

Abstract
The cullin‐4‐based RING‐type (CRL4) family of E3 ubiquitin ligases functions together with dedicated substrate receptors. Out of the ˜29 CRL4 substrate receptors reported, the DDB1‐ and CUL4‐associated factor 1 (DCAF1) is essential for cellular survival and growth, and its deregulation has been implicated in tumorigenesis. We carried out biochemical and structural studies to examine the structure and mechanism of the CRL4DCAF1 ligase. In the 8.4 Å cryo‐EM map of CRL4DCAF1, four CUL4‐RBX1‐DDB1‐DCAF1 protomers are organized into two dimeric sub‐assemblies. In this arrangement, the WD40 domain of DCAF1 mediates binding with the cullin C‐terminal domain (CTD) and the RBX1 subunit of a neighboring CRL4DCAF1 protomer. This renders RBX1, the catalytic subunit of the ligase, inaccessible to the E2 ubiquitin‐conjugating enzymes. Upon CRL4DCAF1 activation by neddylation, the interaction between the cullin CTD and the neighboring DCAF1 protomer is broken, and the complex assumes an active dimeric conformation. Accordingly, a tetramerization‐deficient CRL4DCAF1 mutant has higher ubiquitin ligase activity compared to the wild‐type. This study identifies a novel mechanism by which unneddylated and substrate‐free CUL4 ligases can be maintained in an inactive state.
Keywords: CRL4/DCAF1, E3 ligases, Oligomerization, Ubiquitin, VprBP
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology
Cullin‐RING ubiquitin ligases are known to be regulated by neddylation or at the level of substrate recognition. Here, cryo‐EM reconstructions of the human CUL4‐RBX1‐DDB1‐DCAF1 (CRL4DCAF1) reveals a novel mechanism based on oligomerization state switch between tetrameric and dimeric complexes.

Introduction
DCAF1 is an essential substrate receptor for the cullin 4 RING ubiquitin E3 ligase (CRL4) family. DCAF1 is implicated in fundamental cellular processes, ranging from DNA replication (McCall et al, 2008), cell cycle progression (McCall et al, 2008; Guo et al, 2016), transcription (Wang et al, 2017), to zygotic development and reproduction (Yu et al, 2013). CRL4DCAF1 (CUL4‐DDB1‐RBX1‐DCAF1) activity requires the direct interaction between the DCAF1 substrate receptor and various reported substrates such as the replication factor MCM10 (Kaur et al, 2012), p53 (Hrecka et al, 2007; Guo et al, 2016), FoxM1 (Wang et al, 2017), TET methylcytosine dioxygenases (Yu et al, 2013), and protein phosphatase 2A (Yu et al, 2015). DCAF1 is conserved among metazoans and is ubiquitously expressed in different tissues (Zhang et al, 2001). Loss of DCAF1 in mice leads to early embryonic lethality (McCall et al, 2008). DCAF1 ablation is often associated with defects in cell cycle, cell division, and cell survival (Schabla et al, 2019). A comprehensive high‐throughput RNAi screening study in ˜400 different cell lines found that loss of DCAF1 is pan‐lethal (McDonald et al, 2017), with DCAF1 knock‐down induced lethality being the highest among all reported CUL4 DCAF substrate receptors in the study (Fig EV1A). CRL4DCAF1 is also the second most abundant CRL4 complex, accounting for ˜12% of all assembled CRL4 complexes, rendering DCAF1 a critical substrate receptor for the CRL4 system (Reichermeier et al, 2020).
Figure EV1. The CRL4‐DCAF1 oligomerization state changes upon neddylation and is dependent on LisH, the DCAF1 dimerization domain.
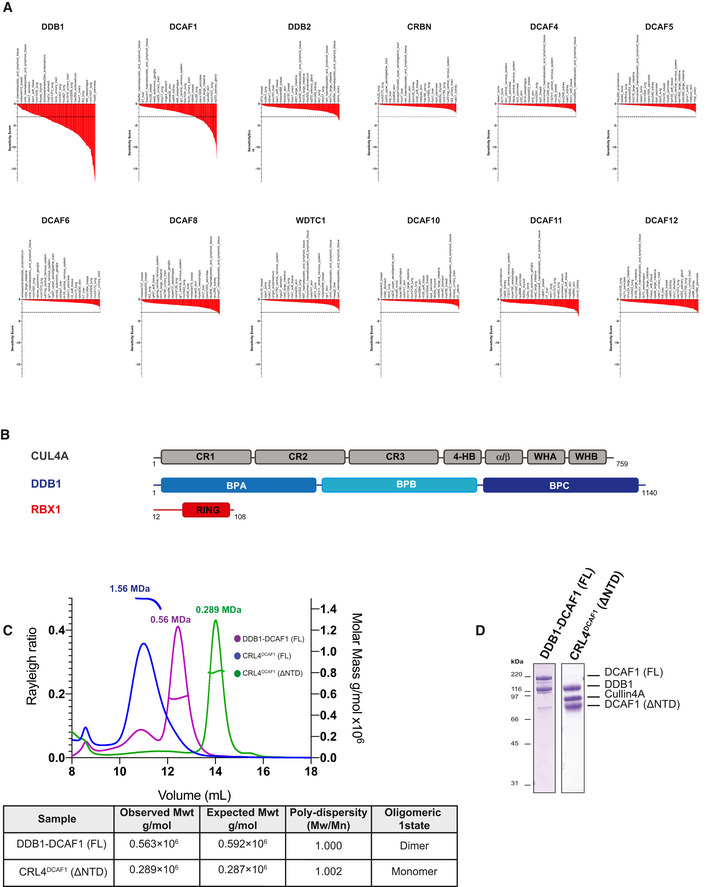
- Sensitivity score profiles adapted from the DRIVE data for DDB1, DCAF1, DDB2, CRBN, DCAF4, DCAF5, DCAF6, DCAF8, WDTC1, DCAF10, DCAF11, and DCAF12. The dropout score in the shRNA screen was converted into log fold‐change (logFC) per shRNA per cell line. The logFC was then normalized per sample to obtain a shRNA level sensitivity score. The shRNA level scores are further aggregated to gene level sensitivity scores using either the ATARiS algorithm (Shao et al, 2013) or the RSA algorithm (König et al, 2007). These give a measure of the statistical significance of the dropout of those 20 shRNAs used per gene compared to the remainder shRNAs in the screen (McDonald et al, 2017).
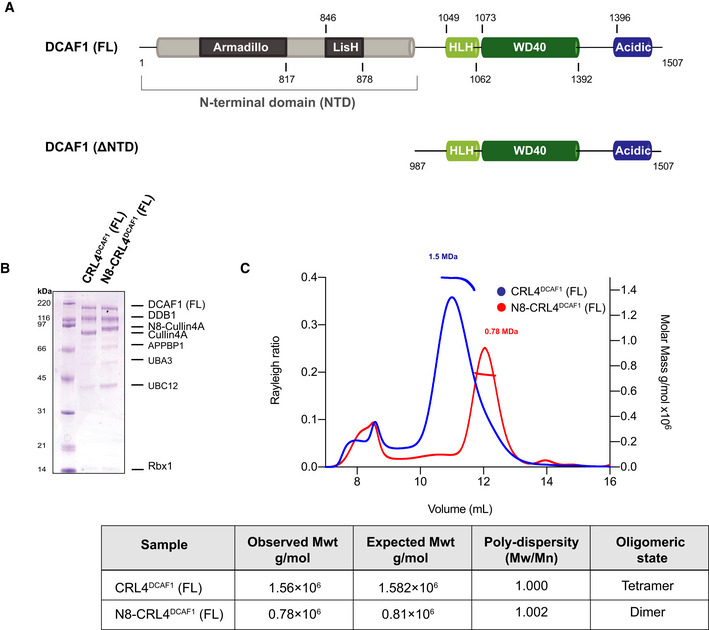
- Domain organization of human CUL4A, DDB1, and RBX1. CR, cullin repeat; UFD, ubiquitin‐fold domain; RING, really interesting new gene; WH, winged helix; BP, β‐propeller.
- SEC‐MALS analysis, the chromatogram shows the Rayleigh ratio curves of CRL4DCAF1 (FL), CRL4DCAF1(ΔNTD), and DDB1‐DCAF1 (FL) together with the molar mass (MDa) of the main peaks determined by MALS. The table summarizes the SEC‐MALS observed molecular weights, the calculated molecular weight, polydispersity values, and oligomeric states of the tested complexes.
- SDS–PAGE of the corresponding complexes.
The CRL4DCAF1 E3 ligase is frequently hijacked by viruses such as the human immunodeficiency virus type 1 and 2 (HIV‐1, HIV‐2) and their viral accessory proteins VPR and VPX (Romani & Cohen, 2012). Efficient replication of HIV in macrophages requires the interaction of VPR/VPX with DCAF1 (Kyei et al, 2015), which redirects the CRL4DCAF1 ubiquitin ligase activity toward degradation of host proteins such as UNG2 (Wu et al, 2016) and SAMHD1 (Schwefel et al, 2014). Viral‐induced degradation of host proteins via CRL4DCAF1 was shown to enhance HIV infection by facilitating viral replication and propagation (Baldauf et al, 2017).
Several lines of evidence further implicate DCAF1 in cancer progression. Hrecka et al (2007) reported that DCAF1 depletion stabilizes the expression of p53 and several of its target genes. Follow‐up studies linked the role of DCAF1 to regulating p53 transcriptional activity in different cell types (Kim et al, 2012; Guo et al, 2016; Wang et al, 2016a). Recent work has also found that MERLIN, a protein that is commonly mutated in several glial cancers, mediates its tumor suppression activities via inactivation of CRL4DCAF1 (Li et al, 2010, 2014).
Despite the growing interest in studying DCAF1 physiological functions, the molecular determinants of DCAF1 regulation are currently poorly understood. CRL systems share common modes of regulation including activation by the ubiquitin‐like protein NEDD8 (neural precursor cell expressed, developmentally downregulated 8) (neddylation) (Duda et al, 2008), inactivation by the multisubunit deneddylase COP9 signalosome (CSN) (Lingaraju et al, 2014), and substrate receptor (SR) exchange mediated by CAND1 (Cullin‐associated Nedd8‐dissociated protein) (Pierce et al, 2013). Besides the standard CRL regulatory mechanisms, DCAF1 has been suggested to be governed by additional modes of activity control. For instance, MERLIN mediates direct binding to DCAF1 and has been linked to CRL4DCAF1 ligase inhibition by an as yet undefined mechanism (Li et al, 2010). Moreover, DCAF1 is known to dimerize in vivo and in vitro, and dimerization is proposed to enhance CRL4DCAF1 ubiquitination activity in vitro (Ahn et al, 2011). The SECexplorer workflow fractionates native protein complexes by size‐exclusion chromatography (SEC) followed by mass spectrometry to identify proteins in each fraction, along with an apparent molecular weight for the eluting complexes (Heusel et al, 2019). The size‐exclusion profile in SECexplorer finds DCAF1 eluting with large native protein complexes of more than 1 MDa.
