 transcript level, we performed reverse transcription real-time quantitative PCR to quantify the amount of mRNA in pancreatic cancer cell line PANC-1. Treatment with increasing concentrations of LanC alone did not significantly alter GRP78 mRNA under non-stressed condition (Fig. 6 A). As expected, treatment with the ER-stress inducer Tg alone resulted in about 15-fold increase in GRP78 mRNA. Combination treatment of LanC and Tg did not reduce GRP78 mRNA (Fig. 6 A). This result indicated that LanC suppression of ER-stress induced GRP78 is at the post-transcriptional level.
Fig. 6.

LanC inhibition of GRP78 expression under ER stress condition is independent of transcription, proteasome degradation, autophagy induction, or calcium flux. (A) PANC-1 cells were treated with increasing concentrations of LanC from 0.1μM to 1μM alone or in combination with the ER stress inducer Tg (300nM) for 24 hr. Total RNA was extracted and the level of GRP78 mRNA was determined by reverse transcription quantitative real-time PCR. Relative level of the GRP78 mRNA is quantified and graphed with β-actin mRNA serving as loading control. (B) PANC-1 cells were either untreated, treated with the ER stress inducer Tg (300nM) alone or in combination with 1μM of LanC and increasing concentrations of the cell-permeable calcium chelator BAPTA-AM (5μM, 10μM, 20μM) for 24 hr. Whole cell lysates were subjected to western blot analysis for GRP78 protein level with β-actin serving as loading control. (C) Same as in (B) except the cells were either untreated, treated with 1μM of LanC alone or in combination with Tg and three different proteasome inhibitors MG101 (10μM), MG115 (10μM), and MG132 (10μM). (D) Same as in (C) except the cells were treated in combination with three different autophagy inhibitors 3-MA (10mM), Chloroquine (CQ, 20μM), and Bafilomycin A1 (BafA1, 100nM). All drugs and small molecules were added to the cells at the same time. Data are presented as mean ± S.E.M.
Cardiac glycosides have been known to cause an increase in cytosolic calcium level by blocking the Na+/K+-ATPase ion pumps on the cell surface [31,32,46,47]. Changes in the cytosolic calcium level can activate many signaling pathways and have wide-ranging effects on cellular activities [48]. To test whether this calcium flux is responsible for LanC effect on GRP78, we treated PANC-1 cells with a cell-permeable calcium chelator called BAPTA-AM. Treatment with increasing dose of BAPTA-AM (5μM, 10μM, 20μM) in concurrence with LanC and Tg did not restore the GRP78 protein level suppressed by LanC following Tg treatment (Fig. 6 B). Next, we examined the role of protein degradation in LanC suppression of GRP78 by utilizing three different inhibitors of proteases and proteasome activity. MG101 (also known as ALLN) is an inhibitor of cysteine proteases such as calpain as well as lysosomal proteases such as cathepsins. MG115 and MG132 are well-known inhibitors of the proteasome degradation complex. Treatment of MG101, MG115, and MG132 in conjunction with LanC and Tg combination did not restore the protein level of GRP78 suppressed by LanC (Fig. 6 C). Autophagy is another major process through which cells can eliminate dysfunctional cellular components or damaged proteins by encapsulating them in a double-membrane structure known as autophagosome which later fused with a lysosome to degrade the contents inside [49]. To block autophagy, we utilized three different autophagy inhibitors 3-methyladenine (3-MA), Chloroquine (CQ), and Bafilomycin A1 (BafA1). Concurrent treatment of 3-MA, CQ, or BafA1 with LanC and Tg combination in PANC-1 cells did not restore the GRP78 protein level suppressed by LanC (Fig. 6 D). Taken together, these results suggest that transcription, calcium flux, proteasome degradation and autophagy induction are likely not the underlying mechanisms for the suppression of GRP78 under ER stress by LanC.
