

Abstract
In targeted protein degradation of kinases, key discoveries have been made specifically involving selective kinase degradation. Structural and biophysical studies on the ternary complex formation have provided a clear understanding of the basis for achieving degradation selectivity which is important in guiding the design of efficient and selective protein degraders.
Keywords: Kinases, Protein degradation, Ternary complex, Cooperativity, Selectivity
Many diseases, specifically cancers, are marked by dysregulation of signaling pathways. Protein kinases are the enzymes within cells that perform phosphorylation reaction of proteins, which is a process central to signaling pathways in controlling both cellular and extracellular processes. The deregulation of phosphorylation can lead to excess cellular proliferation or continued survival of cancerous motifs. Thus, the identification of the essential roles of protein kinases in oncogenic processes has intensified the discovery and development of kinase inhibitors.1 However, despite the increasing growth of kinase inhibitors in cancer treatments, an intimidating issue that persists is resistance to these targeted therapies and subsequent relapse.2,3 In addition to developing new kinase inhibitors, other modalities, including those from targeted protein degradation strategies, are actively being explored for overcoming resistance issues. Protein degradation is a natural process in cells, and the majority occurs through the ubiquitin proteasome system (UPS). It is an emerging form of chemical knockdown whereby molecular degraders can tag specific proteins for degradation and often comes with inherent advantages over those of typical inhibitors.4
Of particular note are two classes of protein degraders: “molecular glue” degraders, e.g. cereblon E3 ligase modulating drugs (CELMoDs),5 and proteolysis targeting chimeras (PROTACs).6 Molecular glue degraders and PROTACs function in a similar manner through their recruitment of an E3 ligase to trigger degradation, but there are key distinctions between the two classes. Molecular glue degraders are small molecules that scaffold protein–protein interactions (PPIs), in essence gluing the two protein surfaces of E3 ligase and protein of interest together. PROTACs, meanwhile, are heterobifunctional molecular constructs for performing ligand-directed degradation. They are composed of an E3 ubiquitin ligase binding ligand and target protein binding ligand, connected to each other via a linker. Both degrader classes form a ternary complex with the E3 ubiquitin ligase and target protein, enabling proximity triggered ubiquitination of the target protein and subsequent proteasome-dependent degradation (Figure 1). This allows for the degradation of the whole protein, which in the case of protein kinases, will erase both enzymatic activity and nonenzymatic functions, e.g. scaffolding roles. Molecular degraders have many intrinsic advantages over conventional inhibitors. Due to their catalytic, substoichiometric nature of action, degraders could achieve efficacy at lower doses as compared to inhibitors, thus minimizing potential toxicity and off-target liability. Furthermore, it has been observed that protein degraders can achieve higher level of selectivity that is difficult to realize via inhibitors, as discussed in the following section. Since the development of degraders as a therapeutic modality, their number and scope have dramatically increased. This Viewpoint highlights recent key advances in the area of targeted degradation of protein kinases, particularly in the understanding of the structural and biophysical basis of degradation selectivity.
Figure 1.
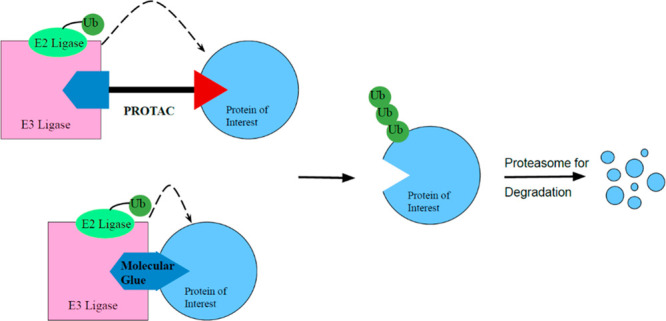
Schematic for the mode of action of molecular glue and PROTAC. These protein degraders bring the protein of interest into close proximity to the ubiquitination system, allowing for polyubiquitination and proteasome dependent degradation.
As noted, kinases are essential to the functioning of basic cellular processes, and cyclin-dependent kinases (CDKs) are one subgroup. Among these kinases are CDK4 and CDK6, which play an important role in the transition between the G1-phase and S-phase of mitosis. CDK4/6 kinases function by association with cyclin D, enabling phosphorylation of retinoblastoma protein (Rb) and subsequent cell proliferation. Deregulation of CDK4/6 can result in malignant Rb phosphorylation that leads to cancer. A specific dependency in acute myeloid leukemia cell lines on CDK6 has been identified. However, there are currently only dual CDK4/6 inhibitor drugs, which have been approved for treatment of breast cancer.7 The dual inhibitors lack the homologue-selective inhibition due to the high amino acid sequence similarity (94%) in the ATP binding pocket of CDK4 and CDK6. It was hypothesized by Gray et al. that a PROTAC could be utilized to induce selective degradation of CDK6 over CDK4 by inducing PPIs that would result in differential ternary complex formation.8 This idea is also known as cooperativity (α). In the case of PROTACs, cooperativity, as first defined by earlier work conducted in the Ciulli lab, is the ternary complex binding affinity relative to that of the binary complex.9 It can be quantitatively defined by the equation α = KDbinary/KDternary. Systems with positive cooperativity (Figure 2, top) have an α value greater than one, meaning the ternary complex is stabilized by protein–protein interactions, and thus, the relative ternary binding affinity is enhanced compared to binary affinity. On the other end of the spectrum is negative cooperativity (Figure 2, bottom) when the relative ternary complex affinity is decreased compared to that of the binary (α < 1) due to protein–protein destabilizing effects. Finally, when cooperativity is equal to one, this is defined as a noncooperative or neutral system where the binary complex has no impact on ternary complex formation. Based on the assumption that differential ternary complex formation could occur, PROTAC BSJ-03-123 (Figure 3A) was developed. BSJ-03-123 is comprised of a cereblon (CRBN) targeting ligand, pomalidomide, a phenoxy acetamide linker, and a CDK4/6 inhibitor, palbociclib. Despite the comparable binary affinity of BSJ-03-123 for CDK4 and CDK6, only selective stabilization of CDK6 ternary complex occurred. Further, BSJ-03-123 was able to selectively disrupt proliferation of CDK6-dependent AML cells while sparing CDK4-dependent cancer cell lines, supporting their hypothesis that structural differences in ternary complex formation could be exploited to achieve selective degradation of highly homologous protein kinases.8
Figure 2.

Ternary complex equilibria showing effects of cooperativity on degradation and selectivity. Complex formation can be enhanced (top) or diminished (bottom) in the ternary complex compared to the binary complex. Enhancement often results from induced PPIs, achieving in an increase in cooperativity, that could lead to selective degradation. Shown above are two homologous protein targets of an inhibitor (represented by the red triangle), but only the protein of interest is selectively degraded by the PROTAC. For clarity, the binary complex of E3 ligase and PROTAC is not shown.
Figure 3.

(A) Structure of BSJ-03-123, a CDK6 targeting PROTAC with a CRBN E3 ligase binding ligand. (B) Structure of SGK3-PROTAC1, a SGK3 targeting PROTAC with a VHL E3 ligase binding ligand. (C) Structure of MZ1, a BRD4 selective PROTAC with a VHL binding ligand.
Gray’s conclusion that the selectivity arises from differential ternary complex formation can be further supported by previous work conducted by the Ciulli lab, specifically the development of an SGK3 selective degrader, SGK3-PROTAC1 (Figure 3B), and the accompanying biophysical and kinetic studies.10 SGK3 is a downstream component of the class 1 phosphatidylinositol 3-kinase (PI3K) pathway and is critical to moderating breast cancer cell resistance to class 1 PI3K or Akt inhibitors. Current inhibitors are unable to discriminate between SGK3 and S6K1, but the PROTAC developed had isoform specificity for SGK3 degradation over SGK1 and SGK2, and further specificity for SGK3 against S6K1. Additionally, by altering the length and hydrophobicity of the linker, they were able to amplify the degradation of SGK3 while decreasing the inhibitor potency against S6K1. To investigate the implications of linker properties and how differential ternary complex formation or cooperativity can impact selectivity, focus was directed to biophysical and kinetic studies. The first crystal structure of a PROTAC ternary complex was determined by Ciulli’s lab in 2017 and showed PROTAC MZ1 (Figure 3C) bound to von Hippel−Lindau (VHL) ligase and BRD4BD2 in a bowl-shaped interface created by PPIs between VHL and BRD4.9 They also observed interactions between the PEG linker and the BC loop of BRD4. The de novo protein–protein and PROTAC–protein interactions that occurred contributed to the isoform selectivity of MZ1 to induce the preferential degradation of BRD4 over BRD2 or BRD3. This was easily observable through the experimental data on dissociation kinetics, half-life, and cooperativity. SPR experiments showed that the ternary complex of VHL:MZ1:BRD4BD2 had the slowest dissociation kinetics with a koff of 0.006 s–1, the longest half-life of 130 s, and a positive cooperativity of 22. Meanwhile, ternary complexes containing BRD2BD2 or BRD3BD2 were more transient in nature with much faster dissociation kinetics and shorter half-lives, which in turn led to less potent degradation.11 In fact, it was discovered that a single amino acid residue was able to influence ternary complex formation by inducing steric clash between MZ1 and VHL. Mutagenesis was conducted on the BRD4BD2 protein to swap the native Gly386 for Glu386 to mimic BRD3BD2. This single amino acid mutation resulted in a substantial decrease in ternary complex cooperativity to 3.5 and a dramatic reduction in ternary complex half-life from 130 to 4 s.11 Ciulli’s research findings demonstrate how subtle differences in protein composition can be exploited by PROTACs and lead to differential ternary complex formation and subsequent selectivity.
The X-ray crystallography studies coupled with kinetic analyses via SPR experiments greatly contributed to the understanding of the structural and biophysical basis for achieving target selectivity of a heterobifunctional protein degrader. It also led to the important recognition that the linker is not a bystander but participates in protein–protein interactions to enhance or destabilize ternary complex stability. Those appreciations are critical in guiding the design of protein degraders that are efficient and selective against desired targets as demonstrated in the degradation of protein kinases as described above.12 Importantly, the principals should also be applicable to other classes of proteins that are of significant therapeutic interest.
Acknowledgments
C.E.G. (Trinity College, Hartford, CT, Class 2021) thanks Bristol Myers Squibb for a 2020 Summer Internship in Cancer Resistance and Neuroscience Chemistry, Small Molecule Drug Discovery. We thank Dr. Robert Borzilleri, Dr. Nicholas Meanwell, and Dr. Paul Scola for helpful comments.
Author Present Address
† Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
Views expressed in this viewpoint are those of the author and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Ferguson F. M.; Gray N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 2018, 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Cross D.; Jänne P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discovery 2021, 20, 551–569. 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Yoon H.; Xiong Y.; Dixon-Clarke S. E.; Nowak R. P.; Fischer E. S. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat. Struct. Mol. Biol. 2020, 27, 605–614. 10.1038/s41594-020-0438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P.; Hamann L. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Burslem G.; Crews C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B.; Finn R. S.; Turner N. C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- Brand M.; Jiang B.; Bauer S.; Donovan K.; Liang Y.; Wang E.; Nowak R. P.; Yuan J. C.; Zhang T.; Kwiatkowski N.; Müller A. C.; Fischer E.; Gray N. S.; Winter G. E. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell. Chem. Biol. 2019, 26, 300–306. 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M.; Testa A.; Lucas X.; Chan K.; Chen W.; Lamont D.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–52. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Zhou H.; Shpiro N.; Crafter C.; Ciulli A.; Alessi D. R. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol. 2019, 14, 2024–2034. 10.1021/acschembio.9b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem. Biol. 2019, 14, 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent report demonstrated selective degradation of PI3Kα mutant over wild-type by PI3K kinase inhibitors in a proteasome-dependent manner, thus suggesting further possibility in achieving selectivity in the degradation of protein kinases; see:; Song K. W.; Edgar K. A.; Hanan E. J.; Hafner M.; Oeh J.; Merchant M.; Sampath D.; Nannini M. A.; Hong R.; Phu L.; Forrest W. F.; Stawiski E.; Schmidt S.; Endres N.; Guan J.; Wallin J. J.; Cheong J.; Plise E.; Philips G.; Salphati L.; Heffron T. P.; Olivero A.; Malek S.; Staben S. T.; Kirkpatrick D. S.; Dey A.; Friedman L. S. RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077 (Inavolisib) efficacy. Cancer Discovery 2021, candisc.0072.2021 10.1158/2159-8290.CD-21-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]