

Abstract
Mutations in VAV1, a gene that encodes a multifunctional protein important for lymphocytes, are found at different frequencies in peripheral T‐cell lymphoma (PTCL), non‐small cell lung cancer, and other tumors. However, their pathobiological significance remains unsettled. After cataloguing 51 cancer‐associated VAV1 mutations, we show here that they can be classified in five subtypes according to functional impact on the three main VAV1 signaling branches, GEF‐dependent activation of RAC1, GEF‐independent adaptor‐like, and tumor suppressor functions. These mutations target new and previously established regulatory layers of the protein, leading to quantitative and qualitative changes in VAV1 signaling output. We also demonstrate that the most frequent VAV1 mutant subtype drives PTCL formation in mice. This process requires the concurrent engagement of two downstream signaling branches that promote the chronic activation and transformation of follicular helper T cells. Collectively, these data reveal the genetic constraints associated with the lymphomagenic potential of VAV1 mutant subsets, similarities with other PTCL driver genes, and potential therapeutic vulnerabilities.
Keywords: angioimmunoblastic T‐cell lymphoma, follicular helper T cells, oncogene, peripheral T‐cell lymphoma, tumor suppressor
Subject Categories: Cancer, Haematology, Signal Transduction
Systematic mapping of non‐passenger mutations in the multifunctional signaling factor VAV1 offers new insights into their signaling consequences and functional outcomes in vivo.
Introduction
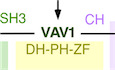
VAV1 is a multifunctional protein that plays signaling amplification and diversification roles downstream of the T‐cell receptor (TCR) (Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). One of the main functions of VAV1 is to catalyze the activation of the RHO GTPase RAC1 (Crespo et al, 1997; Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019) (Fig 1A). This enzyme reaction is mediated by a central cassette of the protein composed of the catalytic Dbl homology (DH) domain and the adjacent pleckstrin homology (PH) and C1‐subtype zinc finger (ZF) regions (Fig EV1A) (Movilla & Bustelo, 1999; Zugaza et al, 2002; Chrencik et al, 2008; Rapley et al, 2008; Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). The activation of RAC1 leads to the remodeling of the F‐actin cytoskeleton and the stimulation of downstream elements such as the cJun N‐terminal kinase (JNK) and the transcriptional factors AP1 and serum responsive factor (SRF) (Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). VAV1 also activates other signal transduction pathways using adaptor, catalysis‐independent mechanisms. One of them is the stimulation via the VAV1 calponin homology (CH) domain of a phospholipase Cγ1 (PLCγ1)‐dependent pathway that leads to the stimulation of the nuclear factor of activated T cells (NFAT) (Fig 1A) (Wu et al, 1995; Kuhne et al, 2000). NFAT is involved in the regulation of proliferation and cytokine production by T cells (Muller & Rao, 2010). More recently, it has been involved in other T‐cell‐specific responses such as the differentiation of follicular helper T (TFH) cells (Martinez et al, 2016). A target of NFAT, the thymocyte selection‐associated HMG‐box protein (TOX) (Scott et al, 2019), is also involved in this latter differentiation process (Xu et al, 2019). Another adaptor‐like function of VAV1, which is associated with tumor suppressor roles in T‐cell acute lymphoblastic leukemia of the TLX+ subtype, is the negative regulation of the active intracellular fragment of NOTCH1 (ICN1) (Fig 1A) (Robles‐Valero et al, 2017). This pathway is mediated by the most C‐terminal VAV1 SH3 (CSH3), which promotes the ubiquitin‐mediated degradation of ICN1 by forming complexes with the E3 ubiquitin ligase CBL‐B (Casitas B‐lineage lymphoma B) (Robles‐Valero et al, 2017). The VAV1 CSH3 can bind to additional partners such as the heterogeneous nuclear ribonucleoprotein K (HNRNPK) and dynamin 2 (DNM2) (Bustelo et al, 1995; Gomez et al, 2005), suggesting that VAV1 might play additional adaptor‐like functions both in normal and cancer cells.
Figure 1. Functional impact of VAV1 mutations in the RAC1 and NFAT pathways.
-
ADepiction of the three main signaling branches of VAV1 in T cells. Additional binding partners of the CSH3 domain that will be studied in this work are also shown. Signaling crosstalk between the NFAT route and parallel TCR‐triggered pathways is also depicted. DAG, diacylglycerol; RAS‐GRP1, Ras GDP releasing protein 1; cNFAT, cytosolic NFAT; nNFAT, nuclear NFAT. Rest of abbreviations have been introduced in the main text.
-
BVav1‐dependent biological readouts and cell types used to test the biological activity of Vav1 mutant proteins. The color of each assay represents the VAV1‐regulated downstream pathway shown in panel (A). Please, note that the GTPase activation and protein–protein interaction experiments were done with smaller subsets of mutants than the JNK, SRF, and NFAT experiments.
-
C, DHeatmap representations summarizing the activity of VAV1 mutants (top) in the indicated assays tested (left). The mutations are clustered according to their specific distribution within the primary structure of the protein (C) and type of behavior in these experiments with the rest of mutants tested (D). Activity scores are depicted on a dark blue (lowest activity) to dark red (highest activity) scale relative to the activity levels found for Vav1WT under nonstimulated conditions (which was given an arbitrary value of 1) (n = 3 independent experiments, each performed in triplicate). The color code used for each mutant is associated with the biological activity exhibited in these assays following the color code indicated in (C) (bottom box). DGOF, double gain of function in RAC1 and NFAT pathways; RSGOF, RAC1 single gain of function; NSGOF, NFAT single gain of function; DLOF, double loss of function; NSLOF, NFAT single loss of function; SG+SLOF, RAC1 gain of function and NFAT loss of function. The laboratory‐made VAV1 mutants used as positive control are indicated by asterisks.
-
EHeatmap representation summarizing the activity of VAV1 mutants (top) in COS (left panel) and Jurkat (right panel) cells on the indicated GTPases. Activity scores are depicted on a white (WT activity) to dark red (highest activity) scales relative to the activity levels found for Vav1WT (which was given an arbitrary value of 1). n = 3 independent experiments, each performed in duplicate. The color code used for each mutant is as in panels (C and D). Activated RHO‐GTPase proteins (Rac1Q61L, RhoAQ63L, and Cdc42Q61L) were used as positive control in the appropriate assay.
-
FTop, heatmap summarizing the results obtained in our GST pull‐down experiments with the indicated CSH3 mutant proteins. Binding partners tested are shown on the left. The mutations are represented in a sequential manner and following the color code used in panels (C and D). Activity scores are depicted on a dark blue (lowest interaction) to dark red (highest interaction) scale relative those obtained with the CSH3WT (which was given an arbitrary value of 1) (n = 3 independent experiments). Bottom, heatmap showing the biological activity of the indicated CSH3 mutants obtained in the experiments described in panels (C and D). This heatmap has been included to facilitate the comparison of the activities of these mutants in all the assays used in this figure.
Source data are available online for this figure.
Figure EV1. Tumor‐associated VAV1 mutations.
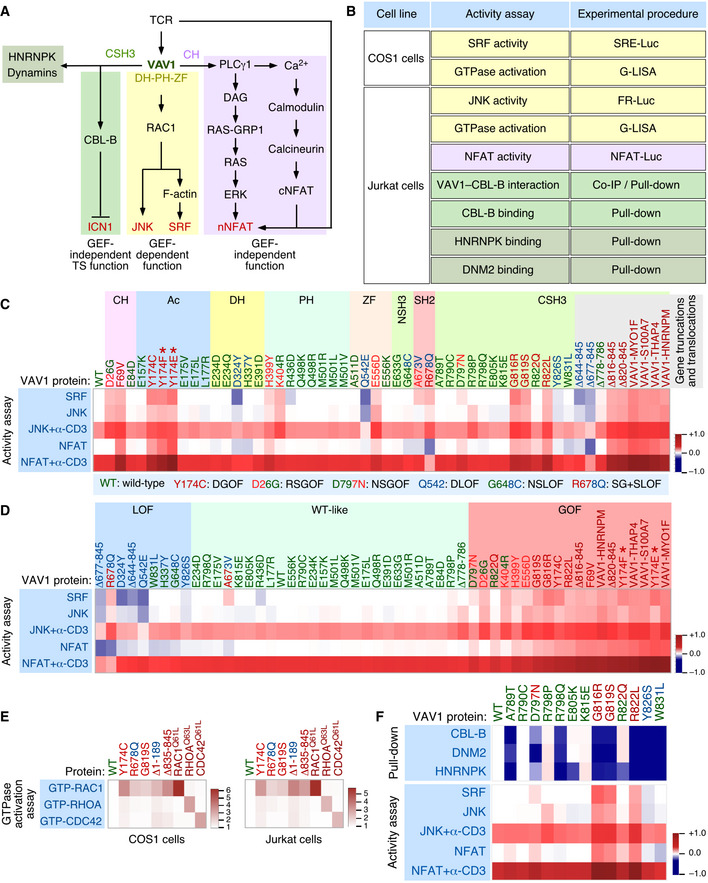
- Localization of VAV1 missense mutations identified in PTCL (top) and NSCLC (bottom) in the primary structure of the protein. Each dot represents one patient. Abbreviations for domains have been described in main text. Mutations analyzed in this study are shown in gray color. The main pathways activated by the VAV1 domains are indicated. CH, calponin homology; Ac, acidic; DH, Dbl homology; PH, pleckstrin homology; ZF, zinc finger; NSH3, most N‐terminal SH3 domain; CSH3, most C‐terminal SH3.
- Examples of C‐terminal truncations (top, proteins 1 to 6), translocations affecting the C‐terminal end (middle, proteins 7 to 11, fused protein indicated as a violet box), and internal gene deletions (bottom, proteins 12 to 14) found in human patients. Mutations analyzed in this study are shown in gray font. Those not tested are in black font.
One of the main regulatory features of VAV1 is that its ability to stimulate both the RAC1 and NFAT pathways is highly dependent on its tyrosine phosphorylation state (Bustelo, 2014). Thus, in nonstimulated cells, nonphosphorylated VAV1 is in an inactive state due to inhibitory interactions established by the N‐terminal (CH and acidic region) domains and the CSH3 region with the central DH‐PH‐ZF cassette. These interactions are eliminated upon the phosphorylation of VAV1 on tyrosine residues located on the acidic (Ac), ZF, and CSH3 regions. This activation step is mediated in trans by the SH2 and most N‐terminal SH3 (NSH3) region that facilitate the interaction of VAV1 with upstream protein tyrosine kinases and adaptor molecules, respectively (Bustelo, 2014).
VAV1 was identified in 1989 due to its oncogenic activity in focus formation assays (Katzav et al, 1989). Despite this, its role in cancer has been circumscribed so far to the upregulation of the wild‐type protein (Bustelo, 2014). This has changed recently, since the characterization of cancer genomes has revealed that VAV1 is frequently mutated in a number of PTCL subtypes such as adult T‐cell leukemia/lymphoma (ATLL, 17% of total cases), PTCL‐not otherwise specified (PTCL‐NOS, 7% of total cases), and angioimmunoblastic T‐cell lymphoma (AITL, 6% of total cases) (Yoo et al, 2014; Crescenzo et al, 2015; Kataoka et al, 2015; Boddicker et al, 2016; Vallois et al, 2016; Abate et al, 2017; Park et al, 2017). Mutations have also been detected at much lower frequencies in anaplastic large cell lymphoma (ALCL), cutaneous T‐cell lymphoma (CTCL), and non‐small‐cell lung cancer (NSCLC) (Crescenzo et al, 2015; Boddicker et al, 2016; Campbell et al, 2016; Abate et al, 2017; Park et al, 2017) (Fig EV1A and B). The actual relevance of these mutations from a functional and pathobiological perspective remains, however, ill‐defined. In this context, recent studies have shown that some tumor‐associated VAV1 mutations lead indeed to gain‐of‐function (GOF) events (Boddicker et al, 2016; Abate et al, 2017; Fukumoto et al, 2020). However, these analyses have been limited to a very small (8%) and overlapping subset of mutations that target obvious regulatory layers of the protein. Moreover, those studies have not tested all the spectrum of downstream signals elicited by this protein (Boddicker et al, 2016; Abate et al, 2017; Fukumoto et al, 2020). As a result, we do not know yet whether most VAV1 mutations found in tumors act as bona fide oncogenic drivers in vivo and, if so, whether they do it autonomously or in combination with ancillary inputs from other genetic lesions and/or cancer cell‐extrinsic events. Likewise, we do not know the spectrum of downstream signaling pathways that have to be engaged to promote full cell transformation.
To address these relevant knowledge gaps, we have measured qualitatively and quantitatively the functional impact of 51 VAV1 mutations found in tumors on all known VAV1‐dependent signaling outputs. This strategy allowed us to generate a comprehensive signaling and functional catalogue of the tumor‐associated VAV1 mutations. Using adoptive cell transfer experiments, we have also demonstrated that the most frequent functional subclass of VAV1 mutations identified in our study can drive AITL formation. Finally, we have identified the main signaling and pathobiological programs involved in the generation of Vav1‐driven tumors using a combination of cellular, signaling, and genome‐wide gene expression approaches. These findings suggest that VAV1 mutations probably play clinical‐relevant roles in AITL and other tumor types. They also pinpoint several VAV1 signaling‐based pharmacological strategies to treat them.
Results
Variegated functional impacts of VAV1 mutations found in tumors
To start building a functional catalogue of the VAV1 mutations present in tumors, we decided to comprehensively analyze the impact of 51 mutations recently found in PTCL and NSCLC (Fig EV1A and B) using several experimental readouts for the main downstream signaling pathways engaged by this protein (Fig 1A and B). The mutations tested encompassed an internal deletion, four fusions, four truncations, and 42 missense changes. In the case of point mutations, we included in the analyses allelic variants found in some codons. First, we investigated the impact of the 51 mutations on the two main catalysis‐dependent (RAC1–JNK and RAC1–SRF) and CH‐dependent (PLCγ1–NFAT) pathways regulated by VAV1 (Fig 1B). To this end, we carried out in the former case luciferase‐based reporter assays to measure the effect of the interrogated mutant proteins on the activity of both JNK and SRF in Jurkat (nonstimulated and TCR‐stimulated) and exponentially growing COS1 cells, respectively (Fig 1B). In the case of the NFAT pathway, we carried out luciferase‐based reporter experiments in nonstimulated and TCR‐stimulated Jurkat cells to measure the impact of the mutants in the transcriptional activity of an Il2 promoter containing three NFAT‐binding sites (Fig 1B). It is worth noting that, unlike the case of RAC1‐dependent pathways, the optimal stimulation of the NFAT pathway by VAV1 requires parallel signaling inputs from the antigen receptor in this assay. As a result, the activation of this transcriptional factor is enhanced upon the engagement of the TCR even in T cells that express fully deregulated, constitutively active VAV1 versions (Barreira et al, 2014). As positive control, we included in these analyses the lab‐made Vav1Y174E and Vav1Y174F mutant proteins that are known to promote high levels of stimulation of both the RAC1 and NFAT pathways (Lopez‐Lago et al, 2000; Barreira et al, 2014). The results obtained for all the interrogated Vav1 mutants in these assays, compared with the reference basal (Vav1WT) and positive (Vav1Y174F, Vav1Y174E) controls, are collectively depicted using a heatmap representation in Fig 1C and D. The wet‐lab data used to generate these heatmaps are shown both in the Appendix Figs S1–S8. Using this approach, we found that 25% of the interrogated mutations elicit a bivalent GOF effect on the RAC1 and NFAT pathways (Fig 1C and D; residues shown in red). These mutants can be further subdivided according to their specific impact on the signaling output of the protein as weak (e.g., E556D), intermediate (e.g., Y174C, G819S), and strong (e.g., Δ820–845) (Fig 1D). Only the latter ones, which are associated with the generation of C‐terminally truncated and fusion proteins, exhibit fully constitutive, phosphorylation‐independent biological activity (Fig 1D). More unexpectedly, we observed that 12% of the VAV1 mutations display signaling branch‐specific effects that cause the specific stimulation of either the RAC1 or the NFAT pathway (Fig 1C and D; residues labeled in red and blue colors at the same time). Finally, 63% of the interrogated mutations show either WT‐like (47%, Fig 1C and D; residues labeled in green) or reduced activity in these two assays (16%, Fig 1C and D; residues labeled in blue). It is worth noting, however, that some of these mutants will induce the elimination of the tumor suppressor pathway regulated by VAV1 and, therefore, must have an impact in Notch1 signaling (see below in this section). As a complementary avenue to the data obtained using the indirect JNK and SRF assays, we used the G‐LISA method to test the direct effect of 3 VAV1 mutants belonging to the bivalent (Y174C, G819S) and signaling branch‐specific (R678Q) subsets on the activation of the three main RHO family GTPases in both COS1 and Jurkat cells. As positive controls, we utilized constitutively active versions of VAV1 (Δ1‐189 and Δ835‐845), RAC1 (Q61L), RHOA (Q63L), and CDC42 (Q61L). When compared to VAV1WT, we found that all the chosen VAV1 mutants could activate the incorporation of GTP onto RAC1 irrespectively of the functional subclass involved (Fig 1E and Appendix Fig S9). By contrast, they exhibited much lower activities on RHOA and CDC42 (Fig 1E and Appendix Fig S9). This RAC1 specificity is consistent with previous biochemical and cell‐based experiments (Crespo et al, 1997; Aghazadeh et al, 2000; Couceiro et al, 2005; Rapley et al, 2008).