The main therapeutic target of cardiac glycosides in patients with heart failure is the Na+/K+-ATPase ion pump on the cell surface [31,32]. Notably, the Na+/K+-ATPase ion pump in mouse is resistant to CG inhibition compared to the human Na+/K+-ATPase due to some amino acid differences in critical positions [31]. To test whether the Na+/K+-ATPase pumps are important for LanC suppressive effect on ER stress induction of GRP78, we utilized two different murine cell lines, one normal and one cancer, and examined their response to LanC treatment. Mouse embryonic fibroblasts and mouse acinar pancreatic cancer cells 266-6 were treated with 1μM of LanC alone or combination with three different ER stress inducers Tg, Tu, or 2-DG for 24 hr. Similar to human cancer cell lines, treatment with Tg, Tu, or 2-DG alone upregulated GRP78 expression. However, LanC treatment in combination with Tg, Tu, or 2-DG failed to suppress ER-stress induced GRP78 expression in these two mouse cell lines (Fig. 7 A and B). These results suggest that LanC suppression of ER stress induction of GRP78 in human cancer cells is dependent on its inhibitory effect on the Na+/K+-ATPase ion pump.
Fig. 7.

Murine cell lines are resistant to LanC inhibition of GRP78 expression under three different ER stress conditions. (A) Mouse embryonic fibroblasts (MEFs) were treated with 1μM of LanC alone or in combination with three different ER stress inducers Tg (300nM), Tu (1.78μM), or 2-DG (10mM) for 24 hr. Whole cell lysates were subjected to western blot analysis for GRP78 protein level with β-actin serving as loading control. (B) Same as in (A) except mouse acinar pancreatic cancer cells 266-6 were used. All drugs were added to the cells at the same time.
Overexpression of GRP78 mitigates apoptotic activation in LanC treated pancreatic cancer cells under ER stress
LanC has been reported to have anti-tumor activities in vitro through a variety of mechanisms. To test whether LanC suppression of GRP78 expression under ER stress conditions could be one key aspect of LanC anti-tumor activities in pancreatic cancer cells, we overexpressed GRP78 in PANC-1 by transiently transfecting a vector expressing a FLAG-tagged human GRP78. After a 24-hr period for the protein to be expressed, the cells were treated with 1μM of LanC alone or in combination with Tg for an additional 24 hr. Probing for GRP78 protein level confirmed that cells overexpressing GRP78 had about 2-3-fold more GRP78 protein compared to cells expressing empty vector pcDNA3 (Fig. 8 A). In cells transfected with empty vector pcDNA3, treatment with LanC alone resulted in some cell death as indicated by cleaved PARP and cleaved Caspase-3 (Fig. 8 B). In LanC and Tg combination treatment, there was a large increase in the expression level of cleaved PARP, cleaved Caspase-3, and cleaved Caspase-7 indicating a substantial increase in apoptotic activities (Fig. 8 B), consistent with our previous observations (Fig. 5 B-D). Importantly, in cells overexpressing GRP78 and treated with LanC and Tg, we observed a significant decrease of about 50% in the cleavage of these apoptotic markers (Fig. 8 B-D). Interestingly, cells overexpressing GRP78 and treated with LanC alone exhibited approximately similar level of apoptotic markers compared to cells expressing empty vector (Fig. 8 B-D). These results suggest that suppression of GRP78 under ER stress conditions is at least in part responsible for the apoptotic effects of LanC in human pancreatic cancer cells. The key findings of our studies are summarized in Fig. 9.
Fig. 8.

GRP78 overexpression alleviates LanC induced apoptotic activities in pancreatic cancer cells under ER stress condition. Human pancreatic cancer line PANC-1 was transfected with empty vector pcDNA3 or vector expressing FLAG-GRP78 for 24 hr. The cells were then treated with 1μM of LanC alone or in combination with the ER stress inducer Tg (300nM) for an additional 24 hr. (A) Whole cell lysates were subjected to western blot analysis for GRP78 and FLAG-GRP78 protein levels with β-actin serving as loading control. The relative protein level of GRP78 were quantified and shown below. (B) Same as in (A) except protein levels of cleaved PARP (C-PARP), cleaved Caspase-3 (C-Cas 3), and cleaved Caspase-7 (C-Cas 7) were probed with β-actin as loading control. The relative levels of the indicated proteins were quantified and shown in (C) and (D). All drugs were added to the cells at the same time. Data are presented as mean ± S.E.M. * P ≤ 0.05, ** P ≤ 0.01 (Student's t test).
Fig. 9.

