

Abstract
Phenylketonuria (PKU; also known as phenylalanine hydroxylase (PAH) deficiency) is an autosomal recessive disorder of phenylalanine metabolism, in which especially high phenylalanine concentrations cause brain dysfunction. If untreated, this brain dysfunction results in severe intellectual disability, epilepsy and behavioural problems. The prevalence varies worldwide, with an average of about 1:10,000 newborns. Early diagnosis is based on newborn screening, and if treatment is started early and continued, intelligence is within normal limits with, on average, some suboptimal neurocognitive function. Dietary restriction of phenylalanine has been the mainstay of treatment for over 60 years and has been highly successful, although outcomes are still suboptimal and patients can find the treatment difficult to adhere to. Pharmacological treatments are available, such as tetrahydrobiopterin, which is effective in only a minority of patients (usually those with milder PKU), and pegylated phenylalanine ammonia lyase, which requires daily subcutaneous injections and causes adverse immune responses. Given the drawbacks of these approaches, other treatments are in development, such as mRNA and gene therapy. Even though PAH deficiency is the most common defect of amino acid metabolism in humans, brain dysfunction in individuals with PKU is still not well understood and further research is needed to facilitate development of pathophysiology-driven treatments.
Phenylketonuria (PKU; also known as phenylalanine hydroxylase (PAH, EC 1.14.16.1) deficiency (OMIM # 261600) and Følling disease) is an inborn error of phenylalanine (Phe) metabolism, which is caused by pathogenetic variants in the PAH gene. PAH is responsible for the conversion of Phe to tyrosine (Tyr), in a reaction that requires the co-substrate tetrahydrobiopterin (BH4). Of note, BH4 can also act as a chaperone to facilitate the proper folding of the PAH monomer, as does DNAJC12 (REFS1–4); consequently, in a small number of cases of hyperphenylalaninaemia (HPA), the HPA is caused by defects in BH4 metabolism or pathogenetic variants in DNAJC12. HPA is the core biochemical abnormality of PKU (FIG. 1), in which normal blood Phe concentrations (35–120 μmol/l)) are exceeded.
Fig. 1 |. Phenylalanine metabolism and PKU.
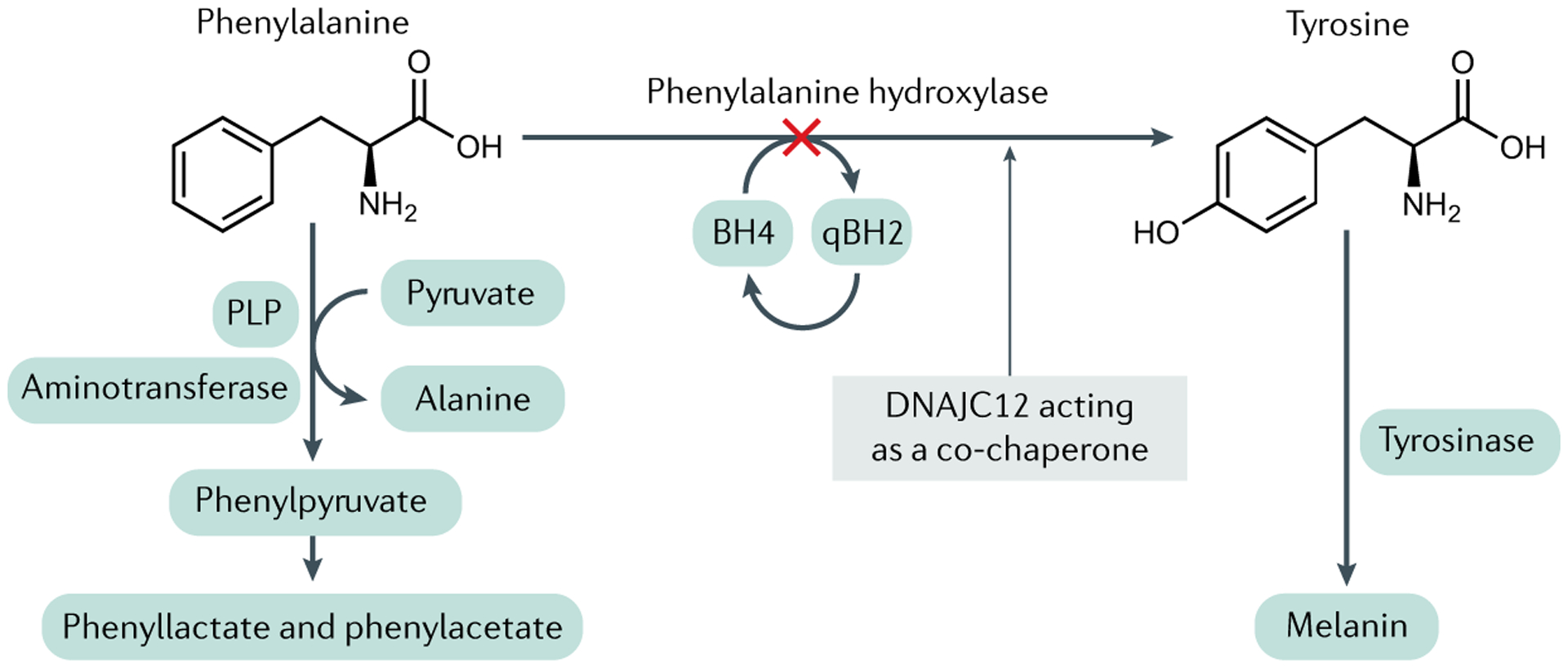
Phenylalanine hydroxylase (PAH) catalyses the hydroxylation of l-phenylalanine (Phe) to l-tyrosine (Tyr), a reaction that occurs pre-dominantly in the liver but also in the proximal renal tubules in the kidneys. The reaction requires the reduced pterin tetrahydrobiopterin (BH4), along with ferrous iron and molecular oxygen (not shown) as cofactors. BH4 is oxidized to quinonoid dihydrobiopterin (qBH2) in the course of the hydroxylation reaction; qBH2 is enzymatically recycled back to BH4 in order to support ongoing Phe hydroxylation. In individuals with recessively inherited pathogenetic variants in the PAH gene, PAH enzymatic activity is either entirely lacking or severely diminished. As approximately 90% of the daily dietary intake of Phe must be metabolized through this pathway, PAH deficiency causes the accumulation of Phe in the body, most readily measured as extreme elevations of Phe concentrations in the blood (hyperphenylalaninaemia). Blood Tyr concentration is also diminished relative to that in PAH-sufficient individuals, but hypotyrosinaemia is not typically severe, perhaps because of dietary Tyr intake. In the setting of hyperphenylalaninaemia, deamination of Phe forms phenylpyruvate and other phenylketones, which are readily excreted in the urine and are the source of the colloquial name phenylketonuria (PKU). PLP, pyridoxal 5′-phosphate.
In untreated patients with PKU, blood Phe concentrations are markedly increased, resulting in the formation of phenylketone bodies that are excreted in urine; conversely, Tyr concentrations are usually somewhat low. Clinically, untreated patients develop severe intellectual disability, epilepsy and behavioural, psychiatric and movement problems, as well as light pigmentation of skin, eyes and hair, eczema and a musty odour5. Less severe forms of PAH deficiency are variously referred to as moderate PKU, mild PKU, mild HPA or benign HPA, whereas severe forms are referred to as classic PKU. Historically, the different severities of PAH deficiency were differentiated by the untreated Phe concentration5, which has provided data regarding the global prevalence of these different severities of PAH deficiency (FIG. 2). However, these prevalence data must be interpreted with care, as establishing the severity of PAH deficiency is difficult because patients are now diagnosed before the biologically highest untreated Phe concentration is reached. Therefore, this classification is no longer valid6. Tolerance for Phe (measured or estimated by prescribed intake, or 3-day dietary history from a diary or by asking the patient to estimate the intake) has long been seen as an alternative criterion for PKU classification7, but this measure also depends on patient age, the target therapeutic Phe concentration, the current bodily growth rate and patient health status with the possibility of protein catabolism, the accuracy of the dietary intake and the adherence to dietary control. Therefore, in line with the first European guidelines for PKU, we classify PAH deficiency into mild HPA (Phe concentrations 120–360 μmol/l; no treatment necessary) and PKU (>360 μmol/l), which can be further categorized as BH4-responsive PKU or BH4-non-responsive PKU8. Here, we use PKU to refer to all severities of PAH deficiency and only differentiate between them where necessary.
Fig. 2 |. The prevalence of PAH deficiency and different PAH deficiency phenotypes worldwide.
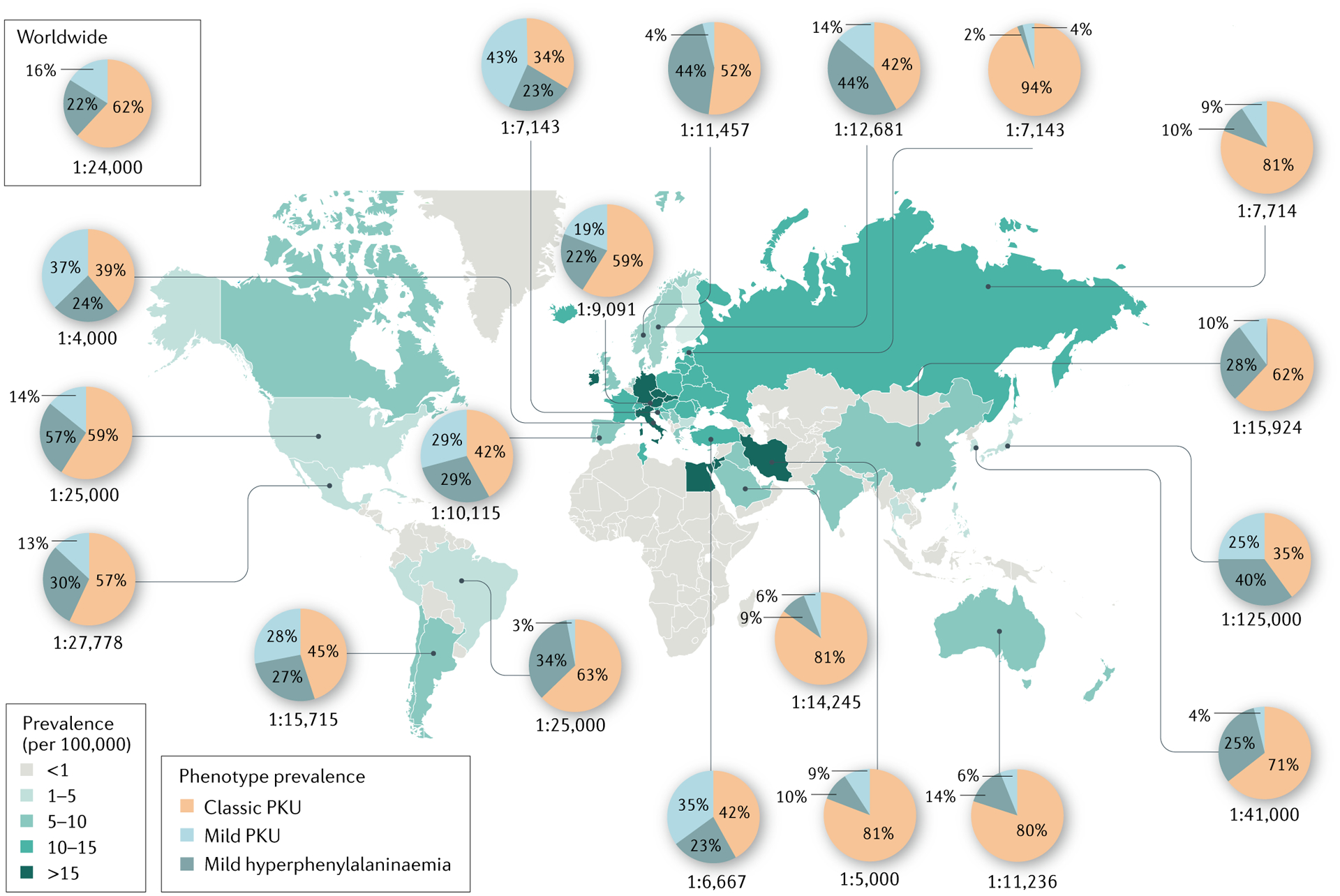
The global prevalence of phenylalanine hydroxylase (PAH) deficiency and that of the different severities of PAH deficiency are depicted. The prevalence of PAH deficiency varies considerably between different geographic regions (coloured map), with the highest prevalence reported in the Republic of Ireland, Germany, Italy, Iran and Jordan. The proportions of cases of classic PKU, mild phenylketonuria (PKU) and mild hyperphenylalaninaemia in representative countries are indicated (pie graphs). Data are from Hillert et al. (2020)28. As a clear definition of variants of PAH deficiency is lacking, prevalence data for the different PAH severities should be interpreted with care.
In the historical efforts to achieve ‘normal outcomes’ in individuals with PKU, researchers and clinicians had to overcome various hurdles, and these experiences provided a proof of principle and a general guide for the development of diagnostic and therapeutic solutions for many inherited metabolic diseases (IMDs) (BOX 1). Furthermore, the success of these efforts started to counter the perception that IMDs in general are untreatable, although they may also have hampered further innovation in therapy in PKU for some decades.
Box 1 |. Lessons from PKU in the treatment of IMDs.
Phenylketonuria (PKU) has for a long time been the foremost example of inherited metabolic diseases (IMDs). Compared with other IMDs, PKU seems a rather simple, straightforwardly diagnosed and treated IMD, but still it has hidden complexity. The reasons that PKU has been such an example for other IMDs are as follows (in chronological order):
The first disease for which a biochemical explanation for severe intellectual disability was found. Together with the later gained knowledge on inheritance, this meant that families knew the inherited cause of their child’s disability.
The first disease in which a theoretically simple (albeit burdensome in daily practice) dietary restriction treatment is enough to prevent the sequelae of the biochemical abnormality, if diagnosed and treated in time.
The first disease for which a reliable and cheap method for population-based (neonatal) screening was developed, by which patients with the condition could be identified in time to prevent development of the clinical entity of PKU.
The first disease in which dietary restriction therapy was replaced by a drug in some patients.
A disease in which variability in disease severity means that not every patient needs the same strict dietary treatment.
Screening for abnormality of a biochemical marker such as phenylalanine (Phe) rather than a disease such as phenylalanine hydroxylase deficiency will also find patients with increased Phe due to another IMD, such as those with defects in tetrahydrobiopterin metabolism, who cannot easily be diagnosed in time in another way, but are (officially) not within the scope of the newborn screening programme.
There have been important milestones1,9–25 in the history of PKU research and treatment to improve outcomes (FIG. 3). PKU was first described in two Norwegian siblings in 1934 by Følling9, was first treated (with dietary control) in 1953 by Bickel et al.14, and population-based newborn screening using dried blood spot (DBS) testing to assess Phe concentration was introduced in 1963 by Guthrie and Susi17, and enabled early diagnosis and initiation of treatment. Despite marked improvement in PKU management in the first decades after the introduction of dietary control, this theoretically simple treatment has not completely remedied the manifestations of PKU, and keeping blood Phe concentrations within the target range is burdensome in practice, especially in children after the first decade of life26. In this Primer, we summarize current knowledge of PKU epidemiology and review the diagnosis of PKU (PAH deficiency) and the other known genetic aetiologies of HPA, including defects in BH4 metabolism and DNAJC12 deficiency1. In addition, we discuss the present understanding of PKU pathophysiology, current management approaches and the challenges in further improving treatment of PKU. Finally, we provide an outlook on new treatments that will hopefully be effective in improving outcomes in individuals with PKU.
Fig. 3 |. Timeline of milestones in the understanding and treatment of PKU.
