

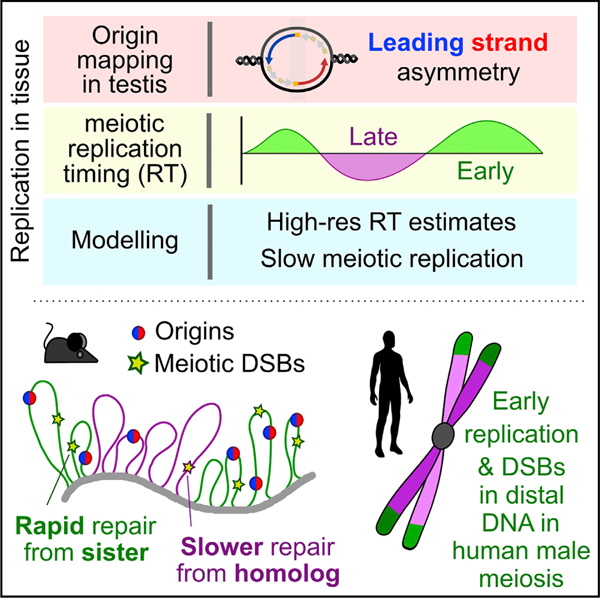
SUMMARY
Genetic recombination generates novel trait combinations, and understanding how recombination is distributed across the genome is key to modern genetics. The PRDM9 protein defines recombination hotspots; however, megabase-scale recombination patterning is independent of PRDM9. The single round of DNA replication, which precedes recombination in meiosis, may establish these patterns; therefore, we devised an approach to study meiotic replication that includes robust and sensitive mapping of replication origins. We find that meiotic DNA replication is distinct; reduced origin firing slows replication in meiosis, and a distinctive replication pattern in human males underlies the subtelomeric increase in recombination. We detected a robust correlation between replication and both contemporary and historical recombination and found that replication origin density coupled with chromosome size determines the recombination potential of individual chromosomes. Our findings and methods have implications for understanding the mechanisms underlying DNA replication, genetic recombination, and the landscape of mammalian germline variation.
Graphical Abstract
In brief
Combining a new approach for locating replication origins in mammalian cells and tissues with maps of replication timing and a computational model for genome-wide replication reveals that differences in the male pre-meiotic replication program can predict the recombination landscape during human and mouse meiosis.
INTRODUCTION
Sexual reproduction uses a specialized cell division called meiosis, in which a single round of DNA replication is followed by two cell divisions to create haploid gametes. Genetic recombination in meiosis assures faithful segregation of chromosomes and establishes patterns of genetic linkage and inheritance. Recombination is initiated by the formation of hundreds of programmed DNA double-strand breaks (DSBs). In mice and humans, DSBs are targeted by DNA sequence-specific binding of a meiosis-specific histone methyltransferase, PRDM9 (Baudat et al., 2010; Myers et al., 2010; Parvanov et al., 2010). Hundreds of PRDM9 alleles exist (Berg et al., 2010; Buard et al., 2014) that primarily differ in the DNA-binding domain. Thus, each variant may yield a distinct patterning of meiotic DSBs (Smagulova et al., 2016). Nonetheless, megabase-scale similarities between individuals demonstrate that a broad-scale, PRDM9-independent layer of control also shapes meiotic recombination (Davies et al., 2016; Myers et al., 2005; Smagulova et al., 2011). The clearest manifestation of this is seen in the elevation of meiotic DSBs in subtelomeric DNA of human males, independent of PRDM9 genotype (Pratto et al., 2014).
Because DNA replication determines genome structure (Klein et al., 2021) and immediately precedes DSB formation in meiosis, we hypothesized that DNA replication may drive the broad-scale regulation of recombination in mammals. Replication and recombination are correlated in yeast (Borde et al., 2000; Murakami and Nurse, 2001) and in plants (Higgins et al., 2012; Osman et al., 2021). A mechanistic link has been demonstrated in Saccharomyces cerevisiae where a component of the DSB machinery is activated by passage of the replication fork (Murakami and Keeney, 2014). This results in a spatiotemporal coordination between replication and DSB formation.
Mammalian DNA replication is exclusively studied in cell culture. This presents difficulties in adapting techniques for studying replication in tissue, which have been mostly designed and optimized for cell culture. The paucity of studies of DNA replication in mammalian meiosis stems from the requirement to study meiosis in vivo, in the context of a complex tissue (Handel et al., 2014) and all current knowledge of DNA replication in mammalian meiosis stems from classical papers using early molecular and cytological techniques (for review, see Chandley, 1986). To address this shortcoming, we devised a method to map origins of replication in mammalian tissue (in this case, testis), developed a cell-type-specific method to interrogate replication timing in meiotic S-phase, and designed an in silico modeling strategy to parameterize DNA replication in vivo. This tripartite approach has generated a comprehensive, parameterized description of DNA replication genome-wide in meiosis, or, for that matter, in any mammalian tissue. We found that aspects of DNA replication in the germline are distinct from replication in other cell types. This has the potential to shape megabase-scale patterns of meiotic recombination and genome diversity.
RESULTS
Highly specific replication origin mapping in mammalian testis
To identify origins of replication, we adapted a method to sequence the RNA-primed short nascent leading strands (SNS) (Bielinsky and Gerbi, 1998; Figure 1A). Briefly, RNA-primed leading strands are isolated by using lambda exonuclease to digest DNA that lacks an RNA primer. Okazaki fragments, although also RNA-primed, are excluded by size selection. A key improvement upon small nascent strand sequencing (SNS-seq) (Cayrou et al., 2012; Fu et al., 2014; Jodkowska et al., 2019; Picard et al., 2014) is that we retain the directionality of the nascent strand (origin-derived single-stranded DNA sequencing; Ori-SSDS) (Figure 1A). Ori-SSDS works by ligating a sequencing adaptor directly to ssDNA; this is possible because ssDNA spontaneously forms hairpin-loop structures where the double stranded DNA stem can be processed to allow adaptor ligation (Figure 1A; STAR Methods; Khil et al., 2012). SSDS has previously been used to map ssDNA intermediates at meiotic DSB hotspots, where it yields an accurate estimate of DSB locations and frequency (Khil et al., 2012; Lange et al., 2016). Using Ori-SSDS, we can omit the random-priming and second-strand synthesis steps that prevent the capture of leading strand directionality and that introduce biases (Sequeira-Mendes et al., 2019).
Figure 1. Identification of replication origins.
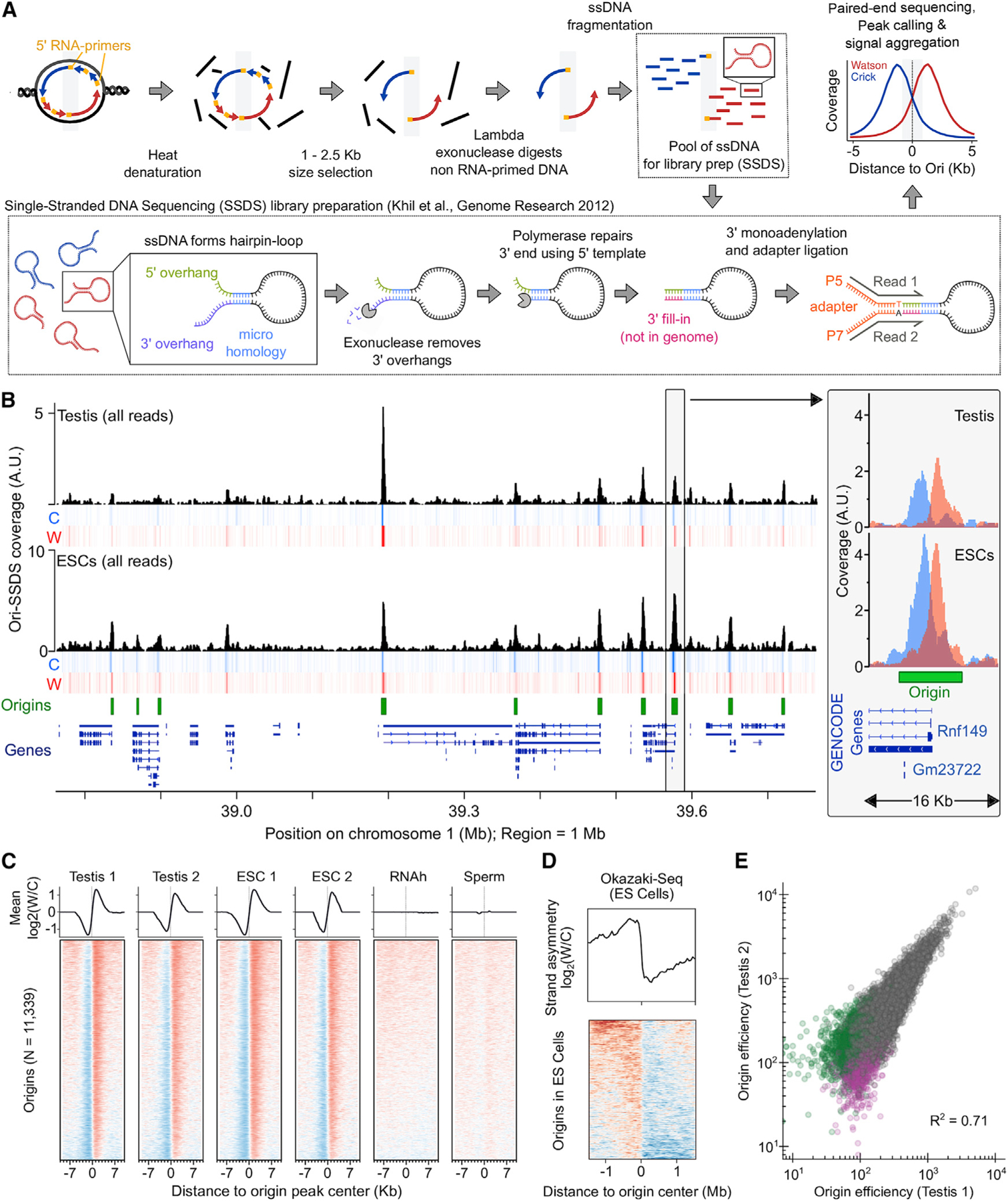
(A) Schematic of the Ori-SSDS protocol to sequence nascent leading strands.
(B) The Ori-SSDS signal in a typical 1 Mb region (black, total coverage; heatmap shows coverage by strand: red, Watson; blue, Crick; 1 kb windows, 147 bp step). Inset: zoom of a typical peak.
(C) Characteristic, reproducible Watson-Crick asymmetry at origins is lost in controls. The log2 ratio of Watson/Crick ssDNA fragments is shown (1 kb smoothing).
(D) Ori-SSDS peaks coincide with a switch in the direction of lagging-strand DNA synthesis (from Okazaki-fragment sequencing).
(E) Origin efficiency is highly correlated among replicates. Origins found only in one replicate are colored purple and green, respectively.
See also Figures S1 and S2 and Data S1.
To facilitate a comparison of Ori-SSDS with other methods, we mapped origins in cultured mouse embryonic stem cells (ESCs), where replication has been extensively studied (Almeida et al., 2018; Cayrou et al., 2015; Petryk et al., 2018). Characteristic bidirectional replication at origins manifests as a strand switch in sequenced reads (Figures 1B and 1C). Thus, distinct from SNS-seq experiments, we can differentiate true origins from non-specific signals such as G-quadruplexes by imposing a requirement for leading-strand asymmetry at origins (Figures S1A–S1C; STAR Methods). 86% of replication origins in ESCs are found in SNS-based origin maps from ESCs (78% overlap origins from Almeida et al. [2018]; 73% overlap origins from Cayrou et al. [2015]). Ori-SSDS peaks also coincide with a switch in the polarity of lagging-strand DNA synthesis (data from Okazaki-fragment sequencing in ESCs) (Petryk et al., 2016; Figures 1D and S1D). This is consistent with replication originating at these loci.
SNS-seq experiments typically start from 1 × 109 exponentially growing cultured cells of which ~30% are replicating (3 × 108 cells) (Almeida et al., 2018). Nonetheless, we performed Ori-SSDS from 2 × 108 cells (from two mouse testes), among which ~2% of cells are replicating (4 × 106 replicating cells, 67% are meiotic) (Figure S2; Kojima et al., 2019); this is 3 times fewer cells than in our successful Ori-SSDS experiments in ESCs, (4 × 107 cells; ~1.2 × 107 replicating cells), 3- to 5-fold fewer than methods that immunoprecipitate pre-replication complexes (i.e., ORC1/ORC2 chromatin immunoprecipitation sequencing [ChIP-seq]; Miotto et al., 2016) and orders of magnitude fewer than are required for the sequencing of Okazaki fragments (Petryk et al., 2016). We detected 11,209, 12,406, and 13,476 origins in replicate experiments from testes of individual male mice, demonstrating that our method is sufficiently sensitive to map replication origins from individual animals. 97% of Ori-SSDS peaks in the smallest testis set were found in another experiment (Figure S1F). No origins were detected in control experiments by using non-replicating tissue (sperm) or by removing the leading strand RNA-primer (STAR Methods; Figures 1C and S1A–S1C). Most testis origins are also found in ESCs; however, fewer origins are detected in testis than in the better ESC sample (Figures S1E and S1F). Origin efficiency is a measure of the frequency with which an origin is used. We use the sequencing depth of ‘‘correctly’’ oriented Ori-SSDS read-pairs to infer origin efficiency. This efficiency varies ~100-fold and is highly correlated both between replicates (Figures 1E and S1G–S1L; Spearman R2 = 0.65–0.74) and between ESCs and testis origins (Spearman R2 = 0.63–0.76). Thus, we do not find high efficiency origins unique to either testis or ESCs. The 11,339 high-confidence origins, found in at least two of three testis replicates (Figure S1E), were used for subsequent analyses. Together, these data show that Ori-SSDS can identify the origins of replication genome-wide from either cultured cells or from mammalian tissue.
Ori-SSDS peaks represent sites at which replication preferentially initiates in a population of cells. The overlap of the Watson and Crick signals (Figures 1B, 2A, S1B, and S1C) implies that the exact initiation point within each peak may vary by several hundred nucleotides. SNS-seq identifies up to 65,000 putative origins in mice, however, by clustering peaks within 1 kb, the number of origins inferred more closely approximates the number from Ori-SSDS (Figures 2A–2C). Alternative peak-calling for SNS-seq identified kilobase-scale peaks that encompassed many local SNS-seq summits within what appear to be single Ori-SSDS peaks (Figures 2A and 2B; IZ; Cayrou et al., 2015). Thus, the structure revealed by Ori-SSDS suggests that the resolution of population-scale methods that capture the leading strand for origin mapping is ~1 kb.
Figure 2. Clustering of replication origins in the genome.
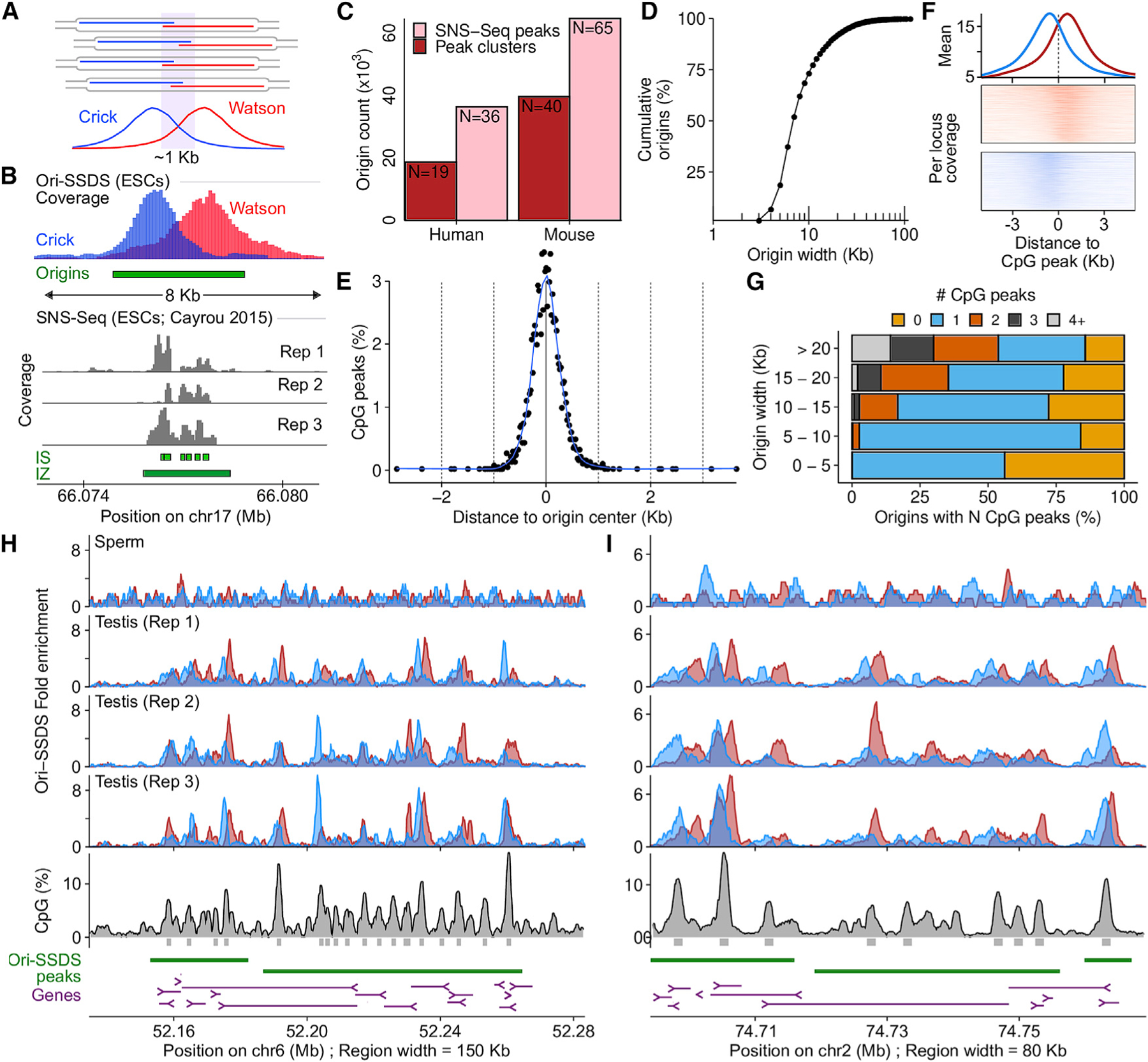
(A) Stochastic replication initiation at Ori-SSDS peaks.