Summary of the key findings. Under ER stress condition, cancer cells upregulate GRP78 to cope with the stress and enhance their survival. Treatment of LanC suppresses the induction of GRP78 protein level under stress conditions via inhibition of the Na+/K+-ATPase ion pump and deprives the cells of a key pro-survival molecule leading to increased cell death and reduced cell viability.
Discussion
GRP78 is a major ER chaperone and the major regulator of the UPR. As such, GRP78 has been widely reported to be up-regulated in many cancers and is critical for cancer cell survival, metastasis, and chemoresistant [1,2,4,14]. Thus, it is important to explore novel therapeutic approaches to target GRP78 in development of new anti-cancer therapies. As GRP78 protective effects on cancer involve not only the ER form, but also stress-induced cytosolic isoforms, secreted and cell surface forms [2,20,50], small molecule inhibitors of GRP78 expression have the advantage that they can simultaneously suppress the multiple forms of GRP78 induced under stress. To facilitate rapid translation into the clinic, we screened libraries of clinically relevant compounds and identified a class of compounds known as Cardiac Glycosides which exhibited inhibitory effects on ER-stress induced GRP78 expression in a wide range of human cancer cells. Two potential candidates from this family, Digoxin and LanC, are FDA-approved drugs to treat heart conditions in humans. In this proof-of-concept study, we focused on LanC and pancreatic cancer as model systems.
First, we found that LanC can effectively suppress GRP78 expression under ER stress triggered by distinct modes of action and that CGs action on GRP78 is a class effect since all three CGs tested can efficiently block GRP78 up-regulation under ER stress and this can be observed in a wide variety of human cancers in vitro. Interestingly, among the chaperone family of proteins, the effect of LanC suppression is most prominent with GRP78, with moderate reduction of GRP94, while having no effect on calnexin, PDI or HSP70, the closely related cytosolic counterpart of GRP78. One possible explanation is that GRP78 and GRP94 are up-regulated following ER stress, whereas these other chaperones are not. Thus, LanC may target proteins acutely induced by ER stress. We note that in our studies where we combined LanC treatment with ER stress inducers, a 24-hour treatment period was chosen since cells, as visualized by brightfield microscopy and analysis of apoptosis markers, showed considerable distress past this treatment period. Longer treatment period with LanC alone did result in a dosage-dependent decrease in GRP78 protein level since GRP78 is a stable protein with long half-life of over 24 hr [51,52], suggesting LanC is capable of reducing both the basal level and ER stress induction of GRP78. In probing for biochemical markers of apoptosis in these cells, we observed that LanC treatment in ER stressed cells showed enhanced activation of apoptosis, including caspase-7 which is regulated by GRP78 and is an indicator of ER-stress mediated onset of apoptosis [53]. These results were confirmed by FACS analysis which detected higher levels of apoptotic cells in ER-stressed cells treated with LanC.
Towards understanding how LanC can inhibit ER-stress induced GRP78 expression, we examined the effect of LanC on ER-stress induced GRP78 mRNA level and observed that LanC did not affect GRP78 mRNA level under non-stressed condition and even appeared to increase it under ER stress condition, suggesting that ER stress may be further exacerbated in these cells due to GRP78 suppression with UPR signaling intensify leading to more GRP78 transcription and this requires more investigation. We further determined that combination treatment with the cell permeable calcium chelator BAPTA-AM, proteasome or autophagy inhibitors did not reverse LanC suppression of GRP78 induction, suggesting that they are not potential mechanisms for LanC inhibition of ER stress induction of GRP78. On the other hand, our studies revealed that the ubiquitous Na+/K+-ATPase ion pump on the cell membrane may be involved. The Na+/K+-ATPase pump is a large heterotrimeric complex consisting of three subunits: α, β, and FXYD [31]. The α subunit is the main catalytic core of the complex with the binding sites for the ions, ATP, and cardiac glycosides [31]. Interestingly, there is a species-specific difference in the sensitivity of the Na+/K+-ATPase pump to cardiac glycoside inhibition. Murine Na+/K+-ATPase pump has several different amino acids in key positions on their α subunit compared to human Na+/K+-ATPase pump which render mouse cells resistant to CGs [31]. Our observation that LanC was not able to suppress ER-stress induced GRP78 expression in mouse cell lines provides an important mechanistic clue that CG effect on GRP78 involves inhibition of the Na+/K+-ATPase pump regardless of the precise molecular mechanism downstream. This observation can also explain the class effect of cardiac glycosides on GRP78 since all cardiac glycosides are able to inhibit the Na+/K+-ATPase pump which is their defining characteristic.