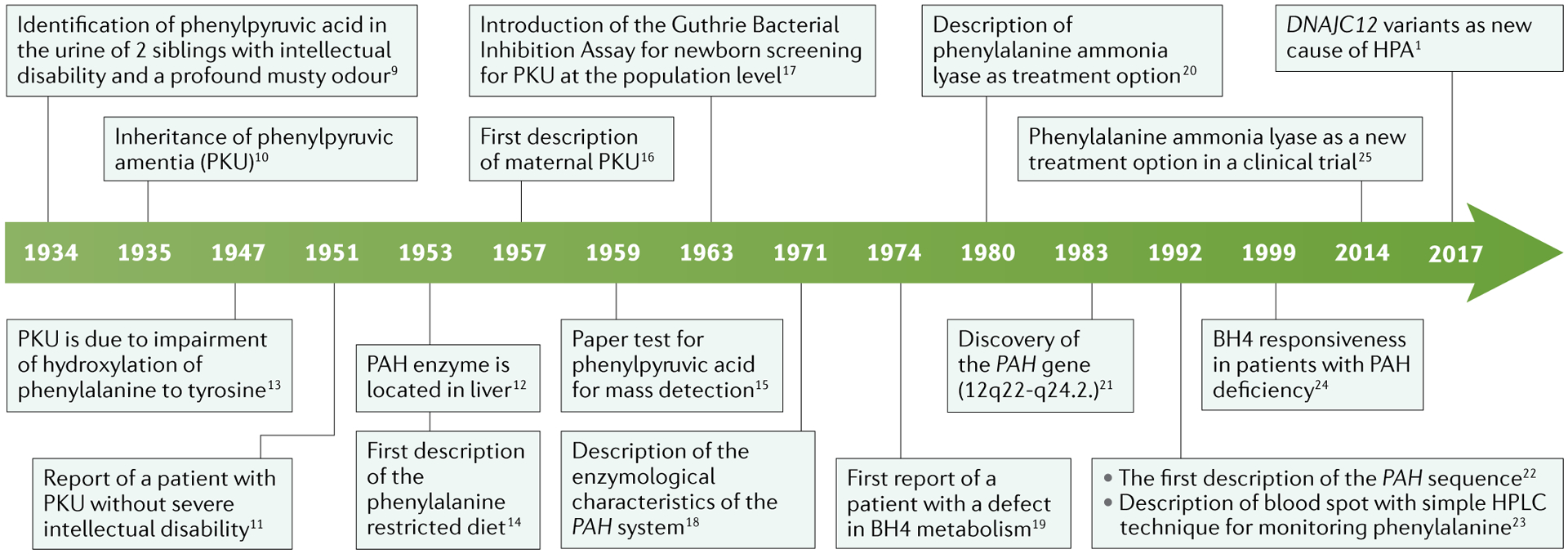
The initial description and major advances in the understanding of hyperphenylalaninaemia (HPA) and phenylketonuria (PKU) pathogenesis are shown. Furthermore, developments in the treatment of PKU are also highlighted. BH4, tetrahydrobiopterin; HPLC, high-performance liquid chromatography; PAH, phenylalanine hydroxylase.
Epidemiology
The prevalence of PKU varies substantially among ethnicities and between different geographic regions worldwide (FIG. 2). PKU prevalence is generally highest in white or East Asian populations (~1:10,000–15,000 live births)27. In Europe, the prevalence ranges widely, from 1:2,700 live births in Italy and 1:4,500 live births in Ireland to <1:100,000 live births in Finland28. Spain differs from other European countries/regions in having a high prevalence of mild HPA and PKU with lower untreated Phe concentrations (blood Phe 360–600 μmol/l), arising from partial inactivation of PAH29. The prevalence of PKU in some countries/regions in the Middle East is comparable to or even higher than that in white or East Asian populations. For example, in Turkey30, the Fars province of Iran31 and the Russian republic of Karachay-Cherkessia32, the prevalence is 1:4,370, 1:4,698 and 1:850 newborns, respectively. This extremely high prevalence might be explained by the more frequent occurrence of consanguineous marriages. PKU prevalence is low in some Asian populations, such as those of Thailand (1:212,535)33 and Japan (1:120,000)34. The prevalence in South America varies from ~1:25,000–50,000 live births, with a lower prevalence in the northern than in the southern part of the continent35. Prevalence data are lacking for some regions of the world, such as parts of Africa, Asia and the Caribbean. The prevalence of PKU in people of African or South Asian descent may be lower than in white populations36. Estimates of prevalence have been made based on prevalence in individuals of South Asian (~1:35,000) and sub-Saharan African (~1:90,000) ancestry living in the UK37.
Mechanisms/pathophysiology
PAH is a tetrameric, iron-containing monooxygenase enzyme that catalyses the hydroxylation of Phe to form Tyr18 (FIG. 1). This reaction requires molecular oxygen as a cofactor and the reduced pterin BH4 as a co-substrate. PAH-mediated hydroxylation is the rate-limiting step in the intermediary metabolism of l-Phe. Even with current advanced research tools, the exact amount of Phe utilized for net protein metabolism is unknown. From the difference in Phe requirements in healthy children and adults compared with those in children and adults with PKU, we estimate that ~10–20% of typical dietary Phe intake is utilized in the course of routine protein turnover; the remainder is converted to Tyr through the action of PAH. Tyr has several metabolic fates, including the production of the neurotransmitters dopamine, adrenaline and norepinephrine, conversion to thyroxine in the thyroid gland and to melanin in melanocytes, and complete catabolism to acetoacetate (a ketone) and fumarate (a Krebs cycle intermediate) to be utilized as fuel. Inherited or functional deficiency of PAH activity leads to HPA, mild Tyr deficiency and, when severe, urinary excretion of phenylpyruvate (the product of spontaneous Phe deamination) and other phenylketone bodies.
Genetic aetiology
HPA is most commonly caused by pathogenetic variants in the PAH gene located on chromosome 12, which are inherited in an autosomal recessive manner, leading to the production of PAH monomers with reduced or no activity or the complete absence of PAH protein. More rarely, functional PAH deficiency is caused by BH4 deficiency due to inherited defects in biopterin synthesis (GTP cyclohydrolase 1 (GTPCH) or 6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiencies) or BH4 recycling (dihydropteridine reductase (DHPR) or pterin-4a-carbinolamine dehydratase (PCD) deficiencies). Interestingly, the autosomal dominant form of GTPCH deficiency as well as sepiapterin reductase (SR) deficiency decrease PAH activity but not by enough to result in HPA38. Folding and assembly of functional PAH monomers is disrupted in the absence of the required chaperone DNAJC12, a recently described additional cause of inherited HPA, with great clinical variability1,4,39.
PKU is genetically very heterogeneous, with >1,000 PAH variants catalogued in individuals with PKU from around the world28,40. Many patients are compound heterozygous for two different PAH variants, leading to more than 2,600 known PKU-causing genotypes. The majority of PAH variants are in-frame missense amino acid substitutions (58.3%), whereas frameshift variants (13.9%), splice variants (13.1%), nonsense variants (6.9%) and synonymous substitutions (4.9%) are less common28. Of pathogenetic variants, 17.9% occur in intronic or untranslated regions of the PAH gene. Missense variants may result in the production of abundant but hypoactive or inactive PAH monomers. However, some variants, such as the relatively common c.1222C>T (p.Arg408Trp) variant that is prevalent in Celtic and Eastern European populations41, are associated with extreme instability and proteolytic degradation of the mutant PAH monomers, leading to severe PAH enzyme deficiency42. However, coexpression of two different variant alleles can alter the stability and PAH activity of the resultant tetramer in comparison to the predicted activity from either individual allele alone; examples of both positive and negative inter-allelic complementation are known43. These features add significant complexity to genotype–phenotype prediction44. Genotypes yielding some residual PAH activity may be associated with a BH4-responsive phenotype, in which oral BH4 supplementation leads to stabilization of the PAH tetramer45, with consequent increases in liver PAH activity and dietary Phe tolerance24.
The BTBR.Cg-Pahenu1 mouse, which was generated in an ethylnitrosourea (ENU)-induced random mutagenesis screen46, has a PAH p.Val106Ala missense variant that results in an unstable PAH protein and mild BH4-responsive HPA47. These mice accurately model BH4-responsive PKU in humans, which is now treated with the BH4 synthetic analogue sapropterin dihydrochloride48. Other animal models of PKU are available (TABLE 1) for investigation of the pathophysiological mechanisms underlying PAH deficiency, including the most widely studied model, the Pahenu2 mouse. This mouse model was also generated by ENU-induced random mutagenesis and its severe BH4-non-responsive phenotype is a model of untreated or late-treated severe PKU in humans49.
Table 1 |.
Animal models of PKU
| Animal | Method of generating model | Genetic background | Comparison with patients with PKU | Ref. | |
|---|---|---|---|---|---|
| Biochemical | Clinical | ||||
| Rat | Give large amounts of phenylalanine in combination with para-chloro-Phe, α-methyl-Phe or both | Various, usually Wistar | Resembles BH4 defects rather than PKU | Resembles BH4 defects rather than PKU | 240 |
| Mouse | Enu-1 (ENU-induced random mutagenesis) | BTBR | Mild HPA with Phe challenge | Models BH4-responsive PAH deficiency | 46 |
| Enu-2 (ENU-induced random mutagenesis) | BTBR | High blood and brain Phe concentrations in line with PKU | Behavioural and memory issues, partly also perhaps in relation with BTBR-specific features, such as lack of corpus callosum | 49 | |
| Enu-2 (ENU-induced random mutagenesis) | C57Bl/6 | High blood and brain Phe concentrations in line with PKU | Fewer behavioural and memory issues considering the high blood and brain Phe concentrations | 222 | |
| Enu-3 (ENU-induced random mutagenesis) | BTBR | High blood and brain Phe concentrations in line with PKU | Severe PKU; not available due to difficult breeding and husbandry | 241 | |
| PAH exon 1 deletion | C57Bl/6 | High blood and brain Phe concentrations in line with PKU | Behaviour not yet assessed | 242 | |
| Minipig | PAH exon 6 deletion | Yucatan minipig | High blood Phe concentrations | Hypopigmentation and ventriculomegaly | 243 |
| Humanized p.R408W PAH allele | Ossabaw minipig or Yorkshire full-size pig | High blood and brain Phe concentrations in line with PKU | Prenatal growth failure and neonatal seizures | 244 | |
BH4, tetrahydrobiopterin; ENU, ethylnitrosourea; HPA, hyperphenylalaninaemia; PAH, phenylalanine hydroxylase; Phe, phenylalanine; PKU, phenylketonuria.
Infants with PKU are phenotypically and functionally normal at birth. Low birthweight and short body length at birth have been reported in two studies50,51, but a meta-analysis of several studies showed normal prenatal growth52. Although PAH is only expressed in the liver and kidneys53, neither organ suffers any apparent significant pathology as a result of PAH deficiency. Left untreated, PKU most profoundly affects the brain (FIG. 4). The few post-mortem neuropathological evaluations of untreated individuals with PKU have documented small brain size and impaired myelination in all cases, although neuron numbers are usually normal54. The complexity of dendritic branching and the number of synaptic connections are reduced in untreated individuals with PKU55. Similar findings have been described in Pahenu2 mice56. The precise cause of brain dysfunction in PKU is unclear. Even after decades of study, the molecular pathophysiology that forms the basis of the neuropathology associated with PAH deficiency remains incompletely understood57.
Fig. 4 |. Pathological manifestations in PKU.

The clinically most important pathological manifestations of phenylalanine hydroxylase (PAH) deficiency are on the brain and are mediated by effects of excessive l-phenylalanine (Phe) concentration. Hypopigmentation (owing to impaired melanin production) is the solitary non-neurological manifestation of PAH deficiency. The large neutral amino acids (LNAAs), including Phe, tyrosine (Tyr) and tryptophan, cross the blood–brain barrier from the circulation by facilitated diffusion down a concentration gradient via the transporter LAT1 (also known as SLCA7A5). Elevated blood Phe competitively inhibits transport of the other LNAAs into the brain, reducing their concentrations; decreased LNAA concentrations may impair cerebral protein synthesis and contribute to monoamine neurotransmitter deficiency. However, the compensatory activity of energy-requiring amino acid exporters (EXPORT) may be able to mitigate amino acid imbalance to some extent and restore homeostasis. Phe competitively inhibits the activities of Tyr hydroxylase (TH) and tryptophan hydroxylase (TPH) in the brain, leading to dopamine and, especially, serotonin deficiencies. This mechanism is probably linked to the high prevalence of anxiety and mood disorders in individuals with hyperphenylalaninaemia. Elevated Phe has also been implicated in epigenetic alterations of gene expression patterns in the brain, and in impaired myelin synthesis, decreased cerebral glucose metabolism (visualized by PET imaging), the formation of amyloid plaque-like fibrils and increased oxidative stress. TYR, tyrosinase.
Biochemical effects of PAH deficiency
Inspection of the Phe hydroxylation pathway (FIG. 1) suggests three different proximal biochemical consequences as potential primary causes of pathology in inherited PAH deficiency: HPA, hypotyrosinaemia or the effects of accumulating phenylpyruvate or related metabolites. Organic acids such as phenylpyruvate are rapidly excreted in urine, and their tissue concentrations are probably too low to be of any clinical consequence, even in completely untreated individuals58. Plasma Tyr concentrations in untreated individuals with PKU are on average lower than in the general population but are usually not below the normal range, as some l-Tyr is ingested from dietary protein in a normal diet, as for all other amino acids. Although l-Tyr deficiency may have a role in brain amino acid balance and neurotransmitter deficiency (as described below), simply supplementing the diet with l-Tyr without any other treatment does not prevent severe cognitive disability in individuals with PKU59. As first demonstrated in 1953 (REF.14) and supported by several other anecdotal reports soon thereafter, and ultimately confirmed in a longitudinal study in 1960 (REF.60), restriction of dietary Phe intake substantially prevents the major manifestations of PKU, implicating Phe itself as the primary neurotoxin in PKU, although HPA does not completely explain the entire cascade to brain dysfunction61.
The mechanisms through which elevated Phe concentrations in the brain cause dysfunction have been and continue to be a fertile field of investigation. It is likely that multiple different mechanisms are involved, depending on the developmental stage of the brain and when treatment commences (early or late).
Mechanisms of neuropathology
White matter disruption.
HPA causes disturbances of neuronal dendritic outgrowth and synaptic connectivity, in both in vitro experiments with cultured neurons62,63 and in animal models64,65, most importantly the Pahenu2 mouse. Some of the most severe manifestations in individuals with PKU (that is, intellectual disability and epilepsy) are considered to have, at least in part, a grey matter component; nevertheless, most studies are about white matter abnormalities and only a few report findings of abnormal grey matter66,67. It has been suggested that HPA alters the phenotype of oligodendrocytes from myelinating to non-myelinating68, but cultured rat oligodendrocytes are capable of laying down normal myelin sheaths in HPA69. Phe may impair the synthesis of cholesterol (through inhibiting 3-hydroxy-3-methylglutaryl-CoA reductase activity70) or other brain lipids and thereby interfere with myelin production. However, the precise molecular mechanisms underlying the white (and grey) matter disturbances associated with elevated brain Phe concentrations remain unknown.
Cerebral metabolism.
The rate of cerebral glucose metabolism is reduced in the frontal cortex of hyperphenylalaninaemic Pahenu2 mice, suggesting that energy production is impaired71, the extent of which correlates with the severity of behavioural abnormalities in these animals72. However, severe memory impairment is the most prominent cognitive deficit in Pahenu2 mice72–74, despite seemingly normal glucose uptake in the hippocampus. Similar alterations of cerebral glucose metabolism have been measured by PET imaging in patients with PKU75–77. The mechanism may be related to Phe-mediated inhibition of pyruvate kinase78 or other enzymes of glycolysis or oxidative phosphorylation.
Large neutral amino acid deficiency.
Movement of the aromatic amino acids (Phe, Tyr and tryptophan) and other large neutral amino acids (LNAA), including leucine, isoleucine, valine, methionine, threonine and histidine, from the circulation into the brain across the blood–brain barrier (BBB) occurs through sodium-independent transfer79. This transfer is facilitated by the amino acid transporter LAT1 (also known as SLCA7A5), which is a member of the amino acid–polyamine–organocation family of transmembrane transport proteins80 and is expressed on both the luminal and abluminal membranes of brain capillary endothelial cells. As the affinity of LAT1 for all LNAAs is high (Michaelis constant (KM) ~20–200 μM) relative to the physiological concentrations of these amino acids in capillary blood, this transporter is saturated at all times. Therefore, in the hyperphenylalaninaemic state, Phe-mediated competition for binding to LAT1 has been suggested to impair the flux of the other LNAAs into the brain, leading to their deficiency in the brain81. These deficiencies are probably responsible for impaired rates of cerebral protein synthesis measured in adults with PKU82,83 and contribute to brain monoamine neurotransmitter deficiencies84. Oral supplementation of LNAAs other than Phe has been promoted as a treatment approach to correct cerebral amino acid imbalance and the attendant physiological consequences84–88. However, most discussions of the LNAA competition at the BBB theory ignore the fact that there are separate sodium-dependent amino acid transporters on the abluminal brain capillary endothelial cell membrane with very high affinity for LNAAs89; these transporters are capable of pumping amino acids out of the brain back to the circulation and may work to modulate any disturbances of amino acid homeostasis in the brain90. The current working model of LNAA transport at the BBB and its attendant consequences during HPA are probably insufficiently developed91.