In the current study, we have used protein biochemistry and cryo‐electron microscopy (cryo‐EM) to characterize the CRL4DCAF1 complex in vitro. We obtained an 8.4 Å cryo‐EM map of CRL4DCAF1, which finds the complex in a tetrameric arrangement. In the tetrameric conformation the DCAF1 WD40 domain mediates the interaction with the cullin C‐terminal domain (CTD) and RBX1 of a neighboring CRL4DCAF1 protomer. This architecture renders the RING domain of RBX1 inaccessible for the E2 ubiquitin‐conjugating enzyme. Upon activation of CRL4DCAF1 by neddylation, the interaction between the cullin CTD and DCAF1 is disrupted and the complex becomes dimeric and active. This dimeric state is also induced when the viral proteins VPR‐UNG2 are bound to CRL4DCAF1, while the presence of MERLIN is compatible with the tetrameric autoinhibited state of the ligase. These results suggest a novel mechanism by which the activity of isolated CRL4DCAF1 ligase is regulated by oligomerization.
Results
CRL4DCAF1 exists in a dimer/tetramer equilibrium
The 1507 amino acid human DCAF1 receptor consists of a HLH (helix‐loop‐helix) and a WD40 domain at the C‐terminus, which together mediate DDB1 binding (Fig 1A), and in turn bridges DCAF1 to the cullin scaffold (Fig EV1B). A highly acidic tail spans the very C‐terminus of DCAF1, which is reported to mediate MERLIN and p53 binding (Mori et al, 2014; Wang et al, 2016a). The large N‐terminal domain of DCAF1 is predicted to assume an Armadillo‐like fold (Zimmermann et al, 2018). A Lis1 homology (LisH) motif spans residues 846–878 at the N‐terminus and has been implicated in DCAF1 dimerization (Ahn et al, 2011).
Figure 1. CRL4DCAF1 exists in a dimer/tetramer equilibrium.
- Predicted domain organization of the human DCAF1 (FL).
- SDS–PAGE Coomassie gel of the neddylation reaction of CRL4DCAF1 at time 0 h and the fully neddylated N8‐CRL4DCAF1 after 2 h.
- SEC‐MALS analysis of CRL4DCAF1 and N8‐CRL4DCAF1. The chromatogram displays Rayleigh ratio curves for CRL4DCAF1 (blue) and N8‐CRL4DCAF1 (red) together with the molar mass (MDa) of the main peaks calculated by MALS. The table summarizes the SEC‐MALS observed molecular weights in the main peaks, the calculated molecular weights, polydispersity values, and oligomeric states of the tested complexes.
Source data are available online for this figure.
To investigate the oligomeric state of CRL4DCAF1, we first purified the wild‐type CRL4DCAF1 complex and performed a neddylation reaction to generate N8‐CRL4DCAF1 complex (Fig 1B). This was followed by multi‐angle light scattering coupled with size‐exclusion chromatography (SEC‐MALS) on the neddylated (N8‐CRL4DCAF1) and unneddylated (CRL4DCAF1) complexes. While the SEC‐MALS analysis of the neddylated state of CRL4DCAF1 gives a molecular weight (MW) consistent with a dimeric complex (observed MW 780 kDa, calculated MW 810 kDa), the unneddylated form of CRL4DCAF1 has an estimated MW equivalent to a tetramer (observed MW 1,561 kDa, calculated MW 1,582 kDa) (Fig 1C). The polydispersity values (Mw/Mn) are 1.002 and 1.000, respectively, in line with these complexes being present as single, mono‐disperse species (Fig 1C). These data suggest that CRL4DCAF1 switches its oligomeric form as a function of neddylation state. SEC‐MALS analysis on an N‐terminally truncated mutant of CRL4DCAF1(ΔNTD) (987–1,507), lacking the LisH motif, finds a MW of 289 kDa, which corresponds to a monomeric complex with a MW of 287 kDa (Fig EV1C and D), linking the LisH motif in DCAF1 to dimerization (Ahn et al, 2011). SEC‐MALS analysis on the isolated DDB1‐DCAF1 (FL) shows this sub‐complex elutes as a dimer (observed MW 563 kDa, calculated MW 592 kDa) (Fig EV1C and D), suggesting a role of the cullin‐RBX1 scaffold in tetramer formation.
The RBX1 interaction with DCAF1 drives tetramer formation
To gain molecular insights into neddylation‐dependent changes in the oligomeric state of CRL4DCAF1, we pursued structural analysis by single particle cryo‐EM. In an effort to increase the stability of CRL4DCAF1 protein sample for freezing cryo‐EM grids, we had to cross‐link the sample using a gradient fixation protocol (GraFix) (Stark, 2010). This enabled us to obtain an 8.4 Å‐resolution cryo‐EM map of the unneddylated CRL4DCAF1 complex (Fig EV2A–C). In the cryo‐EM map, CRL4DCAF1 assembles into a tetrameric arrangement (Fig 2A and B). Intrinsic flexibility between the protomers of this assembly likely prevented us from obtaining higher‐resolution structural insight. The cryo‐EM map was interpreted by rigid body and restrained flexible fitting of crystallographic models of CUL4‐DDB1‐RBX1 (PDB 2HYE), and DDB1‐DCAF1 (WD40) (PDB 5JK7), which allowed defining the overall architecture of the assembly.
Figure EV2. Cryo‐EM data analysis, classification, and refinement procedures for the CRL4DCAF1 complex.
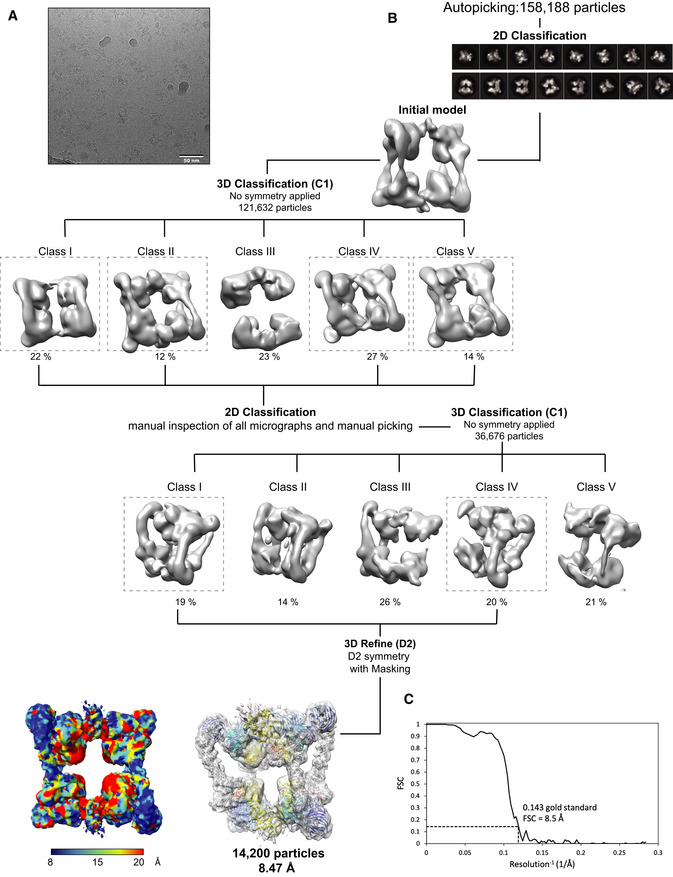
- Representative micrograph for CRL4DCAF1 complex.
- Workflow of cryo‐EM classification and refinement.
- Fourier shell correlation curve (FSC). The resolution values reported for all reconstructions are based on the gold‐standard Fourier shell correlation curve (FSC) at 0.143 criterion and all the related FSC curves are corrected for the effects of soft masks using high‐resolution noise substitution.
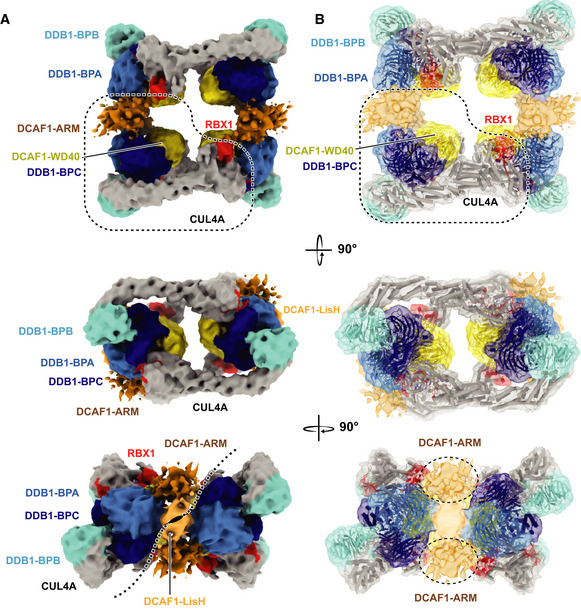
Figure 2. The CRL4DCAF1 assembles a tetrameric complex.
-
A, BDifferent views of CRL4DCAF1 cryo‐EM map (8.4 Å) with fitted crystal structures of CUL4A (gray), RBX1 (red) (PDB 2HYE), DDB1 BPA (light blue), BPB (cyan), and BPC (dark blue), DCAF1 WD40 domain (yellow) (PDB 5JK7), LisH domain (orange), and ARM domain (brown) in (A) surface representation, and (B) cartoon representation.
The CRL4DCAF1 map indicates a potential intermolecular interaction between the DCAF1 (WD40) of one molecule, with the cullin C‐terminal domain (CTD) and RBX1 of a neighboring protomer (Fig 3A). An elementary dimer formed by two CRL4DCAF1 molecules is also evident in the map and is consistent with the interaction of two neighboring LisH domains located proximal to the WD40 domain (Fig EV3A). The tetrameric CRL4DCAF1 assembly in our structure is thus comprised of two CRL4DCAF1 dimers. The structural data are consistent with the results obtained by SEC‐MALS, illustrating that CRL4DCAF1 is tetrameric, and that tetramerization is mediated by the interactions of the cullin CTD‐RBX1 and DCAF1 WD40 domain at one dimerization interface, and the DCAF1 N‐terminal LisH motifs at the other.
Figure 3. The DCAF1 WD40 domain is essential for tetramer formation.