Since cardiac glycosides and LanC have been reported to affect cancer cell viability through a variety of mechanisms [54], [55], [56], [57], [58], [59], to establish the role of GRP78 in LanC anti-tumor activities, we enforced overexpression of GRP78 in pancreatic cancer cells prior to drug treatment to elevate GRP78 protein pool and our results confirmed that higher level of GRP78 protein can reduce the apoptotic effects of LanC under ER stress condition by about 50%, suggesting that GRP78 is a major contributor to protect against apoptosis mediated by the multifaceted anti-cancer activities of LanC. In humans and in vivo studies, it has been acknowledged that solid tumors are frequently under stress from a variety of factors such as rapid proliferation, inadequate vascularization, nutrient deprivation, low pH, hypoxia, or glucose starvation and they need GRP78 upregulation for their survival [1,2,11]. Thus, blocking GRP78 induction under these adverse conditions offers a new strategy to impede their growth and succumb to ER-stress induced cell death. Recently, a high throughput drug screen identified that the hydroxyquinoline analogue YUM70 inhibits GRP78 to induce ER-stress meditated apoptosis in pancreatic cancer both in vitro and in vivo with minimal toxicity to normal tissues in preclinical models [60]. Using human pancreatic cancer cell lines as model systems, our studies provide evidence of the apoptotic activities of CGs, as exemplified by LanC, in pancreatic cancer and that suppression of GRP78 expression may be a major contributor to the anti-tumor activities of this class of compounds. Future studies are warranted to extend these observations in vivo. As several CGs have already been tested in a phase 1 clinical trial to determine dose limiting toxicities and maximum tolerated dose towards cancer treatment [38], our findings support further investigation of this class of drugs as potential anti-neoplastic agents for pancreatic and other cancers that depend on GRP78 for growth, survival, and therapeutic resistance.
Funding Information
National Institutes of Health
Grant/Award Number: R01 CA238029 and R01 CA027607
Author Contributions
D.P.H. and A.S.L. conceived and designed the experiments. D.P.H. and Y.L.T. performed the experiments. D.P.H. and A.S.L wrote the manuscript.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflict of Interests
The authors declare that there is no conflict of interests.
Acknowledgements
We thank Vicky Yamamoto and Hamid M. Said for the gifts of cell lines, Emma Hadley for technical assistance. We thank Yi Zhang and Shenghua Zhou and the Choi Family Therapeutic Screening Facility at the Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research of USC for performing the drug screen. We also thank Kanjin Wu and Kevin Kelly for assistance with Flow Cytometry, Cheng Chang, Wei Li and Axel Schonthal for assistance with hypoxia chamber, and Ze Liu for helpful discussion. This work was supported in part by National Institutes of Health grants R01 CA238029 and CA027607 to A.S.L. The University of Southern California Norris Comprehensive Cancer Center Molecular Genomics Core which provided primer service was supported by NIH grant P30 CA014089.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neo.2021.10.004.