Neurotransmitter deficiency.
Monoamine neurotransmitter deficiencies, initially and most dramatically of serotonin92, but also later of norepinephrine93, in the brains of individuals with PKU were first suggested as possible pathophysiological mechanisms explaining part of the cognitive and behavioural deficits in affected individuals. These deficiencies have been confirmed in post-mortem specimens from untreated individuals with PKU93 and in multiple reports in hyperphenylalaninaemic Pahenu2 mice72,84,88,94–99. All animal studies have detected profound serotonin deficiency, whereas some studies have found a disturbance of dopamine metabolism, albeit less severe than that of serotonin. From the cumulative data, there is evidence that three different mechanisms contribute to the genesis of neurotransmitter deficiencies: first, cerebral deficiency of Tyr and tryptophan (the substrates for dopamine and serotonin synthesis, respectively) in the brain owing to amino acid transport competition84 (described above); second, decreased constitutive expression of Tyr hydroxylase (TH) and tryptophan hydroxylase 2 (TPH2), which catalyse the rate-limiting steps in dopamine and serotonin synthesis, respectively72; and third, Phe-mediated competitive inhibition of TH and TPH2 activities72. Disturbances of monoamine neurotransmitters have been frequently implicated as contributors to the neuropsychiatric symptoms and impaired executive function associated with HPA81,100.
Other cerebral effects of HPA
Phe at millimolar concentrations has been shown to aggregate into amyloid-like fibrils, and deposits of Phe aggregates have been detected in the brain of Pahenu2 mice and a single human PKU brain examined after death101.These amyloid-like fibrils, which are reminiscent of the amyloid plaques associated with Alzheimer disease, have been postulated to have a pathogenetic role in the cognitive deficits associated with PKU. High Phe concentrations have also been shown to alter the methylation pattern of a panel of known methylated genes, including several regulatory microRNAs in the brain tissue and blood of patients with PKU102 and the brain of Pahenu2 mice103, suggesting that elevated Phe has marked effects on the epigenome. Finally, there is evidence for increased oxidative stress in association with HPA, as indicated by increased lipid peroxidation104 and microglial activation105 in the brain of Pahenu2 mice.
Diagnosis, screening and prevention
Manifestations
If untreated, patients with PKU develop severe intellectual disability, epilepsy and behavioural, psychiatric and movement problems, a musty odour and, in some patients, light(er) pigmentation of skin, eyes and hair, and cortical blindness and eczema5. If started immediately after birth, dietary treatment can prevent these sequelae. However, if treatment has been inadequate for long periods of time, adults with PKU may develop clinical issues, including lower extremity spasticity and cerebellar ataxia, tremor, encephalopathy and visual abnormalities106–108. Interestingly, dementia has also been described in patients with PKU first presenting during adulthood109.
Even though the dietary treatment of PKU initiated in the first days after birth prevents the major cognitive and neurological deficits60, the incidence of attention-deficit–hyperactivity disorder and specific learning disabilities, which are probably related to deficits in executive functions, might remain higher in well-treated patients with PKU than in individuals without PKU110. Higher Phe concentrations due to difficulty in adhering to the strict dietary treatment during adolescence and adulthood is associated with emergence of adverse effects on attention, mood, memory and executive function111,112.
Screening
Nowadays, the implementation of newborn screening for PKU in most countries/regions worldwide has resulted in diagnosis typically occurring in the neonatal period. The screening involves collecting a drop of blood from a healthy neonate by a heel prick. Although the exact timing of the heel prick varies among countries/regions, the appropriate time for blood sampling is between 24 hours and 72 hours after birth. The outer or inner side of the baby’s heel is pricked and blood dripped on to a filter paper card (Guthrie card) so that the marked circles on the card are completely saturated. The analytical phase of the screening process consists of the biochemical analysis, and referral of the neonate for confirmatory testing. Different laboratory screening methodologies exist for the assessment of blood Phe concentrations. In the bacterial inhibition assay (BIA), which is the original Guthrie test, the DBSs are placed on agar plates containing a strain of Bacillus subtilis. The agar also contains β−2-thienylalanine, a Phe analogue that inhibits bacterial growth. When high concentrations of Phe are present in the DBS, transport of the analogue into the bacterium is inhibited and bacterial growth occurs, which is easily detectable.
Calibration spots allow rough estimation of the Phe concentration in the patient sample. This assay is simple, inexpensive and suited for screening large numbers of individual specimens; however, it is a semiquantitative method with limited sensitivity (for example, the presence of antibiotics can cause false-negative results). Increased accuracy and sensitivity have been achieved using fluorimetric microassay (FMA) to quantify Phe levels. The method involves chromatography separation followed by derivatization and fluorimeter detection. Both the BIA and FMA only detect a single amino acid, such as Phe, whereas tandem mass spectrometry (TMS) allows the measurement of multiple amino acids. In the past high protein intake could lead to false-positive results, but this is no longer the case with TMS.
Once it was recognized that the concentration of acylcarnitine and amino acids can be determined simultaneously using TMS, screening for many IMDs from a single punched disc from the DBS became possible. Countries that ‘only’ screen for HPA can use the BIA or FMA, which are less expensive than TMS. However, despite the higher cost, TMS is the only choice for screening for multiple IMDs (with other amino acids and/or acylcarnitines). Furthermore, TMS also allows Phe and Tyr to be measured simultaneously, enabling a more sensitive strategy for PKU screening. Whereas 240 μmol/l (4 mg/dl) was a commonly used cut-off Phe concentration for a PKU-positive screening result in the past, TMS has provided more sensitive detection, with a Phe cut-off concentration of 120 μmol/l (2 mg/dl) in combination with a Phe to Tyr ratio >1.5 giving a PKU-positive screening result113.
Identifying the defect underlying HPA
When HPA is detected, further investigations are mandatory to distinguish between PAH deficiency, disorders of BH4 metabolism and DNAJC12 defects. Analysis of pterins and measurement of DHPR activity by DBS testing is important for identifying BH4 defects and should be carried out on all newborns with HPA (FIG. 5). The use of a DBS instead of urine (for analysis of pterins) is more practical and allows measurement of pterins, DHPR activity and amino acids from a single specimen. However, in the USA, pterins are still usually analysed in a urine sample.
Fig. 5 |. Proposed algorithm for screening and diagnosis of PKU and monitoring treatment efficacy.
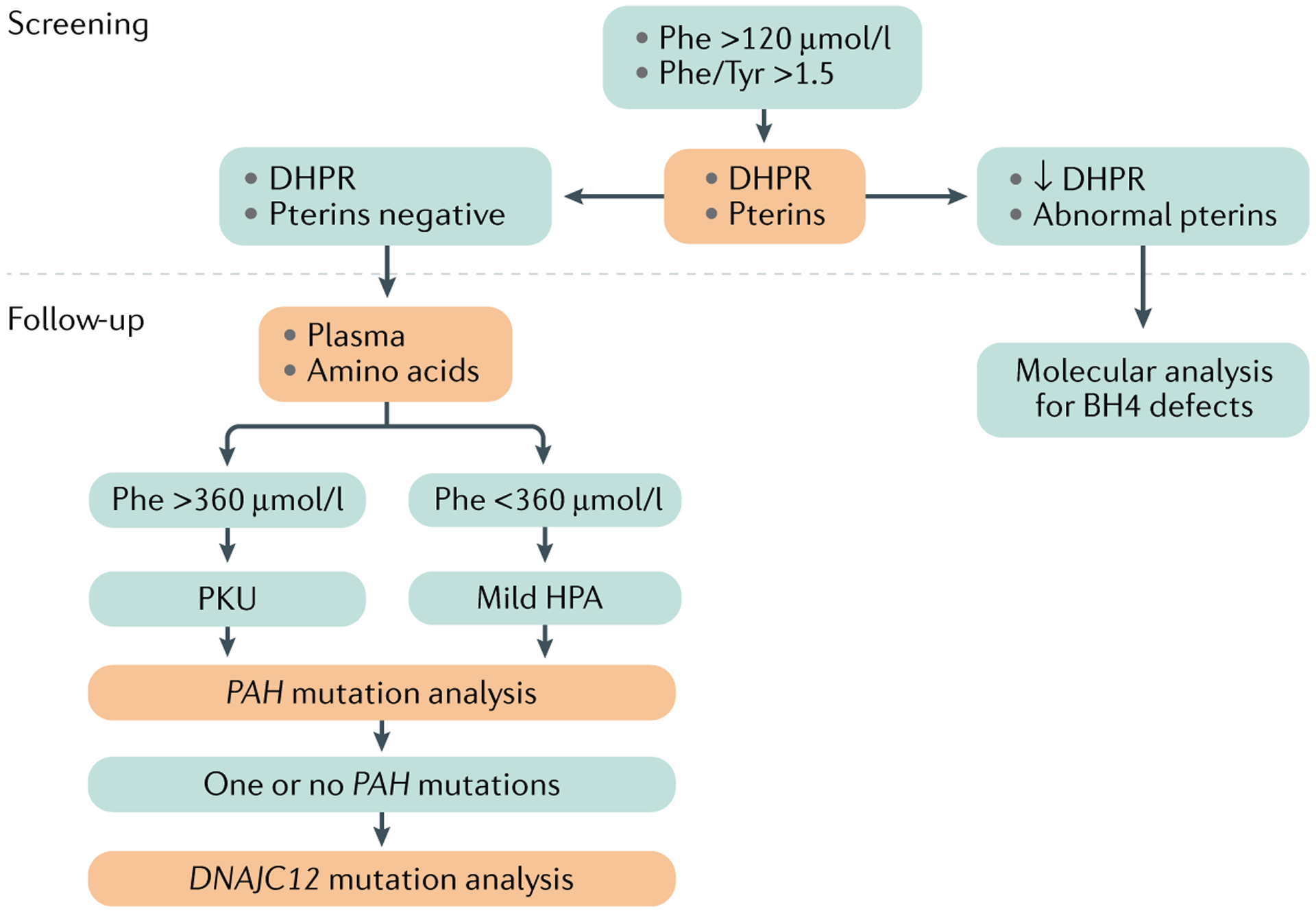
Diagnosis of phenylketonuria (PKU) is made during a neonatal screening programme. Blood obtained with a heel prick from newborns from 12 hours after birth and later is applied to filter paper (that is, a dried blood spot (DBS)), which is used to assess phenylalanine (Phe) concentrations by the Guthrie bacterial inhibition assay, other enzymatic assays or tandem mass spectrometry (TMS). BH4, tetrahydrobiopterin; DHPR, dihydropteridine reductase; HPA, hyperphenylalaninaemia; Tyr, tyrosine.
In cases where the results of pterin and DHPR analysis may be delayed, a 24-hour BH4-loading test can be performed. In this test, 20 mg/kg sapropterin dihydrochloride is given orally and Phe concentration in a DBS is measured before loading and at 4, 8, 16 and 24 hours after loading. Substantial decreases in blood Phe up to 8 hours after BH4 challenge are seen in patients with BH4 defects (especially PTPS, GTPCH and PCD deficiencies) and variants in the DNAJC12 gene (presenting with a normal pterin profile), while patients with BH4-responsive PKU or DHPR deficiency tend to show a much slower decrease in blood Phe. If no decrease in Phe occurs, the patient probably has PAH deficiency, although such a result cannot be used to conclude that a patient has non-BH4-responsive PKU, as some neonates with a negative BH4 loading test in the newborn period are BH4-reponsive when tested at an older age114. Thus, the BH4 loading test enables early diagnosis of BH4 or DNAJC12 deficiency and may also identify BH4-responsive PKU. Although pterins are not altered in patients with DNAJC12 deficiency, treatment is comparable to that for defects in BH4 metabolism, and DNAJC12 deficiency should be considered in individuals who show development different from patients with PAH deficiency.
Of note, newborns with autosomal recessive GTPCH or SR deficiency may present with normal blood Phe in the neonatal screening DBS or in plasma, and thus can be missed if only amino acids are measured. Patient genotyping using a panel of all genes known to cause HPA (including DNAJC12) can provide final diagnostic confirmation and might predict the metabolic phenotype in PAH deficiency40. However, predicting responsiveness to BH4 in patients with PAH deficiency by genotyping is not always conclusive. Genotype analysis seems mainly helpful to exclude patients who are unlikely to respond to BH4 supplementation based on the specific PAH pathogenetic variants115.
The BH4 loading test using 48-hour loading (20 mg/kg sapropterin dihydrochloride every 24 hours) can be useful in identifying the majority of BH4-responsive patients with PAH deficiency (who show a Phe reduction of at least 30% at various time points, but especially at 24 h after the second dose), but may also not be particularly effective in predicting long-term BH4 responsiveness115. All of the proposed loading tests for sapropterin dihydrochloride should include reassessment of the diet and maximizing the Phe tolerance before BH4 loading and after the start of BH4 treatment to measure the gain in Phe tolerance produced by BH4 supplementation, followed by titration of sapropterin dihydrochloride in responders to BH4 treatment113.
Prenatal diagnosis of all variants of BH4 deficiency is possible by measuring the concentration of the pterin metabolites biopterin and neopterin in amniotic fluid (being the fetus’ urine); the pattern of these metabolites mirrors the pattern found in the urine of the same patients after birth and therefore is diagnostic for GTPCH and PTPS deficiency. However, molecular analysis is now the method of choice for all primary BH4 deficiencies116.
Management
Untreated PKU is only seen in those individuals who were born before the advent of newborn screening or in a country in which newborn screening is not yet available for every newborn, and who therefore have been untreated from birth. PKU was the first disease for which a treatment was introduced to prevent intellectual disability. The goals of PKU treatment are: optimal neurocognitive development and functioning, and normal growth, nutritional status, quality of life (QOL) and psychosocial well-being. While diagnostics of HPA is important, dietary treatment of PKU should be initiated in the first days after birth to prevent the major cognitive and neurological deficits60.
Dietary management
It now seems so logical, but initiating dietary control to limit the intake of a specific amino acid has been a tremendous step forward in the understanding of PKU and improving outcomes not only for patients with PKU but also for those with other defects in metabolism of amino acids, carbohydrates or fatty acids. Dietary treatment of PKU has been so successful only because Phe cannot be synthesized in the body and therefore Phe is a so-called essential amino acid, meaning that the blood Phe concentration is highly dependent on dietary Phe intake.
Although the Phe-restricted diet has been shown to be effective, it does not fulfil every treatment goal in an optimal way. For example, the incidence of attention-deficit–hyperactivity disorder and specific learning disabilities, probably related to deficits in executive functions, might remain higher in well-treated children with PKU than in children without PKU110. Loss of metabolic control during adolescence and adulthood is associated with emergence of significant adverse effects on attention, mood, memory and executive function111,112. In a few individuals, lower extremity spasticity and seizures can develop after prolonged non-adherence to therapy during adulthood106. Encephalopathy has been described after long-term lack of metabolic control as well as in patients at first presentation107,108 and is responsive to strict dietary treatment. Visual abnormalities have also been reported to emerge during adulthood when the patient is non-adherent and to disappear after re-introduction of the strict diet107. These data seem consistent with the more subtle visual deficits described in children who do not adhere to metabolic control and with the cortical blindness in untreated adults with PKU117,118.
The dietary treatment comprises three aspects: restriction of natural protein intake, supplementation with a Phe-free amino acid mixture and consumption of low-protein food products. Phe restriction can only be performed by restricting intake of natural protein. The extent of natural protein (Phe) restriction is based on the amount of Phe required for net protein synthesis (for example, age-dependent growth and balance between anabolism and catabolism in periods of illness) and the severity of the PAH deficiency113,119. By restricting natural protein consumption, not only is the intake of Phe reduced but so is that of other (essential) amino acids and other constituents of natural protein, such as vitamins, minerals and carnitine. Natural protein therefore is replaced with a protein substitute, which is an amino acid mixture that lacks Phe, is enriched for Tyr to replace the Tyr that in healthy individuals is produced from Phe, and contains other micronutrients that are present in a standard diet containing natural protein. The third part of the diet is low-protein food products, which consist of carbohydrates and fats that replace basic food items such as bread and pasta. They are especially important for supplying energy and enable patients to have a food profile that to some extent mimics normal food habits119.