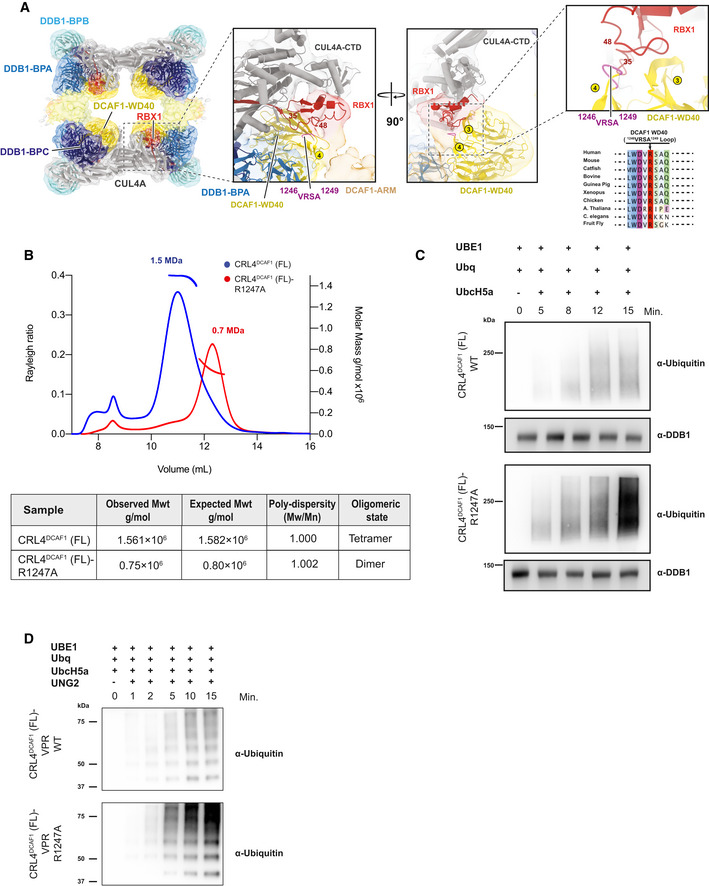
- CRL4DCAF1 cryo‐EM map (8.4 Å) with fitted model (left), and a close‐up view of the CRL4DCAF1 map shows the interaction between DCAF1 (WD40) in yellow and RBX1 in red. The loop in DCAF1 (WD40) that contains the mutated arginine (R1247) is colored in violet. Circled numbers indicate the respective blades in the WD40 β‐propeller. The RBX1 segment potentially contacting the VRSA loop in DCAF1 is indicated by flanking residue numbers. Conservation plot in the region around the residue R1247 in DCAF1 is shown to the lower right.
- SEC‐MALS analysis of CRL4DCAF1 and CRL4DCAF1 (R1247A) mutant. The chromatogram displays Rayleigh ratio curves of CRL4DCAF1 WT (blue) and mutant CRL4DCAF1 (R1247A) (red) together with the molar mass (MDa) of the main peaks calculated by MALS. The table summarizes the SEC‐MALS observed molecular weights in the main peaks, the calculated molecular weight, polydispersity values, and oligomeric states of the tested complexes.
- Autoubiquitination of DCAF1 by wild‐type CRL4DCAF1 (FL) (top) or mutant CRL4DCAF1 (FL) (R1247A) (bottom) observed after incubation with UBA1, UbcH5a, Ubiquitin (WT), at 30°C for 0 to 15 min as indicated (n = 3).
- Ubiquitination of UNG2 by wild‐type CRL4DCAF1‐VPR (FL) (top) or mutant CRL4DCAF1‐VPR (FL) (R1247A) (bottom) observed after incubation with UBA1, UbcH5a, Ubiquitin (WT), at 30°C for 0 to 15 min as indicated (n = 3).
Source data are available online for this figure.
Figure EV3. Modeling the ARM and LisH domains of DCAF1 N‐terminus into the CRL4DCAF1 cryo‐EM map.
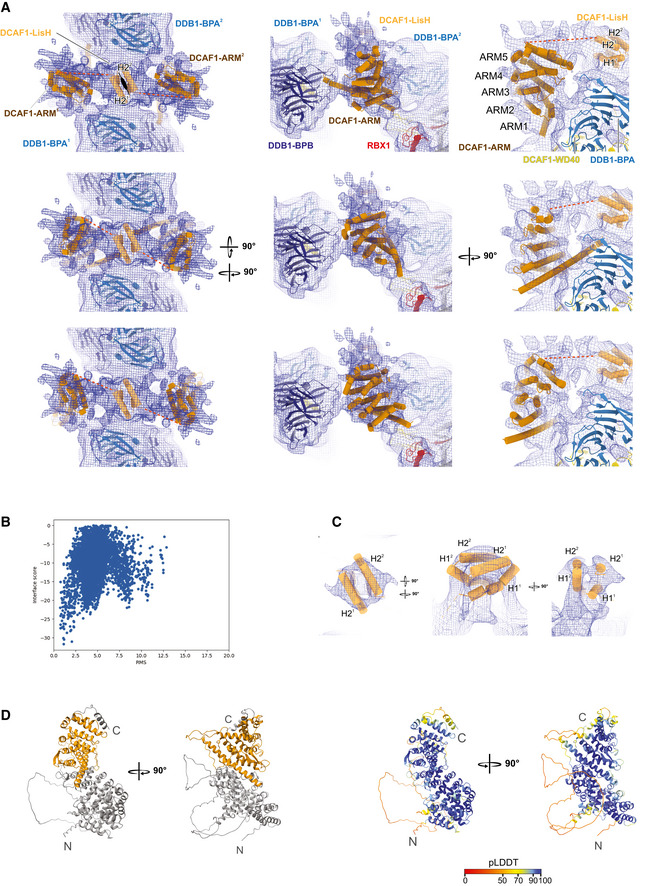
- Different rotation views of DCAF1 ARM domain (brown) and LisH domain (light orange) modeled into the CRL4DCAF1 cryo‐EM map. Three potential docking poses of the ARM domain are shown in the upper, middle, and lower panels. The correlations between simulated 8.4 Å model maps and the experimental map are 0.53, 0.59, and 0.59 (from top to bottom). LisH helices are labeled as H21, H31 from one molecule and H22, H32 from the other protomer. The resolved part of the ARM repeats spanning residues (507–817) and excluding insertions are labeled as ARM (1 to 5). The unmodeled linker between DCAF1‐ARM and LisH domains is indicated by a red dashed line. The map contour level is 0.00934.
- Symmetric docking of LisH homodimer. The root mean square (r.m.s.) deviation was calculated by comparing the decoys against the modeled homodimer based on the LisH domain extracted from coordinates with PDB ID 6IWV.
- Fitting of the LisH domain. The map contour level is 0.0132.
- Initial ARM domain model from the Alphafold pipeline. The part fitted into the density is colored in orange (left). In the right panel, the ARM domain is colored according to pLDDT values (predicted local distance difference test; scale 0–100, low to high confidence, Jumper et al, 2021).
Modeling of the DCAF1 Armadillo (ARM) and LisH domains
The putative LisH density proximal to the WD40 domain showed helical features and strongly suggested the presence of a two‐fold symmetry axis in line with previously observed LisH domain homodimers (PDB 1UUJ, PDB 1VYH) (Kim et al, 2004; Tarricone et al, 2004). The LisH domain fold is characterized by a two‐helix bundle (helices 1 and 2) with a third helix (helix 3) crossing the helical bundle (e.g. PDB 1UUJ, PDB 6IWV). Upon homodimerization, the two‐helix bundles form a four‐helix bundle in which helices 1 and 2 align in an anti‐parallel and slightly diagonal manner, respectively. In several structures, helices 3 of the homodimeric assembly additionally align in a diagonal manner perpendicular to the four‐helix bundle (PDB 1UUJ, PDB 6IWV). The DCAF1 LisH domain structure was predicted by comparative modeling with high confidence (confidence score of 0.67, Song et al, 2013). To obtain a model for the homodimeric complex, the monomeric consensus model from comparative modeling (aa 846–883) was superposed on the dimeric LisH domains from PDB 6IWV. The homodimer interface was independently validated by docking two separated LisH monomer models against each other, imposing twofold symmetry constraints (see Materials and Methods). In these simulations, the LisH dimer interface previously observed in homologous dimeric LisH crystal structures gave the best docking scores (Fig EV3B). The dimeric homology model (obtained from superposition on a template structure) was then docked into the putative LisH density (Fig EV3A and C). The density supports an anti‐parallel alignment of helices 2, while the putative density for helices 1 is fragmented. We observed only uninterpretable density at the expected location for helices 3 and refrained from modeling this helix given the limited local resolution.
A significant portion of uninterpreted density located between DDB1 BPA and DDB1 BPB of different protomers and close to the putative LisH density showed features indicative of several α‐helical bundles. These features would be in agreement with an armadillo fold predicted for the segment N‐terminal to the DCAF1 LisH domain (ARM, 1–817). To obtain a model for this part, we employed the deep learning‐based structure prediction pipeline Alphafold that has been shown to yield highly accurate predictions even in case of targets where no structural templates of close homologs are available (Fig EV3D; Jumper et al, 2021). The part directly N‐terminal to the LisH domain (aa 507–817), which was approximately matching the volume of the remaining density, was extracted from the top‐ranked model of Alphafold. In the predicted model, the extracted portion contains four complete armadillo (ARM) repeats each composed of a short helix that is perpendicular to two somewhat diagonal and anti‐parallel aligned longer helices and an incomplete repeat (third helix only) at the N‐terminus. This particular helix was predicted to be significantly longer. Due to the strong repetitiveness of the α‐helical repeats and the poor map quality in this region, it was not possible to unambiguously dock the model and we cannot exclude that the observed density corresponds to a different part of the ARM domain. In Fig EV3A the three best fitting poses with real‐space correlations between 0.52 and 0.59 (calculated with a simulated model‐map at 8.4 Å) are shown.
In our fitted models, a gap of around 33–46 Å with uninterpretable density remained between the C‐terminal part of the DCAF1 ARM domain and the DCAF1 LisH domain (Fig EV3A and C). These distances would be all consistent with the length of a linker region of 29 aa, which is predicted to be (partially) α‐helical. Some ARM units are interspersed by loop regions and, in particular, the four C‐terminal repeats are connected to the next N‐terminal repeat by a region that has been previously suggested to contain a small chromo‐like domain (Schabla et al, 2019). Owing to the limited local resolution, the presence of a chromo‐like fold could not be verified. No density was observed for the remaining N‐terminal part of the ARM domain that would be consistent with an interspersed casein‐kinase like domain (Kim et al, 2013).