Appendix. Supplementary materials
References
- 1.Luo B., Lee A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013 doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nature Reviews Cancer. 2014 doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature Reviews Cancer. 2014;14(9) doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 4.Ni M., Lee A.S. ER chaperones in mammalian development and human diseases. FEBS Letters. 2007;581(19) doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pobre K., Poet G.J., Hendershot L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. Journal of Biological Chemistry. 2019;294(6):2098–2108. doi: 10.1074/jbc.REV118.002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hetz C., Zhang K., Kaufman R.J. Mechanisms, regulation and functions of the unfolded protein response. Nature Reviews Molecular Cell Biology. 2020;21(8) doi: 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology. 2012;13(2) doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 8.Zhu G., Lee A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. Journal of Cellular Physiology. 2015 doi: 10.1002/jcp.24923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wek R. C. (2018). Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harbor perspectives in biology, 10(7), a032870. https://doi.org/10.1101/cshperspect.a032870 [DOI] [PMC free article] [PubMed]
- 10.Clarke H.J., Chambers J.E., Liniker E., Marciniak S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25(5):563–573. doi: 10.1016/j.ccr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Tameire F., Verginadis I.I., Koumenis C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: Mechanisms and targets for therapy. Seminars in cancer biology. 2015;33:3–15. doi: 10.1016/j.semcancer.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen J., Ha D.P., Zhu G., Rangel D.F., Kobielak A., Gill P.S., Groshen S., Dubeau L., Lee A.S. Proceedings of the National Academy of Sciences of the United States of America. 2017. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casas, C. (2017). GRP78 at the centre of the stage in cancer and neuroprotection. Frontiers in Neuroscience. https://doi.org/10.3389/fnins.2017.00177 [DOI] [PMC free article] [PubMed]
- 14.Roller C., Maddalo D. The molecular chaperone GRP78/BiP in the development of chemoresistance: Mechanism and possible treatment. Frontiers in Pharmacology. 2013 doi: 10.3389/fphar.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook K.L., Clarke R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future medicinal chemistry. 2015;7(12):1529–1534. doi: 10.4155/FMC.15.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wey S., Luo B., Tseng C.C., Ni M., Zhou H., Fu Y., Bhojwani D., Carroll W.L., Lee A.S. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119(3) doi: 10.1182/blood-2011-06-357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangel D.F., Dubeau L., Park R., Chan P., Ha D.P., Pulido M.A., Mullen D.J., Vorobyova I., Zhou B., Borok Z., Offringa I.A., Lee A.S. Endoplasmic reticulum chaperone GRP78/BiP is critical for mutant Kras-driven lung tumorigenesis. Oncogene. 2021;(20):40. doi: 10.1038/s41388-021-01791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shani G., Fischer W.H., Justice N.J., Kelber J.A., Vale W., Gray P.C. GRP78 and Cripto Form a Complex at the Cell Surface and Collaborate to Inhibit Transforming Growth Factor β Signaling and Enhance Cell Growth. Molecular and Cellular Biology. 2008;28(2) doi: 10.1128/mcb.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelber J.A., Panopoulos A.D., Shani G., Booker E.C., Belmonte J.C., Vale W.W., Gray P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene. 2009;(24):28. doi: 10.1038/onc.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni M., Zhang Y., Lee A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochemical Journal. 2011;434(2) doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Tseng C.C., Tsai Y.L., Fu X., Schiff R., Lee A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PloS One. 2013;8(11):e80071. doi: 10.1371/journal.pone.0080071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai Y.L., Ha D.P., Zhao H., Carlos A.J., Wei S., Pun T.K., Wu K., Zandi E., Kelly K., Lee A.S. Proceedings of the National Academy of Sciences of the United States of America. 2018. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng C.C., Stanciauskas R., Zhang P., Woo D., Wu K., Kelly K., Gill P.S., Yu M., Pinaud F., Lee A.S. GRP78 regulates CD44v membrane homeostasis and cell spreading in tamoxifen-resistant breast cancer. Life Science Alliance. 2019 doi: 10.26508/lsa.201900377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tseng C.C., Zhang P., Lee A.S. The COOH-Terminal Proline-Rich Region of GRP78 Is a Key Regulator of Its Cell Surface Expression and Viability of Tamoxifen-Resistant Breast Cancer Cells. Neoplasia. 2019;21(8):837–848. doi: 10.1016/j.neo.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Gronow M., Gopal U., Austin R.C., Pizzo S.V. Glucose-regulated protein (GRP78) is an important cell surface receptor for viral invasion, cancers, and neurological disorders. IUBMB Life. 2021;(6):73. doi: 10.1002/iub.2502. [DOI] [PubMed] [Google Scholar]
