

Abstract
Cells with blocked microtubule polymerization are delayed in mitosis, but eventually manage to proliferate despite substantial chromosome missegregation. While several studies have analyzed the first cell division after microtubule depolymerization, we have asked how cells cope long‐term with microtubule impairment. We allowed 24 clonal populations of yeast cells with beta‐tubulin mutations preventing proper microtubule polymerization, to evolve for ˜150 generations. At the end of the laboratory evolution experiment, cells had regained the ability to form microtubules and were less sensitive to microtubule‐depolymerizing drugs. Whole‐genome sequencing identified recurrently mutated genes, in particular for tubulins and kinesins, as well as pervasive duplication of chromosome VIII. Recreating these mutations and chromosome VIII disomy prior to evolution confirmed that they allow cells to compensate for the original mutation in beta‐tubulin. Most of the identified mutations did not abolish function, but rather restored microtubule functionality. Analysis of the temporal order of resistance development in independent populations repeatedly revealed the same series of events: disomy of chromosome VIII followed by a single additional adaptive mutation in either tubulins or kinesins. Since tubulins are highly conserved among eukaryotes, our results have implications for understanding resistance to microtubule‐targeting drugs widely used in cancer therapy.
Keywords: chromosome segregation, laboratory evolution, microtubule dynamics, resistance to antimitotics
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle; Evolution & Ecology
Laboratory evolution of yeast cells unable to form microtubules shows that their recovery involves a recurring succession of chromosome VIII duplication and adaptive tubulin or kinesis mutations.
Introduction
Microtubules are essential components of the cytoskeleton, formed by alpha‐ and beta‐tubulins (Tub1/3 and Tub2 in budding yeast) (Desai & Mitchison, 1997; Brouhard & Rice, 2018). Early screens in yeast identified a wide array of temperature‐sensitive mutations that either depolymerize or hyperstabilize microtubules (Richards et al, 2000). Cells expressing these alleles are typically very sick and delayed in mitosis. During this phase of the cell cycle, microtubules form the mitotic spindle, which is responsible for the segregation of sister chromatids to the daughter cells. Microtubules of the mitotic spindle via a process of “search and capture” interact with chromosomes at specialized proteinaceous structures called kinetochores (Musacchio & Desai, 2017). As long as there are unattached or improperly attached kinetochores, cells are arrested in pro‐metaphase by a signaling pathway called the mitotic checkpoint or spindle assembly checkpoint (SAC) (Musacchio, 2015). After all chromosomes are properly attached, the checkpoint is silenced, and cells transit into anaphase. Besides the SAC, chromosome segregation also requires proper positioning of the mitotic spindle, so that sister chromatids are segregated along the mother–daughter axis. The spindle position checkpoint (SPOC) delays mitosis when the spindle is not oriented properly (Pereira et al, 2000; Caydasi & Pereira, 2012).
Microtubule dynamics plays a key role both for the search and capture of chromosomes, and for segregating them to the opposite poles of the cell. Not surprisingly, microtubule dynamics is controlled by several different factors. Tubulins can polymerize or shrink, the alternation between the two being heavily affected by the status of the GTP bound to beta‐tubulin on the plus‐end of the filament. At centrosomes, gamma‐tubulins contribute to polymerization by nucleating new filaments. In budding yeast, this requires the gamma‐tubulin small complex (gamma‐TuSC), which is formed by gamma‐tubulin, Tub4 in budding yeast, and two co‐factors called Spc98 and Spc97 (Lin et al, 2015). Tub4 also requires GTP for microtubules polymerization (Gombos et al, 2013). Besides tubulins themselves, other proteins interact with microtubules and control their polymerization. Among them are kinesins, specialized motors that move along filaments, some of which can also depolymerize microtubules (Akhmanova & Steinmetz, 2015). In budding yeast, Kip3, which belongs to the kinesin‐8 family, is such a kinesin with depolymerization activity (Su et al, 2012; Arellano‐Santoyo et al, 2017).
Laboratory evolution experiments have been used to explore how cells recover from the impairment of essential functions. This approach, together with next‐generation sequencing (NGS), allows deciphering the dynamics of genetic changes underlying the emergence of adaptation. “Repair evolution experiments” have shown that cells can recover from the absence of essential components, among them those required for cytokinesis, budding, DNA replication, and many genes originally deemed essential (Rancati et al, 2008; Selmecki et al, 2009; Laan et al, 2015; Fumasoni & Murray, 2020; LaBar et al, 2020). Similarly, it was shown that cells adapt and recover growth following deletions of non‐essential genes which come with a strong reduction in fitness (Szamecz et al, 2014). The mechanisms at place to compensate for growth impairment are quite diverse and occur via genetic changes that often do not restore the function of the inactivated proteins. Rather, they act indirectly, either by inactivating regulators of the impaired proteins, or by re‐purposing cellular components (Liu & Rancati, 2016). Aneuploidy often plays a key role in these processes, especially as a first quick evolutionary response to environmental stress, later followed by mutations (Rancati et al, 2008; Chen et al, 2012; Yona et al, 2012; Ravichandran et al, 2018).
To date, no study addressed the mechanisms of compensatory evolution to the inactivation of tubulins. Given the essential role of these proteins, it is not even clear whether cells can recover at all from their inactivation; and if they do, whether this happens via divergent evolutionary paths or through few reproducible series of genetic changes; whether recovery occurs via the development of alternative means for chromosome segregation or via restoration of tubulin activity; whether compensatory mutations are mostly loss of function; and whether aneuploidy plays any role in this process.
Tubulins are coded by genes highly conserved among eukaryotes. Thus, answering these questions may be also potentially relevant for understanding how cells develop resistance to drugs targeting microtubules, either stabilizers, e.g., taxanes, or destabilizers, e.g., vinca alkaloids. Both types of drugs affect chromosome‐microtubule attachment, activate the mitotic checkpoint, and arrest cells before anaphase. By delaying cell cycle progression, microtubule drugs can promote apoptosis in transformed cells (Taylor & McKeon, 1997). In the long‐term, however, the effects of antimitotics are jeopardized by the emergence of resistance.
Here, we performed a laboratory evolution experiment to study how cells react to forced microtubule depolymerization. We used haploid yeast cells expressing an allele of beta‐tubulin (tub2‐401), which carries four‐point mutations resulting in three amino acid changes (M233V, Y242C, Q245L, Fig EV1A). This is a cold‐sensitive allele, which cannot polymerize microtubules when grown at low temperature (Huffaker et al, 1988; Sullivan & Huffaker, 1992). Several other alleles are available, but tub2‐401 is the one showing the most penetrant phenotype. We opted for a mutation mimicking the effect of drugs, rather than drugs themselves (e.g., benomyl, nocodazole), to avoid generic and well‐characterized multidrug mechanisms of resistance (Prasad & Goffeau, 2012).
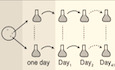
Figure EV1. Characterization of evolved cells.
- The three amino acid changes of the tub2‐401 allele.
- Serial 5‐fold dilution of cells of the indicated genotypes spotted on YPD and incubated at the indicated temperatures.
- Different fates of tub2‐401 cells growing at 18°C (correct chromosome segregation after rebudding, missegregation after rebudding, or death before rebudding). Cells were synchronized in G1, released at 18°C to express the tub2‐401 phenotype, and imaged every hour for 41 h in a microfluidic chamber. The events of segregation were identified by following GFP‐tagged chromosome V. Among the 21.5% of cells correctly segregating, 44.4% died before the end of the movie and 55.5% did not. Among the 50.2% of cell missegregating, 48.5% died before the end of the movie and 51.4% did not. Cells that neither died nor rebudded by the end of the movie (12 out of 221 cells from 2 biological replicates) are not included in this analysis. Scale bars are 5 μm long. Every image, except the leftmost “death”, have the same contrast settings. See Movie EV1.
- Normalized and corrected depth of coverage for the two ancestral strains. Each dot represents the median depth over a 10,000 bp window.
- Ancestral cells and selected Gf populations (G5, D5 and H6) were synchronized in G1 at 30°C and released at 18°C, as explained in Materials and Methods. Cells were collected 3.5 and 5 h after G1 release. DNA content was assessed by Sytox Green staining.
- Selected Gf populations (G5, D4, E5, H6, D5, and B4) and ancestors were synchronized in G1 at 30°C and released at 18°C, as explained in Materials and Methods. The fraction of cells with a large bud was monitored every 30 min from 3 to 5 h after G1 release. Black dots identify the timepoints in which spindle lengths were measured. The kinetics is representative of one biological replicate.
- Representative spindles in Ancestor and selected Gf cells. Tub1 was stained for immunofluorescence, while nuclei were stained with DAPI. Images are examples from one biological replicate.
- mad2Δ, ancestral cells, and selected Gf populations (the same as in Fig 1D and E) were synchronized in G1 at 30°C and released in nocodazole 15 μg/ml. Cells were collected 1, 3, and 4 h after nocodazole addition. DNA content was assessed by Sytox Green staining.
We grew cells for more than one hundred generations at the semi‐restrictive temperature, until they recovered growth. We confirmed that evolved cells regained the ability to assemble regular spindles, and we identified two pathways through which cells can acquire resistance. In both of them, disomy of chromosome VIII (chrVIII 2X) is the likely initial step. Our results may be relevant for understanding principles underlying the emergence of resistance in the context of cancer treatment.
Results
Yeast cells become resistant to stimuli inducing microtubules depolymerization
In order to investigate how yeast cells cope on the long term with tubulin defects, we used a conditional allele of TUB2, tub2‐401. At low temperature, it is non‐functional and results in improper microtubule polymerization (Huffaker et al, 1988; Sullivan & Huffaker, 1992). This leads to the activation of both the mitotic checkpoint (Corno et al, 2019) and the spindle position checkpoint, since deletion of two of their essential genes (MAD2 and BUB2, respectively) impairs growth even at the semi‐permissive temperature of 23°C (Fig EV1B). By using live‐cell imaging at the restrictive temperature (18°C), we confirmed that cells were large and budded, a phenotype typical of mitotic arrest (Movie EV1). Regardless of the prolonged mitotic delay, many cells slipped through the arrest and continued proliferating, as shown previously (Corno et al, 2019). Following the inheritance of GFP‐tagged chromosome V after growing cells at the restrictive temperature, we observed high rates of chromosome missegregation. A large fraction of cells (˜60%) died either before or immediately after segregating chromosomes (Fig EV1C).
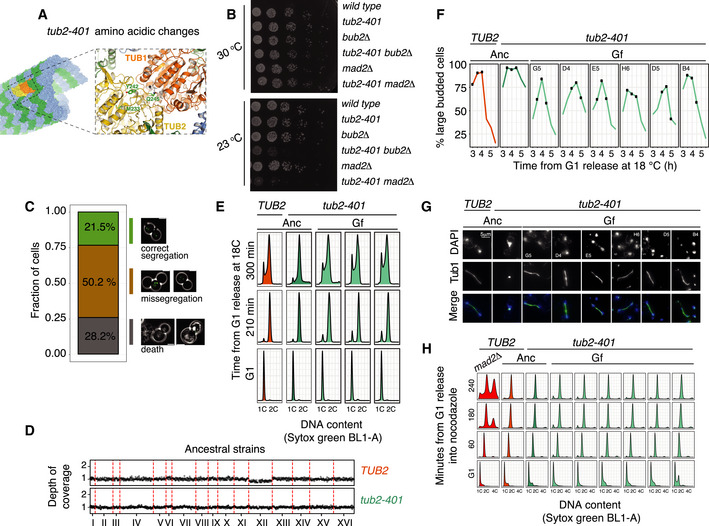
We next performed a laboratory evolution experiment at 18°C using multiple parallel lines of both the tub2‐401 mutant strain (N = 24) and the TUB2 wild‐type (N = 8). Each population started from an individual clone of the same euploid ancestor (Figs 1A and EV1D). Measurements of growth started after 1 day at 18°C (Generation 0 or G0—Fig 1B). Cells expressing tub2‐401 grew slower than control cells expressing wild‐type TUB2. After ˜20 generations, the apparent growth rate (cell division subtracted of cell death) started to increase, and after ˜45 generations (Generation recovery or Gr), growth rate stabilized without ever reaching the wild type. It fluctuated around the same value for another ˜100 generations, when we stopped the experiment (final Generation or Gf). Based on the data gathered from the movie (Fig EV1C and Movie EV1), we interpreted the initial slow growth rate as a consequence of both prolonged activation of the mitotic checkpoints and cell death caused by massive chromosome missegregation.
Figure 1. Cells impaired for microtubule polymerization increase their growth rate during a laboratory evolution experiment.
- Scheme of the evolution experiment. We isolated the two ancestral strains to obtain 8 clonal TUB2 populations and 24 clonal tub2‐401 populations. The first measurement of growth rate was done after 1 day at 18°C (generation 0 or G0). The last measurement was done after 42 days, at generation final (Gf). Between the two, we also analyzed by NGS cells immediately after recovery of growth (generation recovery or Gr).
- Growth rate was measured every 3–4 days by fitting the dynamics of optical density with an exponential (see Materials and Methods). Thus, growth rate is the net output between cell division and cell death. Thick lines mark the medians, and shadowed areas mark the interquartile ranges for the eight wild‐type and 24 tub2‐401 populations that we evolved.