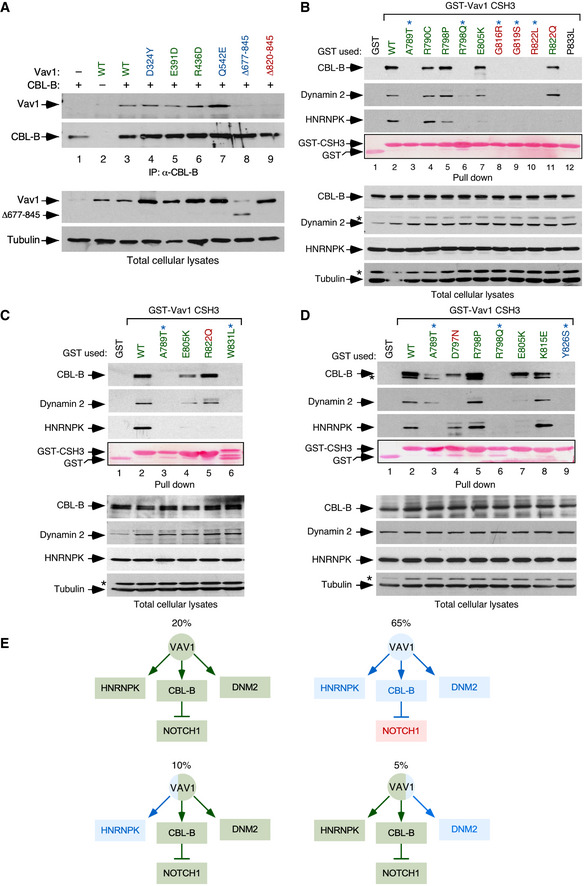
We next investigated the effect of specific mutations on the recently described tumor suppressor pathway regulated by VAV1 (Fig 1A). According to our previous data (Robles‐Valero et al, 2017), this function must be lost in the case of all the truncation and translocation VAV1 mutants that have lost the CSH3 domain. In agreement with this, we found that the Vav1Δ677–845 protein cannot coimmunoprecipitate CBL‐B when expressed in Jurkat cells (Fig EV2A). The role of this mutation must be specifically associated with the elimination of this suppressor pathway, given that Vav1Δ677–845 shows reduced activity when tested in the JNK, SRF, and NFAT assays (see above, Fig 1C). By contrast, VAV1WT and versions of VAV1 with mutations outside the CSH3 that exhibit WT‐like activities in the previous assays (Fig 1C) do associate with this E3 ubiquitin ligase (Fig EV2A). This suggests that these latter mutations are probably bystanders. Since missense mutations targeting residues located in the CSH3 can also lead to the loss of the suppressor function (Robles‐Valero et al, 2017), we next carried out GST pull‐down experiments to evaluate the interaction of the 13 VAV1 CSH3 point mutants with CBL‐B, HNRNPK, and DNM2. As negative control, we used the nonchimeric GST protein and, in some experiments, a GST fusion protein containing a mutant version of the VAV1 CSH3 (P833L) that cannot interact with any known protein partner (Barreira et al, 2014; Robles‐Valero et al, 2017). As positive control, we used a GST‐VAV1 CSH3WT fusion protein as bait. A heatmap summarizing the data obtained in all these experiments is shown in Fig 1F. The raw data can be found in Fig EV2B–D. These analyses revealed that 65% of the Vav1 CSH3 mutants analyzed have impaired physical interactions with CBL‐B and the other two protein partners (Fig 1F, see scheme in Fig EV2E). They also identified a smaller subset (15%) of Vav1 CSH3 mutants with a selective impairment of the interaction with either HNRNPK (E805K, R822Q) or DNM2 (D797N) (Fig 1F, see scheme in Fig EV2E).
Figure EV2. CSH3 mutations alter the interaction between VAV1 and binding partners.
-
ACo‐immunoprecipitation of indicated Vav1 proteins with CBL‐B in Jurkat cells ectopically expressing the indicated combinations of proteins (top). Amount of immunoprecipitated CBL‐B was assessed by reblotting the same filter with antibodies to CBL‐B (second panel from top). Expression of ectopic VAV1 proteins (third panel from top) and endogenous tubulin α (loading control, bottom panel) was determined by immunoblotting using aliquots of the total cellular lysates used in the immunoprecipitation step. The color code of the mutant residues follows the criteria used in Fig 1C and D.
-
B–DRepresentative examples of the interactions found in Jurkat cells among GST‐tagged proteins fused to the indicated CSH3 versions of Vav1 and the endogenous CBL‐B, Dynamin 2, and HNRNPK proteins. In all cases, the relative amount of bait used in the pull‐down assays is shown in the fourth panel from top. Expression of the endogenous proteins was determined by immunoblotting using aliquots of the total cellular lysates used in the pull‐down step. Asterisks in the second and fourth panels in the total cellular lysate panels pinpoint the CBL‐B and HNRNPK immunoreactive bands from the previous immunoblot of the same filter, respectively. In (D), asterisk in the first panel in the pull‐down panels indicates the Dynamin 2 band from the previous immunoblot of the same filter. Similar results were obtained in three additional experiments. GST‐VAV1P833L, negative control of interaction (it bears a mutation that inactivates the binding properties of the domain). The color code of the mutant residues follows the criteria used in Fig 1C and D.
-
ESummary of binding alterations found in the Vav1 CSH3 mutants. Proper binding is shown as green boxes and arrows. Lack of binding is shown as blue boxes and arrows. The activation of NOTCH1 signaling is depicted as a red box. The percentage of each mutant subclass (relative to all CSH3 mutants tested) is indicated at the top of each functional subgroup.
Source data are available online for this figure.
Taken the results from all the experimental readouts together, we conclude that 49% of the interrogated VAV1 mutations are likely passengers according to the currently known functions of VAV family proteins (Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). Many of these mutations, as expected, retain WT‐like activity in all the assays used. However, we also include in this category mutations that cause the inhibition of RAC1 and/or NFAT activities without the elimination of the tumor suppressor activity of VAV1 as they must not significantly contribute to the chronic activation of the downstream signaling of the protein. The remaining VAV1 mutants (51%) interrogated in this work do cause some type of GOF effect in the activity of the protein. Such mutants can be classified in five main functional subtypes according to the spectrum of signaling branches specifically affected by them (Fig 2A). The most prevalent subclass includes mutations that lead to a concurrent activation of the RAC1 and NFAT pathways as well as to the elimination of the tumor suppressor activity of VAV1 (35% of all cases; Fig 2A, trivalent subclass). In terms of incidence, this mutant subset is found in ALCL, PTCL‐NOS, AITL, and lung adenocarcinomas (Fig 2B). This trivalent subclass mostly includes fusion and truncated proteins, with very few cases involving missense mutants (Fig 2C). All of them target, as expected (Barreira et al, 2014; Robles‐Valero et al, 2017), the CSH3 domain (Fig 2A). The second most frequent subclass (27%) corresponds to proteins that have specifically lost the tumor suppressor activity (Fig 2A, monovalent subclass). We include in this category mutants that exhibit either WT‐like or different types of inhibitory effects on the RAC1 and NFAT pathways (Fig 2A). These alterations, which are found in lung tumors, CTCL, AITL, and ATLL (Fig 2B), are associated with the generation of either large truncations (lung tumors, AITL) or single amino acid changes (ATLL, CTCL) in the mutant protein (Fig 2C). These mutations target the CSH3 or the SH2‐CSH3 region (Fig 2A). Mutants that promote a bivalent GOF effect on the RAC1 and NFAT pathways constitute the third most frequent functional category (15% of mutants with deregulated activity; Fig 2A, bivalent subclass). These alterations, which are found in decreasing frequencies in ATLL, AITL, lung adenocarcinoma, and PTCL‐NOS (Fig 2B), are exclusively associated with missense changes (Fig 2C) in the CH, Ac, PH, or ZF domains (Fig 2A). A fourth subclass encompasses mutants showing enhanced GDP/GTP exchange activity and variegated effects on the NFAT pathway (15% of mutants with deregulated activity; Fig 2A; uncoupling subclass 1). This subclass, which is found in ATLL and lung tumors (Fig 2B), is composed of missense mutations (Fig 2C) located on the CH (subtype 4a), PH (subtype 4a), and SH2 (subtype 4b) regions (Fig 2A). Finally, the least frequent subclass (8%) includes mutants that lead to the specific activation of the NFAT pathway while maintaining WT‐like RAC1 activity (Fig 2A, uncoupling subclass 2). Interestingly, these mutants are the only ones that show specific alterations in the binding to either HNRNPK (subtype 5a) or DNM2 (subtype 5b) (Fig 2A). This subclass, which is preferentially found in CTCL, AITL, and ATLL (Fig 2B), is associated with missense mutations targeting the CSH3 region (Fig 2B and C). By far, the most common hot spot region of the protein based on these analyses is the CSH3 domain (Fig 2A). These data also indicate that the tumors with the highest percentage of functionally relevant mutations are ALCL, AITL, and PTCL‐NOS (Fig 2B). By contrast, the tumors with more passenger mutations include ATLL, CTCL, lung adenocarcinoma, and lung squamous cell carcinoma (Fig 2B).
Figure 2. An integrated view of the mutational landscape of VAV1 in tumors.
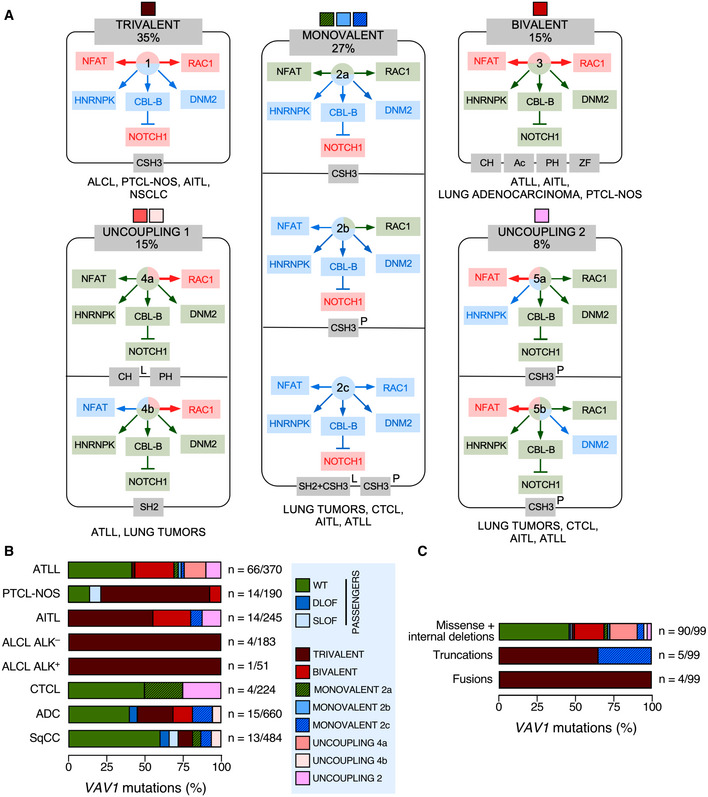
- Main VAV1 mutation functional subclasses according to the data obtained in Fig 1. The color code of each subclass shown at the top will be used in panels (B and C) to facilitate the identification of each of them. The percentage of each mutant subclass (relative to all the deregulated VAV1 mutants tested) is also indicated at the top. The GOF and LOF effects of each mutant subclass are shown in red and blue colors (boxes and arrows). WT‐like activity is depicted as green arrows and boxes. The domains targeted by the mutations are shown in gray boxes at the bottom of each subclass. The tumors where the mutations have been found are shown at the bottom of each subclass. L, lung cancer; P, peripheral T‐cell lymphoma.
- Distribution of VAV1 mutation functional subclasses in the indicated tumor subtypes. The mutation subclasses are indicated following the color codes shown in the inset on the right.
- Type of mutations associated with the indicated functional subclasses (which are indicated following the color codes shown in the inset present in panel B).
New regulatory layers unveiled by the VAV1 mutations
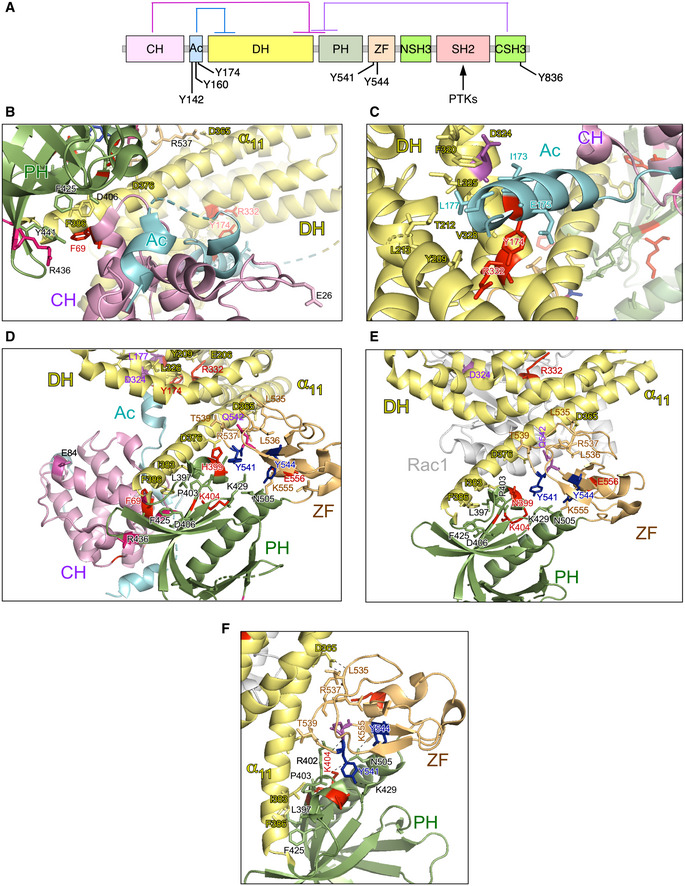
The reason for the high biological activity exhibited by most bivalent and trivalent GOF VAV1 mutations found in this study can be explained well according to the current regulatory model for this protein family (Fig EV3A) (Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). Consistent with this, most of them target common regulatory hot spots of the protein such as the CH, Ac, PH, and CSH3 domains (Fig EV3A). As an example, it is likely that bivalent GOF effect created by the CH domain‐located F69V mutation is generated by the disruption of the interactions that the F69 residue establishes with the PH (D406), the DH α11 helix (F386), and the first PH β strand (Y441) (Fig EV3A and B). These interactions are critical for maintaining the autoinhibited structure of nonphosphorylated VAV1 (Yu et al, 2010). Similarly, the bivalent Y174C GOF mutation targets a phosphorylation site located in the Ac region that contributes to the autoinhibitory structure by making interactions with a hydrophobic pocket present in the DH domain (Yu et al, 2010) (Fig EV3A and C). Hence, these two missense alterations must trigger an opening of the molecule similar to that induced by the laboratory‐made Y174F and Y174E mutations (Lopez‐Lago et al, 2000; Barreira et al, 2014, 2018) (Fig 1C). It is likely that other mutations not surveyed in our analyses could belong to this functional class (Δ151–158, Δ165–174) (Fig EV1B), given that they can either displace or eliminate the critical regulatory phosphosites Y160 and Y174 that are also present in the Ac region of the protein. The GOF effects triggered by most mutations targeting the CSH3 are also consistent with the elimination of the autoinhibitory and/or suppressor functions of this domain (Barreira et al, 2014; Robles‐Valero et al, 2017). However, the present results indicate that the CSH3 autoinhibitory surface is larger than anticipated because the cancer‐associated mutation target residues (G816, G819) located away from the two previously characterized inhibitory interfaces of that domain (Fig 3A, residues shown in raspberry color) (Barreira et al, 2014). Unlike the case of the lab‐made mutations targeting those previously characterized inhibitory sites (Barreira et al, 2014), the mutations of the G816 and G819 residues also impair the interaction with CBL‐B (Fig 1F). As a result, they simultaneously alter the VAV1‐regulated RAC1, NFAT, and tumor suppressor pathways. Hence, the effect of these mutations is analogous to the truncations and translocations that eliminate the entire CSH3 domain. However, the effects found with other VAV1 mutations indicate that there must exist extra regulatory layers involved in the control of the signaling output of this protein. Thus, the bivalent GOF effect induced by mutations in the CH‐located D26 residue (E26 in mouse) is quite unexpected since this residue does not participate in any of the known intramolecular interactions that stabilize the inactive structure of the protein (Aghazadeh et al, 2000). It is possible therefore that this residue could be involved in the formation of interdomain inhibitory interactions with the CSH3 (Barreira et al, 2014) or the SH2 (this work, see below) domains. Likewise, the GOF effects elicited by mutations located in the PH (H399 and K404) and ZF (E556) were also somewhat unexpected, given that the targeted residues do not participate in any of the known intramolecular interactions that contribute to stabilize the inactive state of nonphosphorylated VAV1 (Yu et al, 2010). These residues are located in an area of the catalytic DH‐PH‐ZF cassette (Fig EV3D–F) that harbors residues involved in the phosphorylation‐dependent activation of VAV1 activity (Y541, Y544) (Barreira et al, 2014), the overall spatial organization of the catalytic cassette (Chrencik et al, 2008; Rapley et al, 2008; Yu et al, 2010), and the inhibitory interactions with both the CH and the CSH3 regions (Yu et al, 2010; Barreira et al, 2014). This area also undergoes a significant spatial reorientation during the transition of VAV1 from the inactive to the active state (Yu et al, 2010). It is likely therefore that the bivalent GOF effect induced by the N399Y and E556D mutations is mediated by the displacement of the CH from the autoinhibitory site, leading to the chronic stimulation of the RAC1 and NFAT pathways. Unlike the foregoing residues, we have found that the K404R promotes the stimulation of the RAC1 but not of the NFAT pathway (Fig 1C). Given the multiple direct and indirect interactions established by this residue in the VAV1 structure (Fig EV3F), it is likely that its mutation promotes the exposure of the catalytic site of VAV1 through the displacement of PH α helix away from ZF and DH domains that, in turn, change the spatial orientation of the CH region. However, the lack of activation of NFAT suggests that the displacement of the CH does not involve in this case the full exposure of the effector interface that engages that pathway. Alternatively, this mutation can induce a conformation of the whole protein not compatible with optimal NFAT signaling by that domain. The latter possibility seems more plausible, given that mutations in the SH2 and CSH3 also promote a similar dissociation of the activation of the RAC1 and NFAT (Fig 1E). Signaling uncoupling effects have been previously observed with lab‐made mutations targeting specific tyrosine phosphorylation and lysine acetylation sites of the protein (Barreira et al, 2014, 2018; Rodríguez‐Fdez et al, 2020).