(B) 6 SNS-seq origin calls coincide with a single Ori-SSDS peak (green box). Gray represents replicate SNS-seq coverage tracks (Cayrou et al., 2015). SNS-seq origin calls (IS) or initiation ‘‘zones’’ (IZ) are green.
(C) The number of origins of replication from SNS-seq is reduced if peaks within 1 kb (clusters) are considered as single origins (human: Long et al. [2020], mouse: Cayrou et al. [2015]).
(D) Cumulative width distribution of Ori-SSDS peaks.
(E) Peaks of CpG density (see STAR Methods) coincide with Ori-SSDS peak centers. Origins <6 kb were considered.
(F) CpG peaks within broad initiation zones (peaks >10 kb) exhibit Ori-SSDS Crick-Watson asymmetry.
(G) CpG peak counts as a function of origin width. Most narrow origins contain 0 or 1 CpG peaks.
(H and I) Examples of broad Ori-SSDS peaks.
See also Figures S2 and S3 and Data S1.
Origins identified from mouse testis are unevenly distributed in the genome (Figure S3A) and cluster in gene rich regions; 66% of origins occur within 1 kb of a transcription start site (TSS) (Figure S3B). DNA at the origin center is intrinsically flexible, flanked by relatively rigid DNA. This may reflect a requirement of the ORC complex to bend DNA (Lee et al., 2021; Li et al., 2018; Figure S3C). Origins are GC-rich, with elevated CpG, and to a lesser extent, GpC dinucleotides (Figure S3D). GC content, CpG density, and GpC density are all positively correlated with origin efficiency (Figure S3E) implying that nucleotide content plays a role in origin firing. G-quadruplexes, implicated in origin firing (Valton et al., 2014), are also strongly enriched at origin centers (Figure S3F). 68% of origins coincide with CpG islands (CGIs) in accessible chromatin (as measured by ATAC-seq in spermatogonia) (Maezawa et al., 2018; Figure S3G), yet only 59% of such sites are used as an origin (Figure S3H). CGIs in inaccessible chromatin and open chromatin lacking a CGI are far less predictive of origin locations (Figures S3G and S3H). We did not identify any conserved sequence motif at replication origins; however, the density of CpG dinucleotides alone can predict origin locations (Figure S3I). CpG density is a better predictor of origin locations than consensus motifs are for transcription factors (Figure S3J). Many of these observations are consistent with properties of replication origins defined in cultured cells (Marchal et al., 2019; Miotto et al., 2016) and together suggest that replication in the germline initiates preferentially at CpG islands, near gene promoters, in accessible chromatin.
Broad replication initiation zones—up to 150 kb and comprising ~7% of the genome—have been proposed as a major mode of replication initiation in humans (Petryk et al., 2016; Wang et al., 2021). The methods used to define these zones lacked the resolution to determine sub-structures below 15 kb. To assess if putative initiation zones result from closely spaced, but difficult-to-resolve discrete origins, we examined Ori-SSDS peaks wider than 10 kb (the expected width of the Ori-SSDS signal is approximately ±3 kb around the origin of replication; determined by the size selection step) (Figures 1A and 2D; STAR Methods). CpG peaks coincide with the center of isolated origins (Figure 2E) and we found that within our defined initiation zones, most local CpG peaks exhibited origin-like Ori-SSDS strand asymmetry, implying that these sites are de facto origins (Figure 2F). 620 zones contained multiple CpG peaks (NCpG Peaks = 1,645; Figure 2G), thus, difficult-to-resolve origins account for ~30% of initiation zones (Figures 2H and 2I). Among such zones is the HoxA locus, where ~19 origins of replication occur within just 110 kb (Figure 2H). 816 zones contained a single CpG peak. These may represent origins where we captured longer-than-expected nascent strands. The remaining 490 zones did not contain any CpG peak above our threshold and may represent more amorphous replication initiation regions.
An unambiguous snapshot of replication in meiotic S-phase
Stochastic origin firing and uneven origin density result in distinct earlier and later replicating parts of the genome. We inferred this replication timing (RT) in meiosis. Meiotic S-phase cells constitute ~1% of cells in adult testis (Kojima et al., 2019). Using a variant of fluorescence-activated nuclei sorting (FANS) (Lam et al., 2019), we isolated up to 2 × 105 meiotic S-phase nuclei at ~95% purity (inspection of wide-field images) (Figures 3A–3D) from a single adult mouse; this relied on a combination of DNA content, negative selection for a marker of non-meiotic cells (DMRT1), and positive selection for a meiotic protein (STRA8). A marker not used for sorting, but with expected meiotic expression (SYCP3), validated the purity of the meiotic population (Figure 3C). We then inferred meiotic RT from coverage imbalances in whole-genome sequencing of these nuclei (RT-seq) (STAR Methods; Koren et al., 2014). Replication origin density is high in early replicating regions and low in late-replicating DNA (Figure 3E). This yields a snapshot of replication timing in mammalian meiosis. We also inferred RT from the other major replicating cell types in adult testis—undifferentiated and differentiating spermatogonia—and from published whole-genome sequencing (WGS) of other cell types (Figure S4; STAR Methods). Meiotic RT profiles are highly correlated among replicates and, to a lesser extent, with RT from other cell types (Figures 3E–3G and S4). In particular, meiotic RT is notably similar to RT in the cells that immediately precede meiotic S-phase and to RT in other undifferentiated cells (PGCs, SSCs, and ESCs). RT correlates to varying degrees with GC-content (Spearman R2 = 0.27–0.40) and with the two major nuclear compartments (active [A] and inactive [B]) defined by chromatin conformation capture experiments (Hi-C; data from Patel et al., 2019) (Spearman R2 = 0.57–0.70) (Figure 3H) (for review, see Vouzas and Gilbert, 2021).
Figure 3. Replication timing in meiotic prophase I of male mice.
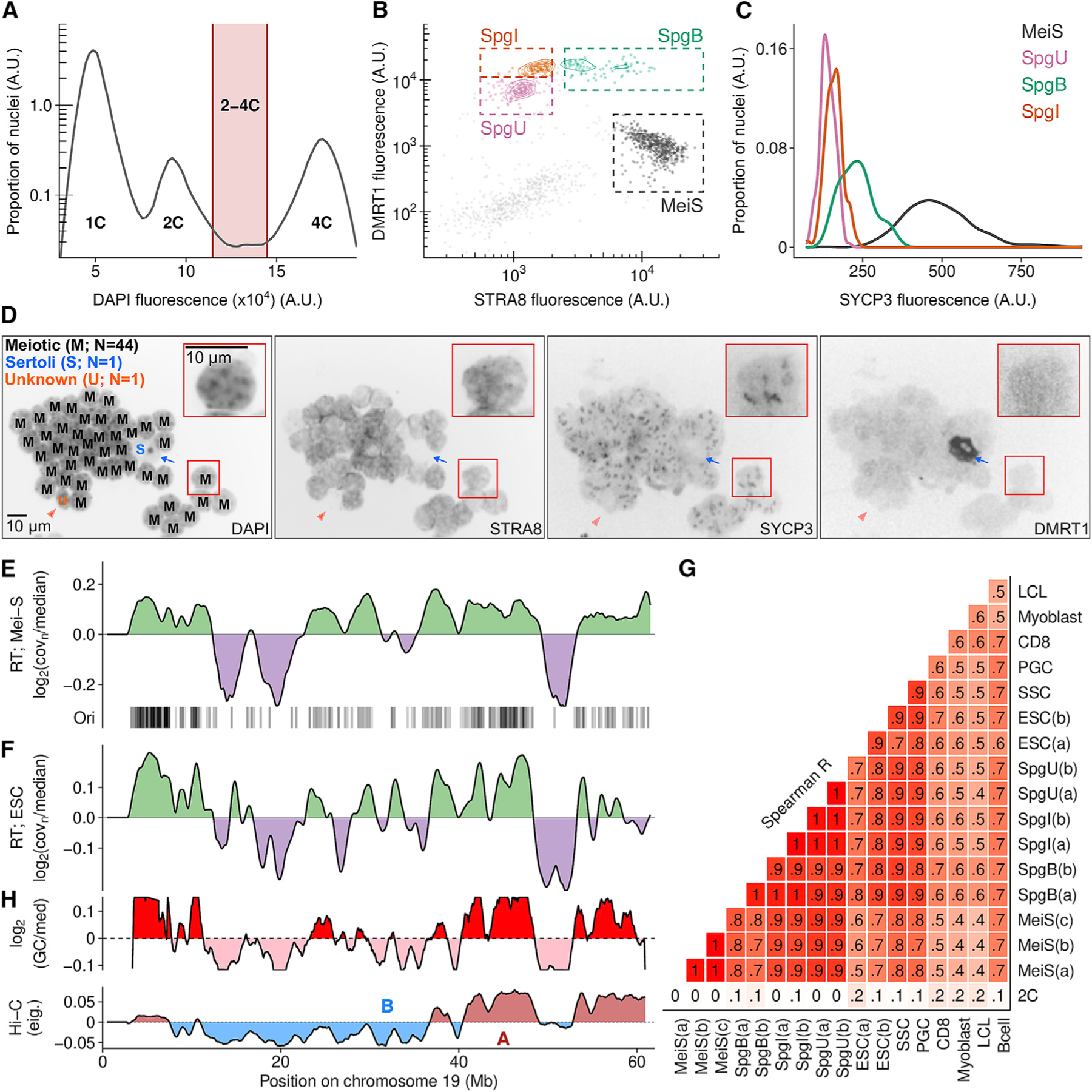
(A–C) Meiotic S-phase and spermatogonia nuclei were isolated using fluorescence-activated nuclei sorting (Lam et al., 2019).
(A) Replicating nuclei were isolated (2–4C).
(B) Isolation of S-phase nuclei (see STAR Methods). MeiS, meiotic; SpgU, undifferentiated spermatogonia; SpgI, intermediate spermatogonia; SpgB, type-B spermatogonia.
(C) SYCP3 is elevated in meiotic nuclei.
(D) Micrograph showing the purity of meiotic S-phase nuclei (403 magnification; insets 1003). Distinctive DAPI morphology is also used to assess purity (Bellvé et al., 1977). Blue arrow, Sertoli cell nucleus (DMRT1+, STRA8−); orange arrow, unknown type (DMRT1−, STRA8+). Letter code indicates nucleus-type (left panel: M, meiotic; S, Sertoli; U, unknown).
(E) Replication timing (RT; log2 of normalized sequencing coverage) in meiotic S-phase correlates with Ori-SSDS peak locations in testis.
(F) RT in ESCs.
(G) RT is correlated across cell-types. RT was inferred from published data for spermatogonial stem cells (SSCs), primordial germ cells (PGCs), embryonic stem cells (ESCs), CD8+ cells (CD8s), and activated B cells (Bcells). Pre-processed RT data were obtained for myoblast and lymphoblastoid cell lines (LCLs). Details of samples in Tables S1, S2, and S3.
(H) GC-content (1 Mb smoothing, 10 kb steps; log2(GC/median)) and genome compartmentalization at zygonema (Patel et al., 2019) correlate with RT. Hi-C track shows the eigenvector values for the first principal component of the Hi-C matrix (100 kb windows, 1 Mb smoothing, 10 kb steps; A, active; B, inactive Hi-C compartments).
In silico modeling recapitulates replication timing from origin locations
RT-seq yields a static snapshot of replication, and these snapshots from different cell types are often remarkably similar. Furthermore, the differences between RT-seq profiles are difficult to interpret because RT-seq is influenced by the stage of S-phase progression and by the relative synchrony of the population (Figure S5A) (Zhao et al., 2020). We hypothesized that subtle differences in the properties of replication among cell types could be captured by in silico modeling.
We built an in silico model of DNA replication (RT-sim) that requires initiation sites as input and that outputs a simulated RT profile. By examining the properties of replication that produce a best-fit between simulated and experimental RT, we can understand the parameters of replication that yield different RT profiles. Modeling parameters are the number of active forks, whether to use origin efficiency estimates as a firing probability, and the total duration of simulated S-phase. Replication fork speed is assumed constant among cell types and throughout S-phase. After an initial round of simultaneous origin firing, further origins fire only when extant forks collide; this simulates the presence of factors that limit the number of active origins (replisome ceiling), deemed important by previous simulations of DNA replication (Gindin et al., 2014; Kelly and Callegari, 2019).
In silico modeling accurately recapitulated experimental RT from both meiotic (Figures 4A, 4B, and S6) and non-meiotic S-phase cells (ESCs) (Figures 4B, 4C, and S6). We could not obtain good-fitting models for non-replicating cells (2C) or when origin locations were randomized (Figure 4B). Our success in modeling RT in all cell types using either testis or ESC-derived origins implies that a common set of high-efficiency origins are used during meiotic and mitotic replication. Indeed, in yeast, origins of replication are common to meiotic and mitotic cells (Blitzblau et al., 2012; Wu and Nurse, 2014). Most origins coincide with CpG islands in open chromatin (Figure S3G), which are broadly similar across cell types (Table S4). RT-sim using these loci as a proxy for origins yields models that are slightly worse than those using de facto origins (AS+CGI) (Figure 4B). Finally, RT-sim does not explicitly distinguish between ‘‘early’’ and ‘‘late’’ firing replication origins, suggesting that the paradigm of early and late origins is not required to explain RT. Instead, early and late replicating regions are defined by high and low origin density, respectively. This is consistent with recent studies in other mammalian cell types (Dileep and Gilbert, 2018; Gindin et al., 2014; Miotto et al., 2016; Takahashi et al., 2019).
Figure 4. In silico modeling recapitulates RT from origin locations.
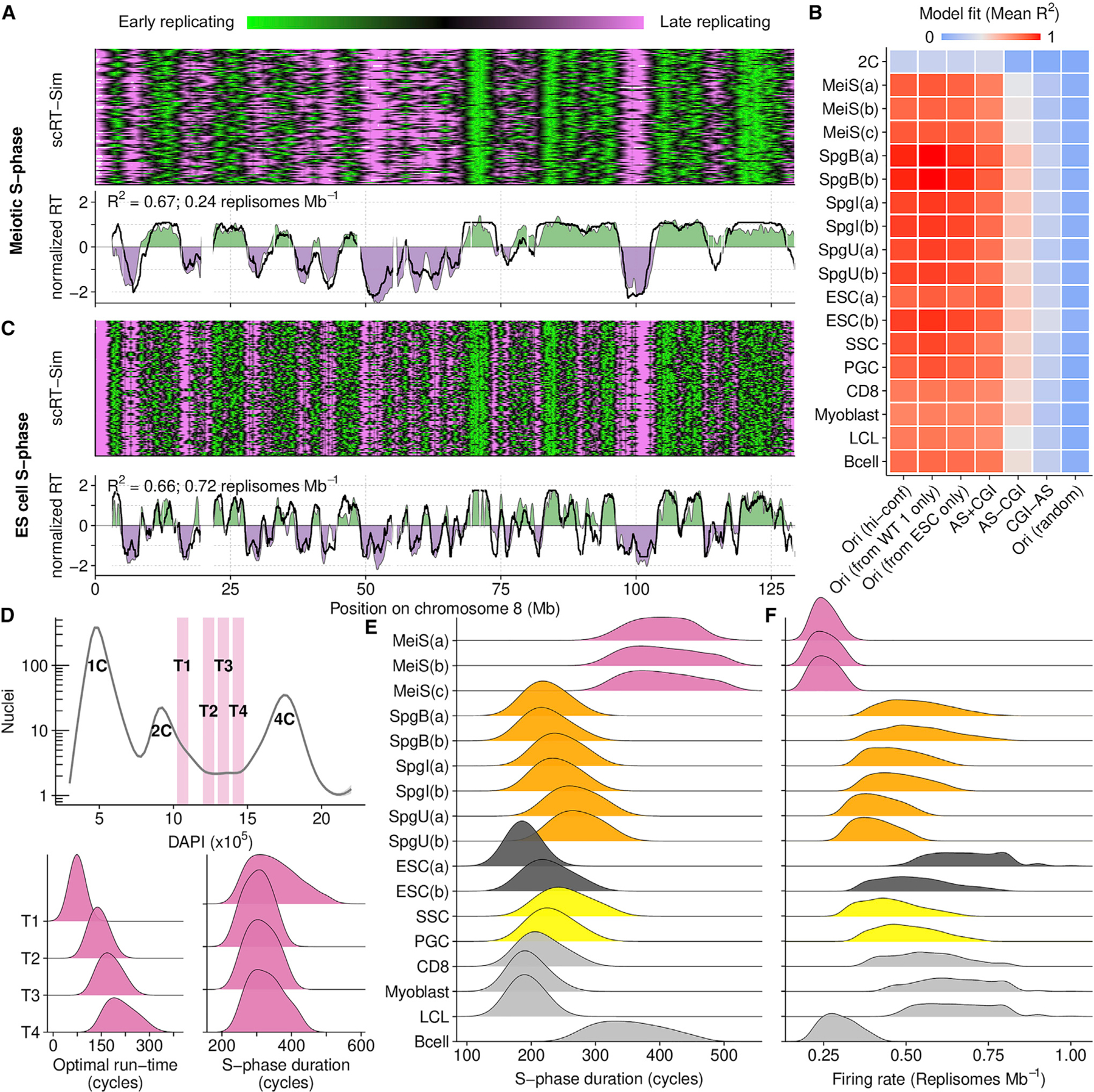
(A) Best fitting RT-sim model for meiotic S-phase RT (MeiS(a)). Heatmap shows RT in individual simulated haploid genomes. Lower panel compares experimentally determined RT (filled area) to simulated RT (black line). Both simulated and experimental RT are normalized by mean and SD ((RT — mean(RT))/SD(RT)).
(B) Fit scores for models built using RT from different cell types and different datasets as a proxy for origins. Mean of the top 0.015% of models is shown. Sample details in Tables S1, S2, and S3. Briefly, RT is from: MeiS, meiotic S-phase; Spg(B,I,U), B-type, intermediate, and undifferentiated spermatogonia; ESCs, embryonic stem cells; SSCs, spermatogonial stem cells; PGCs, primordial germ cells; CD8, CD8 cells; myoblast, myoblast cell line; LCL, lymphoblastoid cell line; Bcells, activated B cells. (a,b,c) designate replicates. ESC(a) is from our ESC culture, ESC(b) is from published whole-genome sequencing data. Ori (hi-conf), high confidence Ori-SSDS peaks; Ori(r), randomized Ori-SSDS peaks; AS+CGI, ATAC-seq peak at a CGI; AS-CGI, ATAC-seq peak not at a CGI; CGI-AS, CGI not at an ATAC-seq peak.