- Evolving cells were treated with the nucleic acid stain Sytox Green, and DNA content was measured with flow cytometry. For each evolving population, the G2 mode of the signal distribution was used to evaluate DNA content (Zhu et al, 2012). DNA content was normalized on the mean value of the TUB2 evolving populations. Thick lines mark the medians, and shadowed areas mark the interquartile ranges for the eight wild‐type and 24 tub2‐401 populations that we evolved. 1.04 is the expected DNA content of a strain disomic for chromosome VIII, since this chromosome accounts for ˜4% of the total DNA of a haploid euploid cell.
- Ancestral cells (Anc) and selected evolved cells collected at the end of the experiment (Gf) were synchronized in G1 at 30°C and released at 18°C. Spindle lengths were measured on Tub1 immunodecorated microtubules across 3 timepoints centered on the maximum proportion of large‐budded cells (dumbell) (see Fig EV1F). Selected evolved populations were chosen among those carrying only one mutation with high frequency in the three most frequently mutated genes—KIP3, TUB2, or TUB1 (populations G5, D4, B4, D5, H6, E5) For each sample, the number of cells (n) and the number of biological replicates (N) is stated. Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (****P‐value < 10−4). The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Individual measures are plotted as dots.
- Ancestral cells and selected Gf populations (the same as in panel (D)) were synchronized in G1 at 30°C and released in nocodazole 2 μg/ml. 1 and 2 h after release cells were collected and DNA content was assessed by Sytox Green staining.
The dynamics of DNA content confirmed high levels of chromosome instability in the early stages of tub2‐401 evolution (Fig 1C). The DNA content of TUB2 and tub2‐401 populations initially overlapped, but quickly diverged. It stayed constant in TUB2 cells, while it rapidly increased in tub2‐401, reaching its peak at around generation 25. After that point, ploidy decreased and kept a constant value, higher than the euploid control. This result is consistent with decreased chromosome missegregation at the end of the experiment.
To explain how cells recovered growth and stabilized their DNA content, we hypothesized that some of them acquired mutations allowing the assembly of more structured mitotic spindles that segregated chromosomes more efficiently. As such, the mitotic checkpoint and the SPOC were lifted, and fitter cells progressed more rapidly. To test this interpretation, we analyzed by FACS the cell cycle dynamics of evolved cells. A subset of Gf populations was synchronized in G1 by α‐factor and released at 18°C. We observed that, compared to the ancestors, evolved cells spent less time with 2C DNA content, in agreement with a shorter mitotic delay (Fig EV1E). We also analyzed mitotic spindles in cells carrying the tub2‐401 mutations at Gf. The length of spindles across three different timepoints, centered on the time with the highest fraction of cells with a large bud (a feature of mitotic arrest; Fig EV1F), was longer in evolved cells than in ancestors (Figs 1D and EV1G). This result is in agreement with increased ability of evolved cells to polymerize microtubules.
This feature of evolved cells was not uniquely related to overcoming the tub2‐401 mutations, but was confirmed also upon treatment with the microtubules depolymerizing drug nocodazole. After synchronization in G1 and release in low concentration of nocodazole at 30°C, wild‐type cells did not arrest, whereas tub2‐401 cells were delayed in mitosis due to increased sensitivity to microtubule depolymerization. Evolved tub2‐401 showed less sensitivity to nocodazole, in‐between ancestors and wild‐type cells (Fig 1E). Under higher concentration of nocodazole, all strains mounted an efficient checkpoint response (Fig EV1H).
In conclusion, we showed that evolved cells became less sensitive to induced microtubule depolymerization, either caused by the tub2‐401 allele or nocodazole.
Evolved strains mutate recurrently a small set of genes
With the aim of understanding the evolutionary dynamics, we addressed the genetic basis of resistance. We sequenced all populations at the final generation and looked for genes that were mutated more than once in different populations with unique mutations. We then collected all mutations occurring in these recurrently mutated genes. Hereafter, we focused our attention only on these mutations (Table EV1).
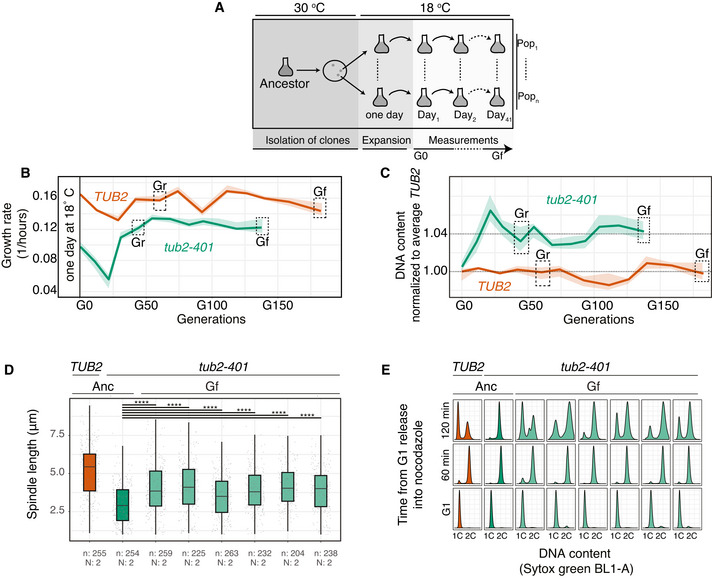
Control cells did not experience impairment of microtubule polymerization, yet they were under stress due to the low temperature. In these cells, genes of the PHO pathway (PHO4 and PHO81, Fig EV2A and B) were recurrently mutated. We did not observe any change of ploidy (Fig EV2C). In cells expressing tub2‐401, we identified recurrently mutated genes that were related to microtubules dynamics, and primarily tubulins themselves. Several mutations affected TUB2 (Fig 2A). They were all missense mutations (neither nonsense nor frameshift), in agreement with TUB2 being essential in budding yeast. None of them were reversions to the original sequence. We also observed mutations in TUB1 (alpha‐tubulin) and components of the gamma‐tubulin complex (SPC98). Like TUB2, these essential genes had missense mutations (Fig 2A). Besides tubulins, we found the gene coding for the kinesin‐8 Kip3 mutated multiple times. Here, mutations were not only missense, but also nonsense and frameshifts, spread all over the gene (Fig 2B). Finally, we observed disomy of chromosome VIII in the large majority of tub2‐401 populations (Figs 2C and EV2D). In only two populations, we observed a slight disomy of chromosome III. This result is in agreement with tub2‐401 having a higher DNA content than TUB2 cells at the end of the experiment (Fig 1C). We did not identify recurrent structural variants.
Figure EV2. Recurrent amino acid changes and disomies in evolved TUB2 .
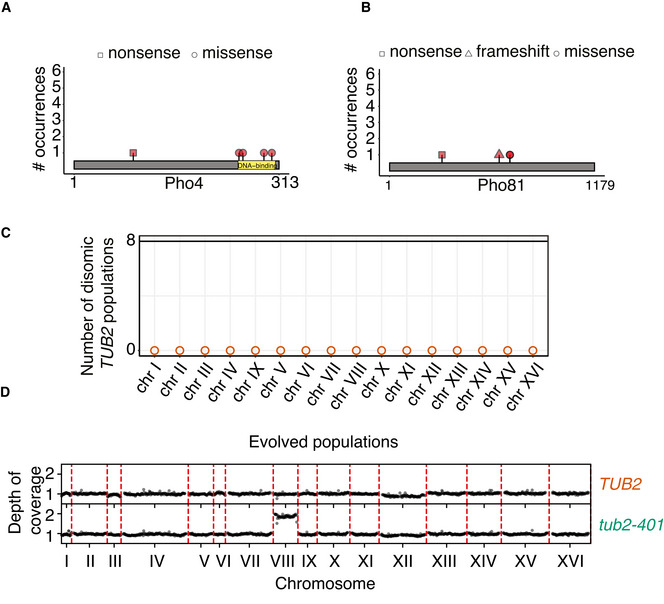
-
A, BAmino acid changes caused by mutations occurring independently multiple times in PHO81 and PHO4. They were detected only in TUB2 cells (Fig 3A)
-
CLack of disomic populations among the eight evolving TUB2 populations. Empty dots are used to mark chromosomes that are monosomic in every population. Chromosome copy numbers were determined by coverage analysis (an example in Fig EV2D).
-
DNormalized and corrected depth of coverage for two representative samples (TUB2, population A1; tub2‐401, population A4). Each dot represents the median depth over a 10,000 bp window.
Figure 2. Amino acid changes in recurrently mutated genes.
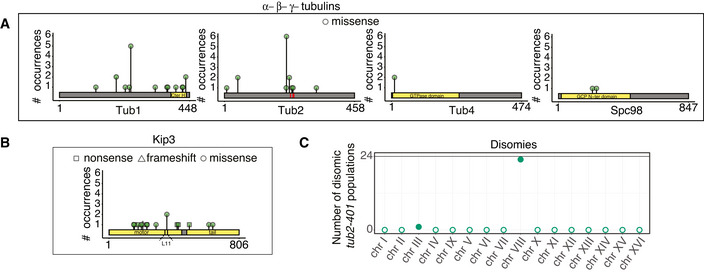
- Amino acid changes caused by mutations occurring independently multiple times in alpha‐, beta‐, and gamma‐tubulin genes. In the analysis, we sum up all mutations that affect the same amino acid residue. Only missense mutations were found. Red stripes in TUB2 mark the three residues mutated in tub2‐401 allele.
- Amino acid changes caused by mutations occurring independently multiple times in KIP3.
- Number of disomic populations among the 24 evolving tub2‐401. Empty dots are used to mark chromosomes that are monosomic in every population. Chromosome copy number was determined by coverage analysis (an example in Fig EV2D). All data were collected at the end of the evolution experiment (Gf in Fig 1B).
Some genes originally identified by our pipeline were not followed up in our analysis. Among them, genes of the ADE pathway, which have already been reported, mutated in evolution experiments of yeast with the same genetic background (W303) but independently from impaired microtubule polymerization and low temperature (Kaya et al, 2020). We also did not follow up the two genes, which have not been characterized yet (YHR033W, YJL070C) and PRR2, as they are present with very low frequency in two populations only (Table EV1). Instead, we followed up TUB4, which is mutated twice with high frequency but always with the same mutation, since its protein product interacts directly with Spc98.
In summary, tub2‐401 cells recurrently mutated genes belonging to two classes: (i) tubulins and members of gamma‐TuSC (TUB1, TUB2, TUB4, SPC98); and (ii) KIP3. Moreover, we showed that the large majority of evolved strains were disomic for chromosome VIII.
Disomy of chromosome VIII precedes acquisition of a single mutation in recurrent genes
Next, we analyzed the genetic composition of the individual populations. All of them but one had at least one recurrently mutated gene (Fig 3A). In several populations, we observed the co‐presence of multiple mutations, some of them affecting the same gene (Fig EV3A). Strikingly, in almost all populations the sum of allele frequencies of mutations approached but did not exceed 100%. This is not the theoretical maximum, since if cells would carry more than one mutation, the sum of frequencies would be higher than 100%. Hence, this result suggests that individual cells are likely to carry only one mutation. To confirm that mutations in recurrently mutated genes are mutually exclusive, we extracted and analyzed clones from populations with more than one mutation at high frequency. By Sanger sequencing, we never found two candidate adaptive mutations in the same clone (Table EV2). The pervasive presence of mutations in the genes we identified, and the fact that the sum of mutation frequencies approaches 100%, suggests that the identified mutations are largely responsible for recovered growth.
Figure 3. Mutation frequencies measured at final (Gf) and intermediate (Gr) timepoints.
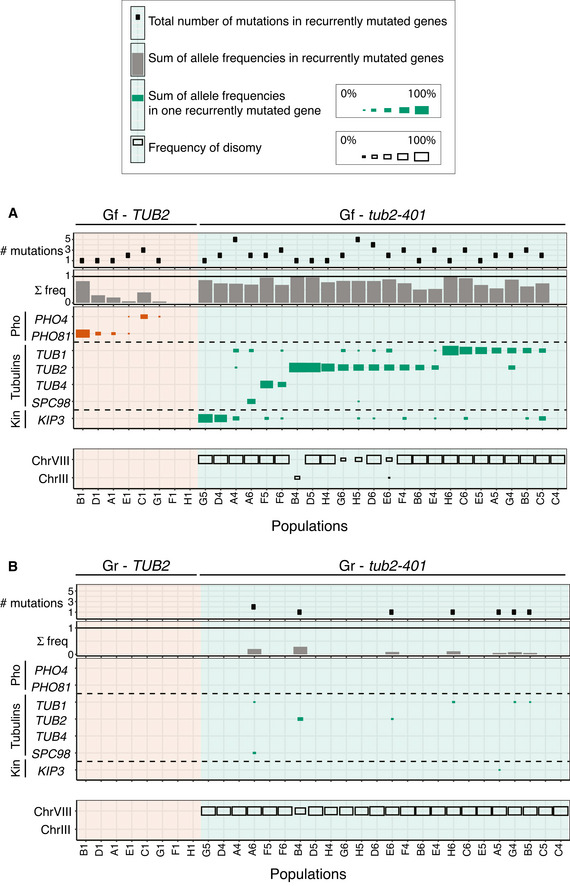
-
A, BThe tables summarize the number and frequencies of mutations in recurrently mutated genes within each population (column). First panel from top: number of mutations in recurrently mutated genes for each population. Notice that there can be more mutations for the same gene. Second panel from top: sum of the frequencies of all mutations in recurrently mutated genes for each population. Third panel from top: frequency of mutations in the indicated genes, for each population. In each population, we group together frequencies of all mutations affecting one specific gene. The size of squares is proportional to this mutation frequency. Bottom panel: frequency of disomic chromosomes within each population. The size of the squares is proportional to the frequency. In (A), we analyzed cells at the end of the experiment (Gf in Fig 1B), in (B) after cells have started recovering growth (Gr in Fig 1B).