The initial experience with dietary Phe restriction revealed two outcomes: a considerable improvement in behavioural rather than intellectual outcome in individuals with PKU who started treatment after damage had already occurred, and enormous improvement when treatment was started before damage had begun120. Furthermore, this initial experience also showed that treatment had to be tailored and monitored, as protein and/or Phe deficiency also had adverse effects, including growth restriction, anorexia, alopecia, lethargy and eczematous eruptions121,122. Of note, such dietary failure may still occur nowadays123, so that growth restriction, anorexia, alopecia, lethargy and skin lesions are red flags.
Developments in protein substitutes.
Although the basis of dietary control has not really changed since its introduction in 1953 (REF.124), protein substitutes have evolved. Initially, protein hydrolysates were used, so it was technically impossible to achieve a completely Phe-free protein substitute, thus requiring that the Phe contribution from natural protein be decreased even more. By contrast, the completely Phe-free amino acid mixtures available nowadays allow patients with PKU to obtain Phe from natural protein only. Despite substantial efforts to improve the quality (adding vitamins, selenium, carnitine, long-chain polyunsaturated fatty acids), texture, taste and ease of use of these mixtures124, the taste is still not optimal. However, the use of glycomacropeptides (GMP), a protein component of whey that is completely devoid of Phe (as well as Tyr, tryptophan, arginine, cysteine and histidine, and is low in methionine and leucine) represents an improvement, including better taste and feeling of satiety as well as improved immunological aspects by decreasing inflammation125. As many patients have problems with the taste of amino acid mixtures, GMP might be a solution especially in patients for whom the intake of protein substitutes is a real issue. However, residual Phe-containing proteins are not easily separable from GMP when it is isolated from whey, so GMP still contains some Phe. Furthermore, GMP is low in some LNAAs and therefore enrichment with LNAAs, such as Tyr, tryptophan, leucine, methionine and histidine, is necessary to provide a protein substitute with the same quality as Phe-free LNAA mixtures84. However, addition of the lacking amino acids negatively affects the taste. Whereas the Phe content of early GMP isolates was ~3–4% (it is 4–5% in natural protein), more recent GMP products have a Phe content of 1–2%126. Therefore, despite these improvements, GMP still has disadvantages, especially for patients with PKU who have a low tolerance of Phe and a high requirement for protein substitutes. Another issue with the Phe-free diet is that the physiological course of absorption of free amino acids in protein substitutes does not follow that of natural protein127. Therefore, a so-called physiomimic formulation was developed, in which a coating was added to the protein substitute128. The coating consists of ethyl cellulose and alginates that encase granules of amino acids (without Phe); this formulation shows a much better release of the essential amino acids to the blood so that the amino acids can be used for protein synthesis rather than energy production.
Pharmacological treatments
Although the Phe-restricted diet is still the cornerstone of treatment, two drugs are available that decrease the blood Phe concentration and thereby that in the brain as well. Some patients with PAH deficiency can forego dietary control when treated with the BH4 synthetic analogue sapropterin dihydrochloride, while others of all ages tend to show a large increase in their Phe tolerance129,130. However, only a subset of patients with PKU respond to this treatment. Consequently, the still rather new therapeutic pegvaliase, an injectable pegylated Phe ammonia lyase (pPAL) approved by the FDA in 2018, was welcomed by the PKU community and has dramatically changed the lives of patients with PKU25,131. This treatment has had a long, complex development (reviewed elsewhere132). Despite pegvaliase proving effective both orally and parenterally in PKU mouse models, the oral formulation was not effective enough due to early degradation133 and the parenteral formulation required pegylation to be effective134. Nevertheless, pegvaliase is highly effective in reducing Phe concentrations in most patients, such that most of them can come off dietary management131. However, pegvaliase is not without drawbacks, including adverse events ranging in severity from local to more general skin reactions, arthralgia and, very rarely, anaphylactic responses135. These adverse events are based on two different immunological responses: the strongest is usually in the first 6 months and mostly involves anti-PEG antibodies, while the anti-pegvaliase antibodies persist and cause some less severe symptoms135. Consequently, patients need to be very persistent with treatment, as it takes months to overcome the antibody response before pegvaliase becomes effective. Based on clinical experience and knowledge of adverse immunological events, guidelines for pegvaliase treatment induction and maintenance in patients with PKU have been proposed136.
Most Phe that is absorbed in the gut is not from dietary protein but instead is derived from the so-called enterorecirculation between the body and the intestine137. In enterorecirculation, large amounts of proteins, enzymes and polypeptides enter the gut by pancreatic and other glandular secretions, which undergo tryptic digestion to release amino acids that are reabsorbed in the intestine. Intestinal Phe was efficiently metabolized by orally administered pegvaliase in a rat model of PKU137, suggesting that oral formulations to metabolize Phe in the intestine might be an effective therapeutic approach.
Breastfeeding
In daily practice, metabolic control is started very early after birth. Breastfeeding was long considered to be impossible due to the risk of uncontrolled Phe concentrations, as the protein content or the amount of human milk consumed by an infant was not known138,139, but this fear was later shown to be unnecessary140. Most PKU clinics advocate giving human milk, although in various ways. One method is to feed infants human breast milk by bottle so that the volume of milk consumed is known. Another, less socially restrictive, approach is to first feed the infant with an amino acid mixture and then allow breast feeding. The amount of amino acid mixture administered is based on the target Phe concentration, assuming that the child will consume more breast milk if a lower volume of amino acid mixture is given and vice versa, with the total ‘protein’ intake remaining the same141. A study investigating the effect of alternating feeds (amino acid supplements and breast milk) over 6 months97 showed that this approach is practical and safe, with the daily number of breast feeds dependent on the Phe concentration142. In the first period after diagnosis, the amount of Phe allowed without exceeding target Phe concentrations (360 μmol/l) can be much higher than at later times. This so-called honeymoon period can last for weeks. Weaning is not different from that in healthy infants but the low natural protein allowance in infants with PKU does not leave room for foods rich in natural protein.
Although the BH4 concentrations in human breast milk have not been extensively studied, one study found that BH4 levels seem quite high, even in healthy mothers not treated with sapropterin143. The Phe content of human breast milk may vary with postpartum age and, in the case of a mother with PKU, with her adherence to dietary control. A mother with PKU may breastfeed her baby whether the baby has PKU or not, as long the Phe concentrations are monitored if the baby has PKU.
Childhood
The first years of life for a patient with PKU are difficult, as Phe requirements can vary considerably owing to the changing growth rate. Furthermore, periods of illness (especially with fever), immunization and teething require energy, which is supplied by protein catabolism for glucogenesis, but as Phe cannot be broken down, its concentration in the circulation will increase.
Theoretically, other risk factors could include a (possibly unconscious) lack of acceptance of the diagnosis or knowledge of aspects of treatment by parents, or a lack of stringent follow up with home sampling and/or timely communication of Phe test results to parents. All those factors are difficult to assess. For example, one study found that the extent of knowledge of PKU has a role in dietary adherence144, while other studies found no association, perhaps because knowledge in itself is not enough to ensure treatment adherence in practice145–147. In fact, a positive attitude to the dietary regimen may help in achieving better metabolic control148. A faster turnaround time of Phe blood test results has not been shown to result in lower Phe concentrations. It has not been studied whether more frequent sampling results in lower Phe concentrations, but, at the same time, patients are sometimes more strict with the diet before a sample is taken, so more frequent testing could be hypothesized to improve their metabolic control149. Importantly, patients can be taught simple aspects of their treatment from an early age.
The PKU dietary handbook is a practical translation of the European guidelines for PKU treatment and provides advice on especially the day to day aspects of all aspects of dietary treatment119, but does not remove the need for follow-up and recommends more intense follow-up that may be clinically relevant in some patients who have difficulty keeping Phe concentrations under control8,113. The European guidelines present an action plan for use in the event that half or more Phe test results are above the target range or control of Phe concentrations is inconsistent, especially in children.
Adolescence
From the last years of the first decade of life onwards, patients should be taught to take control of their own PKU treatment so that they can be in control from around 12 years of age. To the best of our knowledge, there are no data on the optimal transition of responsibility for PKU treatment to the patient, but having a chronic disease without clear acute signals of non-adherence to metabolic control is challenging, especially during adolescence, when a balance is needed between the carer’s responsibility and trust in an adolescent child versus the child’s need for help. Furthermore, the instruments for monitoring (for example, having Phe test results sent directly to a smartphone) and metabolic control must align with the present skills of adolescents150. In patients with PKU, the normally learned executive functions (the higher cerebral processes), such as planning, strategic thinking and organization, may require optimal Phe concentrations and are thus easily negatively affected by high Phe concentrations151,152. In turn, decreased capacity in planning may negatively affect adherence to dietary treatment, resulting in a negative feedback loop.
Although higher blood Phe concentrations are clearly associated with poorer neurocognitive outcomes113, this is not always the case153, especially from the end of the second decade of life. These inconsistencies highlight that the whole pathogenesis of brain dysfunction in PKU is not well understood and that blood Phe concentration is only a surrogate marker of the cerebral processes and only incompletely explains the whole cascade of neurocognitive dysfunction61,154.
Adulthood
Adult patients with PKU may encounter problems with neurocognitive and social functioning155,156. The percentage of patients living a normal life without PKU-related or treatment-related problems is unclear, as it is difficult to have an unbiased population to address this question. Debate is ongoing about the necessity to decrease blood Phe concentrations in adult patients with PKU8,113,157,158, which will probably not be easily resolved, as data from adults and elderly patients are still scarce and only patients with optimal treatment from an early age should be studied. Therefore, we welcome recent studies that add data on the relationship between blood Phe concentration during adulthood and the neurocognitive and social functioning and well-being in adults with PKU112,152,153,159–174. Although it is clear that adults with PKU may encounter brain function-related problems and that these can be related to increased adult blood Phe concentrations, some researchers question whether the effect of these issues is clinically important enough to have a dietary treatment that is socially demanding or a more invasive and expensive pharmacological treatment, as with sapropterin dihydrochloride or pegvaliase113,175.
Of note, this debate about optimal target blood Phe concentrations is not the first point of contention in the history of PKU research and treatment. For example, the value of dietary treatment8,59,113,176–178, the period of life in which treatment remains necessary8,113,157,158,179–181 and the safe blood Phe concentrations in various age groups8,113,157,158,180,182,183 have been the subject of debate in the past.
A special word is necessary for those adult patients with PKU who might consider stopping dietary treatment. These patients may stop taking protein substitutes but then may not resume normal natural protein intake, as they find it hard to become accustomed to and enjoy natural high-protein foods. As a result, they may be at risk of deficiency for some micronutrients and vitamins, especially vitamin B12, zinc and selenium184.
Pregnancy and maternal PKU
Inadequately treated PKU in pregnant women (so-called maternal PKU), but not paternal PKU, may profoundly increase the risk of fetal developmental abnormalities. Maternal PKU was identified as a risk factor in 1957 (REF.16), even before the introduction of the Guthrie method for newborn screening17. Two reports, one in 1963 and another in 1980, were instrumental in fostering recognition that maternal PKU is an important problem185,186. Of note, maternal PKU presents no risk to the mother. Numerous studies have shown that the risk of fetal developmental abnormalities is increased if maternal blood Phe concentrations exceed 360 μmol/l and treatment is not initiated before the start of pregnancy8,113,187,188, although attaining a blood Phe concentration of <600 μmol/l before 10 weeks gestational age may reduce risk to an acceptable level189.
The entire pregnancy and the period of attempted conception should be considered a risky period. Most patients, even those who struggle to keep Phe concentrations within the target range when they are not trying to become pregnant, seem able to get Phe concentrations within the target range if they are trying to become pregnant. Treatment with sapropterin has been slowly introduced to manage Phe concentrations in women with BH4-responsive PKU who aim to become or are pregnant, and leads to increased tolerance for Phe and no excess fetal abnormalities190,191. However, an overall recent registry of the incidence of fetal abnormalities in women with maternal PKU who are treated by diet or BH4 is lacking.
Although problems in pregnancy, such as hyperemesis gravidarum (severe nausea and vomiting of pregnancy), can be very disturbing and distressing, achievement of Phe concentrations below the upper target concentration in the second half of pregnancy is usually not an issue. In fact, starting from ~16–22 weeks gestational age, the growth of the placenta and fetus is so rapid (and thus protein needs are very high) that the Phe intake should be increased considerably to prevent Phe concentrations falling so low that the growth and development of the fetus is at risk of hypophenylalaninaemia192.
Management of maternal PKU depends not only on strict Phe intake restriction but also on controlling other factors, such as total protein, insufficient energy intake, hyperemesis gravidarum, low Tyr, and folic acid supplementation and its resulting concentration. If all these factors are adequately addressed and metabolic control concentrations are within targets, then the risk of fetal issues due to maternal PKU is very low or even absent. In this case, extra procedures during antenatal or perinatal care are not likely to be necessary. However, if blood Phe concentrations are increased or if any of the aforementioned factors are suboptimal, additional monitoring with ultrasonography is needed.
Routine follow-up of patients with PKU is based pre-dominantly on Phe concentrations, so that measurement of blood Phe is crucial. Historically, Phe concentrations were often measured qualitatively, using methods such as the Guthrie method for follow-up, which have now been completely replaced by highly accurate quantitative laboratory techniques, such as high performance liquid chromatography, amino acid analysers and TMS. During the past two decades, home sampling by DBS has taken its place in daily practice with measurement performed in the laboratory, but there are concerns about reliability of the data owing to factors such as variation in sampling material (filter card type) and measurement from the DBS compared with from plasma193–197. A 2020 study showed that DBS measurements can be valid if a laboratory-dependent correction factor based on filter card type is applied and plasma values from lithium heparin tubes are used as the gold standard in extraction and calibration protocols198.
Late-diagnosed PKU
As newborn screening is still not in place in every country, not all patients with PKU are diagnosed preclinically. Some of these patients with severe intellectual disability and other brain dysfunction live in homes for individuals with mental disabilities, others with their family at home, while some are hidden from their environment. Some are refugees who are only diagnosed with PKU in their destination country. In any of these patients, behaviour issues and epilepsy may need attention and it is essential to offer PKU treatment113. An important principle for the introduction of the protein substitutes is ‘gently and slowly’, as becoming accustomed to the protein substitute is challenging and requires time and effort on the part of the patient and the patient’s support network. Sometimes patient behaviour or epilepsy only improve after 6–12 months of dietary control and an increase in neurocognitive function might even be possible199.
International management guidelines
The most recent US and European guidelines for over-seeing all aspects of PAH deficiency are quite consistent8,113,158,200,201. Both guidelines recommend starting treatment when Phe concentrations exceed 360 μmol/l, as in treated patients with PKU higher Phe concentrations have been clearly shown to result in non-optimal outcomes and evidence that it is safe to start treatment when Phe concentrations exceed 600 μmol/l is limited. Both guidelines recommend keeping Phe concentrations below 360 μmol/l in children and in women wishing to bear children. The only real difference between these guidelines is that the US guidelines also recommend keeping blood Phe concentration below 360 μmol/l in adolescents and adults, whereas the European guidelines recommend that Phe concentrations should be kept below 600 μmol/l in those over 12 years of age. While the US guidelines based their advice on the idea that the closer Phe concentrations are to physiological the better the outcome, and that new treatments such as pegvaliase (and gene therapy) will enable patients to achieve these Phe concentrations201. The European guidelines provide no definitive data to suggest that Phe concentrations in the range of 360–600 μmol/l in those over 12 years of age affect outcomes.
Quality of life
Compared with no treatment, early and continuous treatment results in excellent outcomes in patients with PKU. However, the psychological and social burden of having a chronic disorder, the social and practical burden of the dietary treatment, with severe restriction of natural protein and supplementation with unpalatable amino acid supplements, are burdensome and may all affect the QOL of patients and their family (BOX 2).