(C) Best fitting RT-Sim model for RT in ESC(a).
(D) Isolation of S-phase nuclei with increasing DNA content (T1–T4). The optimal simulation run-time for best-fitting models correlates with increasing DNA content. Nonetheless, the predicted S-phase duration is similar among all populations.
(E) S-phase duration estimates from RT-sim.
(F) Replisome firing rate estimates from RT-sim.
See also Figures S5, S6, and S7 and Data S1.
Reduced origin firing lengthens S-phase in meiosis
To validate that RT-sim can explain meaningful properties of replication, we examined model run-time of best-fitting models for very early, early, middle, and late S-phase nuclei (Figure 4D). Optimal model runtime—defined as the time at which simulated RT best fits experimental RT—should reflect the time a population has spent in S-phase. Indeed, optimal runtime got progressively longer from very early through late S-phase populations (Figure 4D). Importantly, differences in experimental RT do not substantially affect model estimates of S-phase duration (Figure 4D); accurate estimates of S-phase duration are difficult to obtain in mammalian meiosis (Kofman-Alfaro and Chandley, 1970). We find that for best-fitting models, median S-phase duration in meiosis is 1.4- to 1.8-fold longer than in spermatogonia (Figures 4E and S7), despite having highly correlated experimentally measured RT (Figure 3G). In Saccharomyces cerevisiae, the slow-down of DNA replication in meiosis may be partly to facilitate recombination, because knocking out Spo11—the protein that makes meiotic DSBs—reduces S-phase duration by 30% (Cha et al., 2000). We found no reduction in meiotic S-phase duration in Spo11−/− mice (Figure S7).
The extended duration of meiotic S-phase results from the use of fewer replisomes; best-fitting models use 0.25 ± 0.04 replisomes Mb−1 in meiosis (mean ± SD; ~675 replisomes per haploid genome; 2,700 Mb genome), compared to 0.69 ± 0.10 replisomes Mb−1 in ESCs (~1,850 replisomes per haploid genome) (Figures 4F and S7). The estimates from other cell types are similar to the replisome count in cultured mouse C2C12 cells (imaging-based estimates; 0.46–0.48 replisomes Mb−1) (Chagin et al., 2016). One consequence of using fewer origins in meiosis is extended replication tract length (Figure S5). This mirrors findings in newt spermatocytes, where replication tracts were notably longer than in mitotic cells (Callan, 1973). Although we do not explicitly model fork speed, universally altering fork speed without changing origin density cannot explain the presence of longer replication tracts in meiosis. Indeed, in other organisms, fork speed does not vary between meiosis and mitosis (Borde et al., 2000; Callan, 1973). By extrapolating from published estimates of S-phase duration in intermediate-stage spermatogonia (12.5 hr) (Monesi, 1962), we estimate that meiotic S-phase in mice takes 21–24 h. Interestingly, the range of S-phase duration estimates for the best-fitting models of meiotic S-phase is larger than that of the other populations (Figures 4E and S7). A similar spread in meiotic S-phase duration is seen in yeast (Cha et al., 2000).
Meiotic recombination is temporally and spatially correlated with DNA replication
Programmed DSB formation in meiosis occurs after DNA replication. In Saccharomyces cerevisiae, passage of the replication fork favors meiotic DSB formation (Murakami and Keeney, 2014). This interplay is unexplored in mammalian genomes. In mice and humans, local DSB patterning is determined by the sequence-specific binding of PRDM9 (Baudat et al., 2010; Myers et al., 2010), yet at megabase scales, DSB density is highly correlated between individuals with different PRDM9 alleles (Davies et al., 2016; Smagulova et al., 2011). Because RT domains occur over megabase scales, we asked whether DNA replication may underlie the megabase-scale control of DSB patterning.
Meiotic DSBs form through a well-documented cascade; PRDM9 tri-methylates H3K4 (Brick et al., 2012; Diagouraga et al., 2018) and/or H3K36 (Diagouraga et al., 2018; Powers et al., 2016) that appear to recruit the DSB complex. A DSB is made by SPO11, which is then released with a short oligonucleotide (Bergerat et al., 1997; Keeney et al., 1997; Neale et al., 2005). The 5′ DNA is resected, and DMC1 loads on exposed ssDNA to facilitate homologous recombination (Bishop et al., 1992). DSBs ultimately repair as a crossover (CO) or as a non-crossover (NCO). H3K4me3 ChIP-seq, SPO11-associated oligo mapping (Lange et al., 2016), DMC1 ChIP-single-stranded DNA sequencing (Brick et al., 2012), and repair outcome mapping (COs/NCOs) provide quantitative readouts of intermediates in this cascade (Figure 5A).
Figure 5. Rapid DSB repair and elevated meiotic recombination in early-replicating regions.
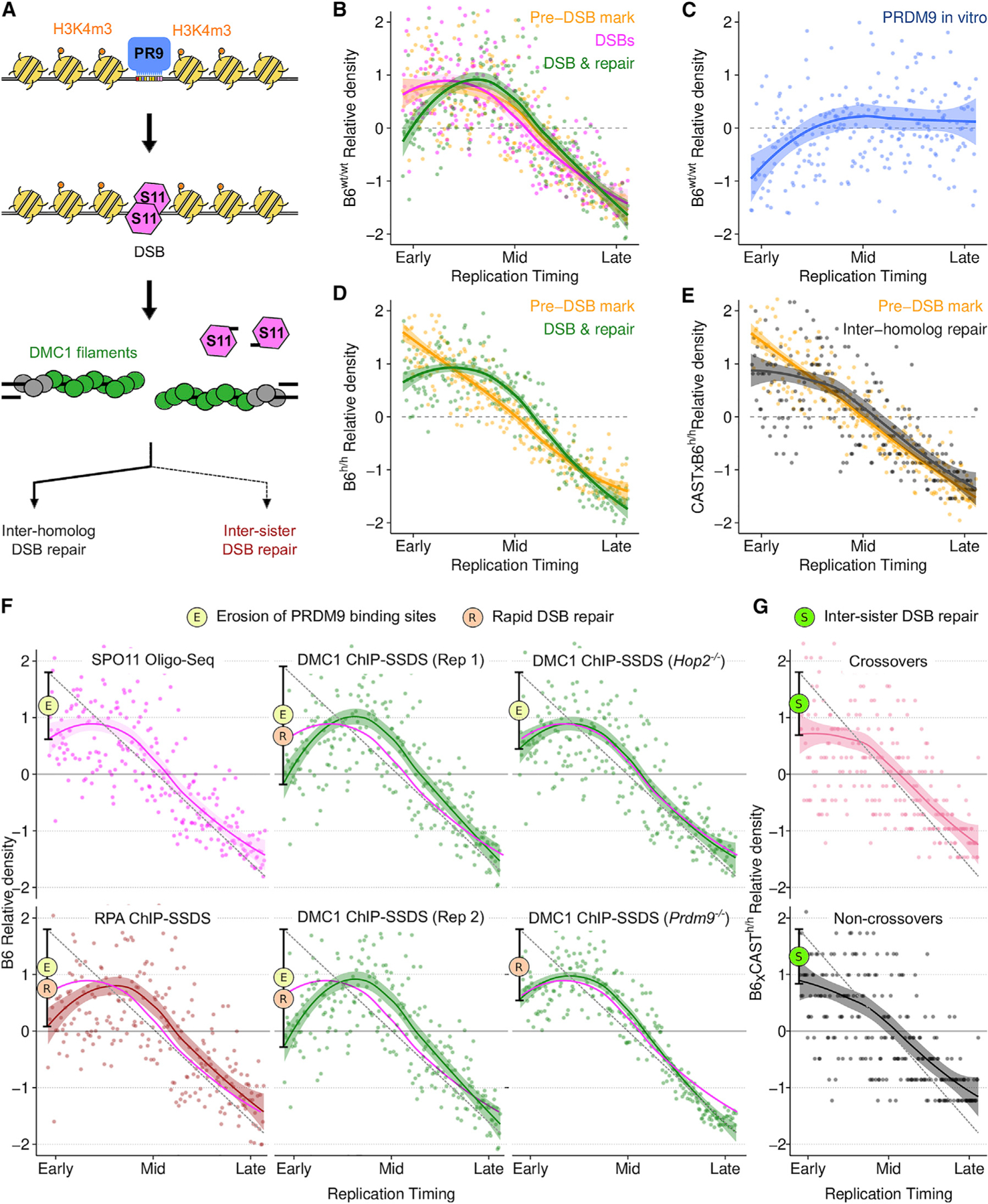
(A) Schematic of meiotic recombination (colors in B–F reflect this schematic; PR9, PRDM9; S11, SPO11).
(B and D) PRDM9-mediated H3K4m3 at DSB hotspots (yellow) is enriched in early replicating DNA in both wild-type B6 male mice (B6wt/wt) (B) and in mice with a humanized PRDM9 allele (B6h/h) (D). DSB formation (SPO11-oligo mapping; pink) follows this Pre-DSB mark (B). The DMC1-SSDS (green) signal decays relative to H3K4me3 in the earliest replicating DNA.
(C) B6 PRDM9 binding sites are depleted in early replicating DNA (measured by Affinity-seq) in the B6wt/wt genome.
(E) Inter-homolog repair products (crossovers + non-crossovers) (Li et al., 2019) are depleted in the earliest replicating DNA relative to DSB-associated H3K4m3 (H3K4m3 data as in D, plotted against B6xCAST RT).
(F) The rate of DSB formation (magenta) is superimposed on all panels. RPA-SSDS signal is depleted in the earliest replicating DNA. Two replicates of DMC1-SSDS are shown (Rep 1 is also in B). Note that PRDM9 binding site erosion does not affect PRDM9-independent DSB hotspots.
(G) Both crossovers (COs) and Non-crossovers (NCO) are depleted in the earliest replicating DNA. For all figures, dots represent the average signal from all autosomal bins for each RT quantile (N = 250). Simulated RT is from the T1 B6 meiocyte population (B–D and F) or from a B6xCAST F1 hybrid (E and G). RT patterns are very similar in B6 and B6xCAST. Solid lines depict a LOESS smoothed signal ± SE (shaded; span = 0.8). The dashed gray line is a projected linear correlation and the deviation from this is shown (black bar) (F and G). Phenomena contributing to each dip are indicated by colored circles.
See also Data S1.
We found that all measures of recombination are enriched in the early replicating regions (ERRs) (Figure 5). Nonetheless, informative differences are apparent. PRDM9-mediated H3K4m3 and DSB frequency are similarly enriched relative to RT (Figure 5B). Both profiles ‘‘flatten’’ in the earliest replicating DNA and this likely reflects hotspot erosion (Boulton et al., 1997; Myers et al., 2010), a process by which strong PRDM9 binding sites are purged from the genome (Figure 5C). Hotspot erosion is indicative of historical recombination, reinforcing the strong association between RT and recombination. Thus, the action of PRDM9 decouples replication and recombination through the erosion of PRDM9 binding sites. In contrast to PRDM9-mediated H3K4me3 and SPO11-oligo density, DSB repair intermediates (DMC1-SSDS and RPA-SSDS) are relatively depleted in the very earliest replicating regions (Figures 5B, 5D, and 5F). SSDS captures both the frequency (Lange et al., 2016) and the lifetime (Pratto et al., 2014) of DSB repair intermediates. Because DSB frequency closely mirrors PRDM9-mediated H3K4me3, we conclude that DSBs forming in ERRs are more rapidly repaired than those elsewhere. A similar, but less pronounced pattern may also implicate rapid repair in late replicating regions (Figures 5B and 5D). The signature of rapid repair is not seen in DMC1-SSDS from a mouse in which all meiotic DSBs remain unrepaired (Hop2−/−) (Figure 5F). Rapid DSB repair in ERRs is seen in mice that lack PRDM9 (Prdm9−/−) (Figure 5F) and is therefore independent of the mechanisms that determine the local patterning of meiotic DSBs.
To simplify the study of repair outcomes, we turned to mice homozygous for a ‘‘humanized’’ PRDM9 allele; this allele has a binding preference not found naturally in mice, and therefore, has left no footprint of hotspot erosion. In these mice, PRDM9-mediated H3K4me3 is linearly correlated with RT (Figure 5D), whereas the DMC1-SSDS signal still shows a relative depletion, indicative of rapid repair. We found that all inter-homolog repair products (COs and NCOs) were depleted in the earliest replicating DNA, where rapid DSB repair is occurring (Figures 5E and 5G). The ‘‘missing’’ repair outcomes likely result from DSBs that use the sister chromatid as a repair template (sister chromatids are genetically identical thus repair products cannot be detected). Inter-sister DSB repair is generally disfavored in meiosis to assure crossover formation between homologs. This inter-homolog bias appears to be gradually established, such that the earliest-forming DSBs can still repair from the sister chromatid (Joshi et al., 2015; Sandhu et al., 2020). Our findings therefore imply that DSBs at the earliest replicating DNA also form early in meiosis.
Chromosome-scale regulation of recombination is predicted by DNA replication
Chromosome-scale regulation of recombination is a PRDM9-independent aspect of recombination patterning. Short chromosomes are statistically less likely to receive a crossover-competent DSB, yet mechanisms exist to assure that they do (Duret and Galtier, 2009; Kaback et al., 1992; Murakami et al., 2020). In mouse males, short chromosomes have a higher crossover density (Figure 6A), however, much of the variance in crossover density remains unexplained. Origin density varies 3-fold among mouse chromosomes (chr11 = 122 Mb, 1,023 origins; chr13 = 120 Mb, 327 origins) and is positively correlated with crossover density (Spearman R2 = 0.4). We derived a more integrated metric of replication from RT-sim models that measures how quickly chromosomes are replicated relative to each other (replication speed) (Figure 6B). This accounts for stochastic origin firing and competition between chromosomes for limiting firing factors. Alone, replication speed can predict crossover density better than origin density (Figures 6C and 6D) and when coupled with chromosome size in a multiple linear regression model can explain 90% of the per-chromosome variance in crossover density (Figures 6E and 6F). This model is only marginally improved by including estimates of DSB frequency (Figure 6G). Strikingly, this implies that the recombination potential of chromosomes is mostly established before DSB formation.
Figure 6. DNA replication predicts per-chromosome crossover rates.
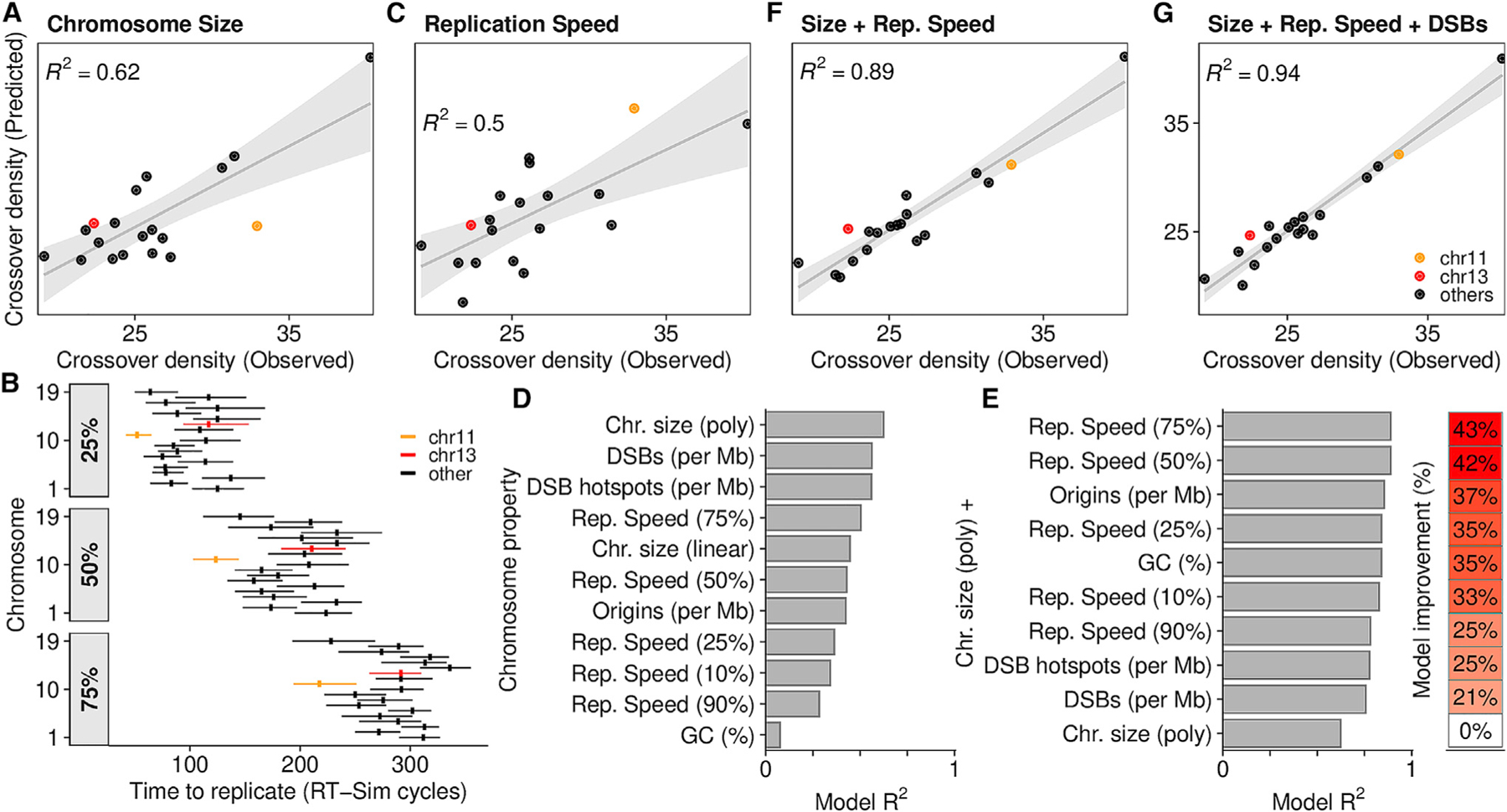
(A) A linear regression model can predict crossover density from chromosome size (polynomial fit). Crossover density = total crossovers per Mb. Data from Yin et al. (2019). R2 is skewed by a single outlier (chr19; top right).
(B) Chromosomes replicate asynchronously. chr11 and chr13 are highlighted. Despite being a similar size, chr11 replicates earlier. The time required to replicate 25%/50%/75% of each chromosome is inferred from the best-fitting RT-sim model. Horizontal lines, interquartile range of times from individual models; vertical line, median.