Figure EV3. Mutation frequencies at final timepoint (Gf) in TUB2 and tub2‐401 evolved populations.

- Bar plot of populations grouped according to the presence of multiple mutations in recurrently mutated genes, detected by NGS at the end of the experiment (Gf).
- Venn Diagram of tub2‐401 populations at Gf, carrying mutations in TUB2, TUB1 or KIP3 (data shown in the third panel from top of Fig 3A).
It is striking that regardless of the variability in mutation frequencies, in almost all populations we detected disomy of chromosome VIII exclusively. This result suggests that chrVIII 2X is an early event in the development of resistance. To test this hypothesis, we performed NGS of “generation recovery”, when cells had largely recovered growth and we did not expect an excessive variability of karyotypes that may have hindered the identification of recurrent aneuploidies. The results (Fig 3B) showed that indeed chromosome VIII was mostly disomic in all populations already at this timepoint. Importantly, the ancestor was euploid, and each population originated from a different clone, ruling out the possibility that disomy of chrVIII was pre‐existent. Unlike the disomy, point mutations at Gr were present at very low frequency. The TUB2 control did not show any relevant disomy at either timepoint, and mutations of PHO genes were entirely missing at the earlier timepoint.
The results shown so far support a scenario where the first step in the development of resistance is the acquisition of an extra copy of chromosome VIII, followed by mutations of recurrently mutated genes. At the end of the experiment, most cells are disomic of chromosome VIII and carry one additional mutation.
Potential adaptive roles of mutations in recurrently mutated genes
We hypothesized that mutations in the identified genes may be adaptive. Before experimentally testing this hypothesis, we confirmed its plausibility based on the available literature.
PHO
In PHO81, two mutations out of three are nonsense or frameshift, and thus could be classified as loss of function. In the case of PHO4, four mutations out of five were missense, clustered in the DNA‐binding region (Fig EV2A). In the hypothesis that they prevent DNA binding, they could also be classified as loss of function. Interestingly, uptake of inorganic phosphate is limiting for growth in the cold (Vicent et al, 2015), and deletion of either PHO81 or PHO4 rescues growth in low phosphate after deletion of the high‐affinity transporter PHO84 (Wykoff et al, 2007).
KIP3
Among the mutations found in KIP3, half are either nonsense or frameshift, again interpretable as loss of function (Laan et al, 2015; Fumasoni & Murray, 2020). The fact that deletion of KIP3 reduces sensitivity to benomyl (DeZwaan et al, 1997) further supports this interpretation and is in line with the depolymerizing activity of Kip3 (Arellano‐Santoyo et al, 2017). It also reinforces the notion that we selected for mutations that generically contrast induced microtubule depolymerization, and not specifically the tub2‐401 allele. The missense mutations found in KIP3 may underlie subtler mechanisms to recover microtubule polymerization. Recent data point at the L11 domain in Kip3 as essential for microtubule depolymerization (Arellano‐Santoyo et al, 2017). Interestingly, a point‐mutation leading to an amino acid change (E359K) was found independently in two evolved populations, and it falls in the L11 domain (Fig 2B). We hypothesize that this mutation disrupts the depolymerase activity of Kip3, while preserving its kinesin function.
gamma‐TuSC
Mutations in TUB4 and SPC98 can be understood in light of increased tubulin nucleation. The amino acid changes in Spc98 are in residues involved in the interaction of Spc98 with Spc97 (P248T) and with Spc110 (H222P), which bridges gamma‐TuSC with the centrosome (Fig 4A). Thus, these mutations may stabilize the formation of the gamma‐TuSC complex, and favor nucleation. The residue mutated twice in Tub4 (Q12) has been described as important for interacting with GTP, and thus affecting nucleation of microtubules and mediating sensitivity to benomyl (Gombos et al, 2013).
Figure 4. Recurrent amino acid changes in Spc98, Tub1 and Tub2.
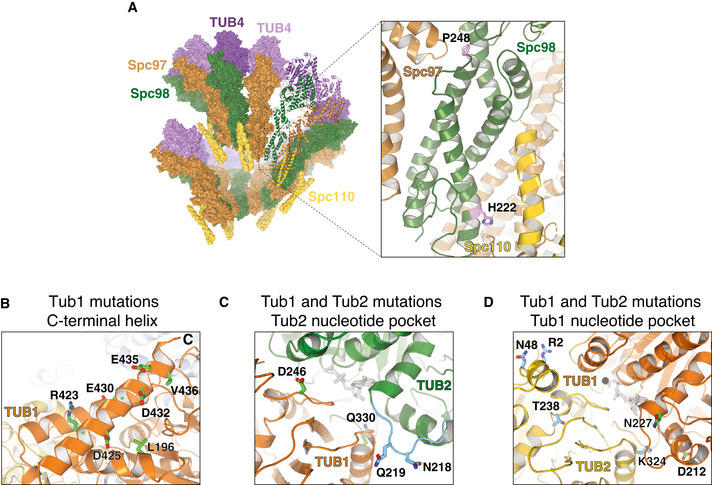
-
ASpc98 residues P248 and H222 are shown in the context of the gamma‐TuSC complex (pdb entry 5flz).
-
BMutated residues on or near the Tub1 C‐terminal helix.
-
C, DResidues mutated close to the Tub2 (in panel (C): Q219H, N218; in panel (D): R2, N48, T238, K324) and Tub1 (in panel (C): D246, Q330; in panel (D): D212, N227) nucleotide pocket, respectively (pdb entry 5W3F).
TUB1
Among the 12 mutations identified in TUB1, we noticed that a large fraction is located on the C‐terminal helix 12 (Fig 4B), a region important for interactions with microtubules binding proteins (Uchimura et al, 2010). Hence, we hypothesize that these mutations may alter the dynamics of interaction with binding partners, ultimately stabilizing microtubules. Along the same line, a mutation that occurs twice, L196W, is situated close to the C‐terminal tail. Its mutation to tryptophan, a residue with a bulky side chain, might misplace the helix, thus inducing a similar effect of the mutants in the helix itself. One of the recurrent mutations is D246Y, which lies at the interface between the Tub1/Tub2 dimer (Fig 4C). Introduction of the bulky tyrosine residue might impair the dynamics of microtubule stability. In agreement with this interpretation, D246A was reported to change sensitivity to benomyl (Richards et al, 2000). The same was observed for Ala substitutions in E435, another site on helix 12 that we find mutated twice (Richards et al, 2000) (Fig 4B).
TUB2
The three amino acid substitutions in ancestral tub2‐401 (M233V, Y242C, G245L) are located in TUB2 (Fig EV1A). We have not identified revertants among mutations identified in the evolved strains. However, in one population in position 242 we find one change from cysteine to tryptophan. Given the similarity between the latter and the original tyrosine, we can interpret this mutation as reversion. All other mutations (Fig 4C and D), instead, recover tubulin function by different means. Mutation of T238 into alanine (Fig 4D) has been shown to decrease sensitivity to benomyl (Richards et al, 2000), with decreasing frequency of catastrophes and slow shrinking (Geyer et al, 2015). We find it mutated into isoleucine, which can be hypothesized to give a similar phenotype. The change to amino acids with similar chemical properties is confirmed in the recurrently observed N48D mutation (two instances; Fig 4D), which is located at the Tub2/Tub2 lateral interface and could affect assembly of protofilaments. Similarly, for V229I (two instances) we find an amino acid change that is not expected to change dramatically the chemical properties of beta‐tubulin. Yet, it is buried inside the tubulin molecule, and the change may affect its structure. Finally, at the interface between Tub2 and Tub1 we find the most recurrently observed amino acid change, Q219H (six instances). Q219 belongs to the loop between H6/H7 (highlighted in light blue in Fig 4C), where other mutations have been reported to alter microtubules dynamics (Gonzalez‐Garay et al, 1999; Yin et al, 2007). The glutamine to histidine mutation may interfere with tubulin assembly favoring its stability.
In summary, for many of the mutations occurring in recurrently mutated genes, we could provide a rationale for their putative effect on microtubule stabilization based on previous studies.
Frequently occurring mutations in recurrently mutated genes are adaptive
To confirm that mutations we identified are adaptive, we introduced some of the most representative ones in the ancestor. In tub2‐401, we mutated the three genes that were mutated in most populations: TUB1 (15/24), TUB2 (12/24), and KIP3 (12/24) (Figs 3A and EV3B). In the TUB2 gene (which already carries the mutations of tub2‐401), we introduced the additional point mutation causing the most frequent amino acid change, Q219H. For brevity, the allele which expresses both the three tub2‐401 amino acid changes and Q219H was called Q219H. In TUB1, the most frequent amino acid change was D246Y (tub1D246Y ). For KIP3, we observed that half of the mutations were nonsense and frameshifts (Fig 2B). In line with (Laan et al, 2015; Fumasoni & Murray, 2020), we mimicked such mutations by deleting the gene. We also analyzed the adaptive role of chrVIII 2X using a disomic strain. As for evolved TUB2 populations, the most often mutated gene was PHO4. Also in this case, we deleted the gene on the assumption that mutations in the DNA‐binding site (Fig EV2A) impair its transcriptional activity.
We grew cells for 24 h at 18°C and measured their growth rate in liquid medium using established protocols (Liu et al, 2015). Q219H, tub1D246Y and kip3Δ partially recovered the growth defect of tub2‐401 (Fig 5A–C). ChrVIII 2X also had a beneficial effect only on tub2‐401 background, while it impaired growth in the WT (Fig 5D). Finally, for cells expressing TUB2, we observed that growth at low temperature was improved by the deletion of PHO4 (Fig EV4A), in agreement with a role for phosphate uptake in adaptation to the cold (Vicent et al, 2015; Smukowski Heil et al, 2019).
Figure 5. Mutations in recurrently mutated genes are adaptive.
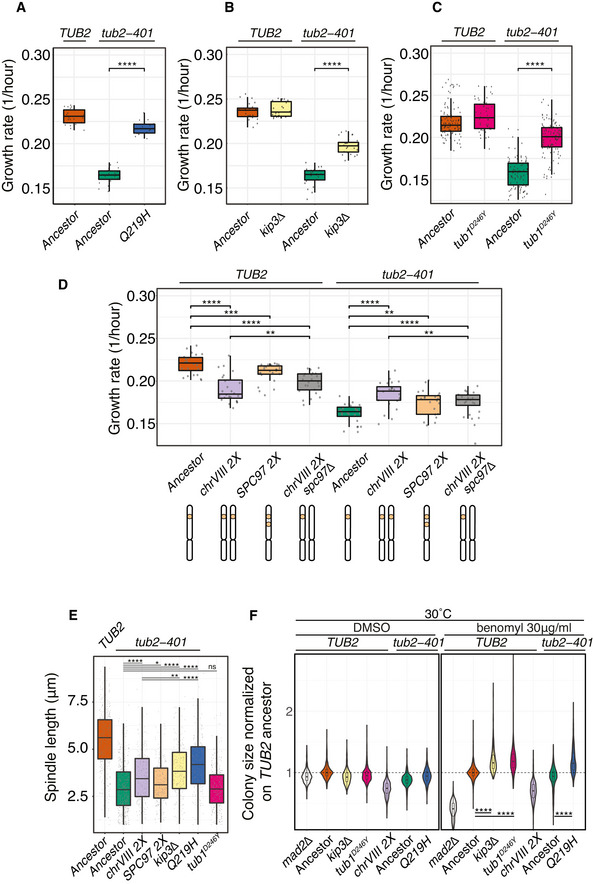
-
A–DGrowth rates measured by in liquid growth assays. Experiments were performed at 18°C after growing cells at 18°C for 24 h. Growth rates result from fitting optical density over time with an exponential (see Materials and Methods for details). Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (**P‐value < 10−2, ***P‐value < 10−3, ****P‐value < 10−4). The plots include three biological replicates, each with ˜7 technical replicates per strain. The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Individual measures are plotted as dots.
-
EAncestors and mutant strains were synchronized in G1 at 30°C and released at 18°C. Spindle lengths were measured on Tub1 immunodecorated microtubules across three timepoints centered on the maximum fraction of large‐budded cells (see Fig EV4B). Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (ns P‐value > 0.5, *P‐value < 0.05, **P‐value < 10−2, **** = P‐value < 10−4). Number of biological replicates (N) and number of cells (n) are: ancestor TUB2 (N = 4, n = 432), ancestor tub2‐401 (4, 530), tub2‐401 chrVIII 2X (2, 222), tub2‐401 SPC97 2X (2, 238), tub2‐401 kip3Δ (2, 203), tub2‐401‐Q219H (2, 221), tub2‐401 tub1D246Y (2, 329). The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Individual measures are plotted as dots.