Overall, the density suggests the presence of a symmetric α‐helical interaction motif and several armadillo‐like repeats in close proximity that likely correspond to the LisH domain and the C‐terminal part of the ARM domain, respectively. Predicted models for these regions of the map can largely explain the density features at the given resolution. Higher resolution experimental data are required to unambiguously dock a model and carry out more detailed residue assignments.
The DCAF1 WD40 domain is essential for tetramer formation
The subnanometer CRL4DCAF1 cryo‐EM map allowed fitting of the DCAF1 WD40 and RBX1 domains into the four copies with reasonable confidence. Within the RBX1/DCAF1 region, we identified a highly conserved loop at the surface of the WD40 propeller that approaches RBX1, namely, loop: 1246VRSA1249 (Fig 3A). We introduced an alanine mutation at the conserved arginine residue 1247 within this loop, and analyzed the mutated CRL4DCAF1 complex by SEC‐MALS. SEC‐MALS analysis showed that this single mutation R1247A is sufficient to disassemble the tetrameric complex into a fully dimeric one (Fig 3B). These data support the model interpretation where the interaction between DCAF1 and the RBX1 RING domain is a key driver in tetramerization, and further confirms the assignment of the two elementary CRL4DCAF1 dimers that together form the tetramer.
The cryo‐EM map finds the RBX1 RING domain shielded by the WD40 domain of DCAF1, rendering it inaccessible for an Ub‐loaded E2 (Fig 3A). To test whether the tetrameric CRL4DCAF1 assembly represents an inactive autoinhibited state, we carried out auto‐ubiquitination assays using the wild‐type and R1247A mutant of CRL4DCAF1 (FL). The wild‐type tetrameric CRL4DCAF1 complex shows a prominent reduction in DCAF1 auto‐ubiquitination as compared to the dimeric mutant CRL4DCAF1 (R1247A) (Fig 3C). Reduced catalytic activity of the tetrameric CRL4DCAF1 compared to the dimeric mutant was also observed toward the viral substrate VPR‐UNG2 (Fig 3D). On the other hand, preventing tetramer formation by incorporating the mutant DCAF1 (R1247A) into the complex overcomes auto‐inhibition and gives rise to a more active E3 ligase (Fig 3C and D). Taken together, these data suggest that the tetrameric CRL4DCAF1 ligase represents a conformation with a significantly reduced catalytic activity.
Tetrameric CRL4DCAF1 counteracts neddylation and deneddylation
In our MALS studies, the neddylated CRL4DCAF1 complex predominantly existed in a dimeric state (Fig 1C). To test whether the tetramer allows for direct access of the neddylation machinery, we used the tetrameric CRL4DCAF1 cryo‐EM map and modeled the position of the putative RBX1‐UBC12 complex (PDB 4P5O) (Fig 4A). The presence of UBC12, the NEDD8 E2, resulted in substantial steric clashes with the neighboring DCAF1 molecule (Fig 4A), suggesting that the tetrameric confirmation of CRL4DCAF1 is inhibitory to neddylation without structural rearrangements. To test this hypothesis, we established a neddylation assay, where we directly compared the amount of neddylated CRL4DCAF1 (WT) and CRL4DCAF1 (R1247A) dimeric mutant. While CRL4DCAF1 (R1247A) is efficiently and fully neddylated by UBC12 within 1 min (Fig 4B, left panel), the tetrameric CRL4DCAF1 is substantially more resistant to neddylation, as evident by longer incubation times, of up to 180 min, required to fully neddylate the CRL4DCAF1 (WT) complex (Fig 4B, right panel). These data illustrate the inhibitory effect of RBX1 sequestration by DCAF1 (WD40) in the tetrameric CRL4DCAF1 conformation. Within the dimer to tetramer equilibrium, it is the dimeric form of CRL4DCAF1 that likely is preferentially neddylated.
Figure 4. The tetrameric conformation renders CRL4DCAF1 refractory toward regulation by neddylation and deneddylation.
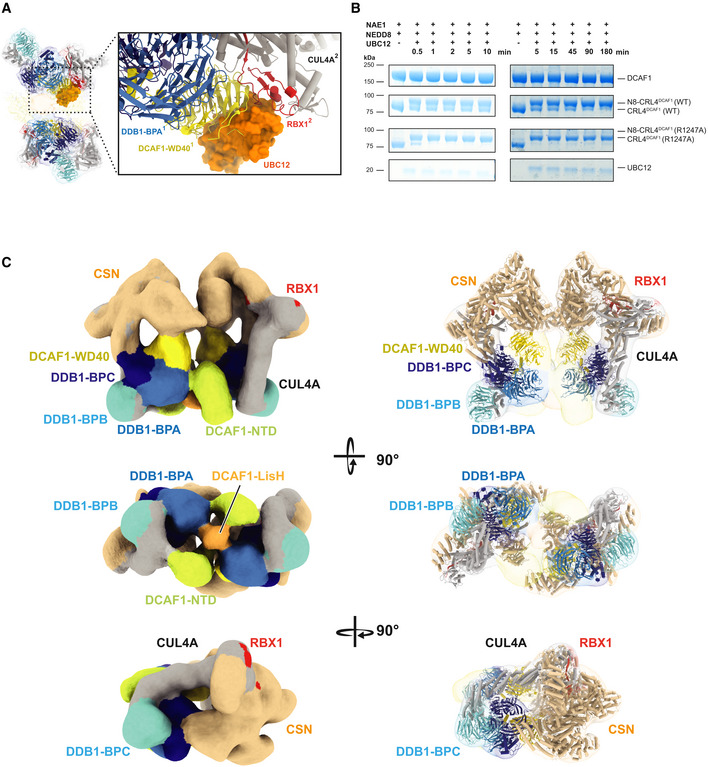
- CRL4DCAF1 cryo‐EM map and a close‐up view of fitted UBC12‐RBX1 structure (PDB 4P5O) that shows steric clashes with DCAF1 (WD40)1 of one CRL4DCAF1 molecule (yellow) (PDB 5JK7) and RBX12 of the second CRL4DCAF1 molecule (red), UBC12 is shown in orange (PDB 4P5O), DDB1 BPA (light blue), BPB (cyan), and BPC (dark blue), CUL4A in gray. Molecules belonging to different protomers are indicated by superscripts (1 or 2).
- SDS–PAGE showing the neddylation reaction time course of CRL4DCAF1 (FL) WT and CRL4DCAF1 (FL) (R1247A) mutant in the presence of 1 μM UBC12 for 0 to 180 min as indicated. Left panel with t = 0 to 10 min, and right panel with t = 5 to 180 min (n = 3).
Source data are available online for this figure.
The impaired neddylation of tetrameric CRL4DCAF1 led us to investigate deneddylation, the opposing regulatory mechanism for CRLs catalyzed by the COP9 signalosome (CSN). Fitting the crystal structure of CSN (PDB 4D10) into the tetrameric CRL4DCAF1 cryo‐EM map suggests that tetramerization and CSN binding are mutually exclusive. This is evident in the steric clashes between CSN and the CRL4DCAF1 tetramer (Fig EV4A). As previously reported, neddylated CRLs (N8‐CRLs) bind CSN with very high affinity (Cavadini et al, 2016; Mosadeghi et al, 2016). The unneddylated CRLs, on the other hand, bind CSN with significantly lower affinities (Cavadini et al, 2016). Pull‐down assays using an active site mutant of CSN (CSN5 E76A) and CRL4DCAF1 (FL) or a CRL4DCAF1(ΔNTD) mutant, which lacks the N‐terminus (973–1,507), shows that N8‐CRL4DCAF1 (FL) and N8‐CRL4DCAF1(ΔNTD) form stoichiometric complexes with CSN (Fig EV4B, lanes 1 and 4). While the unneddylated CRL4DCAF1(ΔNTD) shows residual binding to CSN, this ability is almost completely abolished in the case of the CRL4DCAF1 (FL) tetramer (Fig EV4B, lanes 2 and 3). Consistent with these findings, a 20 Å cryo‐EM map of the N8‐CRL4DCAF1‐CSN complex shows that the complex adopts a dimeric architecture when bound to CSN, revealing the central LisH‐mediated elemental dimer (Figs 4C and EV4C–E). Taken together, tetramerization of CRL4DCAF1 is a conformational state of the ligase that interferes with CSN binding and activation.
Figure EV4. CRL4DCAF1 tetramerization and CSN binding are mutually exclusive.
- Fitting the crystal structure of CSN (PDB 4D10) (wheat) into CRL4DCAF1 cryo‐EM map.
- Strep pull‐down assay of untagged CSN (E76A) complex on strep‐tagged N8‐CRL4DCAF1(FL) (lane1), CRL4DCAF1(FL) (lane2), CRL4DCAF1(ΔNTD) (lane3), and N8‐CRL4DCAF1(ΔNTD) (lane 4) (n = 2).
- A representative cryo‐EM micrograph for CRL4DCAF1‐CSN complex.
- Workflow of cryo‐EM classification and refinement.
- Fourier shell correlation curve (FSC). The resolution values reported for all reconstructions are based on the gold‐standard Fourier shell correlation curve (FSC) at 0.143 criterion and all the related FSC curves are corrected for the effects of soft masks using high‐resolution noise substitution.
Viral substrate binding to CRL4DCAF1 favors the active, dimeric state of the ligase
During its maturation cycle, HIV‐1 hijacks the CRL4DCAF1 ligase via its VPR accessory protein, which binds the WD40 domain of DCAF1 and recruits host UNG2 for ubiquitination and subsequent degradation (Ahn et al, 2010; Wu et al, 2016). We set out to examine this viral‐induced DCAF1 substrate for its ability to break the tetrameric CRL4DCAF1 complex up into dimers. We generated CRL4DCAF1‐VPR and CRL4DCAF1‐VPR‐UNG2 complexes (Fig 5A), and analyzed them in SEC‐MALS. The SEC‐MALS analysis of CRL4DCAF1‐VPR‐UNG2 shows that the complex exists predominantly in its dimeric form when the VPR‐UNG2 substrate is bound (Fig 5B). In contrast, binding to VPR alone is compatible with the tetrameric conformation (Fig 5B). Consistent with the oligomeric states observed in SEC‐MALS, fitting the crystal structure of DDB1‐DCAF1(WD40)‐VPR‐UNG2 (PDB 5JK7) into the EM density obtained for the CRL4DCAF1 tetramer shows steric clashes between VPR‐UNG2 and the WD40 domain of the neighboring DCAF1 molecule (Fig 5C). These data illustrate that the substrate‐like UNG2 protein affects the CRL4DCAF1 oligomeric state similar to neddylation by favoring a dimeric CRLDCAF1 state. Neddylation and substrate binding thus biochemically work hand‐in‐hand to induce and maintain the dimeric form of CRL4DCAF1, a state that represents the active form of the CRL4DCAF1 ligase.