(C) Linear regression to predict crossover density from replication speed.
(D) Individual properties of chromosomes can predict crossover density. The Pearson R2 of predicted versus observed crossover density for each model is shown.
(E) Linear models using chromosome size in addition to other properties better predict crossover density.
(F) Per chromosome crossover density is predicted by a linear model that combines replication speed (75%) and chromosome size.
(G) Adding DSB frequency (DMC1-SSDS in B6xCAST F1 mice) (Smagulova et al., 2016) yields a slightly better model.
See also Data S1.
Distinct subtelomeric patterning of DNA replication in the human male germline
In human males, like in mice, meiotic DSBs exhibit megabase-scale correlations across the genome (Figure 7A). An overt manifestation of this phenotype is seen at the ends of chromosomes, where both DSBs (Pratto et al., 2014) and crossovers (Coop et al., 2008) are grossly enriched in males, independent of PRDM9 genotype (Figure 7B).
Figure 7. Subtelomeric DNA replicates early in human male meiosis.
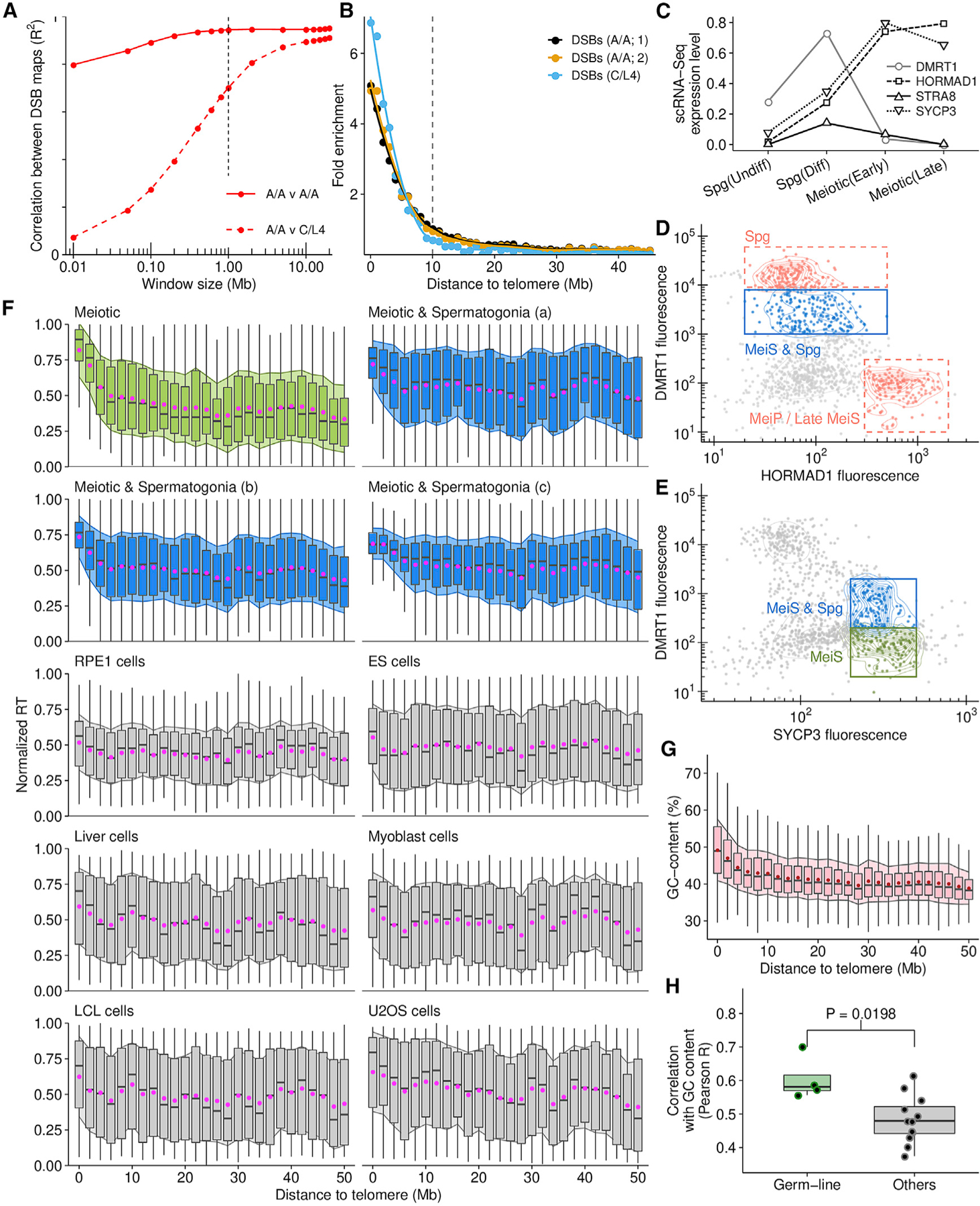
(A) DSB density correlates at megabase-scale in human males with different PRDM9 genotypes (PRDM9A/A homozygotes [A/A] and PRDM9C/L4 [C/L4] heterozygote).
(B) Points represent the average DMC1-SSDS signal in 1-Mb bins, normalized to interstitial regions (>30 Mb).
(C) Stage-specific expression of meiotic markers (Guo et al., 2018).
(D) Putative meiotic S-phase nuclei gated on DMRT1 and HORMAD1 (blue) contain spermatogonia (manual inspection).
(E) A pure meiotic S-phase population (green) is isolated using DMRT1 and SYCP3.
(F) Subtelomeric DNA replicates consistently early in meiotic (green, blue) but not in somatic cells (gray; Table S3). Boxplots depict the interquartile range of replication timing values in 2-Mb regions across the genome; gray bar, median; magenta dot, mean ± 1 SD (filled shadow).
(G) GC content is elevated in subtelomeric DNA.
(H) RT in the germline (all meiotic samples) correlates better with genomic GC content than RT in other cell types.
See also Data S2.
We inferred replication timing from meiotic S-phase nuclei from human testes (Figures 7C–7E). Subtelomeric regions of human chromosomes replicate early and therefore, germline RT is highly correlated with DSB hotspot density (R2 = 0.75) (Figures 7B and 7F). The distal pattern of early replication in male meiosis is highly distinct, as subtelomeric DNA does not replicate notably early in any of the other cell types we studied (Figure 7F). This germline-specific RT patterning may explain why only a weak link was found between RT in a lymphoblastoid cell line (LCL) and meiotic recombination (Koren et al., 2012). GC-content is also elevated in subtelomeric DNA (Figure 7G); thus, RT in the germline is better correlated with genomic GC-content than RT in other cell types (Figure 7H). Importantly, the earliest meiotic DSBs in human males are detected almost exclusively in subtelomeric DNA (Pratto et al., 2014). Thus, early DNA replication in distal regions in the germline appears to underlie the spatiotemporal patterning of meiotic recombination in human males.
DISCUSSION
Aside from a cadre of classical papers in the 1970s, few studies have addressed the intricacies of meiotic DNA replication in mammals at a molecular level. Here, we developed a framework to study DNA replication in vivo.
To map origins of replication in the few S-phase cells found in the mammalian testis, we adapted methods for nascent-strand sequencing to retain the directionality of captured strands. This generated a high-confidence map of ~11,500 origins of replication in the testis, and paves the way for future studies in other cell types. Replication origins appear to be determined by the combination of open chromatin and CpG density and the origins identified in testis and cultured ESCs mostly coincide. These likely represent a core set of cell-type-agnostic replication origins. This does not negate the possibility of low-efficiency, environment dependent or cell-type specific origins (Smith et al., 2016).
Despite using the same population of replication origins, we exposed fundamental differences in S-phase duration among cell-types through in silico modeling. Meiotic S-phase is notably long in a variety of organisms (Bennett et al., 1972; Callan, 1973; Cha et al., 2000; Holm, 1977). We found that meiotic S-phase in mice is ~1.8 times longer than in the germ-cells that precede meiosis; experimental estimates are ~15 h and ~30 h in Spermatogonia-B and Spermatocytes, respectively (Ghosal and Mukherjee, 1971; Monesi, 1962). To accurately model replication, a limiting factor that caps the number of active replisomes is required (Gindin et al., 2014). We found that this replisome ‘‘ceiling’’ differs among cell types, and, in turn, modulates S-phase duration. Investigating the factors that slow down meiotic S-phase may help identify genes that modulate S-phase duration more generally. In yeast, the meiosis-specific Spo11p protein may play a role, however, we found that SPO11 does not regulate S-phase duration in mouse meiosis. Our model only allows alteration of the global properties of replication, however, regional modifiers of replication fork speed such as replication slow zones (Cha and Kleckner, 2002) or common fragile sites (Smith et al., 2006) may impede fork progression differentially in meiosis and other cell types.
Together, these analyses suggest both that the origins captured by Ori-SSDS reflect meiotic replication initiation and that the same origins can yield tangibly different RT profiles if other properties of replication vary. This tripartite approach of origin mapping, RT-seq and in silico modeling, offers an alternative to classical cytogenetic approaches and yields a more comprehensive description of the DNA replication landscape.
Our tripartite approach to describe and parameterize DNA replication enabled us to infer meiotic RT with high temporal resolution in early replicating DNA. This strategy offers an alternative to recently developed experimental methods (Zhao et al., 2020) that would be difficult to apply to replication in mammalian meiosis. We found a strong positive correlation between multiple metrics of recombination and early replicating DNA. Opportunistic binding of PRDM9 to more accessible chromatin in early replicated DNA may establish a link between replication and recombination, however, we also found analogous enrichment in the absence of functional PRDM9. Therefore, broad-scale patterning of recombination acts independent of the factors that determine the local patterning of meiotic DSBs.
We found strong evidence for the rapid repair of meiotic DSBs in early replicating parts of the genome. The rapid repair of DSBs is a correlate of crossover-biased resolution (Hinch et al., 2019), however, we find that all interhomolog repair outcomes are depleted in these regions suggesting increased use of the sister-chromatid as a repair template. In meiosis, DSB repair with the sister chromatid is strongly disfavored and, at least in yeast, this inter-homolog bias is established gradually. Thus inter-sister recombination is more likely at the earliest DSBs (Joshi et al., 2015; Sandhu et al., 2020). We therefore infer that the earliest DSBs occur in the earliest replicating DNA, strongly implicating a mechanistic link between replication timing per se and DSB formation. This would also imply that correlates of replication timing such as GC-content, genome compartmentalization, heterochromatin, or gene density, which lack any temporal component, may indirectly affect recombination by modifying replication patterns. In human males, the earliest meiotic DSBs occur almost exclusively in subtelomeric DNA (Pratto et al., 2014). This coincides with the earliest replicating DNA, reinforcing that there may be a direct link between replication and recombination initiation. It is possible that two phenomena modulate recombination at large scales; one that temporally favors early-forming DSBs in the wake of a passing replication fork (like in yeast), and another that favors recombination in the resultant ‘‘permissive’’ chromatin environment. Because DNA replication establishes the 3D structure of the genome (Klein et al., 2021), these two effects may be one and the same.
It is well established that chromosome size partly determines per chromosome recombination rates (Duret and Galtier, 2009; Kaback et al., 1992; Murakami et al., 2020). We found that in mouse males, the per-chromosome origin density has a similar predictive value; further, the predictive power of chromosome size and replication are additive, implying that they represent uncorrelated aspects of the mechanisms controlling per chromosome recombination rates. Together, almost all of the per chromosome variation in crossover density can be explained by these two properties, suggesting that recombination will follow a deterministic path, established before DSBs are made. Short chromosomes likely benefit from an elevated recombination rate to assure crossover formation. Fast-replicating chromosomes are origin rich, GC-rich, and have elevated gene density. A mechanism that links replication to recombination would assure that such regions benefit from recombination to break linkage blocks, generate diversity, and purge deleterious mutations.
Our finding that subtelomeric DNA replicates notably early in the human male germline has implications for understanding the patterning of de novo variation in the genome and its impact on population genetic structure. In human males, germline DNA replication mirrors GC content more closely than replication in other cell types. Thus, during development, we propose that replication may follow a well-charted course, dictated by the underlying DNA and its biophysical properties. The commitment to differentiation in other cell types may render replication more susceptible to epigenetic regulation (Hiratani et al., 2010). One intriguing possibility is that the link between replication, recombination, and GC content is a self-reinforcing cycle; GC-rich regions replicate early, facilitating recombination that increases GC content via GC-biased gene conversion.
Limitations of the study
We restricted peak calling to a high confidence set of Ori-SSDS peaks in testis. Approximately 40% more peaks can be identified in individual experiments and more by relaxing peak calling thresholds. By examining Watson-Crick asymmetry, a substantial fraction of additional peaks appear to represent low efficiency origins.
Although replication origins can be unambiguously mapped in mammalian tissue, a caveat of this study is that the origins we identified from testis are not uniquely from meiotic cells. Further work is required to adapt our method for sorted populations of cells/nuclei.
In our simulations, the final 5%–10% of the genome takes far too long to replicate because at that point, all origins have either fired or have been passively replicated. Allowing for either an increase in the speed of DNA replication or the firing of diffuse origins in the latest replicating parts of the genome would rectify this. However, we have no experimental data to support either phenomenon.
We identify robust and sensible temporal and spatial correlates of replication and recombination but have not demonstrated a mechanistic link. Genetic perturbations of the replication machinery are challenging (Sima et al., 2019). The data from our experiments and the methods we developed will enable such experiments in the future.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, R. Daniel Camerini-Otero (rdcamerini@mail.nih.gov).
Materials availability
Mouse lines and antibodies generated in this study are available on request, with completion of an MTA.
Data and code availability
Sequencing data are deposited at the GEO (GSE148327). Replication origin locations, replication timing data, and associated data used to generate figures are available in Data S1 (mouse) and Data S2 (human). Custom code is available at Zenodo:
| Analytic pipeline | https://zenodo.org/record/4634002 |
| RT-Seq pipeline | https://zenodo.org/record/4634128 |
| RT-sim | https://zenodo.org/record/4634118 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
C57BL/6J (stock 000664) and CAST/Eij (000928) mice were obtained from The Jackson Laboratory (Maine).
The Spo11 null mice (Spo11−/−) were generated by deleting the 13 kb spo11 genomic locus with CRISPR-Cas9. Briefly, two target sites (upstream of spo11: TAGAGCGCGGAAAGGTTCGC; downstream of spo11: CGAACCTTGAGAGGTTGCGA) were selected using CRISPR.mit.edu. The gRNA-targeting vectors were constructed by incorporating the annealed oligos containing target sites into the PX459 vector (Ran et al., 2013). The vectors carrying each gRNA were injected simultaneously into the pronucleus of zygotes obtained from C57BL/6J (stock 000664) mice. The injected zygotes were implanted into the oviduct of one pseudopregnant foster mother. To produce Spo11−/− mice, heterozygous mice were intercrossed. Mice were genotyped by PCR on tail tip DNA, using the primers Spo11NullF, Spo11NullR and Spo11ex4R. Spo11NullF and Spo11Ex4R give an ~700 bp product indicative of the wild-type allele, whereas Spo11NullF and Spo11NullR give an ~350 bp fragment if the targeted allele is present. The phenotype of these mice is indistinguishable from extensively studied Spo11 knockouts (Baudat et al., 2000; Romanienko and Camerini-Otero, 2000) previously generated.
All animal procedures were approved and performed according to the NIH Guide for the Care and Use of Laboratory Animals.
Cell lines
Mouse 129X1/SvJ-PRX-129X1 #1 Embryonic Stem Cells were obtained from Jackson labs: . These cells were derived from day 3.5 blastocysts of strain 129X1/SvJ (Stock Number 000691) and expanded on primary mouse embryonic fibroblasts (MEFs) in medium containing 1000U/mL LIF and cultured at 7.5% CO2 in humidified air at 37°C.
Human subjects
We obtained human testicular samples from two individuals. Individual 1 was a deceased 24-year-old white male; deidentified donor testes material was obtained with the assistance of the Washington Regional Transplant Community. Individual 2 was a 47-year-old white male; material was normal adjacent tissue from a testicular biopsy provided by a commercial source (Folio, Ohio.
METHOD DETAILS
Mapping origins of replication using short nascent-strand capture followed by single-stranded DNA sequencing (Ori-SSDS)
For the short nascent strands (SNS) preparation we adapted the protocol from Picard et al. (2014) to work with mouse tissue. Mice were euthanized and testes were retrieved. The tunica albuginea was removed and both testes were suspended in 10 mL of DNAzol and transferred to a Dounce homogenizer. Tissue and cells were disrupted with 5–7 strokes of a loose-fitting pestle. The DNA was precipitated with 5 mL of EtOH and then spooled with a pipette tip and rinsed gently twice with EtOH 75% by sequential transfer into 15 mL tubes. DNA was air-dried for 5 min and then resuspended in 500 μL of 1X TEN buffer (Tris 10mM pH = 8; EDTA 1mM; NaCl 100mM) + 80 U of RNase inhibitor (40 U/μl) at 4°C for at least 24h.
DNA was denatured at 95°C for 5 min, chilled on ice for 5 min and size-fractionated on 5 – 30% (w/v) linear sucrose gradients in TEN buffer in a Beckman Ultracentrifuge SW45Ti rotor for 18 h at 26,000 g at 20°C. 500 ul fractions were collected from the top of the gradient, precipitated by adding 1 mL of EtOH 100% and 50 μL of NaOAc 3M pH 5.2 and resuspended in 25 μL Tris Buffer pH = 8. Fractions were analyzed in a denaturing (50 mM NaOH, 1 mM EDTA) 1.2% agarose gel and the ones containing ssDNA ranging in size from 800 to 2000 nt were pooled. The sample was then heat-denatured for 5 min at 95°C and chilled on ice for 5 min and DNA was phosphorylated by Polynucleotide Kinase in 150 ul reactions in PNK Buffer (100 U of PNK; 1X PNK buffer; 1 mM ATP; 40U RNase Inhibitor). The reactions were incubated for 30 min at 37°C and heat-inactivated at 75°C for 15 min. DNA was extracted twice with Phenol/Chloroform/Isoamyl, once with Chloroform/Isoamyl, EtOH precipitated and resuspended in 50 μL Tris Buffer pH = 8. Samples were heat-denatured again for 5 min and digested with lambda-exonuclease in 100 μL of custom lambda buffer (67 mM glycine-KOH pH = 8.8, 2.5 mM MgCl2, 50 μg/ml BSA) with 2.5 – 10 U lambda-exonuclease/μg DNA. Note that these estimates of DNA concentration are affected by the presence of RNA carried over from the sucrose gradient. From the denaturing agarose gels (above) we estimated the ssDNA concentration to be 3–5 ng/μl. This yields ~500 ng in total. Thus, the ratio of lambda-exo: ssDNA may be as high as 100U/μg. 80U RNase inhibitor, 5U of PNK and 1 mM ATP were added to the sample and incubated overnight at 37°C. Reactions were inactivated for 10 min at 75°C and DNA was purified with Qiaquick columns (QIAGEN), as indicated by the instruction manual. RNA primers were hydrolyzed by incubating the eluate for 5 min at 95°C in 0.1 N NaOH, followed by neutralization with HCl and purified again with Qiaquick kit. The sample was recovered in 50 μL of EB buffer; the final concentration is typically below the detection limit for Nanodrop. The sample was then sonicated in a Bioruptor UCD200 for 8–10 min 30 s ON 30 s OFF, ‘‘high’’ setting.