Figure EV3. New regulatory layers unveiled by the VAV1 mutations.
-
ADepiction of the main intramolecular interactions that mediate the inactivation of VAV family proteins in the nonphosphorylated state. The interactions of the CH, Ac, and CSH3 domains with the catalytic cassette are shown in pink, blue, and purple lanes, respectively. The phosphorylation sites involved in the activation step are indicated. PTKs, protein tyrosine kinases involved in the tyrosine phosphorylation‐mediated activation step of VAV family proteins.
-
B, CDepiction of areas of interaction between the CH‐Ac and the catalytic cassette. The side chains of the residues whose mutation leads to GOF effects are shown in red. The main regulatory phosphosites are shown in blue. The CH, Ac, and DH region are colored in purple, blue, and yellow, respectively.
-
D, EDepiction of the area of the catalytic cassette containing the α11 DH helix, the PH, and the ZF regions in the context of the crystallized CH‐Ac‐DH‐PH‐ZF (D) and the CH‐PH‐ZF (E) fragments of human and mouse Vav1, respectively. Residues whose mutation leads to GOF and LOF effects are colored in red and blue, respectively. The CH, Ac, DH, PH, and ZF regions are colored in purple, blue, yellow, green, and brown, respectively.
-
FZoom of the Vav1 α11 DH helix‐PH‐ZF region showing the interactions established by the indicated residues. Residues and domains are depicted as in above panels.
Figure 3. New regulatory layers unveiled by the VAV1 mutations.
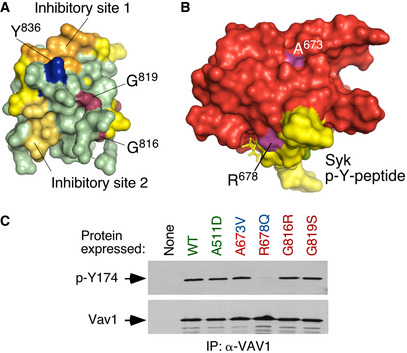
- 3D structure of the Vav1 CSH3 domain. The previously described intramolecular inhibitory sites 1 and 2 of the Vav1 CSH3 domain are depicted in orange and yellow, respectively (Barreira et al, 2014). The regulatory Y836 phosphosite is highlighted in blue (Barreira et al, 2014). The residues present in the new inhibitory interface of the CSH3 are shown in raspberry color.
- 3D structure of the VAV1 SH2 (red) bound to a Syk tyrosine‐phosphorylated peptide (yellow). The mutated residues are shown in purple. p‐Y, tyrosine phosphorylated.
- Levels of phosphorylation of the Y174 phosphosite of the indicated Vav1 mutant proteins immunoprecipitated from Jurkat cells (upper panel). The total amount of Vav1 immunoprecipitated in each sample is shown in the bottom panel (n = 3 independent experiments).
Source data are available online for this figure.
Our studies have also revealed a hitherto unknown regulatory role of the SH2 domain in the intramolecular inhibition of the nonphosphorylated protein. To date, the main function of this domain has been associated with the tyrosine phosphorylation‐mediated activation of the RAC1 and NFAT pathways (Bustelo & Barbacid, 1992; Bustelo et al, 1992; Bustelo, 2014; Rodriguez‐Fdez & Bustelo, 2019). Consistent with this, Vav1 proteins bearing lab‐generated mutations in this domain are always inactive when tested in RAC1 and NFAT assays (Zugaza et al, 2002). Against this well‐established paradigm, we have found two NSCLC‐associated SH2 mutations (R673V and R678Q) that unexpectedly cause the stimulation of the RAC1‐dependent pathways (Fig 1C). Also, against the long‐held consensus regulatory model for the Vav family, one of those mutations targets a residue in the phosphotyrosine‐binding site (R678) that is critical for the interaction with both the upstream protein tyrosine kinases and downstream effectors (Fig 3B). Consistent with this, we have found that Vav1R678Q cannot become tyrosine phosphorylated when ectopically expressed in Jurkat cells (Fig 3C). It is unlikely that the negative regulatory role of the R678 residue is due to the participation of the VAV1 SH2 domain in the interaction of an inhibitory phosphorylation site of the molecule, because other mutations that inactivate this domain (e.g., G691V) trigger a full LOF rather than a GOF effect in the molecule (Zugaza et al, 2002; Rodríguez‐Fdez et al, 2020). In line with this, the second activating SH2 mutation found in this study that maps outside the phosphotyrosine‐binding site does not impair the overall phosphorylation levels of the protein (Fig 3C, residue A673V). Interestingly, these two SH2 mutations show reduced levels of NFAT activation, thus suggesting that the targeted residues are also important for the optimal engagement of the PLCγ1–NFAT axis in lymphocytes (Fig 1C). To our knowledge, this is the first time in which activating mutations in the SH2 domain have been ever described for any VAV family member.
Vav1ΔC drives PTCL formation when expressed in CD4+ T cells
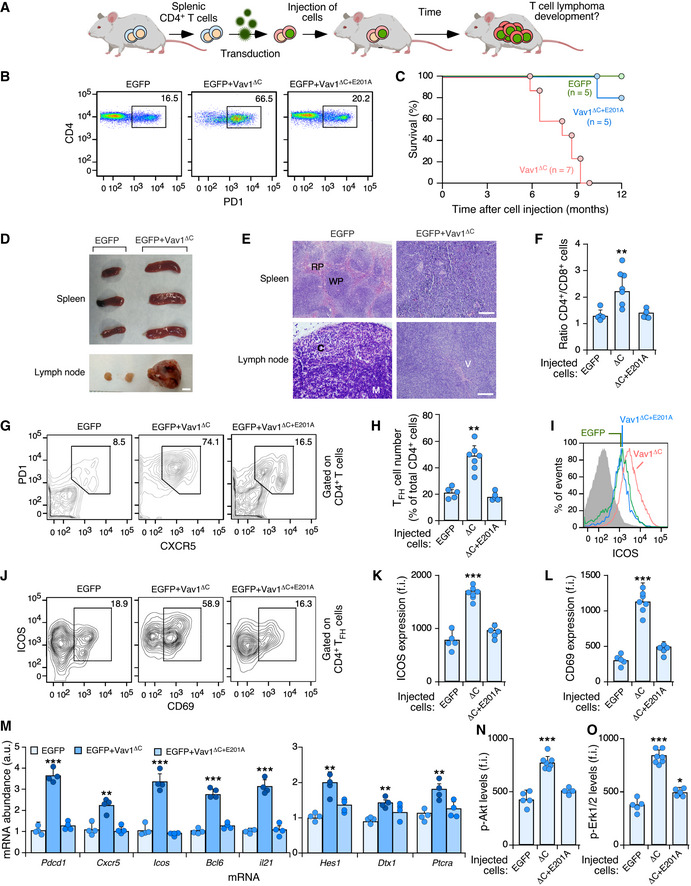
To investigate the relevance of VAV1 mutations and the downstream pathways activated by them in tumorigenic processes in vivo, we next carried out adoptive T‐cell transfer experiments to assess the impact of deregulated Vav1 signaling in T lymphomagenesis. We chose this model because, according to the results from the above sections, ALCL, AITL, and PTCL‐NOS are the tumors that show the highest percentages of functionally relevant VAV1 mutations (Fig 2B). These alterations usually involve the concurrent activation of RAC1, NFAT, and ICN1 signaling (Fig 2B). In line with this, we decided to use in these experiments a mutant version of Vav1 with a truncated CSH3 (amino acids 835‐845, referred to hereafter as Vav1ΔC) that is functionally analogous to the trivalent VAV1 mutant subclass frequently observed in those lymphoma subtypes. We also included in these experiments a catalytically dead version of Vav1ΔC (ΔC+E201A) to assess the specific relevance of the concurrent upregulation of the NFAT and ICN1 pathways to the tumorigenic processes in the absence of RAC1 activation. To this end, we infected TCR plus CD28‐stimulated mouse splenic CD4+ T cells with retroviral vectors encoding bicistronically each mutant Vav1 protein and EGFP and, subsequently, introduced them into Vav1 −/−; Vav2 −/−; Vav3 −/− mice to test the potential development of lymphomas (Fig 4A). These recipient mice are T lymphopenic (Fujikawa et al, 2003; Bustelo & Dosil, 2016), a feature that facilitates their utilization in transplantation experiments involving even cells of human origin (Lorenzo‐Martín et al, 2020). The long‐term follow‐up of these mice indicated that those transplanted with Vav1ΔC‐expressing CD4+ T cells progressively develop an expanded population of CD4+ T cells associated with TFH immunosurface features (PD1+, CXCR5+) (Fig 4B). These animals die within a 3‐month period after the initial detection of this population (t 1/2 = 8 months after the transplantation step) (Fig 4C), indicating that these Vav1ΔC‐expressing cells have been oncogenically transformed. Unlike the case of other mutations found in PTCL cases (e.g., RHOA G17V, FYN‐TRAP3IP2) (Cortes et al, 2018; Moon et al, 2021), the Vav1ΔC‐driven lymphomagenesis does not require the use of CD4+ T cells deficient for the 5‐methylcytosine oxidase Tet2, a suppressor protein that is lost in a large number of human PTCLs (Fiore et al, 2020). Necropsies performed in tumor‐bearing animals showed the recurrent presence of mild splenomegaly as well as enlarged and vascularized lymph nodes (Fig 4D and E). Histological analyses also showed that these tissues display effaced boundaries between the red and the white pulps (spleen) and the cortical and medullary regions (lymph nodes) (Fig 4E). These animals also exhibit an expanded population of PD1+, CXCR5+, and CD4+ T cells (Fig 4F–H) that are highly decorated with ICOS (Fig 4I–K) and CD69 (Fig 4J and L), two surface markers typically associated with an activated TFH state. All these histological and immunophenotypical features are consistent with the development of AITL‐like tumors in these mice. No alterations in CD4+ T cells were observed in mice transplanted with either EGFP‐ or EGFP+Vav1ΔC+E201A‐expressing cell at the same post‐transplantation time (Fig 4F–L).
Figure 4. Vav1ΔC drives PTCL formation when expressed in CD4+ T cells.
-
ADepiction of adoptive T‐cell transfer experiments used in this figure. See Materials and Methods for further details.
-
BFlow cytometry detection of PD1+ CD4+ T cells in the peripheral blood from recipient mice 6 months after being transplanted with CD4+ T cells expressing the indicated proteins (top). Numbers indicate the relative percentage (%) of the EGFP+ population (boxed) found in each case. Same results were obtained in recipient mice transplanted with EGFP‐ (n = 5), Vav1ΔC‐ (n = 7) and Vav1ΔC+E201A‐transduced (n = 5) CD4+ T cells.
-
CSurvival curves of mice transplanted with CD4+ T cells expressing the indicated proteins. The number of animals used is indicated in the graph. Note: The mouse transplanted with EGFP‐transduced cells that has died in these experiments did not show any sign of tumor development according to anatomopathological analyses (data not shown).
-
DRepresentative images of spleen and lymph nodes from recipient mice transplanted with cells expressing the indicated combination of proteins (top) at the time of euthanasia. Scale bar, 1 cm.
-
ERepresentative examples of hematoxylin–eosin‐stained sections of a spleen (top) and a lymph node (bottom) from recipient mice transplanted with the indicated cells (top) at the time of euthanasia. Scale bars, 100 μm. n = 4 animals per class analyzed. RP, red pulp; WP, white pulp; C, cortex; M, medulla; V, venules.
-
FQuantification of CD4+ versus CD8+ T‐cell ratio in peripheral blood from mice transplanted with CD4+ T cells expressing the indicated proteins (bottom). Each point represents the measurement of an individual mouse. n as in (B).
-
GSurface expression of PD1 and CXCR5 in CD4+‐gated splenocytes from recipient mice transplanted with CD4+ T cells expressing the indicated combination of proteins (top). Numbers indicate the relative percentage (%) of the cell population that has been interrogated (boxed).
-
HQuantification of the percentage of TFH cells in CD4+‐gated splenocytes isolated the indicated experimental conditions. Each point represents the measurement of an individual mouse. n as in (B).
-
IExample of a flow cytometry determination of ICOS expression in TFH cells from recipient mice transplanted with cells expressing the indicated proteins. The gray shaded histogram represents the fluorescence obtained with the isotype‐matched control antibody.
-
J–LRepresentative flow cytometry analysis (J) and quantification of the surface levels of ICOS (K) and CD69 (L) in TFH cells from mice transplanted with CD4+ T cells expressing the indicated proteins. In (J), numbers indicate the relative percentage (%) of the cell population that has been interrogated (boxed). In (K and L), each point represents the values obtained with a single experimental mouse. n as in (H).
-
MAbundance of indicated transcripts (bottom) in splenic cells from control (EGFP+), tumor‐bearing mice (expressing EGFP and Vav1ΔC) and mice transplanted with cells transduced with the catalytically dead version of Vav1ΔC (expressing EGFP and Vav1ΔC+E201A). Values are given relative to the expression of each transcript in samples obtained from EGFP controls (which was given an arbitrary value of 1). n = 4 animals per class analyzed. a.u., arbitrary unit.