Figure 5. The effect of DCAF1 substrate versus non‐substrate binding proteins on the tetrameric configuration of CRL4DCAF1 .
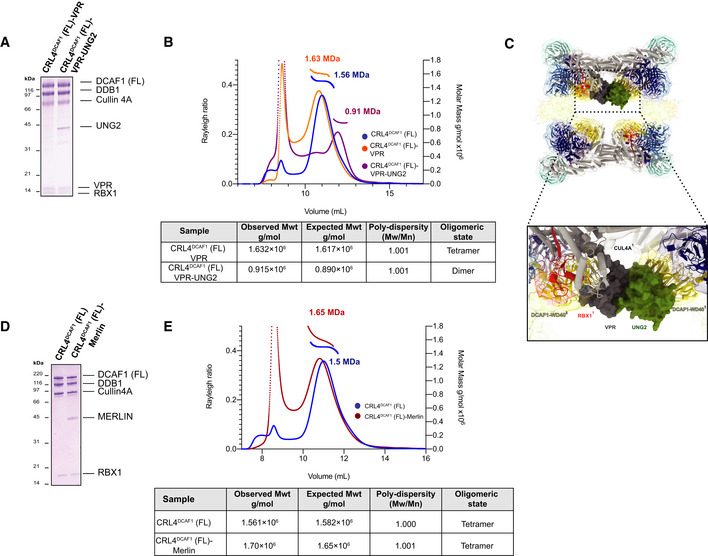
- SDS–PAGE of the purified CRL4DCAF1 (FL)‐VPR and CRL4DCAF1 (FL)‐VPR‐UNG2 complexes.
- SEC‐MALS analysis, the chromatogram shows Rayleigh ratio curves of CRL4DCAF1 (FL), CRL4DCAF1 (FL)‐VPR, and CRL4DCAF1 (FL)‐VPR‐UNG2, together with the molar mass (MDa) of the main peaks determined by MALS. The table summarizes the observed molecular weight in the main peaks by SEC‐MALS, listing the calculated molecular weight, polydispersity values, and oligomeric states of the tested complexes. The peaks eluting close to the void volume were shown to contain heterogeneous large aggregates (Fig EV5A and B).
- SDS–PAGE of the purified CRL4DCAF1 and CRL4DCAF1(FL)‐MERLIN complexes.
- SEC‐MALS analysis, the chromatogram shows the Rayleigh ratio curves of CRL4DCAF1(FL) (blue) and CRL4DCAF1(FL)‐MERLIN (red) together with the molar mass (MDa) of the main peaks calculated by MALS. The table summarizes the SEC‐MALS observed molecular weights, the calculated molecular weights, polydispersity values, and oligomeric states of the tested complexes. The peak eluting close to the void volume contains large unspecific aggregates (Fig EV5C).
Source data are available online for this figure.
We next sought to investigate the effect of a non‐substrate binding protein, such as MERLIN, on the tetrameric conformation of CRL4DCAF1 (Fig 5D). MERLIN was shown to regulate the CRL4DCAF1 ligase activity (Li et al, 2010). We carried out SEC‐MALS analysis on the CRL4DCAF1‐MERLIN complex. MERLIN co‐migration in SEC‐MALS demonstrated that MERLIN is compatible with the tetrameric assembly of CRL4DCAF1 (Fig 5E), which in turn suggests that MERLIN does not act like a DCAF1 substrate.
Previous studies indicate that CSN can distinguish between substrate‐bound and substrate‐free CRL ligases through a steric mechanism (Enchev et al, 2012; Cavadini et al, 2016), where substrate‐bound CRLs clash into CSN, and are thus less effectively deneddylated than substrate‐free ligases. Consistently, fitting the crystal structure of DDB1‐DCAF1(WD40)‐VPR‐UNG2 (PDB 5Jk7) into the CRL4DCAF1‐CSN cryo‐EM map shows significant steric clashes between VPR‐UNG2 and CSN (Fig EV5D). We therefore established a fluorescence‐based CSN deneddylation assay as a complementary approach to examine whether UNG2 and MERLIN function as substrates in regard to CSN regulation. Similar to other substrate‐bound CRL, VPR‐UNG2 binding to the N8‐CRL4DCAF1 ligase inhibits CSN catalysis (Fig EV5E). MERLIN‐bound N8‐CRL4DCAF1, on the other hand, is readily deneddylated by CSN, showing a comparable Kcat value to that of wild‐type N8‐CRL4DCAF1 (Fig EV5F). The CSN catalysis assay, along with the tetramer to dimer switch of CRL4DCAF1 in the presence of substrates, supports the notion that VPR‐UNG2 functions analogous to a substrate protein when bound to CRL4DCAF1, while MERLIN does not. This is consistent with the VPR‐UNG2‐DCAF1 X‐ray structure (Wu et al, 2016), which places UNG2 in a position where it would collide with CSN in the low‐resolution CSN‐CRL4DCAF1 map (Fig EV5D) and where substrates of WD40 containing CUL4 ligases are typically bound. Substrate binding thus is incompatible with the tetrameric state of CRL4DCAF1, and bound substrates instead favor the dimeric, active form of the CRL4DCAF1 ligase.
Figure EV5. CSN catalysis on UNG2‐ and MERLIN‐bound N8‐CRL4DCAF1 .
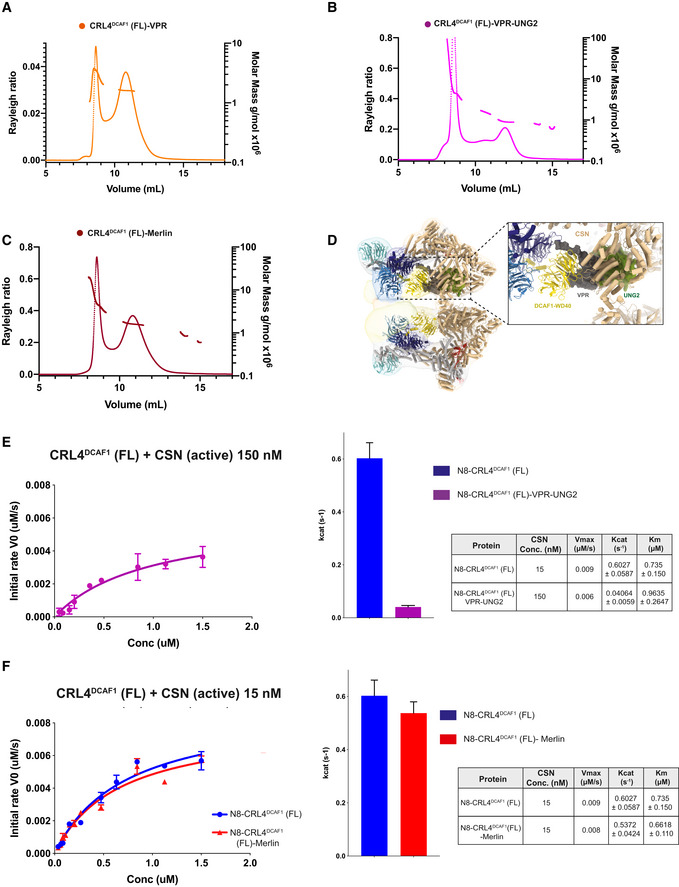
-
A–CSEC‐MALS analysis including the peaks eluting near the SEC column void volume of (A) CRL4DCAF1‐VPR, (B) CRL4DCAF1‐VPR‐UNG2, and (C) CRL4DCAF1‐MERLIN. The chromatogram shows the Rayleigh ratio curves together with the molar mass (MDa) determined by MALS.
- D
-
E, FFluorescence polarization assay investigating the catalytic activity of CSN on N8‐CRL4DCAF1(FL) when bound to (E) VPR‐UNG2 or (F) MERLIN. Initial rates (M/s) are plotted versus concentrations (M) of N8‐CRL4DCAF1(FL)‐VPR‐UNG2 (magenta), N8‐CRL4DCAF1(FL) (blue), and N8‐CRL4DCAF1(FL)‐MERLIN (red). Column representation of Kcat values, and table summarizes KM, Kcat, and Vmax values calculated from the curves (biological replicates, n = 3, mean ± SD).
Discussion
We herein provide the subnanometer map of the CRL4DCAF1 E3 ubiquitin ligase, as well as the mechanistic characterization of this essential ubiquitin ligase. We identify a tetrameric CRL4DCAF1 state that counteracts neddylation (Fig 4B) and auto‐ubiquitination (Fig 3C) and by these measures likely represents the inhibited state of the E3 ubiquitin ligase. Substrate binding to CRL4DCAF1 favors a dimeric, active conformation, which is also the preferred substrate of CSN, further underlining that this is the active conformation of the ligase. Structurally, the mechanism of DCAF1 inhibition by RBX1 sequestration is reminiscent of what has been previously observed for CSN2/RBX1 interactions in CSN structures (Emberley et al, 2012; Cavadini et al, 2016), as well as the arrangement seen in the GLOMULIN‐CRL1‐RBX1 complex (Duda et al, 2012). To our knowledge, this is the first report of a CRL with an inbuilt auto‐inhibitory mechanism.
Several studies have suggested that the cullin CTD is flexible upon neddylation (Angers et al, 2006; Duda et al, 2008; Fischer et al, 2011; Baek et al, 2020; Banchenko et al, 2021). These conformational changes triggered upon neddylation in turn may also alter the interface between DCAF1 WD40 and RBX1. Unlike recent findings for the CUL1 systems (Baek et al, 2020), it is currently unclear how CUL4 ligases interact with ubiquitin loaded E2 enzymes. Given the extensive conformational rearrangements observed for CUL1 CTD, however, it is likely that a neddylated CRL4DCAF1 in its E2 ubiquitin bound form would further impair tetramer formation in cells. Moreover, although our findings are consistent with CUL4DCAF1 being in a dimer‐tetramer equilibrium, we cannot exclude that Ub/Nedd8‐loaded E2 complexes, or in fact also CSN, possess additional molecular mechanisms to open the CUL4DCAF1 tetrameric complex and thereby convert it to a dimer.