Box 2 |. A parent’s perspective on caring for a child with PKU.
In 2003, our first son was born. In the first days of his life we had a phone call from our general practitioner to say that our son tested positive in the newborn screening programme for phenylketonuria (PKU). That was a shock. We went to the hospital and he went on a strict low-protein diet with amino acid supplementation.
In the first 10 years of his life, we were very strict with his diet. Everything he ate during the day was weighed and written down in his personal ‘PKU agenda’. We have always tried to make him responsible for his own diet, which meant that from approximately 10 years of age he took more and more responsibility for managing his own diet. However, he did not weigh all ingredients and we became less strict in the diet. At the age of 9 years, we also started with Kuvan® (sapropterin dihydrochloride, an oral form of tetrahydrobiopterin) after some hesitation. We are very happy that he can have more (natural) protein during the day because of the Kuvan®. It makes life a lot easier.
PKU has given our life a big change. PKU is always something ‘extra’. We cook with an extra pan on the cooker. When planning holiday destinations or when eating out, you always have to think about PKU. But PKU also changed our own eating habits. Because of PKU we learned so much about the food that we eat. Over the years, we eat much healthier (making things ourselves instead of ready-made sauces or food). We eat less meat within our family. So PKU in that way has had a positive impact on the rest of the family.
Our son is 17 years now. He attends the hospital for his PKU check alone. For him, PKU is his life, he does not know anything else. He never had a big issue with it and (as far as we know) never ate a hamburger to try it. His Phe concentrations over the years are OK but of course he is a normal adolescent with age-related challenges for both him and us as parents. But relating to PKU he does very well. We are happy that he is treated in a PKU expert centre. So we know he gets the best care from a team that has the most experience and knowledge and is in direct contact with other European PKU expert centres if questions would arise.
Overall quality of life
The large majority of studies have found an overall QOL fully comparable to that of the general population in children and adults treated early, with some exceptions on specific domains202–209 based on generic health-related QOL (HRQOL) questionnaires, which enable comparison of the QOL of patients with PKU with that of the general population (TABLE 2). A study using the Child Health Questionnaire (CHQ) in 32 Italian children with PKU found lower scores, overall and on specific domains, than in the reference population210. However, in a large international study (202 children and adolescents with PKU) using the CHQ questionnaire, the parents of these children reported scores comparable to those of the general population207. Only one study (in Brazil) found significantly lower overall HRQOL in individuals with PKU, with lower scores on all domains of the Paediatric Quality of Life Inventory (PedsQL) questionnaire211. Importantly, patients in this study differed from patients with PKU in other studies in that cognitive disabilities were detected in this group, despite reported early treatment, probably owing to two severe barriers to optimal adherence to treatment: the health-care system and financial difficulties. These findings emphasize the importance of optimal financial and practical support for patients with PKU to secure the best possible outcomes.
Table 2 |.
Studies of HRQOL in patients with PKU
| Study | Questionnaires | Trial characteristics (number of patients, country, design) | Results | Ref. |
|---|---|---|---|---|
| Bosch et al. (2007) | TAAQoL | 32 adults Netherlands Cross-sectional |
QOL comparable to reference population | 202 |
| Simon et al. (2008) | Profile of QOL in the chronically ill | 67 patients ≥17 years of age Germany Cross-sectional |
QOL comparable to reference population | 203 |
| Thimm et al. (2013) | KINDL-R | 50 children Germany Cross-sectional |
QOL overall comparable to reference population; increased parental concern about school success and success in life at time of poor metabolic control | 204 |
| Demirdas et al. (2013) | PedsQL TAAQoL DISABKIDS |
39 children; 30 adults Netherlands Cross-sectional; baseline, prospective; before and after start of BH4 treatment |
Baseline overall results comparable to reference population. Children with PKU had higher scores for physical and psychosocial functioning; adults with PKU had lower scores for cognitive functioning; no differences in (modified) DISABKIDS scores in BH4-responsive paediatric and adult patients before and after 1 year of BH4 treatment | 205 |
| Cazzorla et al. (2014) | PedsQL WHOQoL 100 |
26 children; 17 patients ≥17 years of age Italy Cross-sectional |
QOL comparable to reference population; HRQOL scores higher in BH4-treated patients than dietary treatment-only patients; HRQOL scores increased with age | 206 |
| Bosch et al. (2015) | PedsQL CHQ SF36 PKU-QoL |
202 children; 104 adults Seven countries Cross-sectional; validation of PKU-QOL questionnaire |
Overall HRQOL comparable to reference population; for PKU-QOL highest scores were for emotional impact, anxiety about blood Phe levels, especially during pregnancy, and guilt regarding poor treatment adherence; patients with mild to moderate PKU or those receiving BH4 treatment reported less impact of dietary treatment on HRQOL | 207 |
| Feldmann et al. (2017) | Ulm QoL Inventory for parents | 38 children Germany Baseline, prospective, before and after start of BH4 treatment |
HRQOL comparable to reference population; HRQOL did not improve with BH4 treatment compared with dietary treatment only | 208 |
| Huijbrechts et al. (2018) | TAPQoL TAAQoL |
32 children; 58 patients ≥16 years of age Netherlands Cross-sectional |
Results overall comparable to reference population; children lower scores on autonomy, adolescents and adults lower scores on domains cognition, depressive mood and anger; adults treated with BH4 had better scores on domains social functioning, happiness and anger | 209 |
| Cotugno et al. (2011) | CHQ SF36 |
32 children; 9 adults Italy Cross-sectional |
Overall CHQ scores and most domain scores lower in children; adherence correlated with global health and family activities; adults had normal scores on SF36 | 210 |
| Vieira Neto et al. (2017) | PedsQL | 49 children Brazil Cross-sectional |
Significantly lower HRQOL than in reference population; included children with cognitive disabilities, probably due to severe (financial) barriers to optimal treatment | 211 |
| Bik-Multanowski et al. (2008) | Psychological General Wellbeing Index |
53 adults Poland Prospective; evaluation of effect of restarting diet |
Without diet, 45% of patients reported moderate to severe distress; from 3 months after restarting diet, significantly improved scores in patients with distress, especially depression and anger | 212 |
| Ziesch et al. (2012) | KINDL-R | 19 children Germany Baseline, prospective; before and after start BH4 treatment |
Overall QOL comparable to reference population; higher scores for physical well-being compared with reference population; no improvement after start of BH4 treatment | 214 |
| Douglas et al. (2013) | PKU-QOL | 19 adolescents; 18 adults USA Prospective; before and after start of BH4 treatment |
No difference in overall HRQOL between responders and non-responders; significant improvement in life impact and life satisfaction scores in responders, associated with less strict diet | 245 |
BH4, tetrahydrobiopterin; HRQOL, health-related quality of life; Phe, phenylalanine; PKU, phenylketonuria; QOL, quality of life.
Age, disease severity and treatment
In one study, HRQOL scores improved with increasing age206. Another study found a negative association between lifetime Phe concentrations and scores on sleep, pain, anger and sexuality domains, while current Phe concentrations were negatively associated with sexuality domain scores209. However, the effects of dietary adherence on the HRQOL of adult patients may vary substantially among individuals212. In a group of non-adherent early-treated adult patients, 45% showed moderate to severe distress on the psychological well-being index. Three months after restarting the diet, these patients had significantly improved scores for distress, especially in the anxiety and depressive mood domains. Negative effects of Phe concentrations on mood, especially on depression and anger, were also demonstrated in a randomized, double-blind, placebo-controlled trial, even though the trial included only a small number of patients213.
HRQOL was studied prospectively at the time of the introduction of BH4 treatment in Europe and the USA. Three studies found no improvement in HRQOL between the baseline measurement (before BH4-responsiveness testing) and up to 1 year after the start of BH4 treatment in responsive patients205,208,214. One study found no overall HRQOL difference between responders and non-responders, but a significant improvement in life impact and life satisfaction scores in responders149.
In addition, no significant differences in HRQOL were found between patients treated with sapropterin dihydrochloride and those treated with diet only, but a trend for higher HRQOL scores, especially on social functioning, happiness and anger domains, was observed in sapropterin dihydrochloride-treated patients209. To date, no studies addressing the effects of treatment with pegvaliase on the HRQOL of patients with PKU have been published. An 8-week randomized, controlled trial did not demonstrate significant differences in mood between patients receiving pegvaliase treatment and those receiving placebo after 8 weeks of treatment215.
PKU-specific questionnaires
Even though overall HRQOL measured with generic questionnaires is mostly normal in patients with PKU, patients do struggle with adherence to treatment. Clearly, generic questionnaires do not sufficiently address the specific problems of patients with PKU. PKU-specific questionnaires have been developed to evaluate the impact of PKU and supportive and therapeutic interventions on HRQOL. In the USA, a PKU-specific PKU-QOL questionnaire was developed from a validated juvenile diabetes QOL questionnaire216.
Since 2015, the PKU-QOL questionnaire has been available in six languages217. A multicentre validation study included 306 patients and 253 parents from seven countries207. The most highly affected PKU-QOL scores were for the emotional impact of PKU and its management (such as anxiety about blood Phe concentrations and guilt regarding poor adherence) and the most highly affected overall impact score was anxiety regarding blood Phe concentrations during pregnancy in women. Furthermore, the impact of the taste of the amino acid supplements was high.
The impact of the severity of PAH deficiency and PKU treatment on HRQOL was evaluated and patients with severe PAH deficiency reported a greater impact of the protein-restricted diet and amino acid supplements on HRQOL and more guilt related to poor adherence than patients with less severe PAH deficiency. The impact of the amino acid supplement on HRQOL and the overall social and practical impact of dietary restriction was lower in sapropterin dihydrochloride-treated patients than in those treated with dietary control only207.
Psychosocial outcomes
The educational attainment, independent living and marital status and employment of a cohort of Dutch adults with PKU was completely normal compared with the general population202. In Germany, a normal educational attainment and career, but a lower or delayed autonomy and a low rate of adult relationships was reported in adults with PKU compared with the general population203.
Outlook
Emerging therapies
PKU was the first IMD for which dietary management was initiated and for which pharmacological therapies were developed. It is not surprising that PKU remains at centre stage for the development of new therapies for IMDs (TABLE 3).
Table 3 |.
Experimental therapies for PKU
| Therapy | Delivery | Mechanism of action | Dose |
|---|---|---|---|
| Gene correction | Systemic | Delivery of base-editing agents to correct variants in the PAH gene | One IV |
| Gene therapy | Systemic | HMI-102: provision of the normal PAH cDNA to hepatocytes (AAV, lentivirus or naked DNA) | One IV |
| mRNA therapy | Systemic | Provision of lipid nanoparticle-encapsulated PAH mRNA | IV, SQ; frequency TBD |
| Enzyme substitution | Systemic | RTX-134: Anabaena variabilis PAL expressed in universal donor red blood cells | IV; frequency TBD |
| Oral | SYNB1618: bacteria overexpressing PAL to metabolize phenylalanine in the gut | Oral; three times daily | |
| Oral | CDX-6114: PAL genetically modified to retain activity after oral administration to metabolize phenylalanine in the gut | Oral; three times daily | |
| Cofactor therapy | Oral | CNSA-001: sepiapterin, a precursor of tetrahydrobiopterin, to stimulate residual enzyme activity of mutant PAH | Oral; once daily |
AAV, adeno-associated virus; cDNA, complementary DNA; IV, intravenous, PAH, phenylalanine hydroxylase; PAL, phenylalanine ammonia lyase; SQ, subcutaneous, TBD, to be determined.
Gene and mRNA therapy.
The ideal therapy would restore PAH activity in the liver and provide a cure for PKU. Gene correction therapy using CRISPR–Cas-associated base editors, which enable nucleotide conversion independent of double-strand DNA break formation and homology-directed repair, was effective in providing sufficient PAH activity (>20% of normal) in Pahenu2 mice to restore physiological blood Phe concentrations218. The correction of PAH enzymatic activity improved with time and was not associated with unwanted DNA changes in genomic regions with homology to the guide RNA utilized. Gene correction in mice has also been achieved using adeno-associated viruses (AAVs) isolated from normal human haematopoietic stem cells that have undergone nuclease-free gene editing through the homologous recombination pathway219. The advantage of this technology is that the corrected gene could be passed to new liver cells, maintaining enzymatic activity even with hepatocyte proliferation. As in gene therapy, the machinery required for gene editing was delivered using AAV vectors, with gene editing limited to the liver by the use of a specific promoter. Although effective in mice, this approach is still far from clinical trials given the possibility of off-target genetic changes.
Gene therapy can also be used to add a functional PAH gene to the one inactivated by biallelic pathogenetic variants. The PAH cDNA is usually delivered using a viral vector. Initial trials had limited success, with a rise in Phe concentrations specifically in female Pahenu2 mice 40 weeks after dosing and lack of efficacy if the administration was not given into the portal vein220,221. Better results were obtained using AAV type 2 serotype 8 (AAV2/8) that is liver-tropic and can provide long-lasting correction of PAH activity in Pahenu2 mice, even when administered systemically at a lower dose222–226. The DNA remains episomal and usually does not integrate in the host genome, limiting the potential for insertional mutagenesis. However, the lack of genomic integration can result in loss of efficacy over time, especially in the case of hepatocyte proliferation. The viral vector also induces an immune response, preventing the possibility of re-administration. Pre-existing antibodies against the viral vector would also limit the population eligible to receive this therapy. Currently, two clinical trials of gene therapy in patients with PKU are ongoing: one using AAVHSC15 (NCT03952156) and the other using AAV2/8 (NCT04480567). No data are yet available on enrolled subjects. Lentiviral gene therapy would allow genomic integration of the PAH gene, providing a stable source of enzyme. There is no pre-existing immunity to lentiviral vectors and thus all patients could receive this therapy. However, insertional mutagenesis remains a possibility. No preclinical data using this approach have yet been published for PKU227.
An alternative to providing the PAH gene is provision of PAH mRNA enclosed within lipid nanoparticles, which would be taken up by the liver and result in the production of the enzyme within hepatocytes, thereby preventing an immune response. Animal studies have demonstrated the efficacy of this approach in methylmalonic acidaemia228, arginase and citrin deficiency229,230, acute intermittent porphyria231, Fabry disease232 and galactosaemia233. Unlike gene therapy, mRNA would need to be administered periodically (frequency still to be determined). No data in humans are yet available and the effectiveness in animal models of PKU has not been reported.
Enzyme substitution therapy.
Based on the approval of injectable pegvaliase for the treatment of PKU by US and European authorities, other approaches are trying to use the same enzyme. Pegvaliase can cause hypersensitivity reactions135. To decrease the frequency and severity of this adverse effect, the enzyme is being produced by red blood cells of a universal blood donor234,235. Mature red blood cells loaded with PAL are then transfused to patients with PKU (with a frequency yet to be determined) to reduce Phe concentrations. The red blood cell should shield the exogenous PAL from the immune system, yet allow the entry of Phe into the cell to be metabolized to trans-cinnamic acid and ammonia. Although no human data have been published, autologous erythrocytes loaded with PAL and administered weekly were effective in reducing Phe concentrations and preventing intellectual disability in Pahenu2 mice170. A clinical trial testing this approach in humans is planned.
Oral therapies.
Old and new approaches are being explored to reduce Phe concentrations using novel oral drugs. Sepiapterin is a natural precursor of BH4 with a higher capacity to enter cells than the current therapy, sapropterin. Oral administration of sepiapterin to healthy volunteers increased plasma BH4 concentrations more than sapropterin and doubled BH4 concentrations in the cerebrospinal fluid236,237. Given these results, more patients with PKU might respond to sepiapterin than to sapropterin and a better response could be seen in responsive patients.
PAL was initially developed as an oral compound to metabolize Phe in the enterorecirculation. SYNB1618, a modified version of the probiotic Escherichia coli Nissle strain, was engineered to overexpress PAL to target gut Phe238. Administration of this probiotic to healthy volunteers and individuals with PKU confirmed production of trans-cinnamic acid but there was no reduction in Phe concentrations at the highest dose used239. Future trials might explore either more effective or higher doses of the probiotic bacteria. A protease-stable PAL (CDX-6114) has also been proposed as an orally administered enzyme therapy. Trials in humans have so far shown safety in healthy volunteers (NCT03577886 and NCT04085666) and further studies in individuals with PKU are necessary.