-
FMeasurement of colony size on agar plates as a proxy for growth. Colonies of the specified genotypes grew on YPD agar plates at 30°C before being pinned onto YPD agar plates supplemented with 1% DMSO with or without benomyl (30 μg/ml final concentration). Plates were incubated at 30°C and imaged after 1 day. Colony size is normalized on the TUB2 ancestor. In every panel, violin plots show the densities along the y‐axis smoothed with a Gaussian kernel, while the boxes span the interquartile range (IQR, from the 25th to the 75th percentiles). The central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. The plot includes seven biological replicates. The number of colonies included in the DMSO panel are, from left to right: 756 mad2Δ, 756 ancestor TUB2, 743 kip3Δ, 753 tub1D246Y, 747 chrVIII 2X, 756 ancestor tub2‐401, 756 tub2‐401‐Q219H. The number of colonies included in the benomyl panel are, from left to right: 645 mad2Δ, 681 ancestor TUB2, 652 kip3Δ, 686 tub1D246Y, 687 chrVIII 2X, 657 ancestor tub2‐401, 665 tub2‐401‐Q219H. For each statistical comparison, the linear model coefficients and the resulting P‐values are, from left to right: 0.193 (P ˜10−90), 0.196 (P ˜10−87), 0.287 (P ˜10−163). See Materials and Materials and Methods for details.
Figure EV4. Characterization of engineered strains.
-
AGrowth rates of wt, tub2‐401 and pho4Δ cells were measured with in liquid growth assays. Experiments were performed at 18°C after growing cells at 18°C for 24 h. Growth rates result from fitting optical density over time with an exponential (see Materials and Methods for details). Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (**P‐value < 10−2). The plot includes three biological replicates, each with ˜12 technical replicates per strain. The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Individual measures are plotted as dots.
-
B–CSpindles measurements in tub2‐401 cells carrying adaptive mutations. Cells were synchronized in G1 at 30°C and released at 18°C as explained in Materials and Methods. (B) The fraction of large‐budded cells (dumbell) was monitored every 30 min from 3 to 5 h after G1 release. Black dots identify the timepoints in which the spindle lengths were measured (Fig 5E). The kinetics is representative of one biological replicate (C) Representative spindles. Microtubules were identified by immunofluorescence on Tub1 while nuclei were stained with DAPI. Images are examples from one biological replicate.
-
DMeasurement of colony size on agar plates as a proxy for growth. Colonies of the specified genotypes grew on YPD agar plates at 30°C before being pinned onto YPD agar plates supplemented with 1% DMSO with or without the spindle poison benomyl (15 μg/ml or 30 μg/ml final concentrations). The plot includes seven biological replicates for every strain but chrVIII spc97Δ, which is present in 3. The number of colonies included in the DMSO panel are, from left to right: 753 mad2Δ, 750 ancestor TUB2, 750 kip3Δ, 753 tub1D246Y, 753 chrVIII 2X, 756 SPC97 2X, 321 chrVIII 2X spc97Δ. The number of colonies included in the benomyl 15μg/ml panel are, from left to right: 611 mad2Δ, 649 ancestor TUB2, 613 kip3Δ, 673 tub1D246Y, 684 chrVIII 2X, 650 SPC97 2X, 291 chrVIII 2X spc97Δ. The number of colonies included in the benomyl 30μg/ml panel are, from left to right: 642 mad2Δ, 669 ancestor TUB2, 627 kip3Δ, 694 tub1D246Y, 665 chrVIII 2X, 645 SPC97 2X, 303 chrVIII 2X spc97Δ. Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. For each statistical comparison, the linear model coefficients and the resulting P‐values are, from left to right: 0.167 (P ˜10−70), 0.143 (P ˜10−55), 0.231 (P ˜10−105), 0.007 (P ˜10−1), 0.037 (P ˜10−5), 0.032 (P ˜10−2). Plates were incubated at 18°C and imaged after 2 days. Colony size is normalized on the TUB2 ancestor. In every panel, violin plots show the densities along the y‐axis smoothed with a Gaussian kernel, while the boxes span the interquartile range (IQR, from the 25th to the 75th percentiles). The central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. See Materials and Methods for details.
We next aimed at identifying whether there is one particular gene on chrVIII whose duplication is adaptive. SPC98, coding for a component of the gamma‐tubulin complex, is mutated in several evolved strains (Fig 2A). Interestingly, another component of the complex, SPC97, is located on chrVIII. Spc97 is the least expressed among the components of Gamma‐TuSC (2.4:1.3:1.0 for Tub4, Spc98, and Spc97) (Erlemann et al, 2012). Hence, we hypothesized that the adaptive effect of the disomy of chrVIII may be due to the duplication of SPC97. This result is not obvious, since large overexpression of Spc97 was reported to decrease viability (Knop et al, 1997), and indeed, we confirmed a limited reduction of growth due to duplication of SPC97 in wild‐type cells (Fig 5D). Accordingly, removing one copy of SPC97 from the disomic strain carrying TUB2 wild type improved slightly its growth rate. However, when we introduced two copies of SPC97 in a strain expressing tub2‐401 and monosomic for chrVIII, we observed an improvement of growth rate. Importantly, deletion of one copy of SPC97 in a strain carrying tub2‐401 and disomic for chrVIII decreased growth compared to the disomic strain. However, it did not revert it completely to that of tub2‐401 alone (Fig 5D), implying that additional genes located on chrVIII contribute to the increased fitness of disomic strains.
To confirm that the adaptive effect is due to the increased stability of microtubules, we analyzed mitotic spindles of cells where we engineered recurrent mutations in the presence of tub2‐401. Cells were synchronized in G1, released at 18°C, and collected at different timepoints when they were arrested in mitosis (i.e., with large buds, Fig EV4B). Mitotic spindles were on average longer in cells carrying the recurrent genetic changes when compared to the ancestor tub2‐401 (Figs 5E and EV4C).
We also tested on solid media the ability of adaptive mutations to recover growth upon treatment with drugs that impair microtubule stability. We grew at 30°C cells expressing the mutations described above in the presence of benomyl. All the strains carried wild‐type TUB2, the only exception being Q219H, which comes with the tub2‐401 mutations. The latter, however, have basically no phenotype at 30°C. In kip3Δ, tub1D246Y and Q219H we observed, as expected, decreased sensitivity to benomyl. The behavior of chrVIII 2X, instead, was different: It was outperformed by the wild type in DMSO and did not improve in benomyl (Fig 5F). Yet, the disomic strain decreased sensitivity to benomyl when the experiment was performed at 18°C (i.e., the temperature of the evolution experiment) (Fig EV4D). In this case, however, duplication of SPC97 did not account for the rescue: two copies of the gene did not improve growth, and the deletion of one copy in the disomic strain did not decrease it. We elaborate on this result in the Discussion. Finally, kip3Δ and tub1D246Y improved growth also at 18°C, as expected.
We conclude that the recurrent mutations we tested suppress the tub2‐401 mutations, and also decrease sensitivity to benomyl. chrVIII 2X suppresses partially the tub2‐401 mutations due to the duplication of SPC97 and makes cells more resistant to benomyl at low temperature.
Epistatic interactions
Next, we aimed at understanding the evolutionary trajectories leading to resistance. To this aim, we combined three of the genetic changes analyzed so far: Q219H, disomy of chrVIII and kip3Δ, always in the presence of the tub2‐401 allele (Fig 6A).
Figure 6. Epistatic effects of adaptive mutations.
- Growth rates measured with in liquid growth assays. Experiments were performed at 18°C after growing cells at 18°C for 24 h. Growth rates result from fitting optical density over time with an exponential (see Materials and Methods for details). For each experiment, growth rates are normalized to the median of the ancestor tub2‐401. Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (ns = P‐value > 0.5). The group comparison among strains with two genomic changes is made using Kruskal‐Wallis test (P‐value = 0.149). Number of biological (N) and technical replicates (n) are, from left to right: tub2‐401 (N = 21, n = 253), tub2‐401 chrVIII 2X (13, 142), tub2‐401 kip3Δ (7, 71), tub2‐401‐Q219H (7, 71), tub2‐401 chrVIII 2X kip3Δ (10, 87), tub2‐401‐Q219H chrVIII 2X (11, 86), tub2‐401‐Q219H kip3Δ (3, 22), tub2‐4101‐Q219H chrVIII 2X kip3Δ (3, 23). The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box.
- Epistatic interactions among adaptive mutations. Epistasis ε is the difference between the observed fitness and the one calculated summing the fitness of individual mutations. Each symbol represents one experiment from panel A where all the strains needed to compute ε are present. Bars are the standard error. is computed as the weighted mean over independent experiments. The calculations were done assuming normalized growth rate as a proxy for fitness. See Materials and Methods for details.
- The median values of the measurements shown in (A). On the x‐axis, we report the number of genetic changes, i.e., point mutations or changes in ploidy. Solid lines identify changes that we propose to have occurred during emergence of resistance. Dotted lines, instead, are changes that have not occurred. For the sake of clarity, Q219H kip3Δ is excluded from this panel.
Q219H was the most effective single mutation in terms of increase of growth rate, followed by deletion of KIP3 and disomy of chrVIII. Any pairwise combination of these three gave a similar increase in growth rate over the ancestor tub2‐401. Hence, additional mutations had the least effect on Q219H, which alone showed a behavior most similar to the wild type (Fig 5A). With the assumption that growth rate is an approximation of fitness, we could quantitatively compute the epistatic interactions. We found that Q219H shows negative epistasis with either chrVIII 2X or kip3Δ (Fig 6B). Both kip3Δ and chrVIII 2X, instead, starting from a lower growth rate benefited from the addition of any of the other two genetic changes (compare column 3, 5, and 7 for kip3Δ and 2, 5, and 6 for chrVIII 2X in Fig 6A). Their interaction showed a slight positive epistasis (Fig 6B). Finally, when we introduced in the same strain all three changes, we observed that the triple mutant fared worse than double mutants (Fig 6A), showing the strongest negative epistasis (Fig 6B). This result further supports our conclusion that evolved strains at the final generation Gf carry disomy of chrVIII and only one additional mutation in the genes we identified.
We then drew the fitness landscape and analyzed the evolutionary trajectories that cells have taken to develop resistance (Fig 6C). The analysis is useful since it allows to compare actual trajectories to all those potentially possible. It is worth noticing that cells seem to have avoided the most direct evolutionary trajectory to Q219H, the fittest solution together with Q219H chrVIII 2X (Fig 6A). We already noticed that at the time of recovery (Gr; Fig 3B) cells show widespread disomy of chrVIII and very limited point mutations. Hence, possibly due to the increased chromosome missegregation rate (Fig EV1C), the first step along the emergence of resistance is disomy of chrVIII. Only later, cells acquired point mutations. Some of these, like deletion of KIP3, increase their fitness when combined with chrVIII 2X, and thus, these cells are unlikely to become euploid. Others, like Q219H, have the same fitness with or without disomy of chromosome VIII, and in fact apparently partially rescue the growth defects coming with disomy of chrVIII (data not shown). Depending on other mutations, however, they may find advantageous becoming euploid again. Accordingly, we only observed a decrease of aneuploidy in the final generation within populations that have mutations in TUB2 (Fig 3A).
We conclude that adaptive genetic changes display epistatic interactions. Mutations in TUB2 give the largest fitness increase. Nevertheless, they are not the first to occur but follow disomy of chrVIII. The same is true for the other point mutations. Whether cells will eventually keep the mutations and loose the disomy will likely depend on the fitness gain coming with the adaptive mutation alone.
Evolved cells have improved microtubule function
Finally, we aimed at verifying that cells carrying mutations typical of Gf have restored microtubule function. tub2‐401 cells can grow at the semi‐permissive temperature of 23°C. However, partial destabilization of microtubules requires the ability of cells to arrest cell cycle progression to correct attachment errors and spindle misalignment. Indeed, at this temperature the cell cycle relies on the mitotic checkpoint and on the spindle position checkpoint (Fig EV1B). We reasoned that if evolved cells improved microtubule functions, their cell cycle should rely less on either of the two checkpoints. Hence, we deleted MAD2 and BUB2 in engineered Gr and Gf, and observed cellular viability by dilution assays at 23°C (Fig 7A). The results confirmed that engineered Gf and Gr grow better than the ancestor. In bub2Δ, deletion of KIP3 gave an advantage on top of disomy of chrVIII, which was not observed in mad2Δ. Apparently, the deletion of KIP3 does not improve much kinetochore/microtubule attachment, but it may restore spindle positioning. It surely has an effect on the mitotic spindle, since we observe further elongation of these cellular structures following KIP3 deletion in disomic strains (Figs 7B and EV5A and B).
Figure 7. Phenotypic characterization of engineered Gf and Gr.
- Serial 5‐fold dilution of ancestors and engineered Gr and Gf cells after deletion of MAD2 and BUB2 spotted on YPD and incubated at the indicated temperatures.