At this stage, we can only speculate as to the enzymatic role of this inhibitory mechanism. We and others have observed spontaneous CRL4 activity and auto‐ubiquitination in the absence of neddylation, in case of CRL4DDB2 (Scrima et al, 2008; Fischer et al, 2011; Cavadini et al, 2016), CRL4CRBN (Fischer et al, 2014; Petzold et al, 2016), and CRL4DCAF15 (Bussiere et al, 2020) using UBCH5a as ubiquitin‐conjugating enzyme. This appears to be in contrast to the CUL1 system, which has been reported to be strictly dependent on neddylation for activity (Baek et al, 2020). Furthermore, studies with CSN inhibitors have reported CRL receptor instability (Schlierf et al, 2016) due to auto‐ubiquitination of the receptor following persistent CRL activation. Although at lower flux, receptor auto‐ubiquitination likely also occurs in cells with an active neddylation/deneddylation machinery. DCAF1 tetramerization may thus provide an additional layer of inhibition preventing futile DCAF1 receptor auto‐ubiquitination and degradation cycles for substrate‐ or neddylation‐free CRL4DCAF1. Given the sensitivity of cells toward DCAF1 loss, this may preserve the cellular CRL4DCAF1 pool. A somewhat different albeit non‐mutually exclusive explanation is that the inactive form of CRL4DCAF1 serves to counteract spurious activation of the enzymes. This would assure that only a tightly bound substrate shifts the equilibrium to a dimeric, active form of the enzyme to then bestow CRL4DCAF1 neddylation and full activity.
We speculate that the CRL4DCAF1 dimer–tetramer transition is primarily regulated through neddylation and substrate binding. However, a possible regulatory mechanism involving post‐translational modifications including phosphorylation can not be excluded at this point. We note that there are a number of residues in the CUL4 C‐terminus expected to be phosphorylated (S642, Y744, and N751), based on the PhosphoSitePlus resource (Hornbeck et al, 2015). When phosphorylated, these residues have the potential to impact tetramerization in light of our structures and would allow activation.
The pronounced pan‐lethality of DCAF1 loss, suggests an important role of DCAF1 for cell survival. As CUL1‐based CRLs may intrinsically be more tightly regulated by neddylation than the CRL4 family, an additional auto‐inhibitory circuitry, as observed for CRL4DCAF1 may thus be required for proper ligase function, targeting and regulation. Future experiments are required to assess the biological role of this inhibitory mechanism, and to examine whether other oligomeric CRLs use a similar mechanism.
Materials and Methods
Cloning, protein expression, and purification
Wild‐type (WT) and mutant constructs of full‐length human DCAF1 (Q9Y4B6) were cloned into pLAF vector, which is derived from the pBacPAK8 system (ClonTech). Cullin 4A (FL) (Q13619) and DDB1 (FL) (Q16531) were assembled into one vector (pBIG1a) using Gibson‐based technique (biGBac) (Weissmann et al, 2016). Recombinant baculoviruses were prepared in Spodoptera frugiperda (sf9) cells using the Bac‐to‐Bac system (Life Technologies). Recombinant protein complexes were expressed in Trichoplusia ni High Five cells by co‐infection of 35 ml of single baculoviruses per 2 l of High Five culture. DCAF1 wild‐type and mutants were expressed with N‐terminal Strep (II) tag, and Cullin 4A and DDB1 were expressed with N‐terminal His6 tags. MERLIN Δ (1–346) was expressed with an N‐terminal glutathione S‐transferase (GST) tag. CRL4DCAF1‐CSN complex was co‐expressed using the CSN4‐RBX1 fusion construct, as described in Cavadini et al (2016).
Cells were harvested 36–48 h after infection and lysed by sonication in 50 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris‐HCl) pH 8.0, 200 mM NaCl, 0.5 mM tris(2‐carboxyethyl)phosphine (TCEP), including 0.1% Triton X‐100, 1x protease inhibitor cocktail (Roche Applied Science), and 1 mM phenylmethanesulfonyl fluoride (PMSF). This was followed by ultracentrifugation at 40,000 g for 45 min to separate the soluble fraction of the lysate (supernatant). The supernatant was loaded on Strep‐Tactin (IBA life sciences) affinity chromatography in buffer containing 50 mM Tris‐HCl pH 7.5, 200 mM NaCl, 0.5 mM TCEP. The Strep (II) elution fractions were further purified via ion exchange chromatography (Poros HQ 50 µm, Life Technologies) and subjected to size‐exclusion chromatography (Superose 6, GE Healthcare) in a buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl, and 0.25 mM TCEP. Pure fractions were collected and concentrated using 10,000 MWT cut‐off centrifugal devices (PALL) and stored at −80°C.
APPBP1‐UBA3, UBC12, and NEDD8 were expressed and purified as described before Duda et al (2008), Huang et al (2007), and Huang et al (2008).
In vitro Ubiquitination of CRL4DCAF1
In vitro ubiquitination was performed by mixing wild‐type or mutant CRL4DCAF1 at 2 μM with a reaction mixture containing E1 (UBA1, Boston Biochem) at 0.1 μM, E2 (UBCH5a, Boston Biochem) at 0.1 μM, wild‐type Ubiquitin (Ubiquitin, Boston Biochem) at 5 μM as indicated. Reactions were carried out in 50 mM Tris pH 7.7, 200 mM NaCl, 10 mM MgCl2, 0.2 mM CaCl2, 3 mM ATP, 2 mM DTT, and 0.1 mg/ml BSA, and incubated for 0–15 min at 30°C. Reactions were then analyzed by Western blot using anti‐ubiquitin (P4D1) primary antibody (Santa Cruz, 1:500) and Horseradish Peroxidase (HRP) conjugated anti‐rabbit secondary antibody (1:10,000). Blots were incubated with SuperSignal™ West Pico PLUS Chemiluminescent Substrate solution (Thermo Fisher) and scanned on Fusion FX7 imaging system (Witec AG).
Pull‐down assay of CRL4DCAF1 and CSN
N8‐CRL4DCAF1 (FL), N8‐CRL4DCAF1(ΔNTD) (987–1,507), CRL4DCAF1 (FL), and CRL4DCAF1(ΔNTD) were loaded on 20 μl Strep‐Tactin Macroprep beads (IBA life sciences), and incubated with untagged and inactive CSN (CSN5 E76A) at 1:1 molar ratio for 1 h at 4°C. Bound complexes were washed three times with 1 ml buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl, and 0.25 mM TCEP. Beads were boiled in SDS loading dye.
Size‐Exclusion Chromatography‐Multi‐Angle Light Scattering (SEC‐MALS)
The molecular masses of CRL4DCAF1 WT and mutant complexes were investigated by SEC‐MALS using an ÄKTA FPLC system (GE Healthcare) equipped with a Superose 6 10/300 size‐exclusion column (GE Healthcare Life Sciences). 50 μl of the protein samples at a concentration of 2 mg/ml was loaded. The light scattering measurements in the SEC‐MALS system used an Optilab T‐rEX refractive index detector and a miniDAWN TREOS 3 angle MALS detector (Wyatt Technology). The samples were loaded on the SEC‐MALS system in a buffer containing 50 mM HEPES pH 7.4, 200 mM NaCl and 1 mM TCEP. The protein fractions at/near the void volume eluted as heterogeneous high molecular weight aggregates, and we therefore did not further investigate their behavior (Fig EV5A–C).
Cryo‐electron microscopy and image processing
In order to increase the stability of the CRL4DCAF1 and CRL4DCAF1‐CSN complexes, gradient fixation (GraFix) protocol was performed (Stark, 2010). The samples were loaded on a glycerol gradient (10–30% w/v) in the presence of the cross‐linker glutaraldehyde (0.25% v/v) followed by ultracentrifugation (SW40Ti rotor) at 30,000 rpm for 19 h at 4°C. Peak fractions containing CRL4DCAF1, or CRL4DCAF1‐CSN complexes were collected and concentrated. For CRL4DCAF1‐CSN, glycerol removal and buffer exchange was performed by several rounds of concentration and dilution using 10,000 MWT cutoff centrifugal devices (PALL). Glycerol removal from the CRL4DCAF1 sample was performed by injecting the sample into Superose 6 3.2/300 in buffer containing 50 mM HEPES pH7.4, 200 mM NaCl, and 1 mM TCEP, and collecting the peak fraction. 4 μl sample (0.1–0.2 mg/ml) was applied on glow discharged Quantifoil holey carbon grids. For the CRL4DCAF1 samples, R1.2/1.3, Cu 400 mesh grids were used for data collection (Quantifoil Micro Tools GmbH, Grosslöbichau, Germany). For the CRL4DCAF1‐CSN sample, Lacey carbon grids (Ted Pella, Inc.) were used. For both samples, CRL4DCAF1 and CRL4DCAF1‐CSN, grids were manually floated with continuous carbon films. Grids were incubated for 30 s at 4°C and 100% humidity, and immediately blotted with Whatman no. 1 filter paper (blotting time 2 s) and vitrified by plunging into liquid ethane (Leica EM GP plunger).
For CRL4DCAF1, micrographs were recorded on a Cs corrected FEI Titan Krios TEM (Thermo Fisher) operated at 300 kV, using a Gatan K2‐summit direct electron detector after a Quantum‐LS energy filter with a slit width of 30 eV (Gatan) (Table EV1). The micrographs were recorded in electron‐counting mode with a dose rate of 5 e−/pixel/s. The raw stacks were recorded with 40 frames and an exposure time of 9 s, yielding a total dose of 45 e−/pixel. Micrographs were recorded with a magnification of ×130,000 resulting in a calibrated pixel size of 0.88 Å. Defocus values varied between −1.5 and −4 μm.
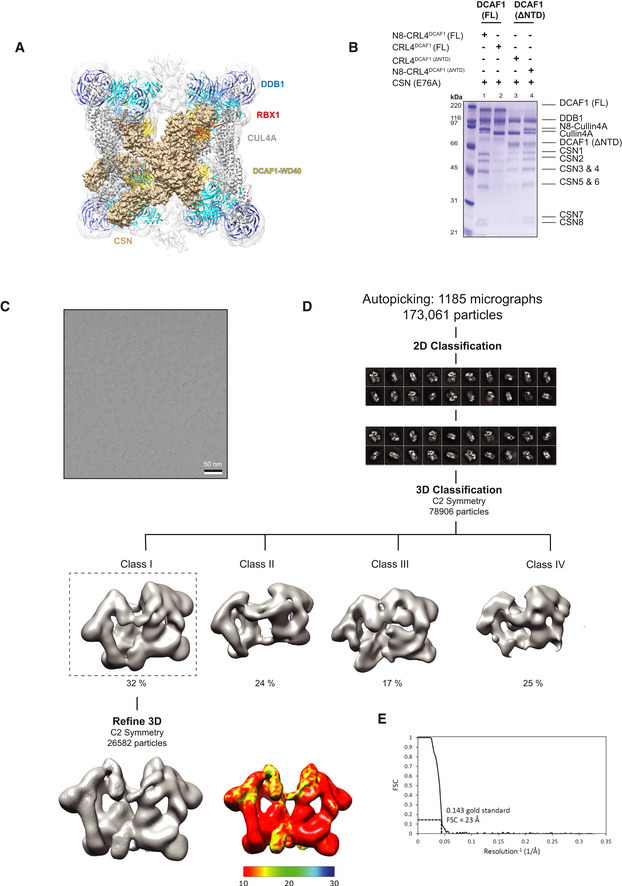
1,280 micrographs were processed. Dose‐fractionated raw stacks were subjected to drift correction using Unblur (Campbell et al, 2012). The contrast transfer function (CTF) was estimated using CTFFIND4 (Rohou & Grigorieff, 2015). Particles were selected by Gautomatch (developed by Kai Zhang, http://www.mrc‐lmb.cam.ac.uk/kzhang/Gautomatch/). 158,188 Particles were imported into RELION and extracted with a box size of 260 × 260 pixels after binning the micrographs by a factor of 2 (1.76 Å/pixel). After four rounds of 2D classification, a subset of particles were used to generate an ab initio model in SPARX (Penczek et al, 1994; Baldwin & Penczek, 2007; Hohn et al, 2007). Two rounds of 3D classification were performed to remove heterogeneous particles (Fig EV2). 3D refinement on 14,000 particles after applying D2 symmetry yielded the final map at 8.4 Å. Post‐processing was done using a mask created in RELION by extending the binary map with three pixels and adding three pixels of soft edge. The binarization threshold was set to 0.0096.