- 26.Carlos A.J., Ha D.P., Yeh D.-W., Van Krieken R., Tseng C.-C., Zhang P., Gill P., Machida K., Lee A.S. The chaperone GRP78 is a host auxiliary factor for SARS-CoV-2 and GRP78 depleting antibody blocks viral entry and infection. Journal of Biological Chemistry. 2021 doi: 10.1016/j.jbc.2021.100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu R., Li X., Gao W., Zhou Y., Wey S., Mitra S.K., Krasnoperov V., Dong D., Liu S., Li D., Zhu G., Louie S., Conti P.S., Li Z., Lee A.S., Gill P.S. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clinical Cancer Research. 2013 doi: 10.1158/1078-0432.CCR-13-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridder G.G., Ray R., Pizzo S.V. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Research. 2012;22(3):225–235. doi: 10.1097/CMR.0b013e32835312fd. [DOI] [PubMed] [Google Scholar]
- 29.D'Angelo S., Staquicini F.I., Ferrara F., Staquicini D.I., Sharma G., Tarleton C.A., Nguyen H., Naranjo L.A., Sidman R.L., Arap W., Bradbury A.R., Pasqualini R. Selection of phage-displayed accessible recombinant targeted antibodies (SPARTA): methodology and applications. JCI insight. 2018;3(9):e98305. doi: 10.1172/jci.insight.98305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niu Z., Wang M., Zhou L., Yao L., Liao Q., Zhao Y. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Scientific Reports. 2015;5:16067. doi: 10.1038/srep16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mijatovic T., Van Quaquebeke E., Delest B., Debeir O., Darro F., Kiss R. Cardiotonic steroids on the road to anti-cancer therapy. Biochimica et Biophysica Acta - Reviews on Cancer. 2007;1776(1) doi: 10.1016/j.bbcan.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Prassas I., Diamandis E.P. Novel therapeutic applications of cardiac glycosides. Nature Reviews Drug Discovery. 2008;7(11) doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- 33.Smith T.W. The fundamental mechanism of inotropic action of digitalis. Therapie. 1989;(6):44. [PubMed] [Google Scholar]
- 34.Newman R.A., Yang P., Pawlus A.D., Block K.I. Cardiac glycosides as novel cancer therapeutic agents. Molecular Interventions. 2008;8(1) doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- 35.Babula P., Masarik M., Adam V., Provaznik I., Kizek R. From Na+/K+-ATPase and Cardiac Glycosides to Cytotoxicity and Cancer Treatment. Anti-Cancer Agents in Medicinal Chemistry. 2013;(7):13. doi: 10.2174/18715206113139990304. [DOI] [PubMed] [Google Scholar]
- 36.Platz E.A., Yegnasubramanian S., Liu J.O., Chong C.R., Shim J.S., Kenfield S.A., Stampfer M.J., Willett W.C., Giovannucci E., Nelson W.G. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discovery. 2011;(1):1. doi: 10.1158/2159-8274.CD-10-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menger L., Vacchelli E., Adjemian S., Martins I., Ma Y., Shen S., Yamazaki T., Sukkurwala A.Q., Michaud M., Mignot G., Schlemmer F., Sulpice E., Locher C., Gidrol X., Ghiringhelli F., Modjtahedi N., Galluzzi L., André F., Zitvogel L.…Kroemer G. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Science Translational Medicine. 2012;(143):4. doi: 10.1126/scitranslmed.3003807. [DOI] [PubMed] [Google Scholar]
- 38.Slingerland M., Cerella C., Guchelaar H.J., Diederich M., Gelderblom H. Cardiac glycosides in cancer therapy: from preclinical investigations towards clinical trials. Investigational New Drugs. 2013;31(4):1087–1094. doi: 10.1007/s10637-013-9984-1. [DOI] [PubMed] [Google Scholar]