- Ancestors and mutant strains were synchronized in G1 at 30°C and released at 18°C. Spindle lengths were measured on Tub1 immunodecorated microtubules across three timepoints centered on the maximum fraction of large‐budded cells (see Fig EV5A). Pairwise strain comparisons were made using a linear model, adjusting for batch effects for experiments performed on different days. Symbols refer to the P‐values of the strain comparison (***P‐value < 10−3, ****P‐value < 10−4). The plot includes two biological replicates, and at least 260 cells per strain overall. The boxes span the interquartile range (IQR, from the 25th to the 75th percentiles), and the central band represents the median. The lower (upper) whisker extends from the box to the smallest (largest) value no further than 1.5*IQR from the box. Individual measures are plotted as dots.
- Missegregation in ancestors and engineered evolved strains, both Gr and Gf. Cells were synchronized and released at 18°C. At different timepoints, they were collected and fixed (see Materials and Methods). Missegregation events were scored as described in Fig EV5C. The plot shows the joined data from 3 biological replicates, with at least 220 cells overall per strain per timepoint.
Figure EV5. Characterization of microtubule function in Gr and Gf.
- Cells were synchronized in G1 at 30°C and released at 18°C, as explained in Materials and Methods. The fraction of cells with a large bud was monitored every 30 min from 3.5 to 5 h after G1 release. Black dots identify the timepoints in which the spindle lengths were measured (see Fig 7B). The kinetics is representative of one the biological replicate.
- Representative spindles of the different strains shown in Fig 7B. Tub1 was stained for immunofluorescence, while nuclei were stained with DAPI. Images are examples from one biological replicate.
- Morphologies of cells that were scored as missegregated in Fig 7C based on the number of chrV‐GFP dots. Budded cells with two dots in the mother cell could not be attributed as missegregated, since they could either end up segregating properly or missegregating, as observed in Movie EV1. In the movie, the proper assignment was based on the pattern of budding (e.g., cells that kept two dots after budding were assigned as missegregated), similarly to what done here.
These data suggest that evolved cells missegregate fewer chromosomes than ancestors. To test this hypothesis, we introduced chrV‐GFP in engineered Gf strains. Cells were synchronized in G1 and released at 18°C. We kept track of chromosome missegregation (Figs 7C and EV5C) and observed a clear decrease of cells with missegregated chromosome V in engineered Gf cells compared to ancestors. An intermediate recovery was observed in disomic strains (i.e., Gr engineered).
In summary, our data show that evolved cells improved microtubule functions: They were less dependent on SAC and SPOC, assembled longer mitotic spindles, and missegregated less chromosomes.
Discussion
We evolved cells expressing mutations impairing microtubules polymerization when grown at low temperature (tub2‐401). Initially, tub2‐401 cells were delayed in mitosis by the mitotic checkpoint and the spindle position checkpoint. Nevertheless, the cell cycle did not stop: cells divided, missegregated chromosomes, and died in large numbers. As a result, growth rate was much reduced compared to wild types. However, mutations emerged that allowed cells to recover microtubule function. Hence, after the initial decrease, the growth rate improved with the selection of beneficiary mutation. At the end of the experiment, after roughly 150 generations, evolved cells gained again the ability to assemble microtubules, displayed fewer errors in chromosome segregation, ploidy became stable and the cell cycle was less delayed in mitosis than in the ancestors. Evolved cells were also capable to survive in the absence of SAC and SPOC at 23°C, further showing the improved microtubule function that characterizes these cells. As a result, growth rate was largely rescued, although it never went back to wild‐type levels.
We evolved in parallel 24 different populations. Remarkably, they were characterized by a simple karyotype with only one disomic chromosome (chrVIII) across most populations. The pattern of recurrently mutated genes was also simple and included only two classes: tubulins and kinesins. By introducing in the ancestors strains the disomy or some of the most frequently observed mutations in these two classes, we could ascertain their adaptive nature and analyze their epistatic interactions. Indeed, the results obtained with engineered strains carrying mutations typical of Gf and Gr mimic what observed in the evolution experiment. Both engineered Gf and Gr outperform the ancestor strains. However, the differences between evolved Gr and Gf are milder than what observed in engineered strains. Probably, this can be explained by the fact that evolved Gf and Gr are mixed populations, and by the additional role played by mutations in non‐recurrently mutated genes.
The two adaptive processes (change of ploidy and point mutations) are not interchangeable, but follow a precise temporal sequence. When we analyzed populations at an earlier timepoint, when they had partially recovered growth, we found chrVIII 2X with high frequency in most of them (> 80%). On the contrary, there was a paucity of single‐nucleotide mutations. Hence, we propose that disomy of chromosome VIII provides the first compensatory solution against the impairing mutations. We further showed that this was partly due to the duplication of SPC97, a member of gamma‐TuSC. At a later time‐point, sequencing confirmed the presence of disomy for chromosome VIII in the large majority of populations. In addition, cells in all populations had mutated one, but not more, of the recurrently affected genes. We speculate that the simultaneous presence of two adaptive mutations is counterselected due to some function that depends on microtubules. The number of mutations we observed was in agreement with the measured fitness coefficient, known mutation rates, population numbers (˜106–107 cells), and generations number, according to a standard Wright‐Fisher model with mutations and selection (see Appendix Supplementary Methods, Appendix Figs S1 and S2).
Hence, disomy of chromosome VIII comes first, mutations of tubulins or KIP3 follow. The repetition of these evolutionary paths of compensatory mutations in several independent experiments shows that the emergence of resistance is potentially predictable.
Aneuploidy is an adaptive solution to impaired microtubule polymerization
The role of aneuploidy as a fast adaptive solution has been reported for several sources of stress (Rancati et al, 2008; Selmecki et al, 2009; Yona et al, 2012; Millet et al, 2015). Indeed, stimuli that increase chromosome missegregation rate generate genetic variability which favors the arising of resistance to other treatments as well (Chen et al, 2012). Whether this solution persists in time, instead, is not obvious. Aneuploidy is a fast but costly solution. As such, in other contexts it was shown to be replaced on the long run by more precise and less detrimental point mutations (Yona et al, 2012). However, our data show that this is not necessarily the case. For example, in the case of KIP3 deletion the combination with an extra copy of chromosome VIII does not show a detrimental effect. In this case, we do not expect a loss of disomy of chromosome VIII as long as Kip3 is dysfunctional. On the contrary, for the tubulin mutations Q219H, the loss of extra chromosome VIII is not a disadvantage. Accordingly, we observed a few euploid populations that carry only mutations in TUB2 at the end of the experiment, while all were disomic at an earlier timepoint.
Is aneuploidy of chromosome VIII a generic adaptive solution for stimuli that destabilize microtubules? Our data show that this is not always the case: disomic strains at 30°C are not resistant to benomyl, although a minor effect was detected at 18°C. The latter, however, could not be explained by the increased levels of Spc97, which could account only partially for the compensatory effect to tub2‐401. Clearly, there are other unknown genes whose copy number increase contributes to the adaptive effect, as shown for other aneuploidies (Bonney et al, 2015). We tested the overexpression of other loci, but we failed to identify other candidates; maybe a combination of genes located on chromosome VIII accounts for the disomy. One possibility is that duplication of some genes increases fitness independently from microtubules. What we observe could be an adaptive response to the low temperature, since disomic strains grow better at 18°C compared to 30°C. This hypothesis fits with the fact that adaptive mutations are all mutually exclusive except for chrVIII 2X. However, none of our controls became diploid for chrVIII, showing that low temperature per se does not suffice to generate disomy. Thus, chrVIII 2X may be a response to the combined stress of low temperature and impaired microtubule dynamics, which tub2‐401 cells experience at 18°C.
This does not imply, however, that aneuploidy is a solution unique to our experimental set‐up. Diploid yeast cells develop aneuploidies upon treatment with benomyl, although without involving extra copies of chr VIII (Chen et al, 2012). We confirmed that also haploids develop aneuploidies when grown in agar in the presence of benomyl (data not shown). Interestingly, aneuploidy as an adaptive response to increased chromosome missegregation was recently analyzed (Ravichandran et al, 2018). In particular, the authors studied the effect of inactivating the chromosome passenger complex via deletion of the survivin homolog BIR1. The adaptive solutions they found are partially different from ours. There are no recurrently mutated genes, and among the genes mutated there are not those that we identify as recurrently mutated. This is maybe not surprising, since microtubule polymerization is not affected by the deletion of BIR1. Aneuploidy, instead, is a major adaptive event in both experiments. The evolved karyotypes partially overlap, but are not identical. Both studies identify disomies in chrVIII and chrIII (the latter being very limited in our study). Ravichandran, Campbell, and co‐workers, however, identified more complex karyotypes, which include disomies of chrII and chrX. Hence, how cells react to chromosomal instability at least in part depends on the stimulus that triggers it. However, it will be important to understand whether specific aneuploidies provide a generic mechanism to cope with high chromosome missegregation rates per se.
Compensatory mutations are largely due to “recovery of functions”
Both high‐throughput studies and specific in‐depth analyses in yeast showed that cells can adopt different strategies to recover from growth defects: aneuploidies, gain‐of‐function mutations, loss‐of‐function mutations, large structural variants (Szamecz et al, 2014). However, not all strategies occur with the same frequency. Previous evolution repair experiments indicated that loss‐of‐function mutations tend to occur most often (LaBar et al, 2020). In our case, we observed something different. Multiple mutations occurred in alpha‐, beta‐, and gamma‐tubulins, as well as in the other elements of the gamma‐TuSC complex. None of them were obvious complete loss of function, i.e., nonsense or frameshift mutations. Only in KIP3 we found such mutations. In this gene, we also identified several missense mutations, which can also be interpreted as partial loss of function, likely partly preserving enzymatic activity (e.g., the two mutations occurring in the L11 domain).
Why are there so few complete loss of function in our evolution experiment? Compared to previous studies (Rancati et al, 2008; Laan et al, 2015; Fumasoni & Murray, 2020), an obvious difference is that we did not start our experiment with a deletion but with three impairing amino acid substitutions. As such, mutations can bring about recovery of function. With this term, we do not mean revertants, which strictly speaking we do not observe. Rather, we refer to mutations that can make use of the mutated and partially dysfunctional tubulin. Such mutations can be intragenic, occurring in TUB2, or in other tubulin genes whose products interact directly with beta‐tubulin to form microtubules (e.g., SPC98, TUB4, TUB1). For all these interactors, mutations that recover growth are unlikely to be loss of function given the essential role of tubulins. The situation is quite different for genes that do not contribute structurally to microtubules but control their stability. In this case, loss‐of‐function mutants are definitely possible, as is the case for mutations affecting KIP3. Such inactivation of negative regulators of microtubule stability is conceptually very similar to what observed, for example, in cell polarity mutants (Laan et al, 2015).
What can we learn from laboratory evolution experiments on the mechanisms leading to resistance to antimitotics?
Several mutations related to resistance against microtubule drugs have been identified, including the differential expression of different isotypes of tubulin, expression levels of microtubule‐associated proteins, and multidrug resistance mechanisms (Ueda et al, 1987; Kavallaris, 2010; Parker et al, 2017). The role of mutations in tubulin genes has been debated. They develop frequently in cell lines, but are harder to find in patients (Berrieman et al, 2004). Recent data, however, have yet again pointed at a potential role in patients for mutations in tubulin genes (Wang et al, 2017). Finally, mutations in proteins interacting with tubulin such as kinesins have also been reported to affect the development of resistance to drug treatment (De et al, 2009; Rath & Kozielski, 2012). However, according to our best knowledge, the time‐resolved series of changes that takes place during the emergence of resistance toward microtubule‐targeting drugs is not explored yet. Moreover, we have limited knowledge about the potential epistatic interactions among adaptive mutations developing in the presence of such drugs.
We impaired microtubule polymerization with mutations mimicking the presence of drugs. The genetic changes we identified recovered robustly growth in the presence of benomyl (with the partial exception of chrVIII 2X). Similar drugs targeting microtubules are routinely used for treating cancer patients. We can then ask whether some of the properties of the adaptive mutations identified in yeast can be extended to mammals as well. One obvious caveat is that the use of microtubule impairing mutations in lieu of real drugs prevented us from finding mechanisms of resistance via direct interaction with drugs. This may actually be an advantage. Some of these mechanisms (e.g., the role of transmembrane transporters) are quite well characterized. Instead, our analysis could unveil properties of mutations in kinesins and tubulins that are less well understood and yet involved in the development of resistance in human cells (De et al, 2009; Rath & Kozielski, 2012; Wang et al, 2017).
To further stress the potential relevance of our approach, we asked whether from an evolutionary viewpoint beta‐tubulin is altered similarly in cancer cells and in our experimental system. To answer this question, we kept track of residues mutated in the two systems. Our eight mutated residues are shown in Fig 2A, while we identified residues mutated in cancer patients (37) using a database of tubulin mutations (https://tubulinmutations.bio.uci.edu/ (Pham & Morrissette, 2019)). We used the same database to estimate an upper limit for the number of beta‐tubulin residues mutated in mammals and yeast (289). We found the enrichment between residues mutated in patients and in our experiment to be statistically significant (P = 0.011; Fig EV6). This result suggests that, given the high level of conservation, under stressful conditions similar selection mechanisms targeting tubulins may take place in yeast and mammals.