For CRL4DCAF1‐CSN complex, micrographs were recorded on a FEI F30 Polara (Thermo Fisher) operated at 300 kV, using a Gatan K2‐summit direct electron detector in electron‐counting mode (magnification of x31,000 and calibrated pixel size of 1.24 Å). The micrographs were exposed for 16 s and dose fractionated into 40 frames. Defocus values varied between −1.5 and −4 μm. 1,185 micrographs were processed. Dose‐fractionated raw stacks were subjected to drift correction using unblur (Campbell et al, 2012). CTF was estimated using CTFFIND (Rohou & Grigorieff, 2015). Particles were auto‐picked in RELION 1.4. 173,061 particles were extracted after binning the micrographs by a factor of 2 (2.48 Å/pixel). Two rounds of 2D classification and one round of 3D classification were performed to remove heterogeneous particles. 3D refinement on 26,582 particles after applying C2 symmetry yielded the final map at 23 Å.
Model building and refinement
The two template structures with PDB IDs 2HYE and 5JK7 were iteratively re‐refined against the crystallographic structure factors using Coot and Phenix (Angers et al, 2006; Emsley et al, 2010; Wu et al, 2016; Liebschner et al, 2019). The template models for CUL4A, RBX1, and DDB1 BPB were extracted from coordinates with PDB ID 2HYE (Table EV2), and the template models for DCAF1 and DDB1 BPA/BPC were extracted from coordinates with PDB ID 5JK7 and docked into the cryo‐EM map using Chimera (Pettersen et al, 2004). The structure was then refined using the Rosetta density‐guided FastRelax protocol (torsional followed by Cartesian space) in combination with constraints to the starting model coordinates and symmetric scoring (Wang et al, 2016b). Before deposition, the model was truncated to poly‐alanine and validation was performed using Phenix and Molprobity (Chen et al, 2009). Figures were generated using ChimeraX (Pettersen et al, 2020).
Modeling of DCAF1 N‐terminal domains
The DCAF1 LisH domain (aa 846–883) was predicted using the Rosetta comparative modeling protocol provided through the Robetta server (Raman et al, 2009; Song et al, 2013). The top‐ranked model was used for superposition on a homologous LisH homodimer (PDB 6IWV, (Wang et al, 2019)), and the resulting dimer was docked into the density using Chimera (Pettersen et al, 2004). The homodimer homology model was validated by docking simulations using the Rosetta symmetric docking protocol (5,000 decoys, André et al, 2007).
The DCAF1 Armadillo domain (aa 1–845) was predicted using Alphafold v2.0 (Jumper et al, 2021). The portion between aa 507 and 817 was extracted and insertions between ARM repeats were removed. The resulting model was rigid body‐docked into the density using Chimera and Coot and the density fit of the α‐helical bundles was evaluated by calculating the correlation between a simulated model‐map (8.4 Å) and the experimental map using the Chimera.
Labeling of NEDD8 with PET22‐maleimide
The purified human NEDD8 mutant (Methionine 1 to Cysteine, M1C) was incubated with 8 mM DTT at 4°C for 1 h to completely reduce the protein sample. Buffer exchange was performed in non‐reducing buffer conditions, 50 mM Tris pH 7.5 and 150 mM NaCl. PT22‐maleimide (TTP Labtech) were dissolved in 100% DMSO and mixed with NEDD8 in 4:1 ratio. The labeling reaction took place at room temperature after 3‐h incubation in a vacuum desiccator, and the reaction continued overnight at 4°C. Labeled NEDD8 was purified on a Superdex 200 16/60 column in 50 mM Tris pH 7.5, 150 mM NaCl, 0.25 mM TCEP, and 10% (v/v) glycerol. The sample was then concentrated and stored at −80°C.
Neddylation of Cullin 4 with wild‐type or fluorophore‐labeled NEDD8
In vitro neddylation was performed by mixing wild‐type or mutant CRL4DCAF1 at 2 μM with a reaction mixture containing 0.15 μM APPBP1‐UBA3, 6 μM wild‐type NEDD8, and 1 μM UBC12. Reactions were carried out in 50 mM Tris‐HCl pH 7.7, 200 mM NaCl, 2.5 mM MgCl2, 5 mM DTT, and 1.25 mM ATP, and incubated at room temperature for 5–180 min as indicated.
The neddylation reaction with the fluorophore‐labeled NEDD8 involves 0.3 μM APPBP1‐UBA3, 1.2 μM UBC12, 4–8 μM CRL4DCAF1, 8–20 μM NEDD8, 2.5 mM MgCl2, and 1.25 mM ATP. The reaction was carried out in HEPES pH7.4, 200 mM NaCl, and 1 mM TCEP buffer at room temperature.
Fluorescence polarization (FP)‐based CSN activity assays
NEDD8 was labeled with PT22‐maleimide (TTP Labtech) and enzymatically conjugated to CRLs as described. 7‐ to 10‐μl reaction mix contains 50 mM Tris‐HCl pH7.5, 200 mM NaCl, and 1 mM TCEP, 0.1% pluronic acid, and different concentrations of PT22‐labeled CRL4DCAF1 (80 nM–1.5μM), and 15 nM CSN wild‐type for CRL4DCAF1 and CRL4DCAF1‐ MERLIN (1–346), and 150 nM CSN wild‐type for CRL4DCAF1‐VPR and CRL4DCAF1‐VPR‐UNG2. Reactions took place at room temperature in 384‐well plates in a BMG PheraStar plate reader (BMG LabTech). Initial rates were calculated by measuring the change in fluorescence polarization at 590 nm after excitation at 540 nm. Product formation was calculated as described in (Marks et al, 2005). Initial rate constants were calculated by plotting product formation (M) over time (s) using GraphPad Prism version 7.00 (GraphPad Software). Km and Kcat were calculated by non‐linear fitting of initial velocities V0 in the Michaelis–Menten equation.
Author contributions
WIM prepared the specimens for EM data collection. WIM, SC, and ADS. collected EM data. WIM performed EM data processing, with help from SC and ADS. Model building and refinement was carried out by GK. WIM performed the activity assays with input from AP and WAR. WIM performed the biochemical assays. JR and KR contributed in data discussion. NHT, WIM, and AB wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Felix Freuler from Novartis Institute of Biomedical Research (NIBR) in Basel for providing the DCAF1 plasmid. Members of the Thomä laboratory for critical discussions. N.H.T was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation program grant agreement (grant no. 666068), the Novartis Research Foundation, and by the SNF (31003A_179541; 310030_201206). W.I.M. was supported by a PhD fellowship of the Boehringer Ingelheim Fonds from August 2013 to September 2015. We are grateful for Matthias Peter at ETH‐Zürich for providing laboratory access to W.I.M. for the completion of this study. We thank C‐CINA for providing time on the Polara microscope for imaging the CRL4DCAF1‐CSN sample.
The EMBO Journal (2021) 40: e108008.
Data availability
The cryo‐EM maps of the CRL4DCAF1 and CRL4DCAF1‐CSN complexes are deposited in the Electron Microscopy databank under the accession codes EMD‐12964 (https://www.ebi.ac.uk/emdb/entry/EMD‐12964) and EMD‐12965 (https://www.ebi.ac.uk/emdb/entry/EMD‐12965), respectively. The model coordinates for the CRL4DCAF1 structure are deposited in the Protein Data Bank under the accession code 7OKQ (https://www.rcsb.org/structure/7OKQ). Table EV1 summaries cryo‐EM data collection, refinement, and validation statistics. Table EV2 includes the validation statistics for re‐refined template models.