Next, we used a library preparation protocol, SSDS, that allows for the direct ligation of sequencing adapters to ssDNA (for details of the molecular intermediates and underlying concepts of SSDS, see Figure 1A and Khil et al., 2012; for a comprehensive protocol, see Brick et al., 2018a). Briefly, Illumina sequencing adapters require a blunt-end for ligation. For most experiments, the end-repair of broken dsDNA creates these blunt-ends. In contrast, SSDS generates a blunt-end from ssDNA. For this, it relies on the intrinsic propensity of ssDNA to form intramolecular hairpin-loop structures. Processing of these hairpins with end-repair enzymes generates a blunt-end that allows the ligation of Illumina sequencing adapters directly to the ssDNA without first converting it to dsDNA. Illumina sequencing adapters are not fully complementary and have distinct sequences that ligate to the 5′ end and the 3′ end of DNA (known as P5 and P7 sequences, respectively; see Figure 1A). When sequencing dsDNA, both ends of a fragment have a 5′ end. Thus, adapters will ligate to both ends but on opposite strands. In contrast, with SSDS, since the blunt-end is created from an intramolecular reaction, the fragment has only a single 5′ end. During paired-end Illumina sequencing, this adaptor orientation assures that the first read is always derived from the 5′ end of the captured ssDNA fragment. One final detail of SSDS is that hairpin processing also introduces a molecular signature of reads derived from ssDNA. This is used to computationally identify reads derived from ssDNA and to exclude those from dsDNA (see Khil et al., 2012).
The first step of end repair was done in 1X T4 DNA ligase buffer with 10 mM ATP in the presence of dNTPs, 0.6 U T4 DNA polymerase, 0.5 U Klenow, 2 U T4 polynucleotide kinase for 30 min at 20°C. This temperature promotes the formation of hairpin loop structures. The stem of the loop is repaired in such a way that adapters can be ligated later on and importantly the polymerase-mediated fill in serves as a molecular signature for ssDNA (Figure 1A). The second step was done in the presence of 1 mM dATP and 1 U Klenow Exo-. Reaction was incubated at 37°C for 30 min and DNA was purified with MinElute kit. To further enrich for ssDNA the sample was denatured for 2 min at 95°C, then cooled to room temperature. The sequencing adaptor mix (TruSeq, Illumina) was diluted 1:100 and added for the ligation step. DNA was purified by MinElute kit and amplified using the KAPA HiFi HotStart Library Amplification Kit according to manufacturer’s instructions. DNA was purified with MinElute kit. Resulting libraries were sequenced with a HiSeqX in PE150 mode.
For one sample (wt_Rep2), separate libraries were made from three fractions from the sucrose gradient (Fraction 10: 800–1,000 nt, Fraction 11: 1,000–1,400 nt and Fraction 12: 1,400–2,000 nt). Sequencing data were subsequently pooled.
To validate that the Ori-SSDS enrichment stems from RNA primed leading strands, we performed experiments where we hydrolyzed the RNA prior to lambda exonuclease treatment. This also necessitated changes to the above Ori-SSDS protocol as follows. Selected fractions from the sucrose gradient were split in two tubes. In one tube, leading strand RNA primers were hydrolyzed by incubating for 30 min at 37°C in 0.25 N NaOH, followed by neutralization with acetic acid. The other tube was incubated for 30 min at 37°C but in Tris Buffer pH = 8. After neutralization, both tubes were heat denatured and spun through a Chromaspin TE-1000 size exclusion column to remove any small degradation products that could interfere with later steps. The eluate was then subjected to PNK treatment and subsequent steps outlined above. Two replicates were performed in parallel, with and without RNA hydrolysis (wt_Rep4, wt_Rep5).
Mouse ESC were obtained from Jackson labs: 129X1/SvJ-PRX-129X1 #1 mES cells derived from day 3.5 blastocysts of strain 129X1/SvJ (Stock Number 000691). The cells were cultured as recommended by Jackson labs in the presence of mitotically inactive MEF feeder cells. Exponentially growing cells were collected and 4x107 were used for origin detection. 8 mL of DNAzol was used for DNA extraction and then we followed the same protocol outlined for testes origin detection. For the RNase control, and aliquot of 4x107 was processed in parallel but before the PNK treatment, DNA was incubated with a mix of RNaseA/T and RNaseI in 1X TEN buffer (Tris 10mM pH = 8; EDTA 1mM; NaCl 100mM) for 30 min at 37°C.
Ori-SSDS in sperm was used as a negative control as there is no DNA replication. Sperm was retrieved from cauda and caput epididymides from 3 adult mice. The mix of somatic cells and sperm was resuspended in 0.1% SDS, 0.5% Triton X-100 to selectively lyse somatic cells. Sperm was recovered by centrifugation and lysed in 6 M guanidinium thiocyanate, 30 mM sodium citrate (pH 7.0), 0.5% Sarkosyl, 0.20 mg/ml proteinase K and 0.3 M Beta-mercaptoethanol, and incubated at 55°C for 1h (Hossain et al., 1997). The DNA was precipitated with 2 volumes of isopropyl alcohol and then spooled with a pipette tip and rinsed gently twice with EtOH 75% by sequential transfer into 15 mL tubes. DNA was air-dried for 5 min and then resuspended in 500 μL of 1X TEN buffer (Tris 10mM pH = 8; EDTA 1mM; NaCl 100mM) + 80 U of RNaseOUT (Thermo-Fisher catalog number 10777019) (40 U/μl) at 4°C for at least 24h.
Identification of origins of replication
Sequencing reads from Ori-SSDS experiments were aligned to the reference genome (mouse = mm10; human = hg38) using bwa 0.7.12 and the single-stranded DNA pipeline (Brick et al., 2018a; Khil et al., 2012). Only fragments derived unambiguously from ssDNA (ssDNA_type1) were used in subsequent analyses. ssDNA fragments where either read1 or read2 had mapping quality < 30 were discarded. Fragments from the mitochondrial chromosome and fragments in blacklisted regions of the genome were also discarded. Duplicate fragments were subsequently discarded.
We used the MACS algorithm for peak calling. To assure that data derived from Ori-SSDS fit the MACS peak model, we performed two operations: 1. The strand orientation of fragments was reversed prior to peak calling. 2. The data were passed to MACS as ssDNA fragment BED files (from SSDS pipeline). Peak calling was performed for each Ori-SSDS sample using MACS 2.1.2 and the following parameters: -g (hs or mm) -q 0.001 -extsize 2000–nomodel–nolambda.
The final origin set for mouse was defined by merging origin calls from wt_Rep1,2,3. Origin intervals overlapping by ≥ 500 bp were merged. Origin efficiency was calculated in each sample using a method analogous to that for DMC1-SSDS hotspot mapping (Brick et al., 2018a). Briefly, each origin is re-centered to the midpoint of the Watson and Crick fragment distributions. Signal is estimated as the Crick-strand signal to the left of the origin center + the Watson-strand signal to the right of the origin center. The background is estimated by counting the remaining fragments at the left and right edges of MACS-defined origins (15%); Watson-strand fragments to the left of the origin center, Crick-strand fragments to the right. This background is then extrapolated to the entire origin and subtracted from the signal. Following this process, origins found in just one sample and origins that did not display the expected Crick/Watson asymmetry (Figure 1A) in any sample were discarded.
Identification of CpG peaks
CpG dinucleotide density was evaluated in all 1 Kb regions in the genome using a 100 bp sliding window. Intervals with > 5% CpGs were expanded ± 100 bp around the center and overlapping windows were merged. The centerpoint of each merged interval was defined as a CpG ‘‘peak.’’
Whole-genome sequencing for replication timing
Nuclei extraction and isolation of specific nuclei types from testis
Nuclei from mouse or human testes were prepared as described in Lam et al. (2019). For experiments with mouse we used between 2 to 6 testes per sort. For humans we used ~200 mg of tissue. To purify the populations of interest, a combination of intranuclear markers were used (Table S1; Figure 3). The following markers were used:
STRA8 is required for meiotic entry (Anderson et al., 2008). It is expressed at low levels in a subset of spermatogonia and expression increases upon meiotic entry (Green et al., 2018; Guo et al., 2018). STRA8 levels are very low in human compared to mouse, therefore we use other markers (HORMAD1 and SYCP3)
DMRT1 is required to regulate meiotic entry (Matson et al., 2010). It is expressed at high levels in spermatogonia and sertoli cells. Expression is greatly reduced upon meiotic entry (Green et al., 2018; Guo et al., 2018).
SYCP3 is a component of the chromosomal scaffold in meiosis (Yuan et al., 2000). It is expressed at low levels in spermatogonia and expression is greatly increased upon meiotic entry (Green et al., 2018; Guo et al., 2018).
HORMAD1 is a component of the chromosomal scaffold in meiosis (Wojtasz et al., 2009). It is expressed at low levels in differentiating spermatogonia and expression increased upon meiotic entry (Green et al., 2018; Guo et al., 2018).
yH2Ax is a marker of DNA damage (Fernandez-Capetillo et al., 2004). Expression is elevated upon meiotic entry (Green et al., 2018; Guo et al., 2018).
REC8 is a meiosis-specific cohesin (Watanabe, 2004). It is expressed at low levels in spermatogonia and expression is greatly increased upon meiotic entry (Green et al., 2018; Guo et al., 2018).
Each population was visually inspected for purity. The meiotic populations are notably high purity (Figure 3D). Assessing the purity of spermatogonia substages is challenging. Undifferentiated and intermediate stage spermatogonia populations (Figure 3C) are reproducibly of high purity. The Spermatogonia B population (Figure 3C) exhibited varying degrees of purity across experiments. A majority of these nuclei are Spermatogonia B, however some A-type spermatogonia are also captured by this gating strategy.
All antibodies are listed in the Key resources table. To raise custom polyclonal antibodies against REC8, a peptide (AEDEKSRTSLIPPEWWAWSEEGQPEPP) from mouse REC8 was synthesized and was used to immunize two rabbits (NeoScientific, Cambridge, MA). The immobilized peptide was subsequently used for affinity purification of REC8 antibodies from the rabbit serum.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Goat polyclonal anti-DMC1 | Santa Cruz Biotechnology | Cat# sc-8973, RRID: AB_2091206 Discontinued |
| Mouse monoclonal anti-DMRT1 (SS6) | Santa Cruz Biotechnology | Cat# sc-101024, RRID: AB_2277252 |
| Mouse monoclonal anti-SYCP3 (D-1) | Santa Cruz Biotechnology | sc-74569, RRID: AB_2197353 |
| Rabbit polyclonal anti-STRA8 | Abcam | Cat# ab49602, RRID: AB_945678 |
| Rabbit polyclonal anti-HORMAD1 | GeneTex | Cat# GTX119236, RRID: AB_11178755 |
| Rabbit monoclonal anti-Phospho-Histone H2A.X (20E3) | Cell Signaling Technology | Cat# 9718, RRID: AB_2118009 |
| Rabbit polyclonal anti-REC8 | This paper | N/A |
|
| ||
| Biological samples | ||
|
| ||
| Human testicular biopsy from cancer patient (Normal adjacent tissue) | Folio Biosciences now Discovery Life Sciences biospecimen bank | https://www.dls.com |
| Human testis | Deidentified deceased donor – Washington Regional Transplant Community | https://www.beadonor.org/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Acetic acid | Mallinckrodt | Cat# 2504 |
| Agarose | Invitrogen | Cat# 16500500 |
| AMPure XP beads | Beckman Coulter | Cat# A63881 |
| ATP solution 100 mM | Thermo Fisher Scientific | Cat# R0441 |
| Chloroform-isoamyl alcohol mixture (24:1) | Sigma-Aldrich | Cat# C0549 |
| cOmplete Mini Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| DAPI | Sigma-Aldrich | Cat# D8417 |
| DNA Polymerase I Large (Klenow) Fragment | New England Biolabs | Cat# M0210 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004D |
| EDTA 0.5 | KD Medical | Cat# RGF3130 |
| EGTA | Sigma-Aldrich | Cat# E3889 |
| Glycine | MP Biomedicals | Cat# 808822 |
| Guanidinium thiocyanate | Sigma-Aldrich | Cat# G9277 |
| HCl | Mallinckrodt | Cat# H613 |
| IGEPAL-CA630 | Sigma-Aldrich | Cat# 18896 |
| Klenow fragment (exo-) | New England Biolabs | Cat# M0212 |
| KOH | Sigma-Aldrich | Cat# 5958 |
| Lambda Exonuclease | Thermo Fisher Scientific | Cat# EN0561 |
| LiCl 8M | Sigma-Aldrich | Cat# L7026 |
| NaCl 5M | KD Medical | Cat# RGF3270 |
| NaHCO3 | Sigma | Cat# S5761 |
| NaOH | Mallinckrodt | Cat# 7708 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Phenol-chloroform-isoamyl alcohol mixture (25:24:1) | Sigma-Aldrich | Cat# 77617 |
| Polynucleotide Kinase | Thermo Fisher Scientific | Cat# EK0031 |
| Proteinase K | New England Biolabs | Cat# P8107S |
| Quick ligation kit | New England Biolabs | Cat# M2200 |
| RNase Inhibitor, Murine | New England Biolabs | Cat# M0314 |
| RNaseA/T1 | Thermo Fisher Scientific | Cat# EN0551 |
| RNaseI | Lucigen | Cat# N6901K |
| Sarkosyl | Sigma-Aldrich | Cat# 61747 |
| SDS 10% | KD Medical | Cat# RGE3230 |
| Sodium Acetate pH = 5.2 3M | Cellgro | Cat# 46033CI |
| Sodium citrate | Mallinckrodt | Cat# 0754–12 |
| Sodium deoxycholate | monohydrate Sigma-Aldrich | Cat# D5670 |
| Sucrose | MP Biomedicals | Catt# 152584 |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | Cat# B0202 |
| T4 DNA polymerase – SSDS library prep | New England Biolabs | Cat# M0203 |
| Tris pH = 8.0 1M | KD Medical | Cat# RGF3360 |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
|
| ||
| Critical commercial assays | ||
|
| ||
| DNAzol | Thermo Fisher Scientific | Cat# 10503027 |
| Qiaquick PCR purification kit | QIAGEN | Cat# 28104 |
| MinElute PCR purification kit | QIAGEN | Cat# 28004 |
| Chromaspin TE-1000 | Takara | Cat# 636079 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32851 |
| Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit | Invitrogen | Cat# C10420 |
| TruSeq Nano DNA Low Throughput Library Prep Kit | Illumina | Cat# 20015964 |
| KAPA HiFi HotStart Library Amplification Kit | Roche | Kit Code KK2620 Cat# 07958978001 |
| KAPA Hyper Prep Kit | Roche | Kit Code KK8502 Cat# 07962347001 |
|
| ||
| Deposited data | ||
|
| ||
| ATAC-Seq in mouse differentiated cKIT+ spermatogonia | Maezawa et al., 2018 | SRA:SRR5956508 |
| ATAC-Seq in mouse embryonic stem cells | GEO: GSE113428 | SRA:SRR7048437,SRR7048438 |
| ATAC-Seq in mouse embryonic stem cells at 6 Days | GEO: GSE113428 | SRA:SRR7048433,SRR7048434 |
| ATAC-Seq in mouse hepatocytes | Li et al., 2019 | SRA:SRR6813698,SRR6813699 |
| ATAC-Seq in mouse hindlimb muscle cells E14.5 | Castro et al., 2019 | SRA:SRR8104383,SRR8104391 |
| ATAC-Seq in mouse mouse embryonic fibroblasts | GEO: GSE113428 | SRA:SRR7048429, SRR7048430 |
| ATAC-Seq in mouse pachytene spermatocytes | Maezawa et al., 2018 | SRA:SRR5956512 |
| ATAC-Seq in mouse undifferentiated THY+ spermatogonia | Maezawa et al., 2018 | SRA:SRR5956504 |
| Crossover and non-crossover data for mouse | Li et al., 2019 | https://idp.nature.com/authorize?response_type=cookie&client_id=grover&redirect_uri=https%3A%2F%2Fwww.nature.com%2Farticles%2Fs41467-019-11675-y |
| Data from this study | This study | GEO:GSE148327 |
| DMC1-SSDS in B10.F-H2pb1/(13R)J (13R) mice | Smagulova et al., 2016 | GEO:GSM1954833 |
| DMC1-SSDS in C57BL6 mice (Rep T1) | Brick et al., 2018 | GEO:GSM2664275 |
| DMC1-SSDS in C57BL6 mice (Rep T2) | Brick et al., 2018 | GEO:GSM2664276 |
| DMC1-SSDS in C57BL6 mice with humanized PRDM9 allele | Davies et al., 2016 | GEO:GSM2049306 |
| DMC1-SSDS in C57BL6 × castaneus F1 hybrid mice | Smagulova et al., 2016 | GEO:GSM1954839 |
| DMC1-SSDS in C57BL6 × castaneus F1 hybrid mice | Davies et al., 2016 | GEO:GSM2049312 |
| DMC1-SSDS in castaneus mice | Smagulova et al., 2016 | GEO:GSM1954846 |
| DMC1-SSDS in Hop2−/− mice | Brick et al., 2018 | GEO:GSM3136743 |
| DMC1-SSDS in Prdm9−/− mice | Brick et al., 2018 | GEO:GSM2664291 |
| DMC1-SSDS in testis of PRDM9A homozygous human male (AA1) | Pratto et al., 2014 | SRA:SRR1528821 |
| DMC1-SSDS in testis of PRDM9A homozygous human male (AA2) | Pratto et al., 2014 | SRA:SRR1528831 |
| H3K4me3 ChIP-Seq in 12 dpp C57BL6 mice | Baker et al., 2014 | GEO:GSM1273023 |
| H3K4me3 ChIP-Seq in C57BL6 mice with humanized PRDM9 allele | Davies et al., 2016 | GEO:GSM1904284 |
| H3K4me3 ChIP-Seq in C57BL6 × castaneus F1 hybrid mice | Baker et al., 2015 | GEO:GSE60906 |
| Hi-C data from Zygonema | Patel et al., 2019 | GEO:GSE122622 |
| Input-SSDS in C57BL6 mice | Brick et al., 2018 | GEO:GSM2664289 |
| Input-SSDS in testis of PRDM9A homozygous human male (AA1) | Pratto et al., 2014 | SRA:SRR1528822 |
| Input-SSDS in testis of PRDM9A homozygous human male (AA2) | Pratto et al., 2014 | SRA:SRR1528832 |
| Okazaki-fragment sequencing in mouse ESCs | Petryk et al., 2018 | SRA:SRR7535256 |
| PRDM9 Affinity-Seq data | Walker et al., 2015 | GEO:GSE61613 |