- 39.Ha D.P., Lee A.S. Insulin-like growth factor 1-receptor signaling stimulates GRP78 expression through the PI3K/AKT/mTOR/ATF4 axis. Cellular Signalling. 2020;75 doi: 10.1016/j.cellsig.2020.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y., Liu R., Ni M., Gill P., Lee A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. Journal of Biological Chemistry. 2010 doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Research. 2019;47(W1):W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Research. 2017;45(W1):W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang S.C., Erwin A.E., Lee A.S. Glucose-regulated protein (GRP94 and GRP78) genes share common regulatory domains and are coordinately regulated by common trans-acting factors. Molecular and Cellular Biology. 1989;9(5):2153–2162. doi: 10.1128/mcb.9.5.2153-2162.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumeister P., Luo S., Skarnes W.C., Sui G., Seto E., Shi Y., Lee A.S. Endoplasmic Reticulum Stress Induction of the Grp78/BiP Promoter: Activating Mechanisms Mediated by YY1 and Its Interactive Chromatin Modifiers. Molecular and Cellular Biology. 2005;(11):25. doi: 10.1128/mcb.25.11.4529-4540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;(4):35. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 46.McConkey D.J., Lin Y., Nutt L.K., Ozel H.Z., Newman R.A. Cardiac glycosides stimulate Ca2+increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Research. 2000;(14):60. [PubMed] [Google Scholar]
- 47.Poindexter B.J., Feng W., Dasgupta A., Bick R.J. Oleandrin produces changes in intracellular calcium levels in isolated cardiomyocytes: A real-time fluorescence imaging study comparing adult to neonatal cardiomyocytes. Journal of Toxicology and Environmental Health - Part A: Current Issues. 2007;(6):70. doi: 10.1080/15287390600882408. [DOI] [PubMed] [Google Scholar]
- 48.Berridge M.J., Lipp P., Bootman M.D. The versatility and universality of calcium signalling. Nature Reviews Molecular Cell Biology. 2000;1(Issue 1) doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 49.Mizushima N., Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147(4) doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Ni M., Zhou H., Wey S., Baumeister P., Lee A.S. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PloS One. 2009;4(8):e6868. doi: 10.1371/journal.pone.0006868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Satoh M., Nakai A., Sokawa Y., Hirayoshi K., Nagata K. Modulation of the phosphorylation of glucose-regulated protein, GRP78, by transformation and inhibition of glycosylation. Experimental Cell Research. 1993;205(1):76–83. doi: 10.1006/excr.1993.1060. [DOI] [PubMed] [Google Scholar]
- 52.Wang J., Lee J., Liem D., Ping P. HSPA5 Gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum. Gene. 2017;618:14–23. doi: 10.1016/j.gene.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy R.K., Mao C., Baumeister P., Austin R.C., Kaufman R.J., Lee A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. Journal of Biological Chemistry. 2003;278(23):20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 54.Badr C.E., Wurdinger T., Nilsson J., Niers J.M., Whalen M., Degterev A., Tannous B.A. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosisinducing ligand and induces an alternative cell death pathway. Neuro-Oncology. 2011;13(11) doi: 10.1093/neuonc/nor067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang M.A., Kim M.S., Kim W., Um J.H., Shin Y.J., Song J.Y., Jeong J.H. Lanatoside C suppressed colorectal cancer cell growth by inducing mitochondrial dysfunction and increased radiation sensitivity by impairing DNA damage repair. Oncotarget. 2016;7(5) doi: 10.18632/oncotarget.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chao M.W., Chen T.H., Huang H.L., Chang Y.W., HuangFu W.C., Lee Y.C., Teng C.M., Pan S.L. Lanatoside C, a cardiac glycoside, acts through protein kinase Cδ to cause apoptosis of human hepatocellular carcinoma cells. Scientific Reports. 2017;7 doi: 10.1038/srep46134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Y., Yu K., Wang G., Zhang D., Shi C., Ding Y., Hong D., Zhang D., He H., Sun L., Zheng J.N., Sun S., Qian F. Lanatoside C inhibits cell proliferation and induces apoptosis through attenuating Wnt/β-catenin/c-Myc signaling pathway in human gastric cancer cell. Biochemical Pharmacology. 2018;150 doi: 10.1016/j.bcp.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 58.Rasheduzzaman M., Yin H., Park S.Y. Cardiac glycoside sensitized hepatocellular carcinoma cells to TRAIL via ROS generation, p38MAPK, mitochondrial transition, and autophagy mediation. Molecular Carcinogenesis. 2019;(11):58. doi: 10.1002/mc.23096. [DOI] [PubMed] [Google Scholar]
- 59.Reddy D., Kumavath R., Ghosh P., Barh D. Lanatoside c induces g2/m cell cycle arrest and suppresses cancer cell growth by attenuating MAPK, wnt, JAK-STAT, and PI3K/AKT/mTOR signaling pathways. Biomolecules. 2019;9(12) doi: 10.3390/biom9120792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samanta S., Yang S., Debnath B., Xue D., Kuang Y., Ramkumar K., Lee A.S., Ljungman M., Neamati N. The Hydroxyquinoline Analogue YUM70 Inhibits GRP78 to Induce ER Stress-Mediated Apoptosis in Pancreatic Cancer. Cancer Research. 2021;81(7):1883–1895. doi: 10.1158/0008-5472.CAN-20-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.