-
N, OFlow cytometry determination of p‐Akt (N) and p‐Erk1/2 (O) levels in CD4+ TFH‐gated splenocytes from recipient mice transplanted with cells expressing the indicated proteins (bottom). In both panels, each point represents the values obtained with a single experimental mouse. n as in (H).
Data information: In panels (F, H, K, L, M, N, and O), data represent the mean ± SEM. Statistical values obtained using the Student’s t are given relative to control EGFP+ cells. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Source data are available online for this figure.
Further buttressing the TFH cell‐like phenotype of these tumor cells, we found using quantitative reverse transcription‐PCR (qRT–PCR) that they express high levels of transcripts encoding typical follicular helper cell markers such as PD1, CXCR6, ICOS, Bcl6, and interleukin 21 (Fig 4M, left panel). In addition, they show elevated levels of canonical Notch1 target genes such as Hes1, Dtx1, and Ptcra (Fig 4M, right panel). Most of these transformed cell isolates also display high levels of p‐Akt (Figs 4N and EV4A) and p‐Erk (Figs 4O and EV4A) when compared to EGFP+ lymphocytes. These molecular and signaling features are not observed in cells expressing the catalytically deficient Vav1ΔC+E201A mutant protein (Fig 4M). The Vav1ΔC‐transformed cells show wild‐type‐like levels of the tumor suppressors Tp53 and Tet2 (Fig EV4A and B). These results indicate that the expression of Vav1ΔC in mouse CD4+ T cells can trigger AITL‐like tumors in a Tp53‐ and Tet2‐independent manner. They also demonstrate that: (i) The catalytic activity of Vav1ΔC is essential to drive this lymphomagenic process. (ii) The activation of the NFAT and ICN1 is not enough per se to drive T lymphomagenesis in these experimental conditions.
Figure EV4. Status of signal transduction pathways in Vav1ΔC‐transformed cells.
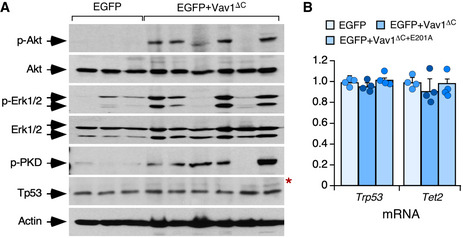
- Western blot showing the levels of phosphorylated and total proteins in spleen extracts from control and tumor‐bearing recipient mice that had been transplanted with the indicated retrovirally transduced CD4+ T cells (top). Total levels of Akt, Erk1/2, and actin were used as loading controls. The red asterisk indicates a panel that has been generated using an independent filter but the same tissue lysates.
- Abundance of indicated transcripts (bottom) in splenic cells from control (EGFP+), tumor‐bearing mice (expressing EGFP and Vav1ΔC), and mice transplanted with cells transduced with the catalytically dead version of Vav1ΔC (expressing EGFP and Vav1ΔC+E201A). Values are given relative to the expression of each transcript in samples obtained from EGFP controls (which was given an arbitrary value of 1). n = 4 animals per class analyzed. a.u., arbitrary unit. Data represent the mean ± SEM. Statistical values obtained using the Student’s t are given relative to control EGFP+ cells.
Source data are available online for this figure.
Vav1ΔC triggers a vast proliferative program in transformed TFH cells
Microarray analyses revealed that Vav1ΔC‐transformed cells contain 2,277 differentially expressed genes (1,554 upregulated, 823 downregulated) when compared to normal CD4+ T lymphocytes (Fig 5A and Dataset EV1). No significant differences were detected between the transcriptomes of cancer cells obtained from the spleen and lymph nodes (Fig 5A), suggesting that the Vav1ΔC‐driven gene expression program is rather tumor niche‐independent. Consistent with our previous analyses, gene set enrichment analyses (GSEAs) indicated that this transcriptome is highly similar to the TFH cell gene expression landscape (Fig 5B). Functional annotation analyses revealed that the Vav1ΔC upregulated transcriptome is mainly composed of proliferation‐related genes (Figs 5C and EV5A, and Dataset EV1). Yet, principal component analyses indicate that this transcriptome is more closely related to PTCLs than to other unrelated proliferative tumor types (Fig EV5B). The downregulated subset of the Vav1ΔC‐induced transcriptome is mainly enriched in genes linked to TCR signaling and activation (Fig 5C and Dataset EV1). Although counterintuitive, this feature is commonly found in previously reported AITL‐associated transcriptomal programs in both mice and humans (Zang et al, 2017). GSEA further revealed that Vav1ΔC promotes the expression of genes linked to heme metabolism, inflammatory processes, the unfolded protein response, and the mTORC pathway (Fig EV5A). From a more molecular perspective, the Vav1ΔC‐driven transcriptome is enriched in gene signatures typically associated with Vav1 downstream signaling elements such as SRF (Fig 5D), AP1 (Fig 5E), the NFAT–Tox axis (Fig 5F), and ICN1 (Fig 5G). Concurring with increased levels of Akt‐mTORC signaling in Vav1ΔC‐transformed cells (Figs 4N and O, and EV4A), we also identified gene signatures linked to the downregulation of Foxo (Fig EV5C), a transcriptional factor negatively regulated by p‐Akt (Manning & Toker, 2017). Unbiased GSEAs further revealed the enrichment in E2F‐ (Figs 5H and EV5A) and Myc‐regulated (Figs 5I and EV5A) gene signatures in this transcriptome.
Figure 5. Vav1ΔC triggers a vast proliferative program in transformed TFH cells.
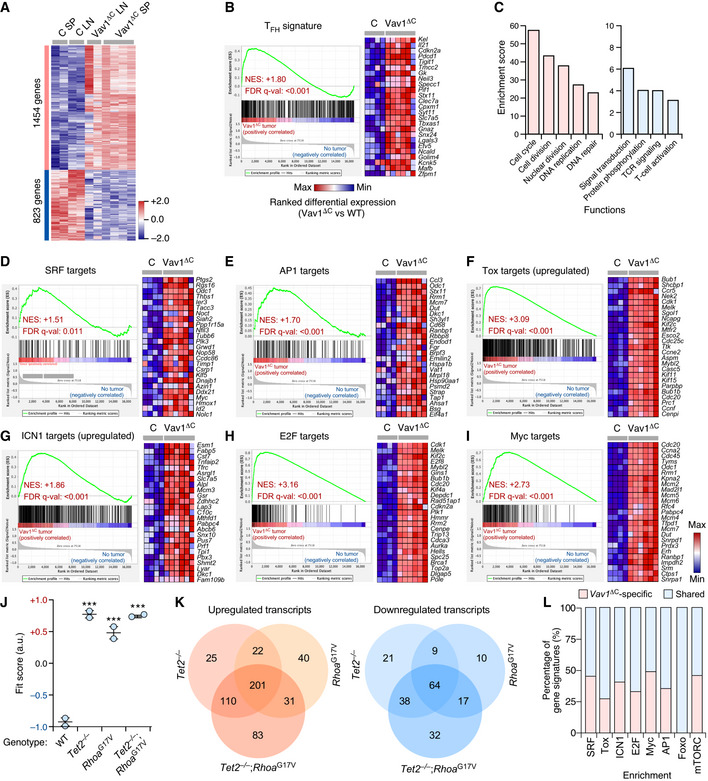
-
AGenes up‐ (red) and downregulated (blue) in the tumor cells from lymph nodes (Vav1ΔC LN) and spleen (Vav1ΔC SP) of recipient mice transplanted with Vav1ΔC‐expressing CD4+ T cells. As comparative control, we used CD4+ T cells from spleen (C SP) and lymph nodes (C LN) of control recipient mice. Rows represent individual genes. Columns represent experimental replicas. Total number of genes is indicated on the left. Relative changes in abundance are shown in color gradients according to the color scale shown on the right.
-
BGSEA showing enrichment of TFH‐associated gene signatures in the transcriptome of Vav1ΔC‐transformed cells. The expression profile of the top 25 leading‐edge genes in the transcriptome of CD4+ T cells from healthy control (C) and Vav1ΔC‐tumor‐bearing mice (Vav1ΔC) is shown. The normalized enrichment scores (NES) and false discovery rate values (FDR, using q values) are indicated inside the GSEA graph. Relative changes in abundance are shown in color gradients according to the color scale shown at the bottom. q‐val, q value.
-
CMain functional categories encoded by the Vav1ΔC‐dependent transcriptome. For all of them, P ≤ 0.001 (Fisher’s exact test).
-
D–IGSEAs showing enrichment of indicated gene signatures (top) in the transcriptome of Vav1ΔC‐transformed cells. In each case, we show the expression profile of the top 25 leading‐edge genes in healthy control (C) and Vav1ΔC‐tumor‐bearing mice (Vav1ΔC). NES and FDR values (FDR, using q values) are indicated inside each GSEA graph. Relative changes in abundance are shown in color gradients according to the same color scale shown in panel (I).
-
JDot plot of the Vav1ΔC‐dependent gene signature fit score in the indicated experimental groups (bottom) that were retrieved from a previous work (Zang et al, 2017). Dots represent values from an individual sample. Bars represent the mean enrichment value ± SEM for the overall sample set. ***P ≤ 0.001 (according to Tukey's honest significance difference test).
-
KVenn diagrams representing the number of up‐ (left) and downregulated (right) genes from the Vav1ΔC‐dependent transcriptome shared with the indicated datasets.
-
LLevel of enrichment of indicated gene signatures that are either Vav1ΔC‐specific (red) or shared with the different datasets (blue) used in panel (J).
Figure EV5. Vav1ΔC triggers a vast proliferative program in transformed TFH cells.
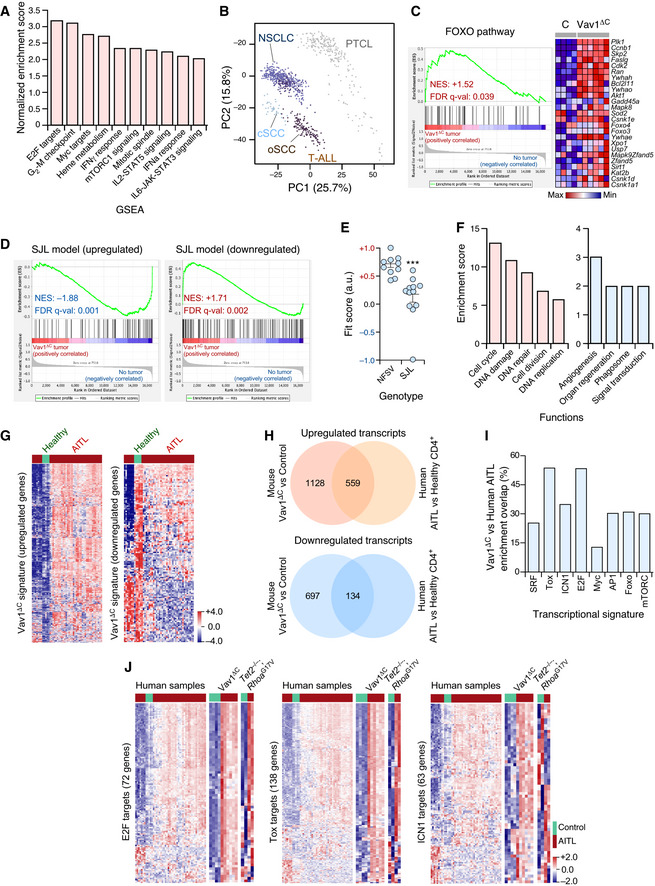
- Main functional pathways upregulated in the Vav1ΔC‐dependent transcriptome according to unbiased GSEAs. For all of them, q‐val < 0.05.
- Principal component analysis showing the enrichment of the Vav1ΔC‐dependent signature across the indicated tumor‐associated transcriptomes. cSCC, cutaneous squamous cell carcinoma; oSCC, oral squamous cell carcinoma; T‐ALL, T‐cell acute lymphoblastic leukemia.
- GSEA for the Vav1ΔC‐tumor gene expression matrix using as gene set the differentially expressed genes by the FOXO pathway. The expression profile of the top 25 leading‐edge genes in the transcriptome of CD4+ T cells from healthy and Vav1ΔC tumor‐bearing mice is shown. The normalized enrichment scores (NES) and false discovery rate values (FDR, using q values) are indicated inside the GSEA graph. Relative changes in abundance are shown in color gradients according to the color scale shown at the bottom. q‐val, q value.
- GSEAs for the Vav1ΔC‐tumor gene expression matrix using as gene set the differentially expressed genes by the SJL mouse model. The NES and FDR (using q values) are indicated inside the GSEA graph as in C. q‐val, q value.
- Dot plot of the Vav1ΔC‐dependent gene signature fit score in the indicated experimental groups (bottom). Dots represent values from an individual sample. Bars represent the mean enrichment value ± SEM for the overall sample set. ***P ≤ 0.001 (according to Tukey’s honest significance difference test).
- Main functional categories encoded by the Vav1ΔC‐specific gene subset described in the main text. Up (red); down (blue). For all of categories, P ≤ 0.001 (Fisher’s exact test).
- Fraction of the upregulated (left) and downregulated (right) Vav1ΔC‐driven transcriptome showing conservation with the expression programs identified in human AITL patients. Relative changes in abundance are shown in color gradients according to the color scale shown on the right. Healthy, CD4+ T cells from healthy patients (green box). AITL, human AITL (red color boxes).
- Venn diagrams representing the number of up‐ (top) and downregulated (bottom) genes from the Vav1ΔC‐dependent transcriptome that are shared with the AITL patient dataset used in panel G.
- Enrichment of indicated Vav1ΔC‐driven gene signatures that are shared with the AITL patient dataset used in panel G.
- Heatmaps showing the expression profiles of human and mouse differentially expressed genes (DEG) belonging to the indicated functional categories (indicated on the left of each panel). Each row represents 1 DEG identified in the indicated AITL models (top). Each column represents 1 sample. The number of genes of each functional category is indicated. Relative changes in abundance are shown in color gradients according to the color scale shown on the bottom right. Green boxes, healthy individuals (in the case of human data), and WT mice. Red boxes, AITL samples from either patients or the indicated mouse models.
Vav1ΔC shares pathobiological programs with PTCL driver genes and human AITL
Given the AITL‐like phenotype exhibited by Vav1ΔC‐transformed TFH cells, we next decided to investigate the level of similarity of the transcriptome of those cells with the gene expression changes previously seen associated with the deletion of Tet2 and/or the expression of a dominant‐negative (G17V mutation) version of the GTPase RhoA in CD4+ T cells (Zang et al, 2017). It has been shown before that the combination of these two genetic events in CD4+ T cells is required for driving the transformation of CD4+ T cells (Zang et al, 2017; Cortes et al, 2018). This cooperativity has been attributed to the downmodulation of Foxo levels that take place upon Tet2 ablation (Zang et al, 2017). In addition, we have included in these in silico comparisons the transcriptome previously described in the AITL‐like condition that spontaneously develops in Swiss Jim Lambert (SJL)/J mice (Jain et al, 2015; Mhaidly et al, 2020). This disease is primarily derived from the exacerbation of IL21 signaling in TFH cells (Jain et al, 2015). In silico analyses revealed a high degree of similarity of the Vav1ΔC‐driven transcriptome with those found deregulated in Tet2 −/− cells (490 genes), RhoAG17V‐expressing WT cells (394 genes), and RhoAG17V‐expressing Tet2 −/− cells (576 genes) (Fig 5J and K, and Dataset EV2). Interestingly, all the genes associated with proliferative processes are found specifically enriched in the transcriptomal subset shared between Vav1ΔC‐expressing cells and RhoAG17V‐expressing Tet2 −/− cells (Dataset EV2). By contrast, the downmodulation of Foxo is found in the overlapping transcriptomes between Vav1ΔC‐expressing cancer cells and the preneoplastic Tet2 −/− cells (Dataset EV2). This further suggests that the Tet2‐independent transformation triggered by Vav1ΔC could be associated with the depletion of Foxo levels in TFH cells. We did not find any statistically significant similarity with the transcriptome previously described in the SJL/J mouse model, indicating that the similarity found between the Vav1ΔC and the RhoAG17V; Tet2 −/− model is specific. In fact, in the case of SJL/J mice, we found quite opposite transcriptomal patterns when using GSEAs (Fig EV5D and E).