Figure EV6. Overlap of residues mutated in human patients and evolved yeast.
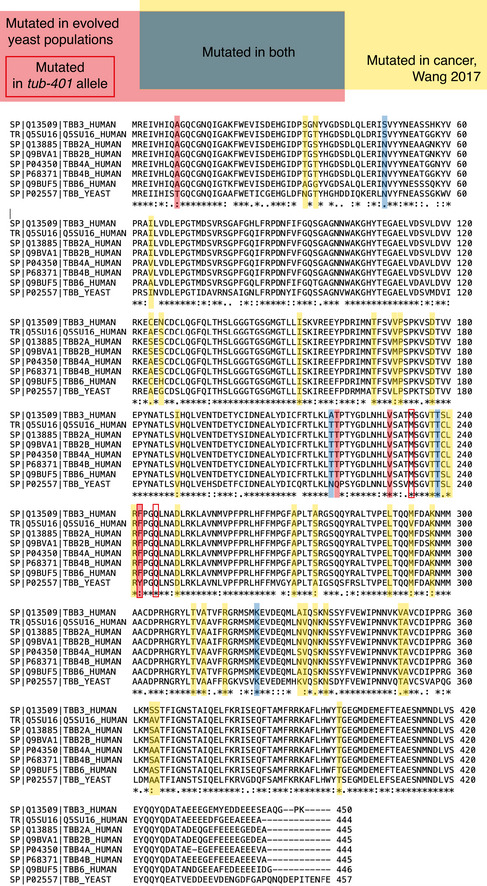
Taking advantage of a database which includes mutations found in beta‐tubulin across eukaryotes (https://tubulinmutations.bio.uci.edu/), we identified beta‐tubulin residues mutated in cancer patients (37). The total number of entries in the database gave us an estimate of the upper limit for the number of residues that can be mutated in yeast and mammals (289). During the evolution experiment, we observed mutations in eight different residues. Of these mutations 4 are among the 37 found in cancer patients, which is a statistically significant overlap (P‐value 0.011, hypergeometric test). Notice that even though not all residues are conserved, superposition of mammalian beta‐tubulin with Tub2 (carried out on 419 C‐alpha's, with a RMSD of 1.01 Å) reveals that all the residues emerging in our study spatially overlap with the corresponding counterparts of mammalian beta‐tubulin highlighted in the sequence alignment. However, the result remains statistically significant including only residues conserved between yeast and at least one human isoform.
An interesting property of the adaptive mutations we identified is that they are mutually exclusive. Our data suggest that they counteract the original tub2‐401 by reinforcing microtubule stability. We did not find mutations that “silenced” tub2‐401 by other means (e.g., activation of chaperons, re‐purposing of other cellular components). As such, coexistence of multiple mutations may become detrimental for some microtubule function. This is reminiscent, for example, of mutations causing resistance to treatment with anti‐EGFR therapy (Misale et al, 2014). Also in that context, mutations conferring resistance were largely mutually exclusive, and it was noted that they all pointed to the same downstream MAPK pathway. This convergence allows in principle the development of effective therapies, bypassing the obvious challenge that the heterogeneity of adaptive mutations poses to treatment. In our instance, for example, resistant cells should be very sensitive to stimuli increasing microtubule stability, regardless of their different genetic basis. Alternatively, our data suggest that a notable exception to mutual exclusivity may be aneuploidy. Hence, targeting aneuploidy during the early stages of emergence of resistance could exhaustively affect the population of resistant cells.
In conclusion, evolutionary studies in yeast may have a yet unappreciated potential to understand the much more complicated emergence of resistance to treatment in mammalian cells.
Material and Methods
Yeast strains
All yeast strains listed in Table EV3 are derivatives of or were backcrossed at least five times with W303. Here, we used a modified W303, prototroph for uracil and tryptophan (ura3::URA3 and trp1::TRP1 (2X)). The evolutionary experiment was performed using MATa haploid strains.
Deletions were obtained by one‐step gene replacement, following S‐primer strategy described in (Knop et al, 1999). For PHO4, KIP3, and SPC97 deletions, PCR was made on plasmid pl67 (pFA6a‐kanMX6 (Longtine et al, 1998)) to obtain KanMX cassette. SPC97 deletion was performed on chrVIII disomic strains, since the gene is essential.
To obtain tub2‐401A657T mutant (Q219H), plasmid pAC149 carrying tub2‐401 allele (a gift from T. Huffaker, Department of Molecular Biology and Genetics, Cornell University—Ithaca, United States) was mutagenized by PCR using primers 455/456 to obtain plasmid pAC173. Then, following (Huffaker et al, 1988), this plasmid was digested with KpnI to transform yeast, directing the integration at TUB2 locus. Transformants were plated on 5‐fluoro‐orotic acid to select clones that excided the URA3 marker, and the resulting colonies were checked by drop test at 18°C and compared with tub2‐401 mutants: The ones that grew better than tub2‐401 were checked by Sanger sequencing to confirm the presence of the tub2‐401A657T allele.
To obtain tub1D246Y mutant, TUB1 ORF and 569 bp downstream the STOP codon were amplified by PCR made on a wt strain (primers 506/507) and cloned into BamHI/PacI sites of plasmid pAC67 (pFA6a‐kanMX6). TUB1 gene was then mutagenized by PCR with primers 517/518 to obtain pAC183. Endogenous TUB1 was swapped with the mutant allele by transformation of yeast with a tub1D246Y::KanMX cassette amplified by PCR on pAC183 using primers 519/520. Transformants were then checked by Sanger sequencing to confirm swapping.
To duplicate SPC97, we followed (Huber et al, 2014). We duplicated the genomic region from the end of ARS820C (first gene upstream SPC97, on the other strand) to the first codon of YHR173C (first gene downstream SPC97, on the other strand). In this way, we kept 5′‐ and 3′UTRs of SPC97, avoiding duplicating the ATG of YHR173C.
Plasmids and oligos described above are listed in Tables EV4 and EV5
Media and growth conditions
Unless otherwise stated, cells were grown in YP medium (1% yeast extract, 2% Bacto Peptone, 50 mg/l adenine) supplemented with 2% glucose (YPD). For the evolutionary experiment, YPD medium was supplemented with Penicillin‐Streptomycin 100X (BioWest).
Live‐cell imaging experiments were performed in synthetic complete medium supplemented with ammonium sulfate and 2% glucose (SCD).
To arrest cells in G1, α‐factor (5 μg/ml) was added to cells growing exponentially at 30°C. After 1 h and 30 min, α‐factor was re‐added at half the concentration (2.5 μg/ml). If cells had to be released from G1 arrest at 18°C, after 2 h from the first α‐factor addition cells were released at 18°C in fresh medium supplemented with α‐factor (5 μg/ml) and left grown at 18°C for 1 h before release from the G1 arrest.
Nocodazole was used at 2 or 15 ug/ml and re‐added at half the concentration 2.5 h after the first addition.
Evolution experiment
Ancestral strains were thawed and isolated on YPD plates at 30°C. 24 independent colonies of tub2‐401 and eight of TUB2 were inoculated at 30°C in 800 μl of YPD in 96‐deepwell plates and shifted at 18°C the day after. The initial population size, after 30°C growth, was roughly 107 cells for each well. During evolution at 18°C, the population size was roughly 5 × 106 cells. During cell growth, plates were covered with aeraseals (Sigma) and incubated at 18°C under constant orbital shaking on deepwell block tilted adaptors. All the clones were cultured in the same 96‐deepwell plate and handled with a TECAN Freedom EVO 150 liquid handler. Cells were diluted twice a day in 800 μl of fresh cold YPD (18°C) to maintain the exponential phase, and assessed for growth rate every 3–4 days. To avoid bottle neck effect during evolution, tub2‐401 and TUB2 cells were diluted, respectively, 1:2/1:6 and 1:8/1:10 (depending on growth rate during evolution and on the OD600 reached during the previous dilution).
To measure growth rate, cells were diluted at OD600 0.2 in 96‐microtiter plates (NUNC—Lifetechnologies) and cultured at 18°C for 8 h while measured every hour using a Cytation 5 (BioTek) OD600 plate reader. For each timepoint, OD600 of each well was acquired.
For each experiment, one or more wells were filled with clean medium, both to check for contamination and to have a measure of background OD600. The background OD600 was subtracted to the raw OD600 to compute the net OD600. For each well, the net OD600 was fitted against the actual time of measurement to estimate apparent growth rate (α), following the equation:
To control for “goodness of fit”, we used Root‐mean‐square deviation RMSD, defined as:
where N is the number of measurements, yi are the measured values, and fi are the fitted values.
Wells with RMSD greater than 0.1 were excluded from the results.
In liquid growth assay (GA) on engineered strains
Cells were inoculated in glass flask in YPD medium at 30°C and grown overnight. The day after, each culture was diluted at OD600 0.1 and divided in eight wells of a deepwell block plate (Greiner‐Bio One) and grown for 24 h at 18°C under constant shaking. Then, cells were diluted in 96‐well microtiter plates (NUNC—Lifetechnologies) at net OD600 0.025 in 200 μl of fresh and cold (18°C) YPD medium and incubated at 18°C under constant shaking. OD600 were measured every hour for 8 h using TECAN 200 M Infinite plate reader. For each timepoint, OD600 of each well was acquired five times, and the mean value was used for further analysis. Measurements with OD600 above 1.5 or below 0.01 were removed, since the linearity range sits between these values (data not shown).
Growth rate was measured as presented above for the evolution experiment.
Growth assay on agar plates
Cell growth in liquid
Cells were inoculated in flasks in YPD medium and grown overnight at 30°C. The day after, cells were aliquoted onto two 96‐well plates, named layout and control plates. In the layout plate, target strains were arranged in a checkboard fashion, each in at least 6 different wells, and the external rows and columns filled with wild‐type control cultures. In the control plate, only wild‐type was present and used to correct for systematic experimental variability.
First pinning
To produce the 384‐colonies plates for incubation, layout and control plates were briefly spun down, then pinned onto an YPD agar plate such that each 2 × 2‐colonies square comprised three colonies of the same target strain and one colony of the control. Because of the layout plate design, the two more external lines of colonies in the incubation plates were wild‐type: These colonies were meant to give to the next‐line target colonies a neighbor that compete for nutrients. Pinning was made using Singer ROTOR, equipped with Repads 96 Long pins, mixing the source cells suspension, and touching only once the target incubation plate. At least three identical incubation plates were produced for each experimental replicate. Incubation plates were then sealed and placed at 30°C to grow for 1–2 days.
Second pinning
To pour target agar plates, melted YPD + 2% agar was cooled to ˜50°C while stirred to prevent solidification. DMSO or DMSO supplied with 100X concentration of benomyl was added to the medium. 45 ml of medium was poured in each plate (Singer PlusPlates). 384‐colonies incubation plates were replicated twice onto cool and dry target plates (768 colonies on each plate). Replication was made using Singer ROTOR, equipped with Repads 384 short pins, and dry mixing the source and touching only once the target plate. Target plates were placed at either 18 or 30°C, depending on the experimental design.
Image acquisition and analysis
The 768‐colonies plates were imaged after 1 day (30°C) or 2 days (18°C) using constant image size with a Chemidoc XRS+System (Bio‐Rad Laboratories). Colonies were identified as regions of interest (ROIs) by thresholding the digital images with a custom ImageJ script. Raw colony size is the area in pixels of each ROI. Each raw colony size was normalized on the closest wild‐type colony, coming from the same 2 × 2‐colonies square in the incubation plate, to correct for spatial effects due to uneven pinning or nutrients.
Epistasis
Epistasis ε is the difference between the measured and the expected fitness of the multiple mutant assuming that the mutations are independent. Following (Krug, 2018) (equation 26), expected fitness is computed as
where and are the fitness of the two mutants, and is the one of the ancestor. By defining as the measured fitness of the multiple mutant, epistasis is computed as
For each experiment, is the median of the normalized growth rate of eight technical replicates of the i‐th strain. Standard error of the epistasis is computed as the root of the sum of the squared standard errors of each .
The final epistasis is the weighted average of the replicates, using the standard errors as weights. Standard error of the weighted mean is
The P‐value is a standard one‐tail P‐value test (right tail for positive epistasis, left tail for negative epistasis) where we have used a Gaussian approximation with mean and standard deviation .
Drop test assay
Cells were grown overnight at 30°C and diluted at the same concentration. Five‐fold serial dilutions were then spotted on YPD + agar plates and incubated at 30°C for 1 day and at 23°C for 2 days.
Tubulin immunofluorescence
Cells were arrested in G1 as described above and released at 18°C. Samples were collected every 30 min from 3 to 5 h after the release. For each strain, the three timepoints with the highest fraction of large‐budded cells (dumbbell) were treated for immunofluorescence as follows.
1 ml of cells was collected and fixed with 3.7% formaldehyde in KPi buffer (0.1 M Kphos pH6.4 0.5 mM MgCl2) and incubated overnight at 4°C.