References
- Ahn J, Vu T, Novince Z, Guerrero‐Santoro J, Rapic‐Otrin V, Gronenborn AM (2010) HIV‐1 Vpr loads uracil DNA glycosylase‐2 onto DCAF1, a substrate recognition subunit of a cullin 4A‐ring E3 ubiquitin ligase for proteasome‐dependent degradation. J Biol Chem 285: 37333–37341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Novince Z, Concel J, Byeon C‐H, Makhov AM, Byeon I‐JL, Zhang P, Gronenborn AM (2011) The Cullin‐RING E3 ubiquitin ligase CRL4−DCAF1 complex dimerizes via a short helical region in DCAF1. Biochemistry 50: 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- André I, Bradley P, Wang C, Baker D (2007) Prediction of the structure of symmetrical protein assemblies. Proc Natl Acad Sci USA 104: 17656–17661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N (2006) Molecular architecture and assembly of the DDB1‐CUL4A ubiquitin ligase machinery. Nature 443: 590–593 [DOI] [PubMed] [Google Scholar]
- Baek K, Krist DT, Prabu JR, Hill S, Klügel M, Neumaier L‐M, Gronau S, Kleiger G, Schulman BA (2020) NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly. Nature 578: 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf H‐M, Stegmann L, Schwarz S‐M, Ambiel I, Trotard M, Martin M, Burggraf M, Lenzi GM, Lejk H, Pan X et al (2017) Vpx overcomes a SAMHD1‐independent block to HIV reverse transcription that is specific to resting CD4 T cells. Proc Natl Acad Sci USA 114: 2729–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin PR, Penczek PA (2007) The transform class in SPARX and EMAN2. J Struct Biol 157: 250–261 [DOI] [PubMed] [Google Scholar]
- Banchenko S, Krupp F, Gotthold C, Bürger J, Graziadei A, O’Reilly FJ, Sinn L, Ruda O, Rappsilber J, Spahn CMT et al (2021) Structural insights into Cullin4‐RING ubiquitin ligase remodelling by Vpr from simian immunodeficiency viruses. Plos Pathog 17: e1009775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere DE, Xie L, Srinivas H, Shu W, Burke A, Be C, Zhao J, Godbole A, King D, Karki RG et al (2020) Structural basis of indisulam‐mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat Chem Biol 16: 15–23 [DOI] [PubMed] [Google Scholar]
- Campbell M, Cheng A, Brilot A, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison S, Potter C, Carragher B et al (2012) Movies of ice‐embedded particles enhance resolution in electron cryo‐microscopy. Structure 20: 1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith REJ et al (2016) Cullin–RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531: 598–603 [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2009) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA (2008) Structural insights into NEDD8 activation of cullin‐RING ligases: conformational control of conjugation. Cell 134: 995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Olszewski JL, Tron AE, Hammel M, Lambert LJ, Waddell MB, Mittag T, DeCaprio JA, Schulman BA (2012) Structure of a glomulin‐RBX1‐CUL1 complex: inhibition of a RING E3 ligase through masking of its E2‐binding surface. Mol Cell 47: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberley ED, Mosadeghi R, Deshaies RJ (2012) Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1‐F‐box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J Biol Chem 287: 29679–29689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enchev RI, Scott DC, da Fonseca PCA, Schreiber A, Monda JK, Schulman BA, Peter M, Morris EP (2012) Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep 2: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F et al (2011) The molecular basis of CRL4. Cell 147: 1024–1039 [DOI] [PubMed] [Google Scholar]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM et al (2014) Structure of the DDB1‐CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512: 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kong Q, Liu C, Zhang S, Zou L, Yan F, Whitmire JK, Xiong Y, Chen X, Wan YY (2016) DCAF1 controls T‐cell function via p53‐dependent and ‐independent mechanisms. Nat Commun 7: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusel M, Bludau I, Rosenberger G, Hafen R, Frank M, Banaei Esfahani A, Drogen A, Collins BC, Gstaiger M, Aebersold R (2019) Complex‐centric proteome profiling by SEC‐ SWATH‐ MS. Mol Syst Biol 15: 24–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ (2007) SPARX, a new environment for Cryo‐EM image processing. J Struct Biol 157: 47–55 [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Gierszewska M, Srivastava S, Kozaczkiewicz L, Swanson SK, Florens L, Washburn MP, Skowronski J (2007) Lentiviral Vpr usurps Cul4–DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci USA 104: 11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA (2007) Basis for a ubiquitin‐like protein thioester switch toggling E1–E2 affinity. Nature 445: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Zhuang M, Ayrault O, Schulman BA (2008) Identification of conjugation specificity determinants unmasks vestigial preference for ubiquitin within the NEDD8 E2. Nat Struct Mol Biol 15: 280–287 [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Khan MM, Kar A, Sharma A, Saxena S (2012) CRL4‐DDB1‐VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res 40: 7332–7346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Cooper DR, Oleksy A, Devedjiev Y, Derewenda U, Reiner O, Otlewski J, Derewenda ZS (2004) The structure of the N‐terminal domain of the product of the lissencephaly gene Lis1 and its functional implications. Structure 12: 987–998 [DOI] [PubMed] [Google Scholar]
- Kim K, Heo K, Choi J, Jackson S, Kim H, Xiong Y, An W (2012) Vpr‐binding protein antagonizes p53‐mediated transcription via direct interaction with H3 tail. Mol Cell Biol 32: 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Kim J‐M, Kim J‐S, Choi J, Lee YS, Neamati N, Song JS, Heo K, An W (2013) VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription. Mol Cell 52: 459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R, Chiang C‐Y, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y et al (2007) A probability‐based approach for the analysis of large‐scale RNAi screens. Nat Methods 4: 847–849 [DOI] [PubMed] [Google Scholar]
- Kyei GB, Cheng X, Ramani R, Ratner L (2015) Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe 17: 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You L, Cooper J, Schiavon G, Pepe‐Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB et al (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4DCAF1 in the nucleus. Cell 140: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cooper J, Zhou L, Yang C, Erdjument‐Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P et al (2014) Merlin/NF2 loss‐driven tumorigenesis linked to CRL4. Cancer Cell 26: 48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ et al (2019) Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Cryst D75: 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thomä NH (2014) Crystal structure of the human COP9 signalosome. Nature 512: 161–165 [DOI] [PubMed] [Google Scholar]
- Marks BD, Qadir N, Eliason HC, Shekhani MS, Doering K, Vogel KW (2005) Multiparameter analysis of a screen for progesterone receptor ligands: comparing fluorescence lifetime and fluorescence polarization measurements. Assay Drug Dev Techn 3: 613–622 [DOI] [PubMed] [Google Scholar]
- McCall CM, Miliani de Marval PL, Chastain PD, Jackson SC, He YJ, Kotake Y, Cook JG, Xiong Y (2008) Human immunodeficiency virus type 1 Vpr‐binding protein VprBP, a WD40 protein associated with the DDB1‐CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol Cell Biol 28: 5621–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K et al (2017) Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large‐scale, deep RNAi screening. Cell 170: 577–592 [DOI] [PubMed] [Google Scholar]
- Mori T, Gotoh S, Shirakawa M, Hakoshima T (2014) Structural basis of DDB1‐and‐Cullin 4‐associated Factor 1 (DCAF1) recognition by merlin/NF2 and its implication in tumorigenesis by CD44‐mediated inhibition of merlin suppression of DCAF1 function. Genes Cells 19: 603–619 [DOI] [PubMed] [Google Scholar]
- Mosadeghi R, Reichermeier KM, Winkler M, Schreiber A, Reitsma JM, Zhang Y, Stengel F, Cao J, Kim M, Sweredoski MJ et al (2016) Structural and kinetic analysis of the COP9‐Signalosome activation and the cullin‐RING ubiquitin ligase deneddylation cycle. eLife 5: e12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczek PA, Grassucci RA, Frank J (1994) The ribosome at improved resolution: New techniques for merging and orientation refinement in 3D cryo‐electron microscopy of biological particles. Ultramicroscopy 53: 251–270 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–A visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE (2020) UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, Thomä NH (2016) Structural basis of lenalidomide‐induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 532: 127–130 [DOI] [PubMed] [Google Scholar]
- Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RLJ, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S et al (2013) Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153: 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O et al (2009) Structure prediction for CASP8 with all‐atom refinement using Rosetta. Proteins 77: 89–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichermeier KM, Straube R, Reitsma JM, Sweredoski MJ, Rose CM, Moradian A, den Besten W, Hinkle T, Verschueren E, Petzold G et al (2020) PIKES analysis reveals response to degraders and key regulatory mechanisms of the CRL4 network. Mol Cell 77: 1092–1106 [DOI] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Cohen ÉA (2012) Lentivirus Vpr and Vpx accessory proteins usurp the cullin4–DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol 2: 755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabla NM, Mondal K, Swanson PC (2019) DCAF1 (VprBP): emerging physiological roles for a unique dual‐service E3 ubiquitin ligase substrate receptor. J Mol Cell Biol 11: 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, Jones M, Hassiepen U, Schaefer M, Kiffe M et al (2016) Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun 7: 13166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwefel D, Groom HCT, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, Taylor IA (2014) Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 505: 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima A, Konícková R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thomä NH (2008) Structural basis of UV DNA‐damage recognition by the DDB1‐DDB2 complex. Cell 135: 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Tsherniak A, Gopal S, Weir BA, Tamayo P, Stransky N, Schumacher SE, Zack TI, Beroukhim R, Garraway LA et al (2013) ATARiS: Computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res 23: 665–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY‐R, Kim D, Miles C, Brunette TJ, Thompson J, Baker D (2013) High‐resolution comparative modeling with RosettaCM. Structure 21: 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark H (2010) Chapter Five GraFix: stabilization of fragile macromolecular complexes for single particle cryo‐EM. Methods Enzymol 481: 109–126 [DOI] [PubMed] [Google Scholar]
- Tarricone C, Perrina F, Monzani S, Massimiliano L, Kim M‐H, Derewenda ZS, Knapp S, Tsai L‐H, Musacchio A (2004) Coupling PAF signaling to dynein regulation structure of LIS1 in complex with PAF‐acetylhydrolase. Neuron 44: 809–821 [DOI] [PubMed] [Google Scholar]
- Wang D, Kon N, Lasso G, Jiang LE, Leng W, Zhu W‐G, Qin J, Honig B, Gu W (2016a) Acetylation‐regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 538: 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY‐R, Song Y, Barad BA, Cheng Y, Fraser JS, DiMaio F (2016b) Automated structure refinement of macromolecular assemblies from cryo‐EM maps using Rosetta. eLife 5: e17219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arceci A, Bird K, Mills CA, Choudhury R, Kernan JL, Zhou C, Bae‐Jump V, Bowers A, Emanuele MJ (2017) VprBP/DCAF1 regulates the degradation and nonproteolytic activation of the cell cycle transcription factor FoxM1. Mol Cell Biol 37: e00609‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang Z, Tang Q, Yan XX, Xu W (2019) Crystal structure of the LUFS domain of human single‐stranded DNA binding Protein 2 (SSBP2). Protein Sci 28: 788–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann F, Petzold G, VanderLinden R, Huis in 't Veld PJ, Brown NG, Lampert F, Westermann S, Stark H, Schulman BA, Peters J‐M (2016) biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc Natl Acad Sci USA 113: E2564–E2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhou X, Barnes CO, DeLucia M, Cohen AE, Gronenborn AM, Ahn J, Calero G (2016) The DDB1–DCAF1–Vpr–UNG2 crystal structure reveals how HIV‐1 Vpr steers human UNG2 toward destruction. Nat Struct Mol Biol 23: 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Zhang Y‐L, Pan W‐W, Li X‐M, Wang Z‐W, Ge Z‐J, Zhou J‐J, Cang Y, Tong C, Sun Q‐Y et al (2013) CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science 342: 1518–1521 [DOI] [PubMed] [Google Scholar]
- Yu C, Ji S‐Y, Sha Q‐Q, Sun Q‐Y, Fan H‐Y (2015) CRL4–DCAF1 ubiquitin E3 ligase directs protein phosphatase 2A degradation to control oocyte meiotic maturation. Nat Commun 6: 8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Feng Y, Narayan O, Zhao L‐J (2001) Cytoplasmic retention of HIV‐1 regulatory protein Vpr by protein‐protein interaction with a novel human cytoplasmic protein VprBP. Gene 263: 131–140 [DOI] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam S‐Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V (2018) A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol 430: 2237–2243 [DOI] [PubMed] [Google Scholar]