The above results indicate that 69% of the 2,277 genes found differentially expressed in Vav1ΔC‐transformed cells are not shared by the rest of datasets used in these in silico analyses (Dataset EV3). This gene subset is basically enriched in proliferation‐related genes (Fig EV5F), suggesting that Vav1ΔC promotes the amplification rather than the rewiring of the transcriptomal changes commonly present in AITL cells. In line with this, we observed using further in silico analyses that this very specific Vav1ΔC transcriptomal subset is associated with a larger enrichment in SRF, AP1, E2F, Myc, the NFAT–Tox axis, mTORC, and ICN1 gene signatures (Fig 5L). By contrast, the Foxo‐related signature is quite similar between Vav1ΔC‐expressing and Tet2‐deficient cells (Fig 5L). This result further emphasizes the convergent action of Vav1 GOF and Tet2 LOF events on the repression of this transcriptional factor.
Further in silico analyses indicated that the Vav1ΔC‐driven transcriptome found in the transformed TFH cells bears high levels of similarity with the differential expression programs present in a large percentage of AITL patients (Fig EV5G and H). Such similarity is significantly higher in the Vav1ΔC upregulated (49.5% of cross‐species overlap) than in the downregulated (19.2% of cross‐species overlap) gene subset (Fig EV5H). The overlapping transcriptomal subsets are enriched in gene signatures linked to the function of E2F and the NFAT–Tox axis, although SRF‐, ICN1‐, AP1‐, Foxo‐, and mTORC‐related gene expression programs are also observed (Fig EV5I and J). As expected (Fig 5J and K), a similar overlap is seen between the transcriptomes of human and mouse RhoAG17V; Tet2 −/− AITL samples (Fig EV5J). However, unlike the case of the Vav1ΔC‐driven transcriptome, we could not observe any consistent enrichment in this case in Notch1‐related gene signatures (Fig EV5J, right panel). Collectively, these data indicate significant levels of similarity of the gene expression program of Vav1ΔC‐transformed CD4+ T cells and human AITL cases.
Vav1ΔC drives proliferation in very early preneoplastic stages
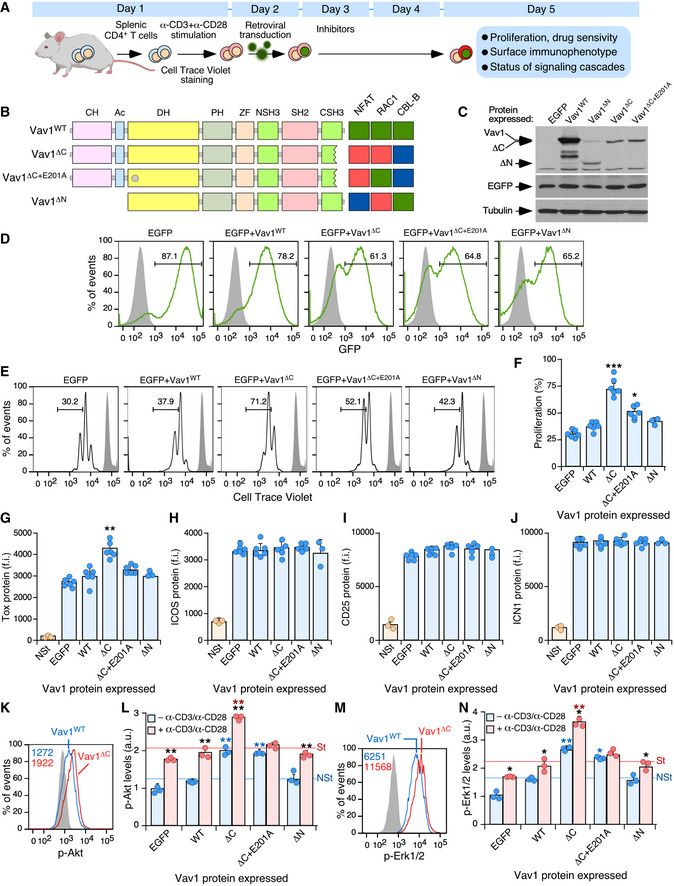
To further dig into the signal transduction pathways and early pathobiological programs triggered by Vav1ΔC, we next analyzed its early effects in primary CD4+ T cells (Fig 6A). To this end, CD4+ lymphocytes were isolated from the spleen of WT mice, stimulated with antibodies to the TCR and CD28, and then infected with retroviral particles encoding bicistronically EGFP and the indicated versions of Vav1 (Fig 6B). Finally, the resulting EGFP+ cells were analyzed three days later using a number of biological and signaling readouts (Fig 6A). In all cases, the effective transduction of cells and Vav1 expression obtained with each viral pool was assessed using both flow cytometry and immunoblotting (Fig 6C and D). These experiments demonstrated that the short‐term expression of Vav1ΔC promotes elevated levels of proliferation (Fig 6E and F) and expression of the NFAT target Tox (Fig 6G) in TCR plus CD28‐primed cells. These two effects can be observed even though the stimulation of lymphocytes carried out before the transduction step already leads to a significant upregulation of these two readouts (Fig 6F and G). By contrast, the expression of Vav1WT or Vav1ΔC+E201A does not upregulate those two responses (Fig 6F and G). None of the interrogated Vav1 proteins changed the levels of ICOS (Fig 6H), CD25 (Fig 6I), or ICN1 (Fig 6J) in the primed cells. It is worth noting, however, that the effects of Vav1ΔC on those three proteins could be obscured by the levels of induction that are already triggered upon the concurrent engagement of the TCR and the CD28 coreceptor in these cells (Fig 6H–J). The activation of ICN1 under these cell stimulation conditions is consistent with previous data in CD4+ T cells (Palaga et al, 2003; Steinbuck et al, 2018; Mitra et al, 2020).
Figure 6. Vav1ΔC‐mediated proliferation requires engagement of several downstream pathways.
-
ASchematic representation of the experiments used in this figure. See details in main text and Materials and Methods.
-
BDepiction of the Vav1 mutants used in these experiments. The E201A mutation is shown as a gray circle. The activity of each mutant is represented on the right. Green box, WT activity; red box, gain of function; blue box, loss of function.
-
CRepresentative immunoblot showing the abundance of the ectopic Vav1 proteins, EGFP, and the endogenous tubulin α (loading control) in CD4+ T cells transduced with retrovirus particles used in these experiments (top).
-
DRepresentative FACS plots showing EGFP epifluorescence in CD4+ T cells transduced with retroviruses encoding the indicated proteins (top). Numbers indicate the relative percentage (%) of the EGFP+ cell population in each case. Gray shaded histograms represent the fluorescence obtained from CD4+ T cells nontransduced with retrovirus particles. Similar results were obtained in CD4+ T cells transduced with virus encoding EGFP (n = 6), Vav1WT (n = 6), Vav1ΔC (n = 6), Vav1ΔC+E201A (n = 6), and Vav1ΔN (n = 3).
-
E, FRepresentative FACS plots (E) and quantification (F) of the proliferation of EGFP+ CD4+ T cells expressing the indicated proteins using the Cell Trace Violet method. In (E), the gray shaded histograms represent the fluorescence obtained from nonstimulated CD4+ T cells before stimulation and retroviral transduction. In (F), each point represents the values obtained with a single experimental mouse. n = 6 per each experimental condition, except in the case of Vav1ΔN (n = 3).
-
G–JFlow cytometry determination of Tox (G), ICOS (H), CD25 (I), and ICN1 (J) expression in EGFP+ CD4+ T cells expressing the indicated Vav1 proteins. NSt, nonstimulated cells (CD4+ T cells before stimulation and retroviral transduction). In all panels, each point represents the values obtained for a single experimental condition. f.i., mean fluorescence intensity relative to the isotype‐matched control antibody. n = 6 per each experimental condition, except for the Vav1ΔN and NSt conditions (n = 3).
-
K–NExample of a flow cytometry analysis (K and M) and final quantification of p‐Akt (L) and p‐Erk1/2 (N) levels in EGFP+ CD4+ T cells expressing the indicated Vav1 proteins in primed (blue bars) and restimulated (red bars) cells. In K and M, the gray shaded histograms represent the fluorescence obtained with the isotype‐matched control antibody. In L and N, NSt, nonstimulated, St, stimulated. n = 3 per each experimental condition.
Data information: In panels (F, G, H, I, J, L, and N), values are shown as means ± SEM from three independent experiments. In panels L and N, P‐values are given relative to nonstimulated (blue asterisks) and stimulated (red asterisks) cells expressing Vav1WT. We also included P‐values for the values exhibited by each Vav1 mutant protein relative to those obtained in nonstimulated condition (black asterisks). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 (Student’s t‐test).
Source data are available online for this figure.
Interestingly, we observed that both Vav1ΔC and Vav1ΔC+E201A promote higher levels of both p‐Akt (Fig 6K and L; blue bars) and p‐Erk (Fig 6M and N; blue bars) in primed cells when compared to the EGFP+ and Vav1WT controls. The restimulation of Vav1ΔC‐expressing cells leads to a hysteresis response in the phosphorylation of those two signaling elements, a phenomenon that does not occur in the case of Vav1ΔC+E201A‐expressing CD4+ T cells (Fig 6L and N; red bars). These results indicate that the short‐term expression of Vav1ΔC in normal lymphocytes recapitulates many biological features that are subsequently observed in Vav1ΔC‐transformed TFH cells.
Vav1ΔC‐mediated proliferation requires engagement of several downstream pathways
The above experiments indicate that both the early signals and the long‐term lymphomagenesis triggered by Vav1ΔC in TFH cells are catalysis dependent. The lack of activity shown by the Vav1ΔC+E201A mutant also suggests that the concurrent engagement of NFAT and ICN1 pathways cannot drive T‐cell transformation per se. However, they do not clarify the issue of whether the catalysis‐dependent pathways work fully autonomously or require the participation of other Vav1 downstream pathways to promote the abnormal proliferation of those cells. To tackle this issue, we included in these short‐term experiments an additional mutant version of Vav1 that lacks both the CH and Ac domains (Vav1ΔN) (Fig 6B). This protein exhibits constitutive, phosphorylation‐independent enzymatic activity due to the removal of the CH‐Ac region (Schuebel et al, 1998; Zugaza et al, 2002). However, due to the lack of the CH domain and the presence of a functional CSH3, it cannot stimulate the PLCγ1‐NFAT and ICN1 pathways (Wu et al, 1995; Robles‐Valero et al, 2017) (Fig 6B). Due to this, this mutant is similar to those belonging to the uncoupling 4a subset described in Fig 2A. Despite being expressed at levels comparable to Vav1ΔC in the transduced cells (Fig 6C), Vav1ΔN cannot trigger the proliferation (Fig 6F) or the elevation of Tox levels (Fig 6G) seen in Vav1ΔC‐expressing CD4+ T cells. Similarly, Vav1ΔN cannot increase the levels of p‐Akt (Fig 6L) and p‐Erk (Fig 6N) in primed and restimulated cells. Vav1ΔN shows in fact less activity than the Vav1ΔC+E201A mutant in primed CD4+ T cells before the restimulation step (Fig 6L and N; blue bars). These results suggest that the full perversion of these signaling responses requires, at least, the concurrent hyperstimulation of the CH‐ and catalysis‐dependent signaling branches of Vav1. In addition, they indicate that those branches cooperate in a stepwise manner to promote (the CH‐mediated pathway) and amplify (the catalysis‐dependent pathway) the phosphorylation of both Akt and Erk in primed and restimulated CD4+ T cells, respectively.
Given that we could not properly assess the role of Vav1ΔC in the upregulation of ICN1 signaling in this experimental setting, we decided to indirectly address whether the activation of this pathway could affect the effects triggered by Vav1ΔC in CD4+ T lymphocytes. To this end, we used Compound E, an inhibitor that eliminates the proteolytic cleavage of NOTCH1 required to generate the active ICN1 fragment by blocking the γ‐secretase endoproteinase (Kopan & Ilagan, 2009). We used as comparative control the treatment of cells with FK506, an inhibitor of the calmodulin‐dependent calcineurin phosphatase (Fig 1A) (Clipstone & Crabtree, 1992). This protein catalyzes the dephosphorylation of cytoplasmic NFAT, an essential step for the nuclear translocation and activation of this transcriptional factor (Muller & Rao, 2010). The γ‐secretase inhibitor abrogates the proliferative advantage (Fig 7A and B) and the elevation of Tox levels (Fig 7C) previously seen with Vav1ΔC in the primed CD4+ T cells. These effects are specific since Compound E does not affect these two readouts in the case of the control EGFP+ cells (Fig 7A–C). As expected, this drug reduces the levels of ICN1 in both control and Vav1ΔC‐expressing cells (Fig 7D). FK506 reduces the proliferation of both control (EGFP+) and Vav1ΔC‐expressing cells (Fig 7A and B). It also eliminates the upregulation of the NFAT‐regulated Tox protein (Fig 7C) while it has no overt effects on the abundance of ICN1 (Fig 7D). We could not observe any effect of those two compounds on ICOS levels (Fig 7E). These results indicate that Vav1ΔC needs to engage at least the downstream Rac1 and NFAT pathways to effectively alter the proliferative state of primary CD4+ T cells. They also indicate that NOTCH1 signals will play a relevant role in this process (Fig 7F).
Figure 7. Vav1ΔC‐driven CD4+ T‐cell proliferation requires ICN1 and proper calcineurin signaling.
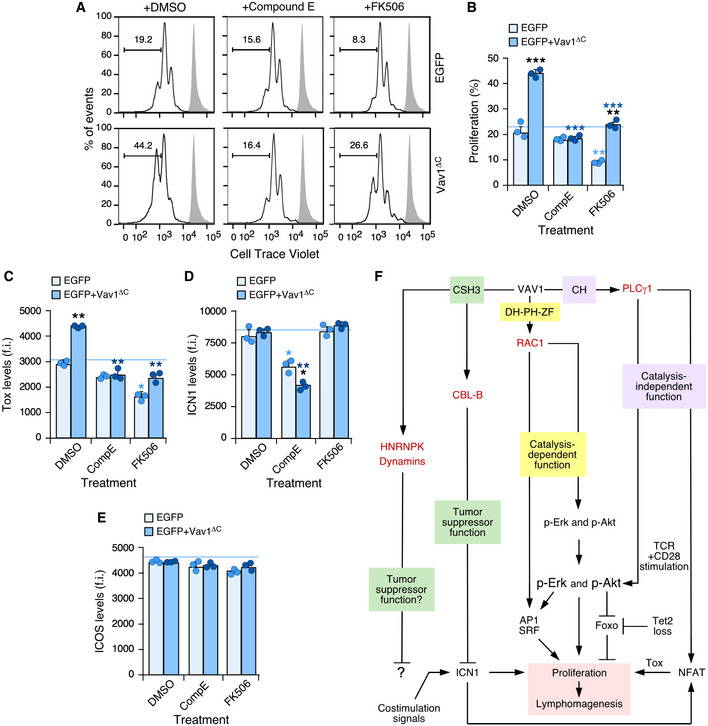
-
A, BRepresentative FACS plots (A) and quantification (B) of the EGFP+ CD4+ T‐cell proliferation in the different experimental groups (right) and indicated experimental conditions (top) using the Cell Trace Violet detection method. In A, gray shaded histograms represent the fluorescence obtained from nonstimulated CD4+ T cells before stimulation and retroviral transduction. n = 3 independent experiments.
-
C–EFlow cytometry determination of intracellular Tox (C), intracellular ICN1 (D), and surface ICOS (E) levels in EGFP+ CD4+ T cells expressing the indicated Vav1 proteins. f.i., mean fluorescence intensity relative to the isotype‐matched control antibody. n = 3 independent experiments.
-
FSummary of the Vav1‐regulated signaling pathways that contribute to promote the proliferation of primary CD4+ T cells that have been unveiled in this work. The other downstream effectors and pathways of Vav1 are indicated. The main primary effectors as shown in red.
Data information: In panels (B, C, D, and E), values are shown as means ± SEM from three independent experiments. P‐values are given relative to nontreated (light blue asterisks) and treated (dark blue asterisks) EGFP+ cells. We also include P‐values for the values exhibited by each experimental group relative to those obtained in nontreated condition (black asterisks). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 (Mann–Whitney U‐test).
Source data are available online for this figure.