Clinical benefits of new therapies
While concentrations of Phe clearly reflect metabolic control in PKU, the effects of metabolic changes on executive function, QOL and mood of adults with PKU are more difficult to assess. Although there are many studies that have found such effects, it is still unclear if a better tool needs to be developed, with more objective, rather than subjective measurements. The NIH toolbox might serve this purpose and allow similar measurements across all sites caring for individuals with PKU. This computerized, inexpensive and easy to use tool could represent a major advance in our capacity to monitor the efficacy of new therapies.
Acknowledgements
The authors thank D. Abeln for giving his thoughts from the perspective of a parent.
Competing interests
F.J.v.S. has been a member of scientific advisory boards for defects in amino acid metabolism of APR, Agios, Arla Food International, BioMarin, Eurocept Int, Lucana, Moderna TX, Nutricia, Rivium, Homoly and Nestlé-Codexis; his institute has received research grants from Alexion, Biomarin, Codexis, Nutricia, SoBi and Vitaflo; his institute has received grants from patient organizations ESPKU, Metakids, NPKUA, Stofwisselkracht, Stichting PKU research and the Tyrosinemia Foundation; and his institute has received honoraria as consultant and speaker from APR, Pluvia, Biomarin, MendeliKABS and Nutricia. N.B. has received honoraria and/or consulting fees from BioMarin, Pharmaceuticals, Censa, Nestlé Pharmaceuticals and Homology Medicines. C.H. has received consulting fees, speaker fees, and travel and research support from BioMarin, Cydan Development Inc., Dimension Therapeutics, Horizon Pharma, Pfizer, Rubius Therapeutics, StrideBio and Synlogic. A.B. has received advisory board honoraria, speaker fees and travel support from Biomarin Pharmaceuticals, Nutricia, Cambrooke, PIAM, APR, Sanofi Genzyme and Takeda. N.L. has received consulting fees from Aeglea, BioMarin, Censa Pharmaceuticals, Dimension Therapeutics, Genzyme/Sanofi, Hemoshear, Horizon, Lumos Pharma, Moderna, Mitobridge, Pfizer, Retrophin and Stealth Therapeutics, and has conducted contracted research for Aeglea, BioMarin, Genzyme/Sanofi, Horizon, Lumos Pharma, Protalix, Retrophin, Shire, Stealth Therapeutics and Ultragenyx. A.M.B. has received a speaker fee from Nutricia and has been a member of advisory boards for Biomarin.
References
- 1.Anikster Y et al. Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am. J. Hum. Genet 100, 257–266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blau N Sapropterin dihydrochloride for the treatment of hyperphenylalaninemias. Expert. Opin. Drug. Metab. Toxicol 9, 1207–1218 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Dhondt J-L Lessons from 30 years of selective screening for tetrahydrobiopterin deficiency. J. Inherit. Metab. Dis 33, S219–S223 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Straniero L et al. DNAJC12 and dopa-responsive nonprogressive parkinsonism. Ann. Neurol 82, 640–646 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Blau N, van Spronsen FJ & Levy HL Phenylketonuria. Lancet 376, 1417–1427 (2010). [DOI] [PubMed] [Google Scholar]
- 6.van Spronsen FJ et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J. Inherit. Metab. Dis 32, 27–31 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Güttler F & Hansen G Different phenotypes for phenylalanine hydroxylase deficiency. Ann. Clin. Biochem 14, 124–134 (1977). [DOI] [PubMed] [Google Scholar]
- 8.van Spronsen FJ et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 5, 743–756 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Følling IA Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Hoppe-Seyler’s Z. für Physiologische Chem 227, 169–181 (1934). [Google Scholar]
- 10.Penrose LS Inheritance of phenylpyruvic amentia (phenylketonuria). Lancet 226, 192–194 (1935). [Google Scholar]
- 11.Cowie VA An atypical case of phenylketonuria. Lancet 1, 272 (1951). [DOI] [PubMed] [Google Scholar]
- 12.Jervis GA Phenylpyruvic oligophrenia deficiency of phenylalanine-oxidizing system. Proc. Soc. Exp. Biol. Med 82, 514–515 (1953). [PubMed] [Google Scholar]
- 13.Jervis GA Studies on phenylpyruvic oligophrenia; the position of the metabolic error. J. Biol. Chem 169, 651–656 (1947). [PubMed] [Google Scholar]
- 14.Bickel H, Gerrard J & Hickmans EM Influence of phenylalanine intake on phenylketonuria. Lancet 265, 812–813 (1953). [DOI] [PubMed] [Google Scholar]
- 15.Wortis J & Giancotti AM A new simple test paper for mass detection of phenylketonuria. Am. J. Public Health Nations Health 49, 463–464 (1959). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dent C Discussion of Armstrong MD: Relation of biochemical abnormality to development of mental defect in phenylketonuria in etiological factors in mental retardation. In Report of 23rd Ross Conference, Columbus, Ohio (1957). [Google Scholar]
- 17.Guthrie R & Susi A A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32, 338–343 (1963). [PubMed] [Google Scholar]
- 18.Kaufman S The phenylalanine hydroxylating system from mammalian liver. Adv. Enzymol. Relat. Areas Mol. Biol 35, 245–319 (1971). [DOI] [PubMed] [Google Scholar]
- 19.Bartholome K Letter: A new molecular defect in phenylketonuria. Lancet 2, 1580 (1974). [DOI] [PubMed] [Google Scholar]
- 20.Hoskins JA et al. Enzymatic control of phenylalanine intake in phenylketonuria. Lancet 1, 392–394 (1980). [DOI] [PubMed] [Google Scholar]
- 21.Woo SL, Lidsky AS, Güttler F, Chandra T & Robson KJ Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. Nature 306, 151–155 (1983). [DOI] [PubMed] [Google Scholar]
- 22.Konecki DS, Wang Y, Trefz FK, Lichter-Konecki U & Woo SL Structural characterization of the 5′ regions of the human phenylalanine hydroxylase gene. Biochemistry 31, 8363–8368 (1992). [DOI] [PubMed] [Google Scholar]
- 23.Standing SJ & Taylor RP Phenylalanine: application of a simple HPLC technique to its measurement in dried blood spots. Ann. Clin. Biochem 29, 668–670 (1992). [DOI] [PubMed] [Google Scholar]
- 24.Kure S et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J. Pediatr 135, 375–378 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Longo N et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicentre, phase 1 dose-escalation trial. Lancet 384, 37–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walter JH et al. How practical are recommendations for dietary control in phenylketonuria? Lancet 360, 55–57 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Scriver CR The PAH gene, phenylketonuria, and a paradigm shift. Hum. Mutat 28, 831–845 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Hillert A et al. The genetic landscape and epidemiology of phenylketonuria. Am. J. Hum. Genet 107, 234–250 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desviat LR et al. Genetic and phenotypic aspects of phenylalanine hydroxylase deficiency in Spain: molecular survey by regions. Eur. J. Hum. Genet 7, 386–392 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Ozalp I, Coskun T, Tokol S, Demircin G & Mönch E Inherited metabolic disorders in Turkey. J. Inherit. Metab. Dis 13, 732–738 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Senemar S, Ganjekarimi A, Senemar S, Tarami B & Bazrgar M The prevalence and clinical study of galactosemia disease in a pilot screening program of neonates, southern Iran. Iran. J. Public. Health 40, 99–104 (2011). [PMC free article] [PubMed] [Google Scholar]
- 32.Gundorova P et al. Molecular-genetic causes for the high frequency of phenylketonuria in the population from the North Caucasus. PLoS ONE 13, e0201489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sutivijit Y, Banpavichit A & Wiwanitkit V Prevalence of neonatal hypothyroidism and phenylketonuria in Southern Thailand: a 10-year report. Indian. J. Endocrinol. Metab 15, 115–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okano Y, Kudo S, Nishi Y, Sakaguchi T & Aso K Molecular characterization of phenylketonuria and tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Japan. J. Hum. Genet 56, 306–312 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Borrajo GJC Newborn screening in Latin America at the beginning of the 21st century. J. Inherit. Metab. Dis 30, 466–481 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Hitzeroth HW, Niehaus CE & Brill DC Phenylketonuria in South Africa. A report on the status quo. S Afr. Med. J 85, 33–36 (1995). [PubMed] [Google Scholar]
- 37.Hardelid P et al. The birth prevalence of PKU in populations of European, South Asian and sub-Saharan African ancestry living in South East England. Ann. Hum. Genet 72, 65–71 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Opladen T, Hoffmann GF, Kühn AA & Blau N Pitfalls in phenylalanine loading test in the diagnosis of dopa-responsive dystonia. Mol. Genet. Metab 108, 195–197 (2013). [DOI] [PubMed] [Google Scholar]
- 39.van Spronsen FJ et al. Heterogeneous clinical spectrum of DNAJC12-deficient hyperphenylalaninemia: from attention deficit to severe dystonia and intellectual disability. J. Med. Genet 55, 249–253 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Garbade SF et al. Allelic phenotype values: a model for genotype-based phenotype prediction in phenylketonuria. Genet. Med 21, 580–590 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Eisensmith RC et al. Multiple origins for phenylketonuria in Europe. Am. J. Hum. Genet 51, 1355–1365 (1992). [PMC free article] [PubMed] [Google Scholar]
- 42.Waters PJ, Parniak MA, Nowacki P & Scriver CR In vitro expression analysis of mutations in phenylalanine hydroxylase: linking genotype to phenotype and structure to function. Hum. Mutat 11, 4–17 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Shen N et al. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype–phenotype correlation. Mol. Genet. Metab 117, 328–335 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Himmelreich N et al. Relationship between genotype, phenylalanine hydroxylase expression and in vitro activity and metabolic phenotype in phenylketonuria. Mol. Genet. Metab 125, 86–95 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Blau N & Erlandsen H The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol. Genet. Metab 82, 101–111 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Shedlovsky A, McDonald JD, Symula D & Dove WF Mouse models of human phenylketonuria. Genetics 134, 1205–1210 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gersting SW et al. Pahenu1 is a mouse model for tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency and promotes analysis of the pharmacological chaperone mechanism in vivo. Hum. Mol. Genet 19, 2039–2049 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Levy HL et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 370, 504–510 (2007). [DOI] [PubMed] [Google Scholar]
- 49.McDonald JD, Bode VC, Dove WF & Shedlovsky A Pahhph-5: a mouse mutant deficient in phenylalanine hydroxylase. Proc. Natl Acad. Sci. USA 87, 1965–1967 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saugstad LF Birthweights in children with phenylketonuria and in their siblings. Lancet 1, 809–813 (1972). [DOI] [PubMed] [Google Scholar]
- 51.Verkerk PH, van Spronsen FJ, Smit GP & Sengers RC Impaired prenatal and postnatal growth in Dutch patients with phenylketonuria. The National PKU Steering Committee. Arch. Dis. Child 71, 114–118 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ilgaz F et al. Long-term growth in phenylketonuria: a systematic review and meta-analysis. Nutrients 11, 2070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayling JE, Helfand GD & Pirson WD Phenylalanine hydroxylase from human kidney. Enzyme 20, 6–19 (1975). [DOI] [PubMed] [Google Scholar]
- 54.Huttenlocher PR The neuropathology of phenylketonuria: human and animal studies. Eur. J. Pediatr 159 (Suppl 2), 102–106 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Bauman ML & Kemper TL Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol. 58, 55–63 (1982). [DOI] [PubMed] [Google Scholar]
- 56.Horling K et al. Hippocampal synaptic connectivity in phenylketonuria. Hum. Mol. Genet 24, 1007–1018 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Surtees R & Blau N The neurochemistry of phenylketonuria. Eur. J. Pediatr 159 (Suppl 2), 109–113 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Jervis GA Studies on phenylpyruvic oligophrenia; phenylpyruvic acid content on blood. Proc. Soc. Exp. Biol. Med 81, 715–720 (1952). [DOI] [PubMed] [Google Scholar]
- 59.Batshaw ML, Valle D & Bessman SP Unsuccessful treatment of phenylketonuria with tyrosine. J. Pediatr 99, 159–160 (1981). [DOI] [PubMed] [Google Scholar]
- 60.Azen CG et al. Intellectual development in 12-year-old children treated for phenylketonuria. Am. J. Dis. Child 145, 35–39 (1991). [DOI] [PubMed] [Google Scholar]
- 61.van Vliet D et al. Can untreated PKU patients escape from intellectual disability? A systematic review. Orphanet J. Rare Dis 13, 149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hörster F et al. Phenylalanine reduces synaptic density in mixed cortical cultures from mice. Pediatr. Res 59, 544–548 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Hartwig C et al. Elevated phenylalanine levels interfere with neurite outgrowth stimulated by the neuronal cell adhesion molecule L1 in vitro. FEBS Lett. 580, 3489–3492 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Andolina D et al. 5-Hydroxytryptophan during critical postnatal period improves cognitive performances and promotes dendritic spine maturation in genetic mouse model of phenylketonuria. Int. J. Neuropsychopharmacol 14, 479–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schlegel G, Scholz R, Ullrich K, Santer R & Rune GM Phenylketonuria: direct and indirect effects of phenylalanine. Exp. Neurol 281, 28–36 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Christ SE et al. Morphometric analysis of gray matter integrity in individuals with early-treated phenylketonuria. Mol. Genet. Metab 118, 3–8 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Pilotto A et al. Cerebrospinal fluid biogenic amines depletion and brain atrophy in adult patients with phenylketonuria. J. Inherit. Metab. Dis 42, 398–406 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Dyer CA et al. Evidence for central nervous system glial cell plasticity in phenylketonuria. J. Neuropathol. Exp. Neurol 55, 795–814 (1996). [DOI] [PubMed] [Google Scholar]
- 69.Schoemans R et al. Oligodendrocyte development and myelinogenesis are not impaired by high concentrations of phenylalanine or its metabolites. J. Inherit. Metab. Dis 33, 113–120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shefer S et al. Is there a relationship between 3-hydroxy-3-methylglutaryl coenzyme a reductase activity and forebrain pathology in the PKU mouse? J. Neurosci. Res 61, 549–563 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Qin M & Smith CB Regionally selective decreases in cerebral glucose metabolism in a mouse model of phenylketonuria. J. Inherit. Metab. Dis 30, 318–325 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Winn SR, Scherer T, Thöny B & Harding CO High dose sapropterin dihydrochloride therapy improves monoamine neurotransmitter turnover in murine phenylketonuria (PKU). Mol. Genet. Metab 117, 5–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cabib S, Pascucci T, Ventura R, Romano V & Puglisi-Allegra S The behavioral profile of severe mental retardation in a genetic mouse model of phenylketonuria. Behav. Genet 33, 301–310 (2003). [DOI] [PubMed] [Google Scholar]
- 74.Zagreda L, Goodman J, Druin DP, McDonald D & Diamond A Cognitive deficits in a genetic mouse model of the most common biochemical cause of human mental retardation. J. Neurosci 19, 6175–6182 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hasselbalch S et al. Cerebral glucose metabolism is decreased in white matter changes in patients with phenylketonuria. Pediatr. Res 40, 21–24 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Ficicioglu C et al. A pilot study of fluorodeoxyglucose positron emission tomography findings in patients with phenylketonuria before and during sapropterin supplementation. J. Clin. Neurol 9, 151–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wasserstein MP, Snyderman SE, Sansaricq C & Buchsbaum MS Cerebral glucose metabolism in adults with early treated classic phenylketonuria. Mol. Genet. Metab 87, 272–277 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Miller AL, Hawkins RA & Veech RL Phenylketonuria: phenylalanine inhibits brain pyruvate kinase in vivo. Science 179, 904–906 (1973). [DOI] [PubMed] [Google Scholar]
- 79.Choi TB & Pardridge WM Phenylalanine transport at the human blood–brain barrier. Studies with isolated human brain capillaries. J. Biol. Chem 261, 6536–6541 (1986). [PubMed] [Google Scholar]
- 80.Kanai Y et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem 273, 23629–23632 (1998). [DOI] [PubMed] [Google Scholar]
- 81.de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ & van Spronsen FJ Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol. Genet. Metab 99 (Suppl 1), 86–89 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Hoeksma M et al. Phenylketonuria: high plasma phenylalanine decreases cerebral protein synthesis. Mol. Genet. Metab 96, 177–182 (2009). [DOI] [PubMed] [Google Scholar]
- 83.de Groot MJ et al. Phenylketonuria: reduced tyrosine brain influx relates to reduced cerebral protein synthesis. Orphanet J. Rare Dis 8, 133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Vliet D et al. Large neutral amino acid supplementation exerts its effect through three synergistic mechanisms: proof of principle in phenylketonuria mice. PLoS One 10, e0143833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pietz J et al. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J. Clin. Invest 103, 1169–1178 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koch R, Moseley KD, Yano S, Nelson M & Moats RA Large neutral amino acid therapy and phenylketonuria: a promising approach to treatment. Mol. Genet. Metab 79, 110–113 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Schindeler S et al. The effects of large neutral amino acid supplements in PKU: an MRS and neuropsychological study. Mol. Genet. Metab 91, 48–54 (2007). [DOI] [PubMed] [Google Scholar]
- 88.van Vliet D et al. Therapeutic brain modulation with targeted large neutral amino acid supplements in the Pah-enu2 phenylketonuria mouse model. Am. J. Clin. Nutr 104, 1292–1300 (2016). [DOI] [PubMed] [Google Scholar]
- 89.O’Kane RL & Hawkins RA Na+-dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood–brain barrier. Am. J. Physiol. Endocrinol. Metab 285, E1167–E1173 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Hawkins RA, O’Kane RL, Simpson IA & Viña JR Structure of the blood–brain barrier and its role in the transport of amino acids. J. Nutr 136, 218S–226SS (2006). [DOI] [PubMed] [Google Scholar]
- 91.Wegrzyn A In silico patient: systems medicine approach to inborn errors of metabolism. Thesis, Univ. Groningen: (2020). [Google Scholar]
- 92.Pare CM, Sandler M & Stacey RS 5-Hydroxytryptamine deficiency in phenylketonuria. Lancet 272, 551–553 (1957). [DOI] [PubMed] [Google Scholar]
- 93.McKean CM The effects of high phenylalanine concentrations on serotonin and catecholamine metabolism in the human brain. Brain Res. 47, 469–476 (1972). [DOI] [PubMed] [Google Scholar]
- 94.Harding CO et al. Pharmacologic inhibition of L-tyrosine degradation ameliorates cerebral dopamine deficiency in murine phenylketonuria (PKU). J. Inherit. Metab. Dis 37, 735–743 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Puglisi-Allegra S et al. Dramatic brain aminergic deficit in a genetic mouse model of phenylketonuria. Neuroreport 11, 1361–1364 (2000). [DOI] [PubMed] [Google Scholar]
- 96.Pascucci T, Ventura R, Puglisi-Allegra S & Cabib S Deficits in brain serotonin synthesis in a genetic mouse model of phenylketonuria. Neuroreport 13, 2561–2564 (2002). [DOI] [PubMed] [Google Scholar]
- 97.Pascucci T et al. 5-Hydroxytryptophan rescues serotonin response to stress in prefrontal cortex of hyperphenylalaninaemic mice. Int. J. Neuropsychopharmacol 12, 1067–1079 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Pascucci T et al. In vivo catecholaminergic metabolism in the medial prefrontal cortex of ENU2 mice: an investigation of the cortical dopamine deficit in phenylketonuria. J. Inherit. Metab. Dis 35, 1001–1009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Vliet D et al. Large neutral amino acid supplementation as an alternative to the phenylalanine-restricted diet in adults with phenylketonuria: evidence from adult Pah-enu2 mice. J. Nutr. Biochem 53, 20–27 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Christ SE, Huijbregts SCJ, de Sonneville LMJ & White DA Executive function in early-treated phenylketonuria: profile and underlying mechanisms. Mol. Genet. Metab 99 (Suppl 1), 22–32 (2010). [DOI] [PubMed] [Google Scholar]
- 101.Adler-Abramovich L et al. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol 8, 701–706 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Dobrowolski SF et al. Altered DNA methylation in PAH deficient phenylketonuria. Mol. Genet. Metab 115, 72–77 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Dobrowolski SF et al. DNA methylation in the pathophysiology of hyperphenylalaninemia in the PAH(enu2) mouse model of phenylketonuria. Mol. Genet. Metab 119, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ercal N, Aykin-Burns N, Gurer-Orhan H & McDonald JD Oxidative stress in a phenylketonuria animal model. Free Radic. Biol. Med 32, 906–911 (2002). [DOI] [PubMed] [Google Scholar]
- 105.van der Goot E et al. Hippocampal microglia modifications in C57Bl/6 Pahenu2 and BTBR Pahenu2 phenylketonuria (PKU) mice depend on the genetic background, irrespective of disturbed sleep patterns. Neurobiol. Learn. Mem 160, 139–143 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Thompson AJ et al. Neurological deterioration in young adults with phenylketonuria. Lancet 336, 602–605 (1990). [DOI] [PubMed] [Google Scholar]
- 107.Rubin S et al. Sight-threatening phenylketonuric encephalopathy in a young adult, reversed by diet. JIMD Rep. 10, 83–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jaulent P et al. Neurological manifestations in adults with phenylketonuria: new cases and review of the literature. J. Neurol 267, 531–542 (2020). [DOI] [PubMed] [Google Scholar]
- 109.Rosini F, Rufa A, Monti L, Tirelli L & Federico A Adult-onset phenylketonuria revealed by acute reversible dementia, prosopagnosia and parkinsonism. J. Neurol 261, 2446–2448 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Arnold GL, Vladutiu CJ, Orlowski CC, Blakely EM & DeLuca J Prevalence of stimulant use for attentional dysfunction in children with phenylketonuria. J. Inherit. Metab. Dis 27, 137–143 (2004). [DOI] [PubMed] [Google Scholar]
- 111.Bilder DA et al. Systematic review and meta-analysis of neuropsychiatric symptoms and executive functioning in adults with phenylketonuria. Dev. Neuropsychol 41, 245–260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jahja R et al. Long-term follow-up of cognition and mental health in adult phenylketonuria: a PKU-COBESO study. Behav. Genet 47, 486–497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Wegberg AMJ et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J. Rare Dis 12, 162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Anjema K et al. The neonatal tetrahydrobiopterin loading test in phenylketonuria: what is the predictive value? Orphanet J. Rare Dis 11, 10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Evers RAF et al. The first European guidelines on phenylketonuria: usefulness and implications for BH4 responsiveness testing. J. Inherit. Metab. Dis 43, 244–250 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Blau N & van Spronsen FJ in Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases (eds Blau N, Duran M, Gibson KM & Vici CD) 3–21 (Springer, 2014). [Google Scholar]
- 117.Kornguth S, Gilbert-Barness E, Langer E & Hegstrand L Golgi-Kopsch silver study of the brain of a patient with untreated phenylketonuria, seizures, and cortical blindness. Am. J. Med. Genet 44, 443–448 (1992). [DOI] [PubMed] [Google Scholar]
- 118.Leuzzi V et al. Subclinical visual impairment in phenylketonuria. A neurophysiological study (VEP-P) with clinical, biochemical, and neuroradiological (MRI) correlations. J. Inherit. Metab. Dis 21, 351–364 (1998). [DOI] [PubMed] [Google Scholar]
- 119.MacDonald A et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J. Rare Dis 15, 171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Knox WE An evaluation of the treatment of phenylketonuria with diets low in phenylalanine. Pediatrics 26, 1–11 (1960). [PubMed] [Google Scholar]
- 121.Rouse BM Phenylalanine deficiency syndrome. J. Pediatr 69, 246–249 (1966). [DOI] [PubMed] [Google Scholar]
- 122.Hanley WB, Linsao L, Davidson W & Moes CA Malnutrition with early treatment of phenylketonuria. Pediatr. Res 4, 318–327 (1970). [DOI] [PubMed] [Google Scholar]
- 123.Pode-Shakked B et al. Man made disease: clinical manifestations of low phenylalanine levels in an inadequately treated phenylketonuria patient and mouse study. Mol. Genet. Metab 110 (Suppl), 66–70 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Daly A, Evans S, Pinto A, Ashmore C & MacDonald A Protein substitutes in PKU; their historical evolution. Nutrients 13, 484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sawin EA et al. Glycomacropeptide is a prebiotic that reduces Desulfovibrio bacteria, increases cecal short-chain fatty acids, and is anti-inflammatory in mice. Am. J. Physiol. Gastrointest. Liver Physiol 309, G590–G601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pinto A et al. Nutritional status in patients with phenylketonuria using glycomacropeptide as their major protein source. Eur. J. Clin. Nutr 71, 1230–1234 (2017). [DOI] [PubMed] [Google Scholar]
- 127.Gropper SS, Gropper DM & Acosta PB Plasma amino acid response to ingestion of L-amino acids and whole protein. J. Pediatr. Gastroenterol. Nutr 16, 143–150 (1993). [DOI] [PubMed] [Google Scholar]
- 128.MacDonald A, Ashmore C, Daly A, Pinto A & Evans S An observational study evaluating the introduction of a prolonged-release protein substitute to the dietary management of children with phenylketonuria. Nutrients 12, 2686 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Keil S et al. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics 131, e1881–e1888 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Muntau AC et al. Efficacy, safety and population pharmacokinetics of sapropterin in PKU patients <4 years: results from the SPARK open-label, multicentre, randomized phase IIIb trial. Orphanet J. Rare Dis 12, 47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burton BK et al. Pegvaliase for the treatment of phenylketonuria: results of the phase 2 dose-finding studies with long-term follow-up. Mol. Genet. Metab 130, 239–246 (2020). [DOI] [PubMed] [Google Scholar]
- 132.Levy HL, Sarkissian CN & Scriver CR Phenylalanine ammonia lyase (PAL): from discovery to enzyme substitution therapy for phenylketonuria. Mol. Genet. Metab 124, 223–229 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Sarkissian CN et al. A different approach to treatment of phenylketonuria: phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc. Natl Acad. Sci. USA 96, 2339–2344 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gámez A et al. Development of pegylated forms of recombinant Rhodosporidium toruloides phenylalanine ammonia-lyase for the treatment of classical phenylketonuria. Mol. Ther 11, 986–989 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Gupta S et al. Association of immune response with efficacy and safety outcomes in adults with phenylketonuria administered pegvaliase in phase 3 clinical trials. EBioMedicine 37, 366–373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Longo N et al. Evidence- and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet. Med 21, 1851–1867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang TM, Bourget L & Lister C A new theory of enterorecirculation of amino acids and its use for depleting unwanted amino acids using oral enzyme-artificial cells, as in removing phenylalanine in phenylketonuria. Artif. Cell Blood Substit. Immobil. Biotechnol 23, 1–21 (1995). [DOI] [PubMed] [Google Scholar]
- 138.Fisch RO, Jenness R, Doeden D & Anderson JA The effect of excess L-phenylalamine on mothers and on their breast-fed infants. J. Pediatr 71, 176–180 (1967). [DOI] [PubMed] [Google Scholar]
- 139.Oseid B Breast-feeding and infant health. Semin. Perinatol 3, 249–254 (1979). [PubMed] [Google Scholar]
- 140.McCabe ER & McCabe L Issues in the dietary management of phenylketonuria: breast-feeding and trace-metal nutriture. Ann. N. Y. Acad. Sci 477, 215–222 (1986). [DOI] [PubMed] [Google Scholar]
- 141.Rocha JC & MacDonald A Dietary intervention in the management of phenylketonuria: current perspectives. Pediatr. Health Med. Ther 7, 155–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Rijn M et al. A different approach to breast-feeding of the infant with phenylketonuria. Eur. J. Pediatr 162, 323–326 (2003). [DOI] [PubMed] [Google Scholar]
- 143.Weinmann A et al. Tetrahydrobiopterin is present in high quantity in human milk and has a vasorelaxing effect on newborn rat mesenteric arteries. Pediatr. Res 69, 325–329 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Bekhof J et al. Influence of knowledge of the disease on metabolic control in phenylketonuria. Eur. J. Pediatr 162, 440–442 (2003). [DOI] [PubMed] [Google Scholar]
- 145.Ozel HG et al. Does maternal knowledge impact blood phenylalanine concentration in Turkish children with phenylketonuria? J. Inherit. Metab. Dis 31 (Suppl 2), 213–217 (2008). [DOI] [PubMed] [Google Scholar]
- 146.Macdonald A et al. Does maternal knowledge and parent education affect blood phenylalanine control in phenylketonuria? J. Hum. Nutr. Diet 21, 351–358 (2008). [DOI] [PubMed] [Google Scholar]
- 147.Durham-Shearer SJ, Judd PA, Whelan K & Thomas JE Knowledge, compliance and serum phenylalanine concentrations in adolescents and adults with phenylketonuria and the effect of a patient-focused educational resource. J. Hum. Nutr. Diet 21, 474–485 (2008). [DOI] [PubMed] [Google Scholar]
- 148.Crone MR et al. Behavioural factors related to metabolic control in patients with phenylketonuria. J. Inherit. Metab. Dis 28, 627–637 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Bilginsoy C, Waitzman N, Leonard CO & Ernst SL Living with phenylketonuria: perspectives of patients and their families. J. Inherit. Metab. Dis 28, 639–649 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Rudolf I et al. Assessment of a mobile app by adolescents and young adults with cystic fibrosis: pilot evaluation. JMIR Mhealth Uhealth 7, e12442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jahja R, Huijbregts SCJ, de Sonneville LMJ, van der Meere JJ & van Spronsen FJ Neurocognitive evidence for revision of treatment targets and guidelines for phenylketonuria. J. Pediatr 164, 895–899.e2 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Feldmann R et al. Neurocognitive functioning in adults with phenylketonuria: report of a 10-year follow-up. Mol. Genet. Metab 126, 246–249 (2019). [DOI] [PubMed] [Google Scholar]
- 153.Leuzzi V, Chiarotti F, Nardecchia F, van Vliet D & van Spronsen FJ Predictability and inconsistencies of cognitive outcome in patients with phenylketonuria and personalised therapy: the challenge for the future guidelines. J. Med. Genet 57, 145–150 (2020). [DOI] [PubMed] [Google Scholar]
- 154.van Vliet D et al. Untreated PKU patients without intellectual disability: what do they teach us? Nutrients 11, 2572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Burlina AP et al. The neurological and psychological phenotype of adult patients with early-treated phenylketonuria: a systematic review. J. Inherit. Metab. Dis 42, 209–219 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Klimek A et al. Everyday life, dietary practices, and health conditions of adult PKU patients: a multicenter, cross-sectional study. Ann. Nutr. Metab 76, 251–258 (2020). [DOI] [PubMed] [Google Scholar]
- 157.Burgard P et al. Issues with European guidelines for phenylketonuria. Lancet Diabetes Endocrinol. 5, 681–683 (2017). [DOI] [PubMed] [Google Scholar]
- 158.van Spronsen FJ et al. Issues with European guidelines for phenylketonuria – Authors’ reply. Lancet Diabetes Endocrinol. 5, 683–684 (2017). [DOI] [PubMed] [Google Scholar]
- 159.De Felice S, Romani C, Geberhiwot T, MacDonald A & Palermo L Language processing and executive functions in early treated adults with phenylketonuria (PKU). Cogn. Neuropsychol 35, 148–170 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Palermo L et al. Cognitive outcomes in early-treated adults with phenylketonuria (PKU): A comprehensive picture across domains. Neuropsychology 31, 255–267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Palermo L et al. Emotional health in early-treated adults with phenylketonuria (PKU): Relationship with cognitive abilities and blood phenylalanine. J. Clin. Exp. Neuropsychol 42, 142–159 (2020). [DOI] [PubMed] [Google Scholar]
- 162.Romani C, MacDonald A, De Felice S & Palermo L Speed of processing and executive functions in adults with phenylketonuria: quick in finding the word, but not the ladybird. Cogn. Neuropsychol 35, 171–198 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Romani C et al. Adult cognitive outcomes in phenylketonuria: explaining causes of variability beyond average Phe levels. Orphanet J. Rare Dis 14, 273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Romani C et al. Cognitive outcomes and relationships with phenylalanine in phenylketonuria: a comparison between Italian and English adult samples. Nutrients 12, 3033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Romani C et al. The impact of phenylalanine levels on cognitive outcomes in adults with phenylketonuria: effects across tasks and developmental stages. Neuropsychology 31, 242–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Manti F et al. Psychiatric disorders in adolescent and young adult patients with phenylketonuria. Mol. Genet. Metab 117, 12–18 (2016). [DOI] [PubMed] [Google Scholar]
- 167.Nardecchia F, Manti F, De Leo S, Carducci C & Leuzzi V Clinical characterization of tremor in patients with phenylketonuria. Mol. Genet. Metab 128, 53–56 (2019). [DOI] [PubMed] [Google Scholar]
- 168.Nardecchia F et al. Neurocognitive and neuroimaging outcome of early treated young adult PKU patients: a longitudinal study. Mol. Genet. Metab 115, 84–90 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Scala I et al. Large neutral amino acids (LNAAs) supplementation improves neuropsychological performances in adult patients with phenylketonuria. Nutrients 12, 1092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Weglage J et al. Neurocognitive functioning in adults with phenylketonuria: results of a long term study. Mol. Genet. Metab 110 (Suppl), 44–48 (2013). [DOI] [PubMed] [Google Scholar]
- 171.Bartus A et al. The influence of blood phenylalanine levels on neurocognitive function in adult PKU patients. Metab. Brain Dis 33, 1609–1615 (2018). [DOI] [PubMed] [Google Scholar]
- 172.Christ SE et al. Executive function in phenylketonuria (PKU): insights from the Behavior Rating Inventory of Executive Function (BRIEF) and a large sample of individuals with PKU. Neuropsychology 34, 456–466 (2020). [DOI] [PubMed] [Google Scholar]
- 173.Hellewell SC, Welton T, Eisenhuth K, Tchan MC & Grieve SM Diffusion kurtosis imaging detects subclinical white matter abnormalities in phenylketonuria. Neuroimage Clin. 29, 102555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jahja R et al. Cognitive profile and mental health in adult phenylketonuria: a PKU-COBESO study. Neuropsychology 31, 437–447 (2017). [DOI] [PubMed] [Google Scholar]
- 175.Sundermann B et al. Approaching altered inhibitory control in phenylketonuria: a functional MRI study with a Go-NoGo task in young female adults. Eur. J. Neurosci 52, 3951–3962 (2020). [DOI] [PubMed] [Google Scholar]
- 176.Bessman SP PKU–some skepticism. N. Engl. J. Med 278, 1176–1177 (1968). [DOI] [PubMed] [Google Scholar]
- 177.Bessman SP Historical perspective: tyrosine and maternal phenylketonuria, welcome news. Am. J. Clin. Nutr 67, 357–358 (1998). [DOI] [PubMed] [Google Scholar]
- 178.Bessman SP The justification theory: the essential nature of the non-essential amino acids. Nutr. Rev 37, 209–220 (1979). [DOI] [PubMed] [Google Scholar]
- 179.Cabalska B et al. Termination of dietary treatment in phenylketonuria. Eur. J. Pediatr 126, 253–262 (1977). [DOI] [PubMed] [Google Scholar]
- 180.Poustie VJ & Wildgoose J Dietary interventions for phenylketonuria. Cochrane Database Syst. Rev 1, CD001304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Waisbren SE, Schnell RR & Levy HL Diet termination in children with phenylketonuria: a review of psychological assessments used to determine outcome. J. Inherit. Metab. Dis 3, 149–153 (1980). [DOI] [PubMed] [Google Scholar]
- 182.Diamond A Phenylalanine levels of 6–10mg/dl may not be as benign as once thought. Acta Paediatr. 407, 89–91 (1994). [DOI] [PubMed] [Google Scholar]
- 183.Waisbren SE et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol. Genet. Metab 92, 63–70 (2007). [DOI] [PubMed] [Google Scholar]
- 184.Lammardo AM et al. Main issues in micronutrient supplementation in phenylketonuria. Mol. Genet. Metab 110 (Suppl), 1–5 (2013). [DOI] [PubMed] [Google Scholar]
- 185.Mabry CC, Denniston JC, Nelson TL & Son CD Maternal phenylketonuria. A cause of mental retardation in children without the metabolic defect. N. Engl. J. Med 269, 1404–1408 (1963). [DOI] [PubMed] [Google Scholar]
- 186.Lenke RR & Levy HL Maternal phenylketonuria and hyperphenylalaninemia. An international survey of the outcome of untreated and treated pregnancies. N. Engl. J. Med 303, 1202–1208 (1980). [DOI] [PubMed] [Google Scholar]
- 187.Drogari E, Smith I, Beasley M & Lloyd JK Timing of strict diet in relation to fetal damage in maternal phenylketonuria. An international collaborative study by the MRC/DHSS Phenylketonuria Register. Lancet 2, 927–930 (1987). [DOI] [PubMed] [Google Scholar]
- 188.Maillot F, Lilburn M, Baudin J, Morley DW & Lee PJ Factors influencing outcomes in the offspring of mothers with phenylketonuria during pregnancy: the importance of variation in maternal blood phenylalanine. Am. J. Clin. Nutr 88, 700–705 (2008). [DOI] [PubMed] [Google Scholar]
- 189.Koch R et al. The maternal phenylketonuria international study: 1984–2002. Pediatrics 112, 1523–1529 (2003). [PubMed] [Google Scholar]
- 190.Grange DK et al. Sapropterin dihydrochloride use in pregnant women with phenylketonuria: an interim report of the PKU MOMS sub-registry. Mol. Genet. Metab 112, 9–16 (2014). [DOI] [PubMed] [Google Scholar]
- 191.Feillet F et al. Use of sapropterin dihydrochloride in maternal phenylketonuria. A European experience of eight cases. J. Inherit. Metab. Dis 37, 753–762 (2014). [DOI] [PubMed] [Google Scholar]
- 192.Teissier R et al. Maternal phenylketonuria: low phenylalaninemia might increase the risk of intra uterine growth retardation. J. Inherit. Metab. Dis 35, 993–999 (2012). [DOI] [PubMed] [Google Scholar]
- 193.Dhondt JL, Loeber J, Elvers LH & Paux E Preparation of the first European working standard for phenylalanine determination in dried blood spots. J. Med. Screen 5, 63–66 (1998). [DOI] [PubMed] [Google Scholar]
- 194.Gregory CO, Yu C & Singh RH Blood phenylalanine monitoring for dietary compliance among patients with phenylketonuria: comparison of methods. Genet. Med 9, 761–765 (2007). [DOI] [PubMed] [Google Scholar]
- 195.Groselj U et al. Comparison of tandem mass spectrometry and amino acid analyzer for phenylalanine and tyrosine monitoring–implications for clinical management of patients with hyperphenylalaninemia. Clin. Biochem 48, 14–18 (2015). [DOI] [PubMed] [Google Scholar]
- 196.Holub M et al. Influence of hematocrit and localisation of punch in dried blood spots on levels of amino acids and acylcarnitines measured by tandem mass spectrometry. Clin. Chim. Acta 373, 27–31 (2006). [DOI] [PubMed] [Google Scholar]
- 197.Lawson AJ, Bernstone L & Hall SK Newborn screening blood spot analysis in the UK: influence of spot size, punch location and haematocrit. J. Med. Screen 23, 7–16 (2016). [DOI] [PubMed] [Google Scholar]
- 198.van Vliet K et al. Dried blood spot versus venous blood sampling for phenylalanine and tyrosine. Orphanet J. Rare Dis 15, 82 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Koch R et al. Long-term beneficial effects of the phenylalanine-restricted diet in late-diagnosed individuals with phenylketonuria. Mol. Genet. Metab 67, 148–155 (1999). [DOI] [PubMed] [Google Scholar]
- 200.Singh RH et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet. Med 16, 121–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vockley J et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med 16, 188–200 (2014). [DOI] [PubMed] [Google Scholar]
- 202.Bosch AM et al. The course of life and quality of life of early and continuously treated Dutch patients with phenylketonuria. J. Inherit. Metab. Dis 30, 29–34 (2007). [DOI] [PubMed] [Google Scholar]
- 203.Simon E et al. Evaluation of quality of life and description of the sociodemographic state in adolescent and young adult patients with phenylketonuria (PKU). Health Qual. Life Outcomes 6, 25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Thimm E, Schmidt LE, Heldt K & Spiekerkoetter U Health-related quality of life in children and adolescents with phenylketonuria: unimpaired HRQoL in patients but feared school failure in parents. J. Inherit. Metab. Dis 36, 767–772 (2013). [DOI] [PubMed] [Google Scholar]
- 205.Demirdas S et al. Evaluation of quality of life in PKU before and after introducing tetrahydrobiopterin (BH4); a prospective multi-center cohort study. Mol. Genet. Metab 110 (Suppl), 49–56 (2013). [DOI] [PubMed] [Google Scholar]
- 206.Cazzorla C et al. Quality of life (QoL) assessment in a cohort of patients with phenylketonuria. BMC Public. Health 14, 1243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bosch AM et al. Assessment of the impact of phenylketonuria and its treatment on quality of life of patients and parents from seven European countries. Orphanet J. Rare Dis 10, 80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Feldmann R, Wolfgart E, Weglage J & Rutsch F Sapropterin treatment does not enhance the health-related quality of life of patients with phenylketonuria and their parents. Acta Paediatr. 106, 953–959 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Huijbregts SCJ et al. The impact of metabolic control and tetrahydrobiopterin treatment on health related quality of life of patients with early-treated phenylketonuria: a PKU-COBESO study. Mol. Genet. Metab 125, 96–103 (2018). [DOI] [PubMed] [Google Scholar]
- 210.Cotugno G et al. Adherence to diet and quality of life in patients with phenylketonuria. Acta Paediatr. 100, 1144–1149 (2011). [DOI] [PubMed] [Google Scholar]
- 211.Vieira Neto E et al. Quality of life and adherence to treatment in early-treated Brazilian phenylketonuria pediatric patients. Braz. J. Med. Biol. Res 51, e6709 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bik-Multanowski M et al. Quality of life in noncompliant adults with phenylketonuria after resumption of the diet. J. Inherit. Metab. Dis 31 (Suppl 2), 415–418 (2008). [DOI] [PubMed] [Google Scholar]
- 213.ten Hoedt AE et al. High phenylalanine levels directly affect mood and sustained attention in adults with phenylketonuria: a randomised, double-blind, placebo-controlled, crossover trial. J. Inherit. Metab. Dis 34, 165–171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ziesch B et al. Tetrahydrobiopterin (BH4) in PKU: effect on dietary treatment, metabolic control, and quality of life. J. Inherit. Metab. Dis 35, 983–992 (2012). [DOI] [PubMed] [Google Scholar]
- 215.Harding CO et al. Pegvaliase for the treatment of phenylketonuria: a pivotal, double-blind randomized discontinuation phase 3 clinical trial. Mol. Genet. Metab 124, 20–26 (2018). [DOI] [PubMed] [Google Scholar]
- 216.Regnault A et al. Development and psychometric validation of measures to assess the impact of phenylketonuria and its dietary treatment on patients’ and parents’ quality of life: the phenylketonuria – quality of life (PKU-QOL) questionnaires. Orphanet J. Rare Dis 10, 59 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mapi: Merck-Serono. Phenylketonuria impact and treatment Quality Of Life Questionnaire (PKU-QOL). https://eprovide.mapi-trust.org/instruments/phenylketonuria-impact-and-treatment-quality-of-life-questionnaire
- 218.Villiger L et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med 24, 1519–1525 (2018). [DOI] [PubMed] [Google Scholar]
- 219.Ahmed SS et al. Sustained correction of a murine model of phenylketonuria following a single intravenous administration of AAVHSC15-PAH. Mol. Ther. Methods Clin. Dev 17, 568–580 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Oh H-J, Park E-S, Kang S, Jo I & Jung S-C Long-term enzymatic and phenotypic correction in the phenylketonuria mouse model by adeno-associated virus vector-mediated gene transfer. Pediatr. Res 56, 278–284 (2004). [DOI] [PubMed] [Google Scholar]
- 221.Mochizuki S et al. Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 11, 1081–1086 (2004). [DOI] [PubMed] [Google Scholar]
- 222.Harding CO et al. Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria. Gene Ther. 13, 457–462 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ding Z, Georgiev P & Thöny B Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther. 13, 587–593 (2006). [DOI] [PubMed] [Google Scholar]
- 224.Yagi H et al. Complete restoration of phenylalanine oxidation in phenylketonuria mouse by a self-complementary adeno-associated virus vector. J. Gene Med 13, 114–122 (2011). [DOI] [PubMed] [Google Scholar]
- 225.Thöny B, Ding Z, Rebuffat A & Viecelli HM Phenotypic reversion of fair hair upon gene therapy of the phenylketonuria mice. Hum. Gene Ther 25, 573–574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Rebuffat A, Harding CO, Ding Z & Thöny B Comparison of adeno-associated virus pseudotype 1, 2, and 8 vectors administered by intramuscular injection in the treatment of murine phenylketonuria. Hum. Gene Ther 21, 463–477 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wagemaker G Lentiviral hematopoietic stem cell gene therapy in inherited metabolic disorders. Hum. Gene Ther 25, 862–865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.An D et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 21, 3548–3558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Truong B et al. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl Acad. Sci. USA 116, 21150–21159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cao J et al. mRNA therapy improves metabolic and behavioral abnormalities in a murine model of citrin deficiency. Mol. Ther 27, 1242–1251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Jiang L et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med 24, 1899–1909 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Zhu X et al. Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am. J. Hum. Genet 104, 625–637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Balakrishnan B et al. Novel mRNA-based therapy reduces toxic galactose metabolites and overcomes galactose sensitivity in a mouse model of classic galactosemia. Mol. Ther 28, 304–312 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pascucci T et al. A new therapy prevents intellectual disability in mouse with phenylketonuria. Mol. Genet. Metab 124, 39–49 (2018). [DOI] [PubMed] [Google Scholar]
- 235.Rossi L et al. Erythrocyte-mediated delivery of phenylalanine ammonia lyase for the treatment of phenylketonuria in BTBR-Pah(enu2) mice. J. Control. Rel 194, 37–44 (2014). [DOI] [PubMed] [Google Scholar]
- 236.Smith N, Longo N, Levert K, Hyland K & Blau N Exploratory study of the effect of one week of orally administered CNSA-001 (sepiapterin) on CNS levels of tetrahydrobiopterin, dihydrobiopterin and monoamine neurotransmitter metabolites in healthy volunteers. Mol. Genet. Metab. Rep 21, 100500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Smith N, Longo N, Levert K, Hyland K & Blau N Phase I clinical evaluation of CNSA-001 (sepiapterin), a novel pharmacological treatment for phenylketonuria and tetrahydrobiopterin deficiencies, in healthy volunteers. Mol. Genet. Metab 126, 406–412 (2019). [DOI] [PubMed] [Google Scholar]
- 238.Isabella VM et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol 36, 857–864 (2018). [DOI] [PubMed] [Google Scholar]
- 239.Vockley J, Sacharow S, Searle S, Kurtz C & Querbes W A phase 1/2a oral placebo-controlled study of SYNB1618 in healthy adult volunteers and subjects with phenylketonuria [abstract O-027]. J. Inherit. Metab. Dis 42 (Suppl. 1), 13 (2019). [Google Scholar]
- 240.Lane JD, Schöne B, Langenbeck U & Neuhoff V Characterization of experimental phenylketonuria augmentation of hyperphenylalaninemia with α-methylphenylalanine and p-chlorophenylalanine. Biochim. Biophys. Acta 627, 144–156 (1980). [DOI] [PubMed] [Google Scholar]
- 241.Haefele MJ, White G & McDonald JD Characterization of the mouse phenylalanine hydroxylase mutation Pah(enu3). Mol. Genet. Metab 72, 27–30 (2001). [DOI] [PubMed] [Google Scholar]
- 242.Richards DY et al. A novel Pah-exon1 deleted murine model of phenylalanine hydroxylase (PAH) deficiency. Mol. Genet. Metab 131, 306–315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Koppes EA et al. A porcine model of phenylketonuria generated by CRISPR/Cas9 genome editing. JCI Insight 5, e141523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kaiser RA et al. Development of a porcine model of phenylketonuria with a humanized R408W mutation for gene editing. PLoS ONE 16, e0245831 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Douglas TD, Ramakrishnan U, Kable JA & Singh RH Longitudinal quality of life analysis in a phenylketonuria cohort provided sapropterin dihydrochloride. Health Qual. Life Outcomes 11, 218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]