Cells were washed three times with KPi buffer and once with Sorbitol solution (1.2 M Sorbitol, 0.1 M KPi pH 7.4, 0.5 mM MgCl2). Then, cell wall was digested at 37°C with 200 μl of Sorbitol solution supplemented with 5 μl of zymolyase 10 mg/ml and 2 μl of 2‐mercaptoethanol for 15/20 min and washed with Sorbitol solution. 5 μl of spheroplasts were loaded to glass slides (Thermo Scientific) coated with polylysine (Sigma‐Aldrich). After 15/20 min, slides were immersed in cold MeOH (−20°C) for 3 min and in cold acetone (−20°C) for 10 s. Samples were incubated with anti‐Tub1 primary antibody (YOL1/34 Bio‐Rad), for 2 h at room temperature and washed three times with 1% BSA‐PBS. Samples were then incubated with secondary antibody (FITC‐conjugated anti‐rat antibody from Jackson ImmunoResearch Laboratories, pre‐absorbed) for 1 h at room temperature in a dark chamber and washed four times with 1% BSA‐PBS. DAPI/antifade (100 mg p‐phenylenediamine + 10 ml PBS adjust to pH 8.0 ad DAPI 0.05 μg/ml) was added in each sample and slides were closed. Images were acquired using DeltaVision Elite imaging system (Applied Precision) based on an inverted microscope (IX71; Olympus) with a camera (CoolSNAP HQ2; Photometrics) and an UPlanFL N100x oil immersion objective lens (NA 1.4, Olympus).
Spindle length was measured with a custom script (a gift from Rosella Visintin's laboratory) written in Fiji. This script computes the Laplacian of the maximum projection of fluorescence intensities, identifies the spindle using the RidgeDetection plugin on the cell selected by the user, and computes the spindle length. We measured budded cells, as identified by the DAPI signal. To include in the analysis only cells that had developed a mitotic spindle, we analyzed only those with a spindle longer than 1 μm. At least 100 cells per sample were included in the analysis.
Flow cytometry for cell cycle analysis
Cells were treated according to Pavelka et al (2010). Briefly, exponentially growing cells fixed overnight in EtOH 70% were incubated in RNAse A solution (0.05 M NaCitrate, 0.25 mg/ml RNAse A) for 3 h at 37°C then in proteinase K solution (0.05 M Tris–HCl pH8, 0.01 M CaCl2, 0.25 mg/ml Proteinase K) overnight at 55°C. After staining for 20 min in Sytox Green Solution (1 μM Sytox Green, 0.05 M NaCitrate), samples were sonicated and acquired using attune cytofluorimeter (Thermo Scientific). Sytox fluorescence was acquired using BL1 laser. fcs files were analyzed using a customized R script using ggplot for visualization. Singlets were gated on SSC parameters. Cell cycle phases were discriminated based on Sytox Green signal (DNA content).
Apparent ploidy/DNA content
Cells were treated as presented in the paragraph Flow Cytometry for cell cycle analysis. Samples were acquired using MACSquant Analyzer (Miltenyi Biotec) using the same voltage settings in every measurement. Sytox fluorescence was acquired using BL1 laser with a flow rate of 1,000 events/s.
Data analysis was performed using a custom MATLAB script. Singlets were gated using physical parameters (SSC and FSC). DNA content was computed using the G2/M mode of the binned Sytox signal, using bins whose width is ˜1/1,000 of the instrument range.
The mean value of the eight TUB2 evolving populations across every timepoint was used as normalizer, based on the observation that these populations come from an euploid strain (Fig EV1D) and are euploid at Gr and Gf (Fig 3). In Fig 1C, a dashed line marks 1.04: this is the expected DNA content of a strain disomic for chromosome VIII, based on the fact that chromosome VIII's length represents ˜4% of the euploid genome size.
Sanger sequencing of selected genes
The indicated evolved strains were streaked on agar plates and incubated at 18°C until colonies were visible (about 5 days). Then, colonies were picked and amplified on agar at 18°C to make a glycerol stock. PCR to amplify genes to be sequenced was done directly from colonies or after defrosting and growing on agar at 18°C: cells were boiled in 3 μl of 20 mM NaOH for 10 min and directly added to PCR mix. The amplification products were then Sanger‐sequenced.
Evaluation of missegregation in EVO eng cells
Cells were grown overnight at 30°C and synchronized in G1, as described above, and released at 18°C. Cells were fixed after 0, 4, 6, 8, and 12 h in cold pure EtOH. 100 μl of fixed cells was dissolved in 900 μl of 50 mM Tris, pH 7.6. The cells were sonicated, pelleted, and resuspended in the remaining liquid after discarding the supernatant. 5 μl of cells were loaded on a slide coated with 2% agar layer. Samples were imaged for ChrV‐GFP with DeltaVision Elite imaging system (Applied Precision) based on an inverted microscope (IX71; Olympus), a UPlanFL N 60× (1.25 NA) oil immersion objective lens (Olympus), and a camera (Scientific CMOS Camera). GFP was acquired using single bandpass filters (EX475/28 EM523/36) with 25 z‐stack (0.2 μm), exposure time 0.05 s, and power lamp 100%. After acquisition, images were deconvolved using SoftWorx. Missegregation events were determined as described in Fig EV5C.
Live cells imaging experiment
To measure missegregation rate by single cell analysis, cells were grown overnight at 30°C in SCD. The day after, cells were diluted at OD600 0.15, grown for 1 h, supplied with α‐factor (5 μg/ml), shifted at 18°C for 1 h, sonicated, and loaded on a microfluidic chamber (CellASIC), where they grew at 18°C supplied with α‐factor for 30 min. Then, cells were released from G1 arrest and grown for 41 h at 18°C in the microfluidic chamber under the microscope. Missegregation was identified by following the positions of GFP‐tagged chromosomes V after the first cell division or adaptation. Temperature was controlled as in (Corno et al, 2019) using a BOLD LINE Water‐Jacket Top Stage Incubation System (OKOlab).
Time‐lapse movies were recorded using a DeltaVision Elite imaging system (Applied Precision) based on an inverted microscope (IX71; Olympus), a UPlanFL N 60x (1.25 NA) oil immersion objective lens (Olympus), and a camera (Scientific CMOS Camera).
Reference images were taken every 15 min, while fluorescence images every hour. GFP was acquired using single bandpass filters (EX475/28 EM523/36) with 5 z‐stack (0.85 μm), exposure time 0.105 s, and power lamp 10%. To evaluate any phototoxicity of the acquisition settings, cell cycle duration of excited and non‐excited fields of TUB2 strains was compared, and no difference was observed. Z‐stacks were deconvolved with SoftWoRx software and then projected with maximum intensity projection with Fiji.
Cells were segmented and tracked using a custom version of phylocell (Charvin et al, 2008), and missegregation was assessed semi‐automatically using a MATLAB script based on the FindPeaks function.
Whole‐genome sequencing and analysis
DNA extraction
To extract DNA of ancestors or evolved populations (Gr and Gf), cells were inoculated in YPD at 30 or 18°C, respectively, and grown for 1 or 4 days, respectively. Then, DNA was extracted from 10 ml of yeast culture at stationary phase. Cell wall was digested at 37°C with 200 μl of SCE solution (1 M Sorbitol, 0.1 M NaCitrate, 0.06 M EDTA, pH 7.0) supplemented with 2 mg/ml of zymolyase and 8 μl/ml of 2‐mercaptoethanol for 30–60 min. Spheroplasts were lysed SDS solution (SDS 2%, 0.1 M Tris–HCl pH 9.0, 0.05 M EDTA) at 65°C for 5 min. DNA were purified with standard NH4OAc/isopropanol precipitation and resuspended in 200 μl of water supplemented with 1 μl of RNAse 10 mg/ml. RNA was removed by overnight digestion at 37°C. Following, DNA was purified with a second round of NH4OAc/Isopropanol precipitation and resuspended in water.
Sequencing and bam files creation
Sequencing was performed at Beijing Novogene Bioinformatics Technology Co., Ltd., using an Illumina high‐throughput sequencer returning 2 × 150 bp paired‐end reads.
FASTA files were trimmed using trimmomatic v0.36 (Bolger et al, 2014), then aligned to reference sequence (sacCer3) using Burrows‐Wheeler Aligner (BWA) v0.7.17 (Li & Durbin, 2010). Then, duplicates were removed using samblaster v0.1.24 (Faust & Hall, 2014) and sorted using samtools v1.9 (Li et al, 2009). Lastly, bam files were realigned around indels using Picard v2.19.0 (http://broadinstitute.github.io/picard/) and GATK v3.8‐1 (McKenna et al, 2010). Final bam files have an average depth of coverage of 50 reads. Notice that ancestors were clonal, while evolved populations were mixed.
Identification of single‐nucleotide changes and short indels
We aimed at identifying single‐nucleotide changes and short indels (less than ˜50 bp) filtering out variations of noisy genomic regions or specific of the genetic background (W303). To do so, we used IsoMut (Pipek et al, 2017). Briefly, this tool compares a set of isogenic sample sequences and calls a variation when it is unique for one of the sequences. To identify variations in each of our target samples, we fed IsoMut with a set of 11 sequences: the target sample sequence, plus 10 technical replicates of wild‐type yAC4179, which were used to remove W303‐specific variations and noisy genomic regions. As output, IsoMut returns the variations on non‐noisy regions that the sample does not share with the background set of 10 wild‐type. The variations in all the samples (ancestors and evolved populations) are then joined in a unique database.
We then filtered the mutations, using a conservative approach. Using a custom written R script based on VariantAnnotation v1.28.11 (Obenchain et al, 2014), mutations were mapped to genomic regions. Following Zhu et al (2014), we removed regions annotated in the SGD database as simple repeats, centromeric regions, telomeric regions, or LTRs (SGD project; http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.tab, downloaded July 26, 2019). To identify mutations with good signal, we removed those identified by < 5 reads, regardless of their allelic frequency, those with IsoMut cleanliness parameter < 0.98, those with IsoMut score < 0.21.
We analyzed the mutations found in the two ancestors, to assess their isogenicity. They differ by nine mutations (listed in Table EV6), five of which are nonsynonymous.
To identify potentially adaptive mutations, we removed from the evolved strains the mutations inherited from their ancestor and those found in mitochondrial DNA. We then identified genes mutated in more than one population at the end of the experiment (Gf) by unique mutations resulting in nonsynonymous amino acid changes. This definition gave a list of 10 genes (PHO4, PHO81, KIP3, TUB1, TUB2, SPC98, ADE6, PRR2, YHR033W, YJL070C). These genes form the core of the “recurrently mutated genes”, genes whose mutations are potentially adaptive to cold or impaired microtubule polymerization. In the final list (genes shown in Table EV1), we did not include PRR2, YHR033W, ADE6, and YJL070C as they were present only twice in very low frequencies. Instead, we included TUB4, which was mutated twice with high frequency but with identical mutations, and for this reason originally not present in the list. Nevertheless, we decided to include it based on the fact that we had identified SPC98, another component of gamma‐TuSC, which suggests mutations in gamma‐TuSC being potentially adaptive.
Having defined the recurrently mutated genes, eventually, we kept track of all mutations that hit any of them in any population. 61 mutations (listed in Table EV1) belong to this group. There are other 146 mutations which hit other genes and are not reported in Table EV1: either they did not fulfill the conditions to be “recurrently mutated” (unique mutations in more than one population), or were excluded (YHR033W, YJL070C, PRR2, ADE6) as explained above. Mutations of recurrently mutated genes have higher frequencies than the other mutations (median of mutations frequencies 0.218 and 0.086, respectively). In the manuscript, we focused our analysis on mutations hitting recurrently mutated genes.
Analysis of aneuploidy
Depth of coverage was extracted from bam files using a custom script written in python that makes use of the “depth” function contained in the samtools package. Chromosomes were partitioned into bins of 10,000 bp, and the bin‐wise median was used for further analysis.
In order to avoid an influence of outliers (e.g., due to large peaks resulting from repeat elements), we removed values more than 20% below or above the median signal for each chromosome.
We noticed a pronounced bias of depth of coverage apparently related to chromosome length. Plotting chromosome median depth of coverage against chromosome length suggests that this relation can be approximated by an exponential of the form
We fit each sample individually and divided the median depth for each bin by the value corresponding to the respective chromosome resulting from the fit in order to detrend the data. We used robust regression in order to avoid an influence on detrending due to the actual biological signal. Thus, clear aneuploidies are treated as outliers that do not distort the fit.
For individual chromosomes, we noticed a further bias of depth of coverage toward the ends of chromosomes, resulting in a “smile” shape. In order to correct for this bias, we superposed the signal for all chromosomes, normalizing by median and length, and then fitting a quadratic function. The median of the corrected depth of coverage signal was used as a proxy for chromosomal copy number.
Analysis of structural variations
In order to search for structural variations beyond the level of SNVs and small indels, we applied a set of different methods, as there is no single method that detects the full spectrum of possible variants with high sensitivity and specificity. The identification of variants is further complicated by the fact that mixed populations were sequenced.