| Processed data at DMC1-SSDS hotspots in C57BL6 mice | Brick et al., 2018 | https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-018-0492-5/MediaObjects/41586_2018_492_MOESM3_ESM.zip |
| Processed RT data from Human CyT49 Liver cells | Zimmerman and Gilbert, 2018 | ID: Int81158282; https://www2.replicationdomain.com/ |
| Processed RT data from Human FM01–154-001 Myoblast cells | Zimmerman and Gilbert, 2018 | ID: Int58331187 ; https://www2.replicationdomain.com/ |
| Processed RT data from Human GM06990 Lymphoblastoid cells | Zimmerman and Gilbert, 2018 | ID: Ext54054609; https://www2.replicationdomain.com/ |
| Processed RT data from Human H7 ES Cells | Zimmerman and Gilbert, 2018 | ID: Ext35479608; https://www2.replicationdomain.com/ |
| Processed RT data from human RPE1 cells | Takahashi et al., 2019 | GEO:GSM2904948 |
| Processed RT data from Human U2OS Bone epithelial cells | Zimmerman and Gilbert, 2018 | ID: Int66343918 ; https://www2.replicationdomain.com/ |
| Processed RT data from Mouse J185a Myoblast cells | Zimmerman and Gilbert, 2018; Hiratani et al., 2010 | ID: Int61896107; https://www2.replicationdomain.com/ |
| Processed RT data from Mouse L1210 Lymphoblastoid cells | Zimmerman and Gilbert, 2018; Hiratani et al., 2010 | ID: Ext49892535; https://www2.replicationdomain.com/ |
| SNS-Seq coverage in mouse ESCs (Almeida) | Almeida et al., 2018 | GEO:GSE99741 |
| SNS-Seq coverage in mouse ESCs (rep 1) | Cayrou et al., 2015 | GEO:GSM1668878 |
| SNS-Seq coverage in mouse ESCs (rep 2) | Cayrou et al., 2015 | GEO:GSM1668879 |
| SNS-Seq coverage in mouse ESCs (rep 3) | Cayrou et al., 2015 | GEO:GSM1668880 |
| SNS-Seq in human HELA cells | Long et al., 2020 | GEO:GSE134988 |
| SNS-Seq peaks and initiation zones in mouse ESCs | Cayrou et al., 2015 | GEO:GSE68347 |
| Spo11-oligo mapping data in mouse | Lange et al., 2016 | GEO:GSM2247727 |
| WGS in activated mouse B cells | Tubbs et al., 2018 | GEO:GSM3227969 |
| WGS in CD8+ T cells cells (S-phase) | Yehuda et al., 2018 | SRA:SRR7249814,SRR7249815,SRR7249816 |
| WGS in mouse E14 ES-Cells (S-phase) | Dey et al., 2015 | SRA:SRR1639635 |
| WGS in mouse primordial germ cells (S-phase) | Yehuda et al., 2018 | SRA:SRR6638995,SRR6638997,SRR6638999,SRR6639001,SRR6639003 |
| WGS in mouse spermatogonial stem cells (S-phase) | Yehuda et al., 2018 | SRA:SRR6639005,SRR6639007,SRR6639009 |
| WGS in non-replicating mouse B Cells | Tubbs et al., 2018 | GEO:GSM3227968 |
| WGS in non-replicating mouse primordial germ cells | Yehuda et al., 2018 | SRA:SRR6639002 |
| Crossovers in B6xCAST mice | Yin et al., 2019 | SRA: PRJNA511715 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: Passage 10 129X1/SvJ-PRX-129X1 #1 mES cells | The Jackson Laboratory | RRID: CVCL_2H79 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: CAST/Eij | The Jackson Laboratory | JAX: 000928 |
| Mouse: Spo11−/− in C57BL/6J | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Guide RNA: Upstream spo11: TAGAGCGCGGAAAGGTTCGC | This paper | N/A |
| Guide RNA: Downstream spo11: CGAACCTTGAGAGGTTGCGA | This paper | N/A |
| Primer: Spo11NullF: CCTCCCTGAAGGGTAGTGTG | This paper | N/A |
| Primer: Spo11NullR: GAACGGAGCAGAAGAAGACG | This paper | N/A |
| Primer: Spo11Ex4R: CTCCCGGTGCTGAAATTAAA | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: pSpCas9(BB)-2A-Puro (PX459) | Ran et al., 2013 | Addgene Plasmid #48139 |
|
| ||
| Software and algorithms | ||
|
| ||
| BEDtools v.2.27.1 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| BWA 0.7.12 | Li, 2013 | https://sourceforge.net/projects/biobwa/files/bwa-0.7.12.tar.bz2/download |
| DeepTools v.3.0.1 | Ramírez et al., 2014 | https://github.com/deeptools/deepTools |
| Juicer v.1.19.02 | Durand et al., 2016) | https://github.com/aidenlab/juicer |
| MACS v.2.1.2.1 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Nextflow v.20.01.0 | Di Tommaso et al., 2017 | https://github.com/nextflow-io/nextflow/releases |
| Picard v.2.9.2 | Broad Institute of MIT and Harvard, 2018 | https://broadinstitute.github.io/picard/ |
| R v.3.6.0 | R Core Team, 2014 | https://www.r-project.org/ |
| SAMtools v.1.9 | Li et al., 2009 | http://samtools.github.io/ |
| SRA toolkit v.2.9.2 | Leinonen et al., 2011 | https://github.com/ncbi/sra-tools |
| UCSC toolkit v.396 | Kent et al., 2010 | https://github.com/ucscGenomeBrowser/kent |
| Analytical pipeline | This paper | https://zenodo.org/record/4634002 |
| RT-Seq pipeline | This paper | https://zenodo.org/record/4634128 |
| RT-Sim | This paper | https://zenodo.org/record/4634128 |
To assess the identity of replicating cells from whole testes, mice were injected intraperitoneally (i.p.) with 100–200 μg of 5-ethynyl-2’-deoxyuridine (EdU, a thymidine analog) in PBS and mouse testes were harvested at 30 min after injection. We then combined the same sorting strategy we used to isolate MeiS nuclei (Table S1) plus the EdU detection for quantification.
Purity assessment of sorted nuclei using immunofluorescence microscopy
Nuclei were concentrated in a small volume after centrifugation. The nuclei suspension was then pipetted onto silane coated slides and mounted with Vectashield.
Nuclei extraction and isolation of replicating ES cells
Exponentially growing mouse ES-cells were cross-linked by adding fresh 1% paraformaldehyde solution to the ES media for 10 minutes at room temperature. The cells were washed twice with 1X PBS and scraped off the plates, pelleted and nuclei were prepared as described in Lam et al. (2019). Replicating cells were sorted based DNA content.
Library preparation for whole-genome sequencing (WGS)
Pure nuclei populations were resuspended in 300 ul of lysis buffer (SDS 1%, 10mM EDTA, 50mM Tris pH = 8) and then sonicated in a Bioruptor UCD200 for 20 min 30 s ON 30 s OFF, ‘‘high’’ setting. NaCl was added to a final concentration of 0.2M and the sample incubated overnight at 65°C to reverse DNA-protein crosslinks. 5U of Proteinase K) was added and the sample further incubated for 1h at 45°C. DNA was purified with MinElute columns (QIAGEN), as indicated by the instruction manual. Whole-genome sequencing libraries were prepared from 100 ng of purified DNA using the KAPA Hyper Prep Kit following the manufacturer’s instructions.
Inferring replication timing from whole-genome sequencing data
The mem algorithm of bwa 0.7.12 (Li, 2013; Li and Durbin, 2009) was used to align each WGS dataset to the reference genome (mm10 for mouse; hg38 for human). WGS from a non-replicating population was used to calibrate the expected GC-biases from different sequencing instruments (Illumina HiSeq 2000, Illumina HiSeq 2500, Illumina HiSeq X, Illumina NextSeq 500). Briefly, these calibration files were generated by examining a subset of perfectly mappable genomic loci. Each locus is a 101 bp region with the following properties:
Full mappability of all bases within the 101 bp window. To determine mappability, we generated a fastq file of pseudoreads of the requisite length from the reference genome fasta file. One pseudo-read was generated per bp; all bases were assigned a q-score of 60. Pseudo-reads were mapped to the reference genome using bwa mem 0.7.12 and the coverage at each base in the genome was assessed.
The window does not overlap with sequencing gaps (UCSC Gaps track), segmental duplications (UCSC SegDups track), high copy repeats (UCSC repeatmasker track) or otherwise blacklisted genomic regions (Amemiya et al., 2019; Kent et al., 2002).
The samples used to generate GC-correction files were the 2CMMC and 2CHSC samples (Table S1) for Hi-Seq X and Hi-Seq 2500, respectively. Samples used to generate GC-correction files for published data are detailed in Table S2 below.
The GC content (rounded to 1%) and WGS sequencing coverage of each 101 bp window was assessed. The median coverage for all windows of a given GC content was used as a correction factor. These correction factors vary substantially across the different sequencing platforms used. 2% of all qualifying 101 bp autosomal windows were used to calculate the GC correction coefficients.
For each RT-Seq experiment, the WGS coverage in 101-bp windows in the genome was calculated and corrected for GC biases using the correction coefficient for the appropriate sequencing platform. Replication timing was calculated as the log2 ratio of corrected sequencing coverage at a locus to the genome-wide median (method was adapted from (Koren et al., 2014)). RT-Seq coverage was smoothed in 500 Kb windows with a 50 Kb step.
Inferring replication timing from published data
Whole-genome sequencing data were obtained from the sources listed in Table S2. Sequencing data were aligned and replication timing profiles were inferred as described above.
Modeling DNA replication
DNA replication was modeled in-silico using custom R code (Brick, 2020). Replication was simulated in a haploid genome. Non-autosomal chromosomes were excluded. The initial wave of origin firing was assumed to be synchronous. k replication origins were selected at random and without replacement, using the origin efficiency as a selection weight. At each origin, a left-ward and a right-ward replication fork was established, and replication was then simulated bi-directionally outward from each origin in 10 Kb steps. After each step, we assessed which replication forks have collided with another fork and which forks reached the end of the chromosome. These forks were terminated. For every two terminated forks, we allowed replication to begin from a new, randomly chosen origin (as above, weighted by origin efficiency). Origins that have previously fired and origins in regions that have already been replicated were not considered for selection. This iterative process continued until the genome was fully replicated or for a defined number of cycles. To simulate replication in a population, this simulation was performed in n haploid genomes and the results were combined.
Hyperparameter search for replication models
To assess the best-fitting parameters of DNA replication in-silico, we performed a grid-hyperparameter search. A hyperparameter search was performed for each pairwise combination of RT and replication origins. We optimized for a scoring function defined as the Pearson correlation coefficient between the experimentally-determined replication timing and the in-silico simulated replication timing. A population of 250 replicating haploid genomes was used for the hyperparameter search. The grid-search generated an RT-Sim model for each combination of the following parameters:
Origin density; Rd = 0.02 to 0.4 in 0.01 steps; 0.45 to 0.6 in 0.05 steps
Modeling runtime; Nepochs = 10 to 7500 in steps of 10
Replicating cells in population; P (%) = 10,30,50,70,90
Use origin efficiency; E = TRUE / FALSE (if FALSE, all origins were equally weighted for ‘‘random’’ selection)
The fine-grained search yielded multiple models with similar scores, therefore in assessing results, the top g% of models were examined.
Generating simulated replication timing
The hyperparameters of the top-scoring RT-Sim model were used to generate an RT-Sim model from 500 haploid genomes and run it to completion. Simulated replication timing was then defined as the mean replication time across all simulations for each 10 Kb window in the genome.
Sources and processing of annotation data for the mouse genome
CpG islands in the mouse genome were obtained from the Irizarry lab at Harvard University, USA (http://www.haowulab.org/software/makeCGI/model-based-cpg-islands-mm9.txt) (Wu et al., 2010). UCSC liftover was used to convert mm9 to mm10 genome coordinates.
Meiotic DSB hotspots in C57BL6J mice were obtained from columns 1,2,3,6 of Supplementary File 41586_2018_492_MOESM3_ESM.zip (Brick et al., 2018b). H3K4m3 ChIP-Seq signals at DSB hotspots in C57BL6J mice (Baker et al., 2014) was obtained from columns 1,2,3,50 of Supplementary File 41586_2018_492_MOESM3_ESM.zip (Brick et al., 2018b). Genome-wide Spo11-associated oligo mapping data in C57BL6 mice (Lange et al., 2016) were obtained in processed form from columns 1,2,3,55 of Supplementary File 41586_2018_492_MOESM3_ESM.zip (Brick et al., 2018b). DSB hotspots in Prdm9-/- mice (Brick et al., 2012) were obtained from GSM2664291 (Brick et al., 2018b). DSB hotspots from mice homozygous for a humanized PRDM9 zinc-finger array (B6h/h) were obtained from the GEO (accession: GSM2049306; file: GSM2049306_dmc1hotspots_B6.PRDM9hh.txt.gz) (Davies et al., 2016). B6h/h hotspots overlapping hotspots from Prdm9−/− were discarded. H3K4m3 ChIP-Seq peaks in B6h/h mice were obtained from the GEO (accession: GSM1904284; file GSM1904284_H3K4me3.ForceCalledValues.B6.PRDM9hh.txt) (Davies et al., 2016). Crossovers and non-crossovers defined by the humanized variant of mouse PRDM9 were obtained from supplementary file 3 (41467_2019_11675_MOESM6_ESM.csv) and 4 (41467_2019_11675_MOESM7_ESM.csv), respectively, from Li et al. (2019). Affinity-Seq data describing the in-vitro binding preferences of the C57BL6J PRDM9 variant were obtained from the Sequence Read Archive (accession: SRR1976005) (Walker et al., 2015). Control data used for Affinity-Seq peak calling was obtained from the Sequence Read Archive (accession: SRR1976003 & SRR1976004) (Walker et al., 2015). Affinity-Seq and control reads were aligned to the reference mm10 genome using bwa mem 0.7.12. Affinity-Seq peak calling was performed using MACS 2.1.2 with the following arguments: -g mm -bw 1000–keep-dup all–slocal 5000 -q 0.05. Affinity-Seq peak strength was determined as Affinity-Seq coverage less Control DNA coverage at Affinity-Seq peaks. Hi-C data from mouse zygotene cells was downloaded from the GEO (GEO accession: GSE122622; file: GSE122622_zygotene_overall.hic) (Patel et al., 2019). The Juicer tools 1.09.02 eigenvector program (Durand et al., 2016) was used to extract the first eigenvector from the .hic file, using the following arguments: KR, BP, binsize = 100000, -p. H3K9m2 (accession: SRR1975998) and H3K9m3 (accession: SRR1585300) ChIP-Seq data (Walker et al., 2015) from spermatocytes were obtained from the SRA. Reads were aligned to the reference mm10 genome using bwa mem 0.7.12.
ATAC-Seq data were obtained from the published datasets described in Table S4. Each dataset was aligned to the mouse mm10 reference genome using bwa mem 0.7.12. Peak calling was performed for each dataset using MACS 2.1.2 without a control and using default arguments. ATAC-Seq peaks coinciding with CpG-islands were defined for use on Table S4.
Mapping Okazaki-fragment sequencing data
Data from Okazaki-fragment sequencing in mouse ES-cells were obtained from SRA accession SRR7535256 (Petryk et al., 2018). Sequencing reads were aligned to the mm10 reference genome using the mem algorithm of bwa 0.7.12 (Li, 2013; Li and Durbin, 2009).
Obtaining published SNS-Seq replication origins data
Processed peak calls for ESCs were obtained from GEO accession GSE99741 (Almeida et al., 2018); GSM2651111_mES_WT_repl_I_peaks.bigBed. Processed peak calls for ESCs from Cayrou et al. (2015) were obtained from GEO accession GSE68347; GSE68347_Initiation_Sites.bedGraph.gz. Processed initiation zones for ESCs from Cayrou et al. (2015) were obtained from GEO accession GSE68347; GSE68347_Initiation_Zones.bed.gz. The UCSC liftover tool was used to convert mm9 to mm10 coordinates.
Mapping DNA DSB hotspots from DMC1-SSDS experiments
A testicular biopsy was obtained from a commercial source (Folio Biosciences, Ohio). Genotyping for PRDM9 alleles using Sanger sequencing (Pratto et al., 2014) revealed that this individual did not carry a copy of the most common PRDM9 variant in humans (PRDM9A). Instead, this individual was heterozygous for the PRDM9C and PRDM9L4 alleles (Berg et al., 2010). A DMC1-SSDS library was prepared using the protocol in Brick et al. (2018a) and Pratto et al. (2014). Published DMC1-SSDS data were also obtained for two human PRDM9A-homozygous males (GEO accessions: GSM1447325 & GSM1447328) (Pratto et al., 2014). Sequencing reads were aligned to the human hg38 reference genome using bwa 0.7.12 and the single-stranded DNA pipeline (Brick et al., 2018a; Khil et al., 2012). Hotspots were called using the DMC1-SSDS DSB hotspot calling pipeline (Brick et al., 2018a).
Crossover and non-crossover data for humans
Crossover data for human males and females were obtained from Data S1 and Data S2, respectively, from Halldorsson et al. (2019). Non-crossover data were obtained from Data S2 from Halldorsson et al. (2016). NCOs from the ChIP-based approach and the sequencing-based approach were merged.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses were performed in R. Statistical parameters, statistical test used, error bar definitions and sample sizes are reported in the figures and corresponding figure legends. No data from experiments were excluded from analysis. Where outliers were removed for plotting purposes, the removed data points were still used for statistical analyses.