Discussion
Our findings indicate that the VAV1 mutations found in tumors can generate a complex panoply of signaling outcomes (Fig 2A). They have also provided new and, in some instances, quite unexpected information regarding the regulatory and effector layers of this protein. Thus, in addition to targeting well‐established regulatory VAV1 hot spots, these mutations have unveiled: (i) An additional surface of the CSH3 domain involved in the intramolecular autoinhibition of the protein (encompassing residues G816 and G819) (Fig 3A). (ii) New intramolecular regulatory areas associated with the DH α11 helix‐PH‐ZF region (H399, K404, E556). (iii) A quite unsuspected role the SH2 domain in the intramolecular inhibition of the protein that has been unveiled by the catalytic GOF effect found by mutations targeting the A673 and R678 residues (Fig 1C). (iv) Mutations causing signaling branch‐specific effects in the RAC1 and NFAT pathways (Fig 2A, uncoupling subclasses 1 and 2). Those mutations target residues present in the CH (D26G), PH (K404R), NSH3 (G648C), SH2 (A673V, R678Q), and CSH3 (D797N, R822Q) regions. Similar uncoupling effects have been observed before with mutations targeting specific tyrosine phosphorylation and lysine acetylation sites of the protein (Barreira et al, 2014, 2018; Rodríguez‐Fdez et al, 2020). In those cases, this effect has been associated with the freezing of the molecule in intermediary conformational states that precede the fully phosphorylated and activated condition (Barreira et al, 2014, 2018; Rodríguez‐Fdez et al, 2020). It would be important to solve the structure of the some of these mutant proteins to figure out the exact conformational and signaling problems associated with all these uncoupling effects. (v) Mutations that preferentially eliminate the binding of either HNRNPK or DNM2 to the VAV1 CSH3 (Figs 1F and EV2). The relevance of this change in the binding to downstream protein partners is currently unknown. However, it is intriguing that, similarly to CBL‐B, HNRNPK and some DNM family members (e.g., DNM3) have been associated with tumor suppression functions (Gallardo et al, 2015; Zhang et al, 2016) (Fig 7F). Another possibility is that the main advantage created by these alterations is just the stimulation of the NFAT pathway that is commonly observed in all the mutants of this functional subtype (Fig 2A, uncoupling subclass 2). Addressing these possibilities, which are not mutually exclusive, deserves further attention in the near future.
Our adoptive T‐cell transfer experiments with our collection of Vav1 mutants indicate that only the most potent mutations that lead to the concurrent hyperstimulation of the RAC1 and NFAT pathways will be able to autonomously drive TFH cell lymphomagenesis in patients (see scheme in Fig 7F). Our short‐term in vivo experiments also indicate that the NOTCH1‐derived signals will be important to provide an extra proliferative boost to the cells bearing VAV1 mutations (Fig 7F). These results agree with the observation that the most prevalent VAV1 mutations found in AITL belong to the trivalent and bivalent subclasses. These data also suggest that the VAV1 mutants that deregulate single signaling branches could only drive AITL when the pathways that retain WT‐like activity in those mutants become hyperactivated by convergent pathways from other mutant proteins (e.g., NOTCH1, PLCγ1) or from strong costimulation signals from adjacent lymphoid or stromal cells.
Our work has also generated a new experimental model that can be used to better understand the etiology of AITL cases lacking TET2 LOF mutations from the earliest preneoplastic to the full‐blown transformed states. An inference from our study is that most PTCL‐NOS cases that are VAV1 mutation+ will likely correspond to the recently characterized TFH subtype (Chiba & Sakata‐Yanagimoto, 2020; Fiore et al, 2020). These two diseases are still poorly understood due to the relatively small number of cases available for study, their complex genetic make‐up, and the multiple cancer cell‐extrinsic processes affecting its evolution (Fiore et al, 2020). The results obtained with our mouse model support, in agreement with previous observations (de Leval et al, 2007; Piccaluga et al, 2007; Cortes et al, 2018; Fiore et al, 2020), that these tumors sprout from TFH cells. They also indicate that the preneoplastic phase of the disease is associated with a population of chronically activated TFH cells that can stay in stasis for long periods of time until shifting into a phase of rapid expansion and terminal disease. This long latency is in syntony with the usual detection of this disease in elderly people (Chiba & Sakata‐Yanagimoto, 2020). It is likely, however, that this process can be accelerated by concurrent genetic lesions that could develop during this latency phase of the disease. Our data also suggest that VAV1 mutant+ patients could benefit from therapies based on the inhibition of the RAC1 and NFAT signaling elements and VAV1 itself. Interestingly, we have observed that the transcriptional program found in Vav1ΔC‐transformed cells is quite similar to that previously observed in RHOAG17V‐expressing Tet2 −/− AITL cells. This suggests that AITL patients with these distinct mutational packages will exhibit similar clinical features and overlapping therapeutic vulnerabilities such as, dependency on exacerbated Akt, Erk, and ICOS signaling.
We have not detected the development of lymphomas with TH1 or TH2 features in our adoptive transfer experiments even though Vav1WT does play roles in these lineages (Gulbranson‐Judge et al, 1999; Tanaka et al, 2005). The reason for this selectivity is unknown, although it can be connected to recent observations indicating that the polarization toward the TFH lineage is associated with a Bcl6‐ and ICOS‐dependent block in the specification of both TH and Treg cells (Hatzi et al, 2015). The lack of detection of other PTLC subtypes in our mouse models is also unclear, although it might reflect the need of cooperating genetic events that could favor the rewiring of the TFH phenotype toward other lymphomagenic states (e.g., viral integrations in the case of ATLL) (Fiore et al, 2020). Alternatively, these subtypes might require more genetic lesions to take off. In fact, in humans, the genomic complexity of AITL cases is much lower than in the case of non‐TFH GATA3+ PTCL‐NOS subtype (Heavican et al, 2019). However, Chiba and coworkers have reported during the elaboration of this work that transgenic mice expressing oncogenic Vav1 can develop GATA3+ PTCL‐NOS‐like tumors when combined with full Trp53 ablation (Fukumoto et al, 2020). The reason for the different spectra of tumors found in the transgenic mice and our adoptive transfer experiments is at present unclear. A plausible explanation is that the GATA3+ PTCL‐NOS tumors could have arisen by the combination of very high levels of expression of the Vav1 mutants and the Trp53 null background in the transgenic animals. In this context, it is worth noting that the loss of TP53 is usually associated with GATA+ PTCL‐NOS in humans (Heavican et al, 2019). Although with very limited numbers of patients scored, VAV1 mutations have not been found so far in GATA3+ PTCL‐NOS patients (Heavican et al, 2019).
Future studies will be required to expand our understanding of the role of VAV1 oncogenic mutations both autonomously and in synergy with other genetic lesions in AITL, PTCL, and other tumors such as NSCLC. Given that VAV1 is not expressed under physiological conditions in the lung epithelium (Bustelo et al, 1993), it is likely that the contribution of the mutant protein to tumorigenic processes in these cells will require genetic or epigenetic events that will trigger the spurious expression of the VAV1 locus as previously described in cell lines (Ilan & Katzav, 2012). This might explain the low frequency of VAV1 mutations in NSCLC when compared to PTCL cases. The adoptive transfer experiments described here will also represent an ideal tool for testing preclinically new therapies against AITL, a disease whose poor prognosis has not significantly improved in the last 30 years (Chiba & Sakata‐Yanagimoto, 2020; Fiore et al, 2020).
Materials and Methods
Ethics statement
All mouse experiments were performed according to protocols approved by the Bioethics Committee of the University of Salamanca and the animal experimentation authorities of the autonomous Government of Castilla y León (Spain). We have not utilized patients or patient‐derived samples in this work. No statistical methods were used to determine sample size. In all cases, we have used at least five mice per experiment. The exact sample size used in each experiment is indicated in the appropriate figure legend of the manuscript. Experimental groups were allocated randomly by a technician that was blind to the hypothesis to be tested.
3D structures
Three‐dimensional structures were generated and analyzed in MacPyMol software using the Protein Data Bank‐stored structure files for the Vav1 CH‐Ac‐DH‐PH‐C1 (PCD ID: 3KY9), the Vav1 DH‐PH‐ZF cassette bound to Rac1 (PCD ID: 2VRW), the Vav1 SH2 bound to the Syk phospho‐peptide (PCD ID: 2MC1), and the Vav1 CSH3 (PCD ID: 2KBT) regions.
Plasmids
Most constructs used in this work used the mouse version of Vav1 when interrogating mutations in conserved residues between the human and mouse homologs. When this conservation did not occur (D26 and H399), we used the plasmids encoding the human VAV1 protein. Plasmids encoding human VAV1WT (pJRV58), mouse Vav1WT (pJLZ52), and mouse Vav1Y174E (pMB123) have been previously described (Zugaza et al, 2002; Abate et al, 2017; Barreira et al, 2018). Plasmids encoding human versions of the activated forms of RHO GTPases (RAC1Q61L, RHOAQ63L, and CDC42Q61L) were a gift from P. Crespo (IBBTEC, CSIC‐University of Cantabria, Santander, Spain). Mammalian expression vectors encoding the appropriate mutants were generated using either the pJLZ52 or the pJRV58 vectors as a template utilizing in situ mutagenesis techniques (QuikChange Mutagenesis Kit, Cat. No. 200518, Agilent Technologies). Oligonucleotides used in the mutagenesis steps are listed in Appendix Table S1. To generate the retroviral vectors encoding the different mouse Vav1 mutants plus EGFP protein, the Vav1 cDNA was PCR amplified (NZYProof DNA polymerase, Cat. No. 14601; NZYTech) using the pJLZ52 plasmid as template, digested with BamHI and XhoI, and ligated into the pMIG‐II vector (Cat. No. 52107, Addgene). The oligonucleotide primers used included those for Vav1WT (pLFN119): 5′‐CTG ACA GGA TCC GCC ACC ATG GAG CTC TGG CGA CAG TGC ACC CAC‐3′ (forward, BamHI site underlined) and 5′‐CTG ACA CTC GAG CGC TCA GCA ATA TTC GGA ATA GTC TTC CTC‐3′ (reverse, XhoI site underlined); Vav1Δ1‐189 (pLFN120): 5′‐CTG ACA GGA TCC GCC ACC ATG ACA GAG TAT GAT AAG CGC TGC TGC‐3′ (forward; BamHI site underlined) and 5′‐CTG ACA CTC GAG CGC TCA GCA ATA TTC GGA ATA GTC TTC CTC‐3′ (reverse; XhoI site underlined) and Vav1Δ835‐845 (pLFN121): 5′‐CTG ACA GGA TCC GCC ACC ATG GAG CTC TGG CGA CAG TGC ACC CAC‐3′ (forward; BamHI site underlined) and 5′‐CTG ACA CTC GAG CGC TCA AGA AGG GAA CCA GCC GAT CCG GCC‐3′ (reverse; XhoI site underlined). All newly generated plasmids were subjected to DNA sequence analysis at the CIC Genomics Unit to confirm both the presence of the targeted mutations and absence of off‐target ones.
The pNFAT‐Luc plasmid was obtained from Addgene (Cat. No. 17870) whereas the pSRE‐luc (Cat. number 219081), the pFR‐Luc (Cat. number 219050), and pFA2‐cJun (Cat. number 219053) plasmids were obtained from Stratagene. The pRL‐SV40 plasmid was obtained from Promega (Cat. number E2231). The pGEX‐4T3 plasmid was a gift from C. Guerrero (Cancer Research Center, CSIC, Salamanca, Spain). The pCL‐Eco (Cat. No. 12371) and pMIG‐II (Cat. No. 52107) plasmids were obtained from Addgene.
The pGEX vectors encoding the GST‐CSH3WT (pMB33) and GST‐CSH3P833L (pMB35) have been described before (Barreira et al, 2014). The rest of pGEX vectors encoding Vav1 CSH3 mutant versions were generated using pMB33 as template. Oligonucleotides used for these mutagenesis steps are also listed in Appendix Table S1.
Cell lines
All cells were obtained from the ATCC and authenticated periodically in the lab. COS1, HEK293, and NIH3T3 cells were grown in DMEM supplemented with 10% fetal calf serum, 1% L‐glutamine, penicillin (10 μg/ml), and streptomycin (100 μg/ml). Jurkat cells were grown in RPMI‐1640 medium supplemented with 10% fetal calf serum, 1% l‐glutamine, penicillin (10 μg/ml), and streptomycin (100 μg/ml). All tissue culture reagents were obtained from Gibco‐Thermo Fisher Scientific. All cell lines were maintained at 37°C in a humidified, 5% CO2 atmosphere.
Luciferase reporter assays
In the case of SRF experiments, COS1 cells exponentially growing in 10‐cm plates were transfected with liposomes (Lipofectamine 2000, Cat. No. 11668019, Thermo Fisher Scientific) with 1 μg of the pSRE‐Luc, 1 ng of pRL‐SV40 encoding the Renilla luciferase, and 1 μg of the appropriate experimental vectors. After 24 h, cells were lysed with Passive Lysis Buffer (Cat No. E1960, Promega) and luciferase activities determined using the Dual Luciferase Assay System (Cat No. E1960, Promega). In the case of JNK assays, 2 × 107 of exponentially growing Jurkat cells were electroporated (250 mV, 950 μF) with 20 μg of the appropriate expression vectors, the pRL‐SV40 (5 ng) plus 10 and 5 μg of the pFR‐Luc and pFA2‐cJun, respectively. 36 h post‐transfection, cells were either left nonstimulated or stimulated with antibodies to human CD3 (UCHT1 clone, Cat. No. 217570, Calbiochem, 5 μg/ml) for 7 h. Cells were then lysed with Passive Lysis Buffer (5×) and luciferase activities determined using the Dual Luciferase Assay System. In the case of NFAT assays, 2 × 107 of exponentially growing Jurkat cells were coelectroporated with 20 μg of the appropriate Vav1‐encoding experimental vectors, the pNFAT‐luc reporter vector (10 μg) and pRL‐SV40 (5 ng). 36 h post‐transfection, cells were either left nonstimulated or stimulated with antibodies to human CD3 and luciferase activities determined as above. In all cases, the values of firefly luciferase activity obtained in each experimental point were normalized considering the activity of the Renilla luciferase obtained in each experimental sample. In addition, we analyzed aliquots of the same lysates by Western blot to assess the appropriate expression of the ectopically expressed proteins interrogated in the experiments. Values are represented in graphs as the n‐fold change in the experimental sample relative to the activity shown by mock‐transfected cells (which was given an arbitrary value of 1 in each case). For heatmap representation, we used the heatmap3 R package including all Vav1‐dependent activities analyzed in this study (assigning to the values obtained with Vav1WT an arbitrary number of 1). Adjusted P‐values for multiple comparisons were calculated applying the Benjamini–Hochberg correction (FDR).
Western blot analyses
To determine abundance of proteins, cells were extensively washed with phosphate‐buffered saline solution and broken in lysis buffer 1 (10 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1% Triton X‐100, 1 mM Na3VO4, 10 mM β‐glycerophosphate, and a cocktail of protease inhibitors [Cømplete, Cat. No. 05056489001, Roche]). Cellular extracts were precleared by centrifugation at 20,000 g for 10 min at 4°C, denatured by boiling in 2× SDS–PAGE sample buffer, separated electrophoretically, and transferred onto nitrocellulose filters (Cat. No. 2022‐04‐26, Thermo Fisher) using the iBlot Dry Blotting System (Thermo Fisher). Membranes were blocked in 5% bovine serum albumin (Cat. No. A4503, Sigma‐Aldrich) in TBS‐T (25 mM Tris–HCl [pH 8.0], 150 mM NaCl, 0.1% Tween‐20) for at least 1 h and then incubated overnight with the appropriate antibodies. Membranes were then washed three times with TBS‐T, incubated with the appropriate secondary antibody (GE Healthcare) for 30 min at room temperature, and washed twice as above. Immunoreacting bands were visualized using a chemiluminescent method (ECL, Cat. No. RPN2209, Amersham). Primary antibodies used included those to the Vav1 DH (homemade, 1:10,000 dilution), tubulin α (Cat. No. CP06‐100UG, Calbiochem; 1:2,000 dilution), CBL‐B (Cat No. 9498, Cell Signaling Technologies; 1:1,000 dilution), DNM2 (Cat. No. ab65556, Abcam; 1:1,000 dilution), HNRNPK (Cat. No. sc‐28380, Santa Cruz; 1:1,000 dilution), phospho‐Vav1 (p‐Tyr174) (homemade, 1:10,000 dilution), phosphotyrosine residues (Cat. No. sc‐7020, Santa Cruz; 1:1,000 dilution), Tp53 (Cat. No. 2524, Cell Signaling Technologies; 1:1,000 dilution), phospho‐PKD/PKCμ (p‐Ser744/748) (Cat. No. 2054, Cell Signaling Technologies; 1:1,000 dilution), phospho‐Akt (p‐Ser473) (Cat. No. 4051, Cell Signaling Technologies; 1:1,000 dilution), Akt (Cat. No. 2920, Cell Signaling Technologies; 1:1,000 dilution), phospho‐Erk1/2 (p‐Thr202 and p‐Tyr204) (Cat. No. 4370, Cell Signaling Technologies; 1:1,000 dilution), Erk1/2 (Cat. No. 4695, Cell Signaling Technologies; 1:1,000 dilution), β‐actin (Cat. No. sc‐47778, Santa Cruz; 1:2,000 dilution), and GFP (Cat. No. MMS‐118P, Covance; 1:2,000 dilution).