We used approaches based on read pair (BreakDancer (Chen et al, 2009)), split read (Pindel (Ye et al, 2009)), and read count (CNVnator (Abyzov et al, 2011)). The read count method was strongly affected by the “smile” bias mentioned above. In order to remove the bias, we downsampled the bam files using samtools “view” proportional to the quadratic fit. To remove false positive calls and variants expected due to genetic differences between the strain used in the experiments (W303) and the reference genome (S288C), we filtered out variants shared by multiple wells using mutual overlap as a criterion for the identification of two variants. Promising candidates were checked by eye using IGV (Robinson et al, 2011). To convince ourselves of the sensitivity of our methods for discovering larger structural variants, we verified that the amplifications of URA3 and TRP1 were actually detected in the strains. However, all our candidates turned out to be either artifacts (in one case due to contamination by a human sequence) or already detected as indels using the basic alignment.
Author contributions
MP performed and contributed to design the evolution experiment, performed and analyzed compensatory mutations experiments, and commented the manuscript; PB contributed to design the evolution experiment, performed and analyzed compensatory mutation experiments and analyzed NGS data, and commented the manuscript; EC contributed to design the evolution experiment, performed and analyzed compensatory mutation experiments, and commented the manuscript; FG analyzed NGS data; FN performed a preliminary evolution experiment; FM performed compensatory mutations experiments; ÁP did preliminary analysis of NGS data; SeP identified the structural role of recurrent tubulin mutations; ZF provided support for the analysis of fitness and epistatic data, and commented the manuscript; SiP and MCL provided the mathematical modeling framework and estimated the expected mutations number; GR supervised the evolution experiment, and commented the manuscript; DS supervised the analysis of NGS data, and commented the manuscript; AC supervised the project, was responsible for funding, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Movie EV1
Acknowledgements
This paper is dedicated to John J. Tyson, mentor and friend, on the occasion of his retirement. We thank Silke Hauf, Claudio Vernieri, and Andrea Musacchio for constructive comments on the manuscript. Camilla Cancrini helped us in the final stages of this work. We thank Marco Foiani, Michael Knop, Roberta Fraschini, Simonetta Piatti, and Tim Huffaker for sharing reagents with us. Work in the group of AC is financed by AIRC, the Italian association for cancer research (Grant AIRC‐IG 21556); MP benefits of a AIRC fellowship; SeP receives supports from the Italian Ministry of Health (Ricerca Corrente and 5x1000 funds); SiP benefits from a Fondazione Umberto Veronesi fellowship; ZF is supported by the National Research, Development and Innovation Office of Hungary (FK 128775), a Janos Bolyai Research Fellowship from the Hungarian Academy of Sciences (BO/779/20) and the New National Excellence Program of the Hungarian Ministry of Human Capacities (Bolyai+, UNKP‐20‐5‐SZTE‐646 and UNKP‐21‐5‐SZTE‐562); the group of MCL is supported by AIRC, the Italian association for cancer research (Grant AIRC‐IG 23258); DS lab is supported by the National Research, Development and Innovation Fund of Hungary (grants K_124881 and FIEK_16‐1‐2016‐0005); GR lab is supported by a Singapore NRF Investigatorship (NRF‐NRFI05‐2019‐0008); the Hungarian Academy of Sciences sponsored the collaboration between the groups of AC and DS by awarding a visiting fellowship to AC.
The EMBO Journal (2021) 40: e108225.
Data availability
Whole‐genome sequencing data of ancestral clones and evolved mixed populations at both timepoints (Gr and Gf) are publicly available at Sequence Read Archive PRJNA757251 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA757251.
References
- Abyzov A, Urban AE, Snyder M, Gerstein M (2011) CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16: 711–726 [DOI] [PubMed] [Google Scholar]
- Arellano‐Santoyo H, Geyer EA, Stokasimov E, Chen GY, Su X, Hancock W, Rice LM, Pellman D (2017) A tubulin binding switch underlies Kip3/Kinesin‐8 depolymerase activity. Dev Cell 42: 37–51.e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrieman HK, Lind MJ, Cawkwell L (2004) Do β‐tubulin mutations have a role in resistance to chemotherapy? Lancet Oncol 5: 158–164 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonney ME, Moriya H, Amon A (2015) Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage‐sensitive genes. Genes Dev 29: 898–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, Rice LM (2018) Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 19: 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Pereira G (2012) SPOC alert–when chromosomes get the wrong direction. Exp Cell Res 318: 1421–1427 [DOI] [PubMed] [Google Scholar]
- Charvin G, Cross FR, Siggia ED (2008) A microfluidic device for temporally controlled gene expression and long‐term fluorescent imaging in unperturbed dividing yeast cells. PLoS One 3: e1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP et al (2009) BreakDancer: an algorithm for high‐resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corno A, Chiroli E, Gross F, Vernieri C, Matafora V, Maffini S, Lagomarsino MC, Bachi A, Ciliberto A (2019) Cellular response upon proliferation in the presence of an active mitotic checkpoint. Life Sci Alliance 2: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Cipriano R, Jackson MW, Stark GR (2009) Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res 69: 8035–8042 [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83–117 [DOI] [PubMed] [Google Scholar]
- DeZwaan TM, Ellingson E, Pellman D, Roof DM (1997) Kinesin‐related KIP3 of Saccharomyces cerevisiae is required for a distinct step in nuclear migration. J Cell Biol 138: 1023–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlemann S, Neuner A, Gombos L, Gibeaux R, Antony C, Schiebel E (2012) An extended γ‐tubulin ring functions as a stable platform in microtubule nucleation. J Cell Biol 197: 59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, Hall IM (2014) SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30: 2503–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M, Murray AW (2020) The evolutionary plasticity of chromosome metabolism allows adaptation to constitutive DNA replication stress. Elife 9: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer EA, Burns A, Lalonde BA, Ye X, Piedra FA, Huffaker TC, Rice LM (2015) A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. eLife 4: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombos L, Neuner A, Berynskyy M, Fava LL, Wade RC, Sachse C, Schiebel E (2013) GTP regulates the microtubule nucleation activity of gamma‐tubulin. Nat Cell Biol 15: 1317–1327 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Garay ML, Chang L, Blade K, Menick DR, Cabral F (1999) A beta‐tubulin leucine cluster involved in microtubule assembly and paclitaxel resistance. J Biol Chem 274: 23875–23882 [DOI] [PubMed] [Google Scholar]
- Huber F, Meurer M, Bunina D, Kats I, Maeder CI, Stefl M, Mongis C, Knop M (2014) PCR duplication: a one‐step cloning‐free method to generate duplicated chromosomal loci and interference‐free expression reporters in yeast. PLoS One 9: e114590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker TC, Thomas JH, Botstein D (1988) Diverse effects of β‐tubulin mutations on microtubule formation and function. J Cell Biol 106: 1997–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M (2010) Microtubules and resistance to tubulin‐binding agents. Nat Rev Cancer 10: 194–204 [DOI] [PubMed] [Google Scholar]
- Kaya A, Mariotti M, Tyshkovskiy A, Zhou X, Hulke ML, Ma S, Gerashchenko MV, Koren A, Gladyshev VN (2020) Molecular signatures of aneuploidy‐driven adaptive evolution. Nat Commun 11: 588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Pereira G, Geissler S, Grein K, Schiebel E (1997) The spindle pole body component Spc97p interacts with the γ‐tubulin of Saccharomyces cerevisiae and functions in microtubule organization and spindle pole body duplication. EMBO J 16: 1550–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999) Epitope tagging of yeast genes using a PCR‐based strategy: more tags and improved practical routines. Yeast 15: 963–972 [DOI] [PubMed] [Google Scholar]
- Krug J (2018) Population Genetics and Evolution, Lecture notes of the 49th IFF spring school "Physics of Life", Forschungszentrum Jülich 2018, https://arxiv.org/abs/1803.08474
- Laan L, Koschwanez JH, Murray AW (2015) Evolutionary adaptation after crippling cell polarization follows reproducible trajectories. eLife 4: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBar T, Phoebe Hsieh YY, Fumasoni M, Murray AW (2020) Evolutionary repair experiments as a window to the molecular diversity of life. Curr Biol 30: R565–R574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T‐C, Neuner A, Schiebel E (2015) Targeting of γ‐tubulin complexes to microtubule organizing centers: conservation and divergence. Trends Cell Biol 25: 296–307 [DOI] [PubMed] [Google Scholar]
- Liu G, Yong M, Yurieva M, Srinivasan K, Liu J, Lim J, Poidinger M, Wright G, Zolezzi F, Choi H et al (2015) Gene essentiality is a quantitative property linked to cellular evolvability. Cell 163: 1388–1399 [DOI] [PubMed] [Google Scholar]
- Liu G, Rancati G (2016) Adaptive evolution: don't fix what's broken. Curr Biol 26: R169–R171 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet C, Ausiannikava D, Le Bihan T, Granneman S, Makovets S (2015) Cell populations can use aneuploidy to survive telomerase insufficiency. Nat Commun 6: 8664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Di Nicolantonio F, Sartore‐Bianchi A, Siena S, Bardelli A (2014) Resistance to anti‐EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov 4: 1269–1280 [DOI] [PubMed] [Google Scholar]
- Musacchio A (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 25: R1002–R1018 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Desai A (2017) A molecular view of kinetochore assembly and function. Biology 6: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M (2014) VariantAnnotation: a bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30: 2076–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker AL, Teo WS, McCarroll JA, Kavallaris M (2017) An emerging role for tubulin isotypes in modulating cancer biology and chemotherapy resistance. Int J Mol Sci 18: 1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R (2010) Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G, Hofken T, Grindlay J, Manson C, Schiebel E (2000) The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell 6: 1–10 [PubMed] [Google Scholar]
- Pham CL, Morrissette NS (2019) The tubulin mutation database: a resource for the cytoskeleton community. Cytoskeleton (Hoboken) 76: 186–191 [DOI] [PubMed] [Google Scholar]
- Pipek O, Ribli D, Molnar J, Poti A, Krzystanek M, Bodor A, Tusnady GE, Szallasi Z, Csabai I, Szuts D (2017) Fast and accurate mutation detection in whole genome sequences of multiple isogenic samples with IsoMut. BMC Bioinformatics 18: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Goffeau A (2012) Yeast ATP‐binding cassette transporters conferring multidrug resistance. Annu Rev Microbiol 66: 39–63 [DOI] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling‐Hampton K, Seidel CW, Li R (2008) Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath O, Kozielski F (2012) Kinesins and cancer. Nat Rev Cancer 12: 527–539 [DOI] [PubMed] [Google Scholar]
- Ravichandran MC, Fink S, Clarke MN, Hofer FC, Campbell CS (2018) Genetic interactions between specific chromosome copy number alterations dictate complex aneuploidy patterns. Genes Dev 32: 1485–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KL, Anders KR, Nogales E, Schwartz K, Downing KH, Botstein D (2000) Structure‐function relationships in yeast tubulins. Mol Biol Cell 11: 1887–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J (2009) Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukowski Heil CS, Large CRL, Patterson K, Hickey AS, Yeh CC, Dunham MJ (2019) Temperature preference can bias parental genome retention during hybrid evolution. PLoS Genet 15: e1008383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Ohi R, Pellman D (2012) Move in for the kill: motile microtubule regulators. Trends Cell Biol 22: 567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan D, Huffaker TC (1992) Astral microtubules are not required for anaphase B in Saccharomyces cerevisiae . J Cell Biol 119: 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szamecz B, Boross G, Kalapis D, Kovács K, Fekete G, Farkas Z, Lázár V, Hrtyan M, Kemmeren P, Groot Koerkamp MJA et al (2014) The genomic landscape of compensatory evolution. PLoS Biol 12: e1001935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, McKeon F (1997) Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 89: 727–735 [DOI] [PubMed] [Google Scholar]
- Uchimura S, Oguchi Y, Hachikubo Y, Ishiwata SI, Muto E (2010) Key residues on microtubule responsible for activation of kinesin ATPase. EMBO J 29: 1167–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Cardarelli C, Gottesman MM, Pastan IRA (1987) Expression of a full‐length cDNA for the human "MDRI" gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA 84: 3004–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent I, Navarro A, Mulet JM, Sharma S, Serrano R (2015) Uptake of inorganic phosphate is a limiting factor for Saccharomyces cerevisiae during growth at low temperatures. FEMS Yeast Res 15: 1–13 [DOI] [PubMed] [Google Scholar]
- Wang W, Zhang H, Wang X, Patterson J, Winter P, Graham K, Ghosh S, Lee JC, Katsetos CD, Mackey JR et al (2017) Novel mutations involving βI‐, βIIA‐, or βIVB‐tubulin isotypes with functional resemblance to βIII‐tubulin in breast cancer. Protoplasma 254: 1163–1173 [DOI] [PubMed] [Google Scholar]
- Wykoff DD, Rizvi AH, Raser JM, Margolin B, O'Shea EK (2007) Positive feedback regulates switching of phosphate transporters in S. cerevisiae . Mol Cell 27: 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Cabral F, Veeraraghavan S (2007) Amino acid substitutions at proline 220 of β‐tubulin confer resistance to paclitaxel and colcemid. Mol Cancer Ther 6, 2798–2806 [DOI] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O (2012) Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci USA 109: 21010–21015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Pavelka N, Bradford WD, Rancati G, Li R (2012) Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet 8: e1002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA (2014) Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci USA 111: E2310–E2318 [DOI] [PMC free article] [PubMed] [Google Scholar]