Supplementary Material
Figure S1. Reproducible mapping of replication origins using Ori-SSDS, related to Figure 1
(A) RNA hydrolysis or RNase treatment prior to lambda exonuclease treatment abolishes Watson/Crick asymmetry at origins of replication. This is because RNA hydrolysis / RNase will remove the nascent strand RNA primer and thus render nascent-strands susceptible to lambda exonuclease digestion (see STAR Methods). RNase was performed as a control in parallel to the ESC 2 experiment and in testis, two replicate experiments were performed in parallel using RNA hydrolysis; WT_Rep4 & WT_Rep5 (see STAR Methods) (B,C) Watson/Crick strand asymmetry at origins of replication. Residual asymmetry is seen in RNase treated ESCs though the magnitude of the signal is greatly reduced. (C) Asymmetry is lost following RNA hydrolysis (RNAh samples), however residual enrichment at origins, without Watson/Crick asymmetry is still seen. This is likely because there is more DNA in the population at these sites as a result of DNA replication. For each sample, the mean Watson (red) & Crick (blue) coverage are shown above heatmaps for reads derived from each strand. Heatmaps depict binned coverage from low (white) to high (colored); the same coverage range is used for all heatmaps in each panel. G-quadruplexes (G4s) are secondary structures that have been hypothesized to impede lambda exonuclease in vitro and may result in SNS-Seq peak artifacts (Foulk et al., 2015). We detect small peaks at putative G4 sequences in Ori-SSDS data. Ori-SSDS sequencing can distinguish G4-signals from origin-derived signals because G4s yield ssDNA on just one strand whereas reciprocal Watson/Crick asymmetry is seen around the center of origins. This differentiates Ori-SSDS from other SNS-Seq methods, which cannot distinguish G4s from origins. At origins that coincide with putative G4s (Huppert and Balasubramanian, 2005) it is not possible to resolve these signals however, since the G4 signal is far weaker than that at origins, the contribution of G4s to true origin signal is a minor concern. Curiously, the signal at G4s is not seen in non-replicating tissue (sperm) or in samples where the RNA-primer is degraded. Thus, the G4-associated signal is replication-dependent and not simply the result of G4 resistance to lambda exonuclease. The stranded-ness of the signal is consistent with the capture of ligated Okazaki-fragments at the G4 sites. This implies that G-quadruplexes are an impediment to fork passage in-vivo. (D) Okazaki fragments, from lagging strand DNA synthesis, are distributed asymetrically around Ori-SSDS-defined origins of replication. Okazaki fragment sequencing (Ok-Seq) data in Embryonic Stem Cells (ESCs) are from Petryk et al. (2018). Note that OK-Seq exhibits the opposite polarity to the Ori-SSDS signal as expected from capture of lagging and leading strands, respectively (see Figure 1). Data are shown in 50 Kb windows. Heatmaps depict the signal at Ori-SSDS peaks in ESCs (dark blue), at SNS-Seq peaks that coincide with Ori-SSDS peaks (light blue), at SNS-Seq peaks that do not coincide with Ori-SSDS peaks (green), and at Ori-SSDS peaks that do not coincide with an SNS-Seq peak (orange). Analyses were performed separately using ESC SNS-Seq data from either Almeida et al. (2018) or Cayrou et al. (2015). The mean signal for each set is shown above heatmaps that depict the signal at individual sites. Heatmaps are scaled to reflect the number of origins in each subset. A large proportion of peaks detected in SNS-Seq experiments, but not in Ori-SSDS do not exhibit OK-Seq asymmetry. This is more pronounced in the Almeida than in the Cayrou data. Ori-SSDS peaks not found in SNS-Seq still exhibit OK-Seq asymmetry. (E) Most Ori-SSDS peak calls are found in both ESCs and Testis and are shared among replicates. Peaks were called for each sample and peaks overlapping by at least 500 bp were merged. Sets with fewer than 50 peaks are not shown. Our consensus set of origins includes all origins found in at least two Ori-SSDS experiments and with Watson/Crick asymmetry in at least one. (F) Most origins are detected in multiple samples. Peaks found in > 1 sample are considered shared (gray). The number of unique origins detected scales with sample quality (# of peaks). (G-I) Ori-SSDS gives a reproducible readout of origin efficiency. Origin efficiency is a measure of the frequency at which each origin is used. This is inferred from the sequencing read coverage in each experiment (see STAR Methods). Grey dots represent origins found in both samples. Magenta and green dots represent origins found only in the x axis designated and y axis designated samples, respectively. R2 is the squared Spearman correlation coefficient of log transformed values. N indicates the number of origins for each pairwise comparison. (G) All Ori-SSDS peaks found in either sample. (H) Only the 11,339 high confidence origins are shown. (I,J) Origin efficiency is highly correlated between testis and ESCs. All peaks found in either sample are shown. (K,L) Origin efficiency is correlated between ESC Ori-SSDS replicates. (K) All origins found in either experiment. (L) Origins found in both experiments.
Figure S2. Most replicating cells in testis are meiotic, related to Figures 1 and 2
To identify replicating cells in testis, the thymidine analog 5-Ethynyl-2’-deoxyuridine (EdU) was injected intraperitoneally to live mice, then mice were sacrificed and testes harvested after 30 min. Nuclei were stained with DMRT1 and STRA8 antibodies to distinguish spermatogonia from preleptotene nuclei. (A) From FANS sorting, 1.8% of nuclei from whole testis were inferred to be replicating (EdU positive; above dashed red line). (B) Most putatively replicating (2–4C; DAPI content) spermatogonia and preleptotene cells were EdU positive (red). (C) A majority of replicating cells in mouse testis are in meiotic S-phase.
Figure S3. Origins of replication in mouse testis occur at GC-rich, accessible sites, related to Figure 2
(A) Origins of replication are more closely spaced than expected in the genome. The expected distances between randomly chosen intervals is shown in gray. (B) Most origins occur near gene promoters; 66% occur within 1 Kbp of a transcription start site (TSS), far more than expected (random; gray line). (C) Replication origins coincide with local changes in DNA bendability (calculated from Goodsell and Dickerson [1994]). The mean (top) and standard deviation (bottom) of the DNA bendability at all origins is shown. Each point represents a 100 bp window. Solid line is a LOESS fit. (D) Origin centers are GC-rich and enriched in CpG and GpC dinucleotides. The log2 ratio of the central 500 bp compared to the region flanking this center is shown. (E) GC-content (GC%), CpG density and GpC density correlate positively with origin efficiency. (F) Putative G4-forming sequences are highly enriched at the center of origins. 68% of origins have a putative G4-forming sequence in the central ± 1 Kb. (G) Origins of replication occur predominantly at CpG islands (CGIs) that coincide with open chromatin as measured by ATAC-Seq (Maezawa et al., 2018). (H) Origins are only detected at about half of the CGIs at ATAC-Seq peaks. (I) Origin locations can be identified from CpG density. CpG dinucleotide density was evaluated in all 1 Kb regions in the genome using a 100 bp sliding window. Intervals at each CpG threshold were expanded ± 100 bp around the center and overlapping windows were merged. The centerpoint of each merged interval was defined as a CpG peak. CpG peaks with CpG content ≥ 7% overlap 6,233 origins (53% of total). 1,853 CpG peaks at this threshold do not occur at an origin (FP). (J) Origins are better predicted by CpG density than transcription factors (TFs) are by transcription factor binding sites (TFBS). Receiver operating curve (ROC) for predicting origin locations using CpG peaks (green) or for predicting peaks in ChIP-Seq datasets for 179 transcription factors (gray). For origins, at each CpG threshold (1% to 15% in 1% steps), peaks were called (as in G). All mouse TFs with ChIP-Seq data (in GTRD database) (Yevshin et al., 2017) and with a designated TFBS in the HOCOMOCO database (Kulakovskiy et al., 2018) were used. SPRY-SARUS (Kulakovskiy et al., 2018) was used to identify TFBS motifs genome-wide with p < 0.005. For each TF, the score at which ≤ 12,000 peaks were found was used as a lower threshold (this is to assure a fairer comparison with origins). Motifs with scores less than this were discarded. TFs with < 12,000 peaks not used. Hits inside ChIP-Seq peaks were considered true positives.
Figure S4. RT-seq yields similar RT profiles across varied cell types, related to Figure 3
RT for all cell types shown on Figure 2. RT is expressed as the log2 enrichment over genomic median coverage for all panels except for Myoblast and LCL data. These data were obtained in processed form from the Replication Domain database. All panels depict chromosome 12. Sample names are explained briefly in Figure 3 and in detail in Tables S1, S2 and S3.
Figure S5. Experimentally derived RT varies as a function of the starting population, related to Figure 4
(A) Four sub-populations of meiotic S-phase were isolated from early S (T1) through late S (T4). RT was inferred from WGS (see STAR Methods). The earliest and latest replicating regions exhibit substantial differences in RT across these four populations. (B-G) Properties of DNA replication inferred from best-fitting models for Meiotic (pink) and ESCs (gray). (B) The cumulative amount of replicated DNA at each simulation cycle is shown. We infer S-phase duration from the time at which 90% of the genome is replicated (magenta line). The rate of replication is reduced in the final 10%–15% of the genome. (C) The final 10%–15% of the genome is slow to replicate because the number of active forks decreases rapidly. (D) The total number of replication origins fired per simulated S-phase is shown. Each simulation models replication of a single haploid genome. (E) Replication tract length is longer in meiotic cells compared to ESCs. Note that meiotic cells use fewer origins. (F) Model estimates of the per S-phase frequency of origin firing. (G) The rate at which new origins fire throughout simulation cycles.
Figure S6. Modeling recapitulates experimental RT genome-wide, related to Figure 4
Experimental RT is shown as filled green (early replicating) and violet (late replicating) regions. The best-fitting RT-Sim predicted RT is shown as a solid black line. Per-chromosome Pearson correlation coefficients are shown. Both (A) Meiotic and (B) ESC RT can be accurately modeled in silico. Both experimental and predicted RT are normalized by mean and standard deviation (s.d.); (RTnorm = (RT-mean(RT)) / s.d.(RT)).
Figure S7. Meiotic S-phase duration is unaffected in Spo11−/− male mice, related to Figure 4
Meiotic S-phase nuclei, B-type spermatogonia and undifferentiated spermatogonia were isolated from a Spo11−/− male mouse testis (SPO11 in sample names) and a wild-type littermate (wtLM in sample names). Details for each sample are in Tables S1, S2, and S3. (A) 2–4C nuclei were sorted and (B) gated by DMRT1 and STRA8 immunofluorescence signal. Gates were defined while sorting nuclei from the Spo11−/− mouse and retained, unchanged for sorting nuclei from the littermate. (C) Summary statistics for best-fitting RT-Sim models for all samples. (D) Comparison of S-phase duration across cell types. The median S-phase duration of good models for each cell-type is compared to meiotic S-phase. (E) The ‘‘fit’’ of RT-Sim models for Spo11−/− samples is comparable to the fit in other cell types. (F) The duration of meiotic S-phase is unaffected in Spo11−/− mice relative to the wild-type littermate control. Notably, both mice exhibit slightly shorter S-phase duration compared to the three wild-type replicates. This is likely because for the Spo11−/− and wild-type littermate, the sorting gates were defined in the Spo11−/− mouse and left unchanged when sorting nuclei from the littermate. Cellularity differences from the loss of Spo11 may result in the capture of a slightly different population than when sorting gates are defined using wild-type. (G) The firing rate is similarly unaffected.
Highlights.
Robust mapping of replication origins and characterization of replication in tissue
Contemporary and historical recombination are elevated in early replicating DNA
Altered template choice for DSB repair in early replicating DNA in meiosis
Consistently early subtelomeric replication in human male meiosis
ACKNOWLEDGMENTS
We thank Galina Petukhova, Michael Lichten, and Peggy Hsieh for critiques; the Flow Cytometry Core of NHLBI; the NIDDK Laboratory of Animal Sciences Section; and the NIDDK genomics core facility. We also thank the Washington Regional Transplant Community for their assistance in obtaining de-identified human testis donations for research. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). This work was funded by NIGMS (R01GM11755 to P.W.J.) and by the NIDDK intramural program (to R.D.C.-O.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.06.025.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Almeida R, Fernández-Justel JM, Santa-María C, Cadoret J-C, Cano-Aroca L, Lombraña R, Herranz G, Agresti A, and Gómez M (2018). Chromatin conformation regulates the coordination between DNA replication and transcription. Nat. Commun 9, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EL, Baltus AE, Roepers-Gajadien HL, Hassold TJ, de Rooij DG, van Pelt AMM, and Page DC (2008). Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. USA 105, 14976–14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Walker M, Kajita S, Petkov PM, and Paigen K (2014). PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, and Keeney S (2000). Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell 6, 989–998. [DOI] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, and de Massy B (2010). PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellvé AR, Cavicchia JC, Millette CF, O’Brien DA, Bhatnagar YM, and Dym M (1977). Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J. Cell Biol 74, 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Smith JB, and Riley R (1972). The effects of polyploidy on meiotic duration and pollen development in cereal anthers. Proc. R. Soc. Lond. B Biol. Sci 181, 81–107. [Google Scholar]
- Berg IL, Neumann R, Lam K-WG, Sarbajna S, Odenthal-Hesse L, May CA, and Jeffreys AJ (2010). PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet 42, 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, and Forterre P (1997). An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386, 414–417. [DOI] [PubMed] [Google Scholar]
- Bielinsky AK, and Gerbi SA (1998). Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science 279, 95–98. [DOI] [PubMed] [Google Scholar]
- Bishop DK, Park D, Xu L, and Kleckner N (1992). DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456. [DOI] [PubMed] [Google Scholar]
- Blitzblau HG, Chan CS, Hochwagen A, and Bell SP (2012). Separation of DNA replication from the assembly of break-competent meiotic chromosomes. PLoS Genet 8, e1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, Goldman AS, and Lichten M (2000). Direct coupling between meiotic DNA replication and recombination initiation. Science 290, 806–809. [DOI] [PubMed] [Google Scholar]
- Boulton A, Myers RS, and Redfield RJ (1997). The hotspot conversion paradox and the evolution of meiotic recombination. Proc. Natl. Acad. Sci. USA 94, 8058–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K (2020). Simulating Replication Timing in the Mouse and Human Genomes (GitHub) [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, and Petukhova GV (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Pratto F, Sun C-Y, Camerini-Otero RD, and Petukhova G (2018a). Analysis of Meiotic Double-Strand Break Initiation in Mammals. Methods Enzymol 601, 391–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Thibault-Sennett S, Smagulova F, Lam KG, Pu Y, Pratto F, Camerini-Otero RD, and Petukhova GV (2018b). Extensive sex differences at the initiation of genetic recombination. Nature 561, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard J, Rivals E, Dunoyer de Segonzac D, Garres C, Caminade P, de Massy B, and Boursot P (2014). Diversity of Prdm9 zinc finger array in wild mice unravels new facets of the evolutionary turnover of this coding minisatellite. PLoS ONE 9, e85021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan HG (1973). DNA replication in the chromosomes of eukaryotes. In Molecular Cytogenetics, Hamkalo BA and Papaconstantinou J, eds. (Springer New York; ), pp. 31–47. [Google Scholar]
- Cayrou C, Grégoire D, Coulombe P, Danis E, and Méchali M (2012). Genome-scale identification of active DNA replication origins. Methods 57, 158–164. [DOI] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau J-C, van Helden J, and Méchali M (2015). The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res 25, 1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, and Kleckner N (2002). ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297, 602–606. [DOI] [PubMed] [Google Scholar]
- Cha RS, Weiner BM, Keeney S, Dekker J, and Kleckner N (2000). Progression of meiotic DNA replication is modulated by interchromosomal interaction proteins, negatively by Spo11p and positively by Rec8p. Genes Dev 14, 493–503. [PMC free article] [PubMed] [Google Scholar]
- Chagin VO, Casas-Delucchi CS, Reinhart M, Schermelleh L, Markaki Y, Maiser A, Bolius JJ, Bensimon A, Fillies M, Domaing P, et al. (2016). 4D Visualization of replication foci in mammalian cells corresponding to individual replicons. Nat. Commun 7, 11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandley AC (1986). A model for effective pairing and recombination at meiosis based on early replicating sites (R-bands) along chromosomes. Hum. Genet 72, 50–57. [DOI] [PubMed] [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. (2016). Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagouraga B, Clément JAJ, Duret L, Kadlec J, de Massy B, and Baudat F (2018). PRDM9 Methyltransferase Activity Is Essential for Meiotic DNA Double-Strand Break Formation at Its Binding Sites. Mol. Cell 69, 853–865.e6. [DOI] [PubMed] [Google Scholar]
- Dileep V, and Gilbert DM (2018). Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat. Commun 9, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, and Galtier N (2009). Biased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genomics Hum. Genet 10, 285–311. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, and Nussenzweig A (2004). H2AX: the histone guardian of the genome. DNA Repair (Amst.) 3, 959–967. [DOI] [PubMed] [Google Scholar]
- Foulk MS, Urban JM, Casella C, and Gerbi SA (2015). Characterizing and controlling intrinsic biases of lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res 25, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Besnard E, Desprat R, Ryan M, Kahli M, Lemaitre J-M, and Aladjem MI (2014). Mapping replication origin sequences in eukaryotic chromosomes. Curr. Protoc. Cell Biol 65, 20.1–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal SK, and Mukherjee BB (1971). The chronology of DNA synthesis, meiosis and spermiogenesis in the male mouse and Golden hamster. Can. J. Genet. Cytol 13, 672–682. [Google Scholar]
- Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, and Bilke S (2014). A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol 10, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell DS, and Dickerson RE (1994). Bending and curvature calculations in B-DNA. Nucleic Acids Res 22, 5497–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CD, Ma Q, Manske GL, Shami AN, Zheng X, Marini S, Moritz L, Sultan C, Gurczynski SJ, Moore BB, et al. (2018). A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev. Cell 46, 651–667.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Grow EJ, Mlcochova H, Maher GJ, Lindskog C, Nie X, Guo Y, Takei Y, Yun J, Cai L, et al. (2018). The adult human testis transcriptional cell atlas. Cell Res 28, 1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson BV, Hardarson MT, Kehr B, Styrkarsdottir U, Gylfason A, Thorleifsson G, Zink F, Jonasdottir A, Jonasdottir A, Sulem P, et al. (2016). The rate of meiotic gene conversion varies by sex and age. Nat. Genet 48, 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson BV, Palsson G, Stefansson OA, Jonsson H, Hardarson MT, Eggertsson HP, Gunnarsson B, Oddsson A, Halldorsson GH, Zink F, et al. (2019). Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, eaau1043. [DOI] [PubMed] [Google Scholar]