RAC1, RHOA, and CDC42 GTPase activation assays
Exponentially growing COS1 and Jurkat cells were transfected with 1 μg and 20 μg of the appropriate experimental expression vector as above, respectively. Thirty‐six hours post‐transfection, the cells were washed with chilled phosphate‐buffered solution and lysed in RIPA buffer [10 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% Triton X‐100, 1 mM Na3VO4 (Sigma‐Aldrich, Catalog No. S6508), 1 mM NaF (Sigma‐Aldrich, Catalog No. S7920), and Cømplete] at 4°C. Extracts were precleared by centrifugation at 20,000 g for 10 min at 4°C and snap‐frozen. Upon thawing and determination of total protein concentration (Bradford reactive, Cat. No. 5000006, Bio‐Rad), extracts were analyzed using G‐LISA Assay Kits according to the manufacturer’s instructions (Cat. No. BK135, Cytoskeleton). Values are represented as the n‐fold change in the experimental sample relative to the activity shown by mock‐transfected cells (which was given an arbitrary value of 1 in each case). For heatmap representation, we used the heatmap3 R package including all GTPase activities analyzed in this study (assigning to the values obtained with Vav1WT an arbitrary number of 1). Adjusted P‐values for multiple comparisons were calculated as above.
Purification of GST proteins
The expression of indicated GST proteins was induced E. coli using isopropyl β‐D‐1‐thiogalactopyranoside (Cat. No. I5502, Sigma) and purified with glutathione‐coated Sepharose beads (Cat. No. 17‐0756‐01, GE Healthcare Life Biosciences) (Barreira et al, 2014). Eluted proteins were dialyzed in phosphate‐buffered saline solution and stored until use at −70°C.
Immunoprecipitations
A 2 × 106 of exponentially growing Jurkat cells were transfected with 20 μg of the appropriate expression vector (diluted in 100 μl of R buffer; Cat No. MPK10096, Life Technologies) using two 20‐ms electroporation cycles at 1.7 mV in the Neon system (Life Technologies). Electroporated cells were then maintained in standard culture media for 36 h and disrupted in lysis buffer 1. Upon elimination of cell debris by centrifugation, cellular extracts were incubated for 2 h at 4°C with a primary antibody to Vav1. Immunocomplexes were collected with Gammabind G‐Sepharose beads (Cat. No. 17‐0885‐01, GE Healthcare), washed three times in lysis buffer 1, resuspended in SDS–PAGE buffer, boiled for 5 min, electrophoresed, and subjected to immunoblot analyses with the appropriate antibodies.
GST pull‐down experiments
A 1 × 107 of exponentially growing Jurkat cells were washed with phosphate‐buffered saline solution and disrupted in lysis buffer 2 (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 5 mM MgCl2, 0.5% Triton X‐100, 5 mM β‐glycerophosphate [Cat. No. 50020, Sigma‐Aldrich], 1 mM dithiothreitol, and Cømplete). After eliminating cellular debris by centrifugation at 20,000 g for 10 min at 4°C, lysates were incubated for 2 h at 4°C with ≈ 20 μg of the indicated GST proteins. Protein complexes were trapped with glutathione‐coated Sepharose beads, washed thrice in lysis buffer, resuspended in SDS–PAGE sample buffer, fractionated electrophoretically, and subjected to immunoblotting using antibodies to CBL‐B, DNM2, and HNRNPK. To detect the GST proteins used in the pull‐downs, the nitrocellulose filters containing the complexes were stained with a Ponceau solution before the Western blotting step. Aliquots of the same cell lysates that were used for the pull‐down experiments were fractionated electrophoretically and subjected to immunoblot analyses to determine the total amount of endogenous protein present in each sample.
CD4+ T‐cell isolation and activation
Single cell suspensions were generated by mechanical homogenization of spleen and lymph nodes in 3 ml of phosphate‐buffered saline solution supplemented with 2% bovine serum albumin and 0.5 mM EDTA (referred to hereafter as cell extraction buffer). Cells obtained were washed once by low‐speed centrifugation, resuspended in cell extraction buffer, subjected to a 0.17 M NH4Cl lysis step to eliminate erythrocytes, and filtered with a cell strainer (nylon mesh with 40‐μm pores, Cat. No. 352340, Falcon) to remove debris. Naïve CD4+ T cells were then purified by negative selection using the EasySep™ Mouse CD4+ T Cell Isolation Kit (Cat. No. 19852, StemCell Technologies) according to the manufacturer’s protocol. 1 × 106 of purified CD4+ T cells were then cultured on goat‐anti‐hamster IgG (Cat. No. 31115, Invitrogen)‐coated plate for 16–24 h in RPMI‐1640 media containing 10% fetal bovine serum, glutamine, 50 μM 2‐mercaptoethanol, antibodies to CD3 (Cat. No. 100202, BioLegend; 1 μg/ml), and antibodies to CD28 (Cat. No. 102102, BioLegend; 0.5 μg/ml). The quality of the purification step was always confirmed by flow cytometry.
Production of retroviral particles
To generate infectious retroviral particles, we transfected the retroviral plasmids together with the pCL‐Eco packaging vector into HEK293T cells using the JetPEI transfection reagent (Cat. No. 101‐10N, Polyplus). Retrovirus‐containing supernatants were collected and concentrated using Lenti‐X™ concentrator (Cat. No. 631231, Takara) by centrifugation at 2,000 g for 1 h at 4°C. Concentrated retroviruses were resuspended and tittered using infection of NIH3T3 cells and scoring GFP‐positive cells by flow cytometry. Viruses with high titers were aliquoted and stored in −80°C for up to 2 months.
Ectopic expression of Vav1 mutants in CD4+ T cells
CD4+ T cells stimulated with antibodies to CD3 and CD28 for 16 h as indicated above were infected in the presence of polybrene (6 μg/ml; Cat. No. H9268‐5G, Sigma‐Aldrich) with retroviruses encoding the indicated proteins, pelleted by centrifugation (2,000 g at 32°C for 90 min), and treated with interleukin 2 (Cat. No. 200‐02, PeproTech; 50 U/ml). The transduction efficiency was examined by flow cytometry 48 h after transduction. Transduced cells were then used either for adoptive T‐cell transfer experiments or for short‐term cell cultures as indicated below.
Adoptive T‐cell transfer experiments
A 24–48 h after the retroviral transduction step, the EGFP+ CD4+ T cells were collected by centrifugation (2,000 g for 10 min) and resuspended in phosphate‐buffered saline solution at a concentration of 1 × 104 cells/μl. A 100 μl of the cell resuspension was then introduced by retro‐orbital injection into 6‐ to 8‐week‐old Vav1 −/−; Vav2 −/−; Vav3 −/− recipient mice (Menacho‐Marquez et al, 2013; Lorenzo‐Martín et al, 2020). This lymphopenic mouse strain was used to minimize the potential rejection of the transplanted cells due to neoantigen expression (Lorenzo‐Martín et al, 2020). Mice were then examined weekly until showing obvious physical signs of sickness. Aliquots of peripheral blood (100 μl) were analyzed by flow cytometry to detect changes in immune populations. Upon euthanasia, the indicated tissues and peripheral blood were collected for histological processing, flow cytometry analyses, and extraction of either total cellular proteins or RNAs.
Flow cytometry determinations of surface and intracellular proteins
Isolated cells were washed once in cell extraction buffer, resuspended in standard phosphate‐buffered saline solution, and stained following standard procedures with combinations of fluorescein isothiocyanate‐ (FITC, Cat. No. 553729), allophycocyanin‐ (APC, Cat. No. 553051), APC‐Cy7‐ (Cat. No. 560181), or V500‐labeled (Cat. No. 560783) antibodies to CD4; FITC‐ (Cat. No. 553031), Pacific blue‐ (PB, Cat. No. 558106), or phycoerythrin‐labeled (PE, Cat. No. 553032) antibodies to CD8; APC‐ (Cat. No. 558643) or PE‐Cy7‐labeled (Cat. No. 552880) antibodies to CD25; peridinin chlorophyll‐cyanin 5.5‐labeled (PerCP‐Cy5.5) antibody to B220 (Cat. No. 45‐0452‐82; eBiosciences); PE‐labeled antibody to TCRβ (Cat. No. 12‐5961‐82, eBiosciences); FITC‐labeled antibody to GL7 (Cat. No. 144603; BioLegend); PE‐labeled antibody to PD1 (Cat. No. 12‐9985‐82; eBiosciences); PE‐Cy7‐labeled antibody to CXCR5 (Cat. No. 25‐9185‐42; eBiosciences); APC‐labeled antibody to ICOS (Cat. No. 17‐9949‐82; eBiosciences); APC‐labeled antibody to CD95 (Cat. No. 17‐0951‐82; eBiosciences), and a V450‐labeled antibody to CD69 (Cat. No. 560690).
For intracellular Tox and ICN1 staining, cells were fixed with Cytofix/Cytoperm (Cat. No. 554714, BD Bioscience) for 10 min and stained with PE‐labeled antibodies to Tox (Cat. No. 12‐6502‐82; eBiosciences; 1:50 dilution) or ICN1 (mN1A, Cat. No. 552768; 1:50 dilution) for 1 h at room temperature in phosphate‐buffered saline solution supplemented with 5% fetal bovine serum and 10% saponin. For flow cytometry detection of phosphorylated intracellular proteins, cells were fixed with 2% formaldehyde, permeabilized with 90% ice‐cold methanol, and incubated with antibodies to p‐Erk1/2 (p‐Thr202/p‐Tyr204) (Cat. No. 4377, Cell Signaling Technologies; 1:200 dilution) and p‐Akt (p‐Ser473) (Cat. No. 4060, Cell Signaling Technologies; 1:400 dilution). Cells were then stained with an Alexa‐648‐labeled secondary antibody to rabbit immunoglobulins (Cat. No. A21443, Invitrogen; 1:500 dilution). Unless otherwise stated, the antibodies used were from BD Biosciences. Antibody‐stained cells were run in a FACSAria III flow cytometer (BD Biosciences) and data analyzed using the FlowJo software.
Determination of mRNA abundance
Total RNA was extracted from cells using NZYol (Cat. No. MB18501, NZYtech) and mRNAs purified using RNeasy Mini Kit (Cat. No. 74106, QIAGEN) following the manufacturer's protocol. mRNAs were analyzed by qRT–PCR using the iScript One‐Step RT–PCR kit with SYBR green (Cat. No. 1708892, Bio‐Rad) and the StepOnePlus Real‐Time PCR System (Applied BioSystems). Raw qRT–PCR data were analyzed using the StepOne software v2.1 (Applied Biosystems), using the abundance of the endogenous Actb as internal normalization control. Primers used were 5′‐ATG TGG GTC CGG CAG GTA CCC TGG‐3′ (forward, Pdcd1), 5′‐TCA AAG AGG CCA AGA ACA ATG TCC‐3′ (reverse, Pdcd1), 5′‐CTC GGC CGA TCA TAG GAT GT‐3′ (forward, Icos), 5′‐CTC CAC TAA GGT TCC TTT CTT‐3′ (reverse, Icos), 5′‐TGG CCT TCT ACA GTA ACA GCA‐3′ (forward, Cxcr5), 5′‐GCA TGA ATA CCG CCT TAA AGG AC‐3′ (reverse, Cxcr5), 5′‐CCT GCA ACT GGA AGA AGT ATA AG‐3′ (forward, Bcl6), 5′‐AGT ATG GAG GCA CAT CTC TGT AT‐3′ (reverse, Bcl6), 5′‐TCA TCA TTG ACC TCG TGG CCC‐3′ (forward, il21), 5′‐ATC GTA CTT CTC CAC TTG CAA TCC C‐3′ (reverse, il21), 5′‐GGC CAG CTG ATA TAA TGG AGA AAA‐3′ (forward, Hes1), 5′‐TCC ATG ATA GGC TTT GAT GAC TT‐3′ (reverse, Hes1), 5′‐CAC TGG CCC TGT CCA CCC AGC CTT GGC AGG‐3′ (forward, Dtx1), 5′‐ATG CGA ATT CGG GAA GGC GGG CAA CTC AGG‐3′ (reverse, Dtx1), 5′‐TCA GGT GTC AGG CTC TAC CA‐3′ (forward, Ptcra), 5′‐ACC AGA CAG GGT TGT CAA GG‐3′ (reverse, Ptcra), 5′‐AGT AGG ACT GAG AAG GGA AAG T‐3′ (forward, Tet2), 5′‐CGG TTG TGC TGT CAT TTG TTT‐3′ (reverse, Tet2), 5′‐GTA TTT CAC CCT CAA GAT CC‐3′ (forward, Trp53), 5′‐TGG GCA TCC TTT AAC TCT A‐3′ (reverse, Trp53), 5′‐GTC CCT CAC CCT CCC AAA AG‐3′ (forward, Actb), and 5′‐GCT GCC TCA ACA CCT CAA CCC‐3′ (reverse, Actb).
Gene expression profiling
These experiments included cells purified from both the spleen and lymph nodes to assess the potential contribution of each niche to the transcriptome of the cancer cells. To this end, total RNAs from control and Vav1ΔC‐expressing EGFP+ CD4+ T cells from those two tissues were isolated using the RNAeasy Mini Kit (Qiagen, Catalog No. 74104) to be analyzed using Affymetrix platform (Clariom™ S Assay HT) at the CIC Genomics Core Facility according to the manufacture's recommendations.
Bioinformatics of microarray data
R version 3.6.3 was used for statistical analyses along with Python version 3.9 for text file processing. Signal intensity values were obtained from expression microarray CEL files after robust multichip average (RMA). Principal component analyses were generated using the factoextra R package. Differentially expressed genes were identified using linear models for microarray data (limma). Adjusted P‐values for multiple comparisons were calculated applying the Benjamini–Hochberg correction (FDR). Gene Ontology and KEGG pathways enrichment analyses were performed using DAVID (https://david.ncifcrf.gov). Expression heatmaps were generated using the heatmap3 R package. Volcano plots were generated using the Glimma R package. GSEAs were performed with described gene sets using gene set permutations (n = 1,000) for the assessment of significance and signal‐to‐noise metric for ranking genes. The datasets used were those for: AP1 (Gustems et al, 2014), SRF (Selvaraj & Prywes, 2004), ICN1 (Li et al, 2008), the Molecular Signatures Database (MSigDB v7.2, for E2F, G2/M checkpoint, Myc and Foxo), TFH cells (Weinstein et al, 2014), Tox (Scott et al, 2019), and SJL mice (Jain et al, 2015). To evaluate the Vav1ΔC‐associated gene signature fitness across the mouse Rhoa G17V and Tet2 mutant CD4+ T‐cell signatures (Zang et al, 2017), SJL mice (Jain et al, 2015), and human PTCL (Maura et al, 2019), the enrichment scores for both the upregulated and downregulated signatures found in Vav1ΔC‐transformed AITL cells were calculated using single‐sample GSEA. The difference between the two normalized enrichment scores yielded the fit score, a measure of the enrichment and depletion of the upregulated and downregulated signatures, respectively. The Vav1ΔC‐specific gene signature was established by comparing the full Vav1ΔC‐associated gene signature with the differential transcriptomes of Tet2 −/−, Rhoa G17V, and Tet2 −/− ; Rhoa G17V CD4+ T cells (Zang et al, 2017).
Evaluation of short‐term effects of proteins in CD4+ T cells
Splenic and lymph node CD4+ T cells purified as above were stained with Cell Trace Violet (Cat. No. C34557, Life Technologies) following the manufacturer’s protocol. T cells were then cultured on goat‐anti‐hamster IgG‐coated plates for 16–24 h in RPMI‐1640 media as above to stimulate them. Upon activation, cells were infected with the indicated retroviral particles in the presence of interleukin 2 (Cat. No. 200‐02, PeproTech; 50 U/ml) and, three days later, were processed to measure the indicated biological and signaling parameters by flow cytometry. In the case of experiments using drug inhibitors, the CD4+ T cells were isolated, activated, and infected as above and, 24 h after infection, treated with 200 nM Compound E (Cat. No. ALX‐270‐415‐C250, Enzo Life Sciences), 10 μM FK506 (Cat. No. tlrl‐fk5, Invitrogen), or solution control (DMSO). Three days later, cells were collected for flow cytometry experiments as indicated above.
Statistics
Calculations were performed using Microsoft Excel 2020 and GraphPad Prism software (version 6.0). The number of biological replicates (n), the type of statistical tests performed, and the statistical significance are indicated for each experiment in the figure legends as well as the Results section of this document. Parametric and nonparametric distributions were analyzed using Student’s t‐test and Mann–Whitney test, respectively. Chi‐squared tests were used to determine the significance of the differences between expected and observed frequencies. Tukey’s honest significance difference test was used to identify groups showing differential enrichment of the indicated signatures. Statistical analyses of the immunoblot‐generated data were carried out using the GraphPad Prism software (version 6.0). In all cases, values were considered significant when P ≤ 0.05. Data obtained are given as the mean ± SEM.
Author contributions
JR‐V participated in all experimental work, analyzed data, and contributed to both artwork design and manuscript writing. LF‐N and ENA‐S carried out experimental work related to the characterization of the biological activity of Vav1 mutants. MC and AA helped in animal model‐based experiments. LFL‐M and RC carried out the in silico analyses. IF‐P collaborated in the adoptive transfer experiments. SR‐F contributed to the characterization of the SH2 mutants. XRB conceived the work, analyzed data, wrote the manuscript, and carried out the final editing of figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View/Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank M.C. García‐Macías and the personnel of both the CIC Flow Cytometry and Genomics Units for expert histological analyses, cell characterization, and microarray work, respectively. The X.R.B.’s project leading to these results has received funding from “la Caixa” Banking Foundation (HR20‐00164), the Castilla‐León autonomous government (CSI252P18, CSI145P20, CLC‐2017‐01), the Spanish Ministry of Science and Innovation (MSI) (RTI2018‐096481‐B‐100), and the Spanish Association against Cancer (GC16173472GARC). J.R.‐V. received funding from the Carlos III Health Institute (PI20/01724). X.R.B.’s institution is supported by the Programa de Apoyo a Planes Estratégicos de Investigación de Estructuras de Investigación de Excelencia of the Castilla‐León autonomous government (CLC‐2017‐01). J.R‐V. is supported by a senior postdoctoral contract of the Spanish Association against Cancer. L.F.‐N contract has been supported by the Salamanca local section of the Spanish Association against Cancer. S.R.‐F. and L.F.L.‐M. contracts have been mostly supported by funding from the MSI (BES‐2013‐063573) and the Spanish Ministry of Education, Culture and Sports (L.F.L.‐M., FPU13/02923), respectively. Subsequently, they both were supported by the CLC‐2017‐01 grant. Both Spanish and Castilla‐León government‐associated funding is partially supported by the European Regional Development Fund.
The EMBO Journal (2021) 40: e108125.
Data availability
A Source Data file is provided with this paper. All relevant data are available from the corresponding author upon reasonable request. Microarray data reported in this paper have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE165006.
References
- Abate F, da Silva‐Almeida AC, Zairis S, Robles‐Valero J, Couronne L, Khiabanian H, Quinn SA, Kim M‐Y, Laginestra MA, Kim C et al (2017) Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T‐cell lymphomas. Proc Natl Acad Sci U S A 114: 764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghazadeh B, Lowry WE, Huang XY, Rosen MK (2000) Structural basis for relief of autoinhibition of the Dbl homology domain of proto‐oncogene Vav by tyrosine phosphorylation. Cell 102: 625–633 [DOI] [PubMed] [Google Scholar]
- Barreira M, Fabbiano S, Couceiro JR, Torreira E, Martínez‐Torrecuadrada JL, Montoya G, Llorca O, Bustelo XR (2014) The C‐terminal SH3 domain contributes to the intramolecular inhibition of Vav family proteins. Sci Signal 7: ra35 [DOI] [PubMed] [Google Scholar]
- Barreira M, Rodriguez‐Fdez S, Bustelo XR (2018) New insights into the Vav1 activation cycle in lymphocytes. Cell Signal 45: 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddicker RL, Razidlo GL, Dasari S, Zeng YU, Hu G, Knudson RA, Greipp PT, Davila JI, Johnson SH, Porcher JC et al (2016) Integrated mate‐pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T‐cell lymphoma. Blood 128: 1234–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR (2014) Vav family exchange factors: an integrated regulatory and functional view. Small GTPases 5: e973757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR, Barbacid M (1992) Tyrosine phosphorylation of the vav proto‐oncogene product in activated B cells. Science 256: 1196–1199 [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Dosil M (2016) The Vav family. In: Encyclopedia of signaling molecules, Choi S (ed.), pp. 1–15. New York, NY: Springer; [Google Scholar]
- Bustelo XR, Ledbetter JA, Barbacid M (1992) Product of vav proto‐oncogene defines a new class of tyrosine protein kinase substrates. Nature 356: 68–71 [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Rubin SD, Suen KL, Carrasco D, Barbacid M (1993) Developmental expression of the vav protooncogene. Cell Growth Differ 4: 297–308 [PubMed] [Google Scholar]
- Bustelo XR, Suen KL, Michael WM, Dreyfuss G, Barbacid M (1995) Association of the vav proto‐oncogene product with poly(rC)‐specific RNA‐binding proteins. Mol Cell Biol 15: 1324–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA et al (2016) Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48: 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Sakata‐Yanagimoto M (2020) Advances in understanding of angioimmunoblastic T‐cell lymphoma. Leukemia 34: 2592–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrencik JE, Brooun A, Zhang H, Mathews II, Hura GL, Foster SA, Perry JJP, Streiff M, Ramage P, Widmer H et al (2008) Structural basis of guanine nucleotide exchange mediated by the T‐cell essential Vav1. J Mol Biol 380: 828–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR (1992) Identification of calcineurin as a key signalling enzyme in T‐lymphocyte activation. Nature 357: 695–697 [DOI] [PubMed] [Google Scholar]
- Cortes JR, Ambesi‐Impiombato A, Couronne L, Quinn SA, Kim CS, da Silva Almeida AC, West Z, Belver L, Martin MS, Scourzic L et al (2018) RHOAG17V Induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell 33: 259–273.e257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couceiro JR, Martin‐Bermudo MD, Bustelo XR (2005) Phylogenetic conservation of the regulatory and functional properties of the Vav oncoprotein family. Exp Cell Res 308: 364–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E et al (2015) Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 27: 516–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR (1997) Phosphotyrosine‐dependent activation of Rac‐1 GDP/GTP exchange by the vav proto‐oncogene product. Nature 385: 169–172 [DOI] [PubMed] [Google Scholar]
- de Leval L, Rickman DS, Thielen C, Reynies AD, Huang Y‐L, Delsol G, Lamant L, Leroy K, Brière J, Molina T et al (2007) The gene expression profile of nodal peripheral T‐cell lymphoma demonstrates a molecular link between angioimmunoblastic T‐cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 109: 4952–4963 [DOI] [PubMed] [Google Scholar]
- Fiore D, Cappelli LV, Broccoli A, Zinzani PL, Chan WC, Inghirami G (2020) Peripheral T cell lymphomas: from the bench to the clinic. Nat Rev Cancer 20: 323–342 [DOI] [PubMed] [Google Scholar]
- Fujikawa K, Miletic AV, Alt FW, Faccio R, Brown T, Hoog J, Fredericks J, Nishi S, Mildiner S, Moores SL et al (2003) Vav1/2/3‐null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J Exp Med 198: 1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K, Sakata‐Yanagimoto M, Fujisawa M, Sakamoto T, Miyoshi H, Suehara Y, Nguyen TB, Suma S, Yanagimoto S, Shiraishi Y et al (2020) VAV1 mutations contribute to development of T‐cell neoplasms in mice. Blood 136: 3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo M, Lee H, Zhang X, Bueso‐Ramos C, Pageon L, McArthur M, Multani A, Nazha A, Manshouri T, Parker‐Thornburg J et al (2015) hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell 28: 486–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK et al (2005) Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol 6: 261–270 [DOI] [PubMed] [Google Scholar]
- Gulbranson‐Judge A, Tybulewicz VL, Walters AE, Toellner KM, MacLennan IC, Turner M (1999) Defective immunoglobulin class switching in Vav‐deficient mice is attributable to compromised T cell help. Eur J Immunol 29: 477–487 [DOI] [PubMed] [Google Scholar]
- Gustems M, Woellmer A, Rothbauer U, Eck SH, Wieland T, Lutter D, Hammerschmidt W (2014) c‐Jun/c‐Fos heterodimers regulate cellular genes via a newly identified class of methylated DNA sequence motifs. Nucleic Acids Res 42: 3059–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, Crotty S (2015) BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med 212: 539–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, Zhang W, Li Y, Dave BJ, Nairismägi M‐L et al (2019) Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T‐cell lymphoma. Blood 133: 1664–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilan L, Katzav S (2012) Human Vav1 expression in hematopoietic and cancer cell lines is regulated by c‐Myb and by CpG methylation. PLoS One 7: e29939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Chen J, Nicolae A, Wang H, Shin D‐M, Adkins EB, Sproule TJ, Leeth CM, Sakai T, Kovalchuk AL et al (2015) IL‐21‐driven neoplasms in SJL mice mimic some key features of human angioimmunoblastic T‐cell lymphoma. Am J Pathol 185: 3102–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J‐I, Totoki Y, Chiba K, Sato‐Otsubo A, Nagae G et al (2015) Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47: 1304–1315 [DOI] [PubMed] [Google Scholar]
- Katzav S, Martin‐Zanca D, Barbacid M (1989) Vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J 8: 2283–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137: 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne MR, Ku G, Weiss A (2000) A guanine nucleotide exchange factor‐independent function of Vav1 in transcriptional activation. J Biol Chem 275: 2185–2190 [DOI] [PubMed] [Google Scholar]
- Li X, Gounari F, Protopopov A, Khazaie K, von Boehmer H (2008) Oncogenesis of T‐ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med 205: 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Lago M, Lee H, Cruz C, Movilla N, Bustelo XR (2000) Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol Cell Biol 20: 1678–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo‐Martín LF, Fernández‐Parejo N, Menacho‐Márquez M, Rodríguez‐Fdez S, Robles‐Valero J, Zumalave S, Fabbiano S, Pascual G, García‐Pedrero JM, Abad A et al (2020) VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat Commun 11: 4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169: 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Hu JK, Pereira RM, Crampton JS, Togher S, Bild N, Crotty S, Rao A (2016) Cutting edge: NFAT transcription factors promote the generation of follicular helper T cells in response to acute viral infection. J Immunol 196: 2015–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maura F, Agnelli L, Leongamornlert D, Bolli N, Chan WC, Dodero A, Carniti C, Heavican TB, Pellegrinelli A, Pruneri G et al (2019) Integration of transcriptional and mutational data simplifies the stratification of peripheral T‐cell lymphoma. Am J Hematol 94: 628–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menacho‐Marquez M, Garcia‐Escudero R, Ojeda V, Abad A, Delgado P, Costa C, Ruiz S, Alarcon B, Paramio JM, Bustelo XR (2013) The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol 11: e1001615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhaidly R, Krug A, Gaulard P, Lemonnier F, Ricci JE, Verhoeyen E (2020) New preclinical models for angioimmunoblastic T‐cell lymphoma: filling the GAP. Oncogenesis 9: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Shanthalingam S, Sherman HL, Singh K, Canakci M, Torres JA, Lawlor R, Ran Y, Golde TE, Miele L et al (2020) CD28 signaling drives notch ligand expression on CD4 T cells. Front Immunol 11: 735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon CS, Reglero C, Cortes JR, Quinn SA, Alvarez S, Zhao J, Lin WW, Cooke AJ, Abate F, Soderquist CR et al (2021) FYN‐TRAF3IP2 induces NF‐κB signaling‐driven peripheral T cell lymphoma. Nat Cancer 2: 98–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movilla N, Bustelo XR (1999) Biological and regulatory properties of Vav‐3, a new member of the Vav family of oncoproteins. Mol Cell Biol 19: 7870–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MR, Rao A (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 10: 645–656 [DOI] [PubMed] [Google Scholar]
- Palaga T, Miele L, Golde TE, Osborne BA (2003) TCR‐mediated Notch signaling regulates proliferation and IFN‐gamma production in peripheral T cells. J Immunol 171: 3019–3024 [DOI] [PubMed] [Google Scholar]
- Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, Kim J, Martinez‐Escala E, Amankulor N, Pro B et al (2017) Genomic analysis of 220 CTCLs identifies a novel recurrent gain‐of‐function alteration in RLTPR (p. Q575E). Blood 130: 1430–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S, Gazzola A, Gloghini A, Righi S, Rossi M et al (2007) Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res 67: 10703–10710 [DOI] [PubMed] [Google Scholar]
- Rapley J, Tybulewicz VL, Rittinger K (2008) Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep 9: 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles‐Valero J, Lorenzo‐Martin LF, Menacho‐Marquez M, Fernandez‐Pisonero I, Abad A, Camos M, Toribio ML, Espinosa L, Bigas A, Bustelo XR (2017) A paradoxical tumor‐suppressor role for the Rac1 exchange factor Vav1 in T cell acute lymphoblastic leukemia. Cancer Cell 32: 608–623.e609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Fdez S, Bustelo XR (2019) The Vav GEF family: an evolutionary and functional perspective. Cells 8: E465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Fdez S, Fernández‐Nevado L, Lorenzo‐Martín LF, Bustelo XR (2020) Lysine acetylation reshapes the downstream signaling landscape of Vav1 in lymphocytes. Cells 9: 609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuebel KE, Movilla N, Rosa JL, Bustelo XR (1998) Phosphorylation‐dependent and constitutive activation of Rho proteins by wild‐type and oncogenic Vav‐2. EMBO J 17: 6608–6621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S et al (2019) TOX is a critical regulator of tumour‐specific T cell differentiation. Nature 571: 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj A, Prywes R (2004) Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol Biol 5: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbuck MP, Arakcheeva K, Winandy S (2018) Novel TCR‐mediated mechanisms of notch activation and signaling. J Immunol 200: 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, So T, Lebedeva S, Croft M, Altman A (2005) Impaired IL‐4 and c‐Maf expression and enhanced Th1‐cell development in Vav1‐deficient mice. Blood 106: 1286–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallois D, Dobay MPD, Morin RD, Lemonnier F, Missiaglia E, Juilland M, Iwaszkiewicz J, Fataccioli V, Bisig B, Roberti A et al (2016) Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T‐cell‐derived lymphomas. Blood 128: 1490–1502 [DOI] [PubMed] [Google Scholar]
- Weinstein JS, Lezon‐Geyda K, Maksimova Y, Craft S, Zhang Y, Su M, Schulz VP, Craft J, Gallagher PG (2014) Global transcriptome analysis and enhancer landscape of human primary T follicular helper and T effector lymphocytes. Blood 124: 3719–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Katzav S, Weiss A (1995) A functional T‐cell receptor signaling pathway is required for p95vav activity. Mol Cell Biol 15: 4337–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Zhao X, Wang X, Feng H, Gou M, Jin W, Wang X, Liu X, Dong C (2019) The transcription factor Tox2 drives T follicular helper cell development via regulating chromatin accessibility. Immunity 51: 826–839.e825 [DOI] [PubMed] [Google Scholar]
- Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee J‐E et al (2014) A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet 46: 371–375 [DOI] [PubMed] [Google Scholar]
- Yu B, Martins IR, Li P, Amarasinghe GK, Umetani J, Fernandez‐Zapico ME, Billadeau DD, Machius M, Tomchick DR, Rosen MK (2010) Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell 140: 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang S, Li J, Yang H, Zeng H, Han W, Zhang J, Lee M, Moczygemba M, Isgandarova S, Yang Y et al (2017) Mutations in 5‐methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest 127: 2998–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen C, Guo W, Zheng S, Sun Z, Geng X (2016) DNM3 attenuates hepatocellular carcinoma growth by activating P53. Med Sci Monit 22: 197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zugaza JL, Lopez‐Lago MA, Caloca MJ, Dosil M, Movilla N, Bustelo XR (2002) Structural determinants for the biological activity of Vav proteins. J Biol Chem 277: 45377–45392 [DOI] [PubMed] [Google Scholar]