- Handel MA, Eppig JJ, and Schimenti JC (2014). Applying ‘‘gold standards’’ to in-vitro-derived germ cells. Cell 157, 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JD, Perry RM, Barakate A, Ramsay L, Waugh R, Halpin C, Armstrong SJ, and Franklin FCH (2012). Spatiotemporal asymmetry of the meiotic program underlies the predominantly distal distribution of meiotic crossovers in barley. Plant Cell 24, 4096–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch AG, Zhang G, Becker PW, Moralli D, Hinch R, Davies B, Bowden R, and Donnelly P (2019). Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 363, eaau8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. (2010). Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20, 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm PB (1977). The premeiotic DNA replication of euchromatin and heterochromatin in Lilium longiflorum (Thunb.). Carlsberg Res. Commun 42, 249–281. [Google Scholar]
- Hossain AM, Rizk B, Behzadian A, and Thorneycroft IH (1997). Modified guanidinium thiocyanate method for human sperm DNA isolation. Mol. Hum. Reprod 3, 953–956. [DOI] [PubMed] [Google Scholar]
- Huppert JL, and Balasubramanian S (2005). Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33, 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodkowska K, Pancaldi V, Almeida R, Rigau M, Graña-Castro O, Fernández-Justel JM, Rodríguez-Acebes S, Rubio-Camarillo M, Pau ECS, Pisano D, et al. (2019). Three-dimensional connectivity and chromatin environment mediate the activation efficiency of mammalian DNA replication origins. bioRxiv [Google Scholar]
- Joshi N, Brown MS, Bishop DK, and Börner GV (2015). Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol. Cell 57, 797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback DB, Guacci V, Barber D, and Mahon JW (1992). Chromosome size-dependent control of meiotic recombination. Science 256, 228–232. [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, and Kleckner N (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. [DOI] [PubMed] [Google Scholar]
- Kelly T, and Callegari AJ (2019). Dynamics of DNA replication in a eukaryotic cell. Proc. Natl. Acad. Sci. USA 116, 4973–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, and Petukhova GV (2012). Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res 22, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein KN, Zhao PA, Lyu X, Sasaki T, Bartlett DA, Singh AM, Tasan I, Zhang M, Watts LP, Hiraga S-I, et al. (2021). Replication timing maintains the global epigenetic state in human cells. Science 372, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofman-Alfaro S, and Chandley AC (1970). Meiosis in the male mouse. An autoradiographic investigation. Chromosoma 31, 404–420. [DOI] [PubMed] [Google Scholar]
- Kojima ML, de Rooij DG, and Page DC (2019). Amplification of a broad transcriptional program by a common factor triggers the meiotic cell cycle in mice. eLife 8, e43738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, and McCarroll SA (2012). Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet 91, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, and McCarroll SA (2014). Genetic variation in human DNA replication timing. Cell 159, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA, et al. (2018). HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res 46 (D1), D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KG, Brick K, Cheng G, Pratto F, and Camerini-Otero RD (2019). Cell-type-specific genomics reveals histone modification dynamics in mammalian meiosis. Nat. Commun 10, 3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Yamada S, Tischfield SE, Pan J, Kim S, Zhu X, Socci ND, Jasin M, and Keeney S (2016). The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 167, 695–708.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CSK, Cheung MF, Li J, Zhao Y, Lam WH, Ho V, Rohs R, Zhai Y, Leung D, and Tye B-K (2021). Humanizing the yeast origin recognition complex. Nat. Commun 12, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv:1303.3997v1 [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lam WH, Zhai Y, Cheng J, Cheng E, Zhao Y, Gao N, and Tye B-K (2018). Structure of the origin recognition complex bound to DNA replication origin. Nature 559, 217–222. [DOI] [PubMed] [Google Scholar]
- Li R, Bitoun E, Altemose N, Davies RW, Davies B, and Myers SR (2019). A high-resolution map of non-crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat. Commun 10, 3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Zhang L, Lv M, Wen Z, Zhang W, Chen X, Zhang P, Li T, Chang L, Jin C, et al. (2020). H2A.Z facilitates licensing and activation of early replication origins. Nature 577, 576–581. [DOI] [PubMed] [Google Scholar]
- Maezawa S, Yukawa M, Alavattam KG, Barski A, and Namekawa SH (2018). Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Res 46, 593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C, Sima J, and Gilbert DM (2019). Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol 20, 721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, and Zarkower D (2010). The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev. Cell 19, 612–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Ji Z, and Struhl K (2016). Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. USA 113, E4810–E4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monesi V (1962). Autoradiographic study of DNA synthesis and the cell cycle in spermatogonia and spermatocytes of mouse testis using tritiated thymidine. J. Cell Biol 14, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, and Keeney S (2014). Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, and Nurse P (2001). Regulation of premeiotic S phase and recombination-related double-strand DNA breaks during meiosis in fission yeast. Nat. Genet 28, 290–293. [DOI] [PubMed] [Google Scholar]
- Murakami H, Lam I, Huang P-C, Song J, van Overbeek M, and Keeney S (2020). Multilayered mechanisms ensure that short chromosomes recombine in meiosis. Nature 582, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, and Donnelly P (2005). A fine-scale map of recombination rates and hotspots across the human genome. Science 310, 321–324. [DOI] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, and Donnelly P (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Pan J, and Keeney S (2005). Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman K, Algopishi U, Higgins JD, Henderson IR, Edwards KJ, Franklin FCH, and Sanchez-Moran E (2021). Distal bias of meiotic crossovers in hexaploid bread wheat reflects spatio-temporal asymmetry of the meiotic program. Front. Plant Sci 12, 631323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, and Paigen K (2010). Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel L, Kang R, Rosenberg SC, Qiu Y, Raviram R, Chee S, Hu R, Ren B, Cole F, and Corbett KD (2019). Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat. Struct. Mol. Biol 26, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C-L, and Hyrien O (2016). Replication landscape of the human genome. Nat. Commun 7, 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Dalby M, Wenger A, Stromme CB, Strandsby A, Andersson R, and Groth A (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Picard F, Cadoret J-C, Audit B, Arneodo A, Alberti A, Battail C, Duret L, and Prioleau M-N (2014). The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10, e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM, and Paigen K (2016). The Meiotic Recombination Activator PRDM9 Trimethylates Both H3K36 and H3K4 at Recombination Hotspots In Vivo. PLoS Genet 12, e1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, and Camerini-Otero RD (2014). DNA recombination. Recombination initiation maps of individual human genomes. Science 346, 1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanienko PJ, and Camerini-Otero RD (2000). The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 6, 975–987. [DOI] [PubMed] [Google Scholar]
- Sandhu R, Monge Neria F, Monge Neria J, Chen X, Hollingsworth NM, and Börner GV (2020). DNA Helicase Mph1FANCM Ensures Meiotic Recombination between Parental Chromosomes by Dissociating Precocious Displacement Loops. Dev. Cell 53, 458–472.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Vergara Z, Peiró R, Morata J, Aragüez I, Costas C, Mendez-Giraldez R, Casacuberta JM, Bastolla U, and Gutierrez C (2019). Differences in firing efficiency, chromatin, and transcription underlie the developmental plasticity of the Arabidopsis DNA replication origins. Genome Res 29, 784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera-Mulia JC, Trevilla-Garcia C, et al. (2019). Identifying cis Elements for Spatiotemporal Control of Mammalian DNA Replication. Cell 176, 816–830.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Gregoretti IV, Brick K, Khil P, Camerini-Otero RD, and Petukhova GV (2011). Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 472, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero RD, and Petukhova GV (2016). The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DI, Zhu Y, McAvoy S, and Kuhn R (2006). Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 232, 48–57. [DOI] [PubMed] [Google Scholar]
- Smith OK, Kim R, Fu H, Martin MM, Lin CM, Utani K, Zhang Y, Marks AB, Lalande M, Chamberlain S, et al. (2016). Distinct epigenetic features of differentiation-regulated replication origins. Epigenetics Chromatin 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Miura H, Shibata T, Nagao K, Okumura K, Ogata M, Obuse C, Takebayashi S-I, and Hiratani I (2019). Genome-wide stability of the DNA replication program in single mammalian cells. Nat. Genet 51, 529–540. [DOI] [PubMed] [Google Scholar]
- Valton A-L, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintomé C, Riou J-F, and Prioleau M-N (2014). G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33, 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouzas AE, and Gilbert DM (2021). Mammalian DNA Replication Timing. Cold Spring Harb. Perspect. Biol Published online February 8, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M, Billings T, Baker CL, Powers N, Tian H, Saxl RL, Choi K, Hibbs MA, Carter GW, Handel MA, et al. (2015). Affinity-seq detects genome-wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Klein K, Proesmans K, Yang H, Marchal C, Zhu X, Borman T, Hastie A, Weng Z, Bechhoefer J, et al. (2021). Genome-Wide Mapping of Human DNA Replication by Optical Replication Mapping Supports a Stochastic Model of Eukaryotic Replication. Mol. Cell Published online June 17, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y (2004). Modifying sister chromatid cohesion for meiosis. J. Cell Sci 117, 4017–4023. [DOI] [PubMed] [Google Scholar]
- Wojtasz L, Daniel K, Roig I, Bolcun-Filas E, Xu H, Boonsanay V, Eckmann CR, Cooke HJ, Jasin M, Keeney S, et al. (2009). Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet 5, e1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-YJ, and Nurse P (2014). Replication origin selection regulates the distribution of meiotic recombination. Mol. Cell 53, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Caffo B, Jaffee HA, Irizarry RA, and Feinberg AP (2010). Redefining CpG islands using hidden Markov models. Biostatistics 11, 499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevshin I, Sharipov R, Valeev T, Kel A, and Kolpakov F (2017). GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res 45 (D1), D61–D67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Jiang Y, Lam KG, Berletch JB, Disteche CM, Noble WS, Steemers FJ, Camerini-Otero RD, Adey AC, and Shendure J (2019). High-Throughput Single-Cell Sequencing with Linear Amplification. Mol. Cell 76, 676–690.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Liu JG, Zhao J, Brundell E, Daneholt B, and Höög C (2000). The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol. Cell 5, 73–83. [DOI] [PubMed] [Google Scholar]
- Zhao PA, Sasaki T, and Gilbert DM (2020). High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol 21, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reproducible mapping of replication origins using Ori-SSDS, related to Figure 1
(A) RNA hydrolysis or RNase treatment prior to lambda exonuclease treatment abolishes Watson/Crick asymmetry at origins of replication. This is because RNA hydrolysis / RNase will remove the nascent strand RNA primer and thus render nascent-strands susceptible to lambda exonuclease digestion (see STAR Methods). RNase was performed as a control in parallel to the ESC 2 experiment and in testis, two replicate experiments were performed in parallel using RNA hydrolysis; WT_Rep4 & WT_Rep5 (see STAR Methods) (B,C) Watson/Crick strand asymmetry at origins of replication. Residual asymmetry is seen in RNase treated ESCs though the magnitude of the signal is greatly reduced. (C) Asymmetry is lost following RNA hydrolysis (RNAh samples), however residual enrichment at origins, without Watson/Crick asymmetry is still seen. This is likely because there is more DNA in the population at these sites as a result of DNA replication. For each sample, the mean Watson (red) & Crick (blue) coverage are shown above heatmaps for reads derived from each strand. Heatmaps depict binned coverage from low (white) to high (colored); the same coverage range is used for all heatmaps in each panel. G-quadruplexes (G4s) are secondary structures that have been hypothesized to impede lambda exonuclease in vitro and may result in SNS-Seq peak artifacts (Foulk et al., 2015). We detect small peaks at putative G4 sequences in Ori-SSDS data. Ori-SSDS sequencing can distinguish G4-signals from origin-derived signals because G4s yield ssDNA on just one strand whereas reciprocal Watson/Crick asymmetry is seen around the center of origins. This differentiates Ori-SSDS from other SNS-Seq methods, which cannot distinguish G4s from origins. At origins that coincide with putative G4s (Huppert and Balasubramanian, 2005) it is not possible to resolve these signals however, since the G4 signal is far weaker than that at origins, the contribution of G4s to true origin signal is a minor concern. Curiously, the signal at G4s is not seen in non-replicating tissue (sperm) or in samples where the RNA-primer is degraded. Thus, the G4-associated signal is replication-dependent and not simply the result of G4 resistance to lambda exonuclease. The stranded-ness of the signal is consistent with the capture of ligated Okazaki-fragments at the G4 sites. This implies that G-quadruplexes are an impediment to fork passage in-vivo. (D) Okazaki fragments, from lagging strand DNA synthesis, are distributed asymetrically around Ori-SSDS-defined origins of replication. Okazaki fragment sequencing (Ok-Seq) data in Embryonic Stem Cells (ESCs) are from Petryk et al. (2018). Note that OK-Seq exhibits the opposite polarity to the Ori-SSDS signal as expected from capture of lagging and leading strands, respectively (see Figure 1). Data are shown in 50 Kb windows. Heatmaps depict the signal at Ori-SSDS peaks in ESCs (dark blue), at SNS-Seq peaks that coincide with Ori-SSDS peaks (light blue), at SNS-Seq peaks that do not coincide with Ori-SSDS peaks (green), and at Ori-SSDS peaks that do not coincide with an SNS-Seq peak (orange). Analyses were performed separately using ESC SNS-Seq data from either Almeida et al. (2018) or Cayrou et al. (2015). The mean signal for each set is shown above heatmaps that depict the signal at individual sites. Heatmaps are scaled to reflect the number of origins in each subset. A large proportion of peaks detected in SNS-Seq experiments, but not in Ori-SSDS do not exhibit OK-Seq asymmetry. This is more pronounced in the Almeida than in the Cayrou data. Ori-SSDS peaks not found in SNS-Seq still exhibit OK-Seq asymmetry. (E) Most Ori-SSDS peak calls are found in both ESCs and Testis and are shared among replicates. Peaks were called for each sample and peaks overlapping by at least 500 bp were merged. Sets with fewer than 50 peaks are not shown. Our consensus set of origins includes all origins found in at least two Ori-SSDS experiments and with Watson/Crick asymmetry in at least one. (F) Most origins are detected in multiple samples. Peaks found in > 1 sample are considered shared (gray). The number of unique origins detected scales with sample quality (# of peaks). (G-I) Ori-SSDS gives a reproducible readout of origin efficiency. Origin efficiency is a measure of the frequency at which each origin is used. This is inferred from the sequencing read coverage in each experiment (see STAR Methods). Grey dots represent origins found in both samples. Magenta and green dots represent origins found only in the x axis designated and y axis designated samples, respectively. R2 is the squared Spearman correlation coefficient of log transformed values. N indicates the number of origins for each pairwise comparison. (G) All Ori-SSDS peaks found in either sample. (H) Only the 11,339 high confidence origins are shown. (I,J) Origin efficiency is highly correlated between testis and ESCs. All peaks found in either sample are shown. (K,L) Origin efficiency is correlated between ESC Ori-SSDS replicates. (K) All origins found in either experiment. (L) Origins found in both experiments.
Figure S2. Most replicating cells in testis are meiotic, related to Figures 1 and 2
To identify replicating cells in testis, the thymidine analog 5-Ethynyl-2’-deoxyuridine (EdU) was injected intraperitoneally to live mice, then mice were sacrificed and testes harvested after 30 min. Nuclei were stained with DMRT1 and STRA8 antibodies to distinguish spermatogonia from preleptotene nuclei. (A) From FANS sorting, 1.8% of nuclei from whole testis were inferred to be replicating (EdU positive; above dashed red line). (B) Most putatively replicating (2–4C; DAPI content) spermatogonia and preleptotene cells were EdU positive (red). (C) A majority of replicating cells in mouse testis are in meiotic S-phase.
Figure S3. Origins of replication in mouse testis occur at GC-rich, accessible sites, related to Figure 2
(A) Origins of replication are more closely spaced than expected in the genome. The expected distances between randomly chosen intervals is shown in gray. (B) Most origins occur near gene promoters; 66% occur within 1 Kbp of a transcription start site (TSS), far more than expected (random; gray line). (C) Replication origins coincide with local changes in DNA bendability (calculated from Goodsell and Dickerson [1994]). The mean (top) and standard deviation (bottom) of the DNA bendability at all origins is shown. Each point represents a 100 bp window. Solid line is a LOESS fit. (D) Origin centers are GC-rich and enriched in CpG and GpC dinucleotides. The log2 ratio of the central 500 bp compared to the region flanking this center is shown. (E) GC-content (GC%), CpG density and GpC density correlate positively with origin efficiency. (F) Putative G4-forming sequences are highly enriched at the center of origins. 68% of origins have a putative G4-forming sequence in the central ± 1 Kb. (G) Origins of replication occur predominantly at CpG islands (CGIs) that coincide with open chromatin as measured by ATAC-Seq (Maezawa et al., 2018). (H) Origins are only detected at about half of the CGIs at ATAC-Seq peaks. (I) Origin locations can be identified from CpG density. CpG dinucleotide density was evaluated in all 1 Kb regions in the genome using a 100 bp sliding window. Intervals at each CpG threshold were expanded ± 100 bp around the center and overlapping windows were merged. The centerpoint of each merged interval was defined as a CpG peak. CpG peaks with CpG content ≥ 7% overlap 6,233 origins (53% of total). 1,853 CpG peaks at this threshold do not occur at an origin (FP). (J) Origins are better predicted by CpG density than transcription factors (TFs) are by transcription factor binding sites (TFBS). Receiver operating curve (ROC) for predicting origin locations using CpG peaks (green) or for predicting peaks in ChIP-Seq datasets for 179 transcription factors (gray). For origins, at each CpG threshold (1% to 15% in 1% steps), peaks were called (as in G). All mouse TFs with ChIP-Seq data (in GTRD database) (Yevshin et al., 2017) and with a designated TFBS in the HOCOMOCO database (Kulakovskiy et al., 2018) were used. SPRY-SARUS (Kulakovskiy et al., 2018) was used to identify TFBS motifs genome-wide with p < 0.005. For each TF, the score at which ≤ 12,000 peaks were found was used as a lower threshold (this is to assure a fairer comparison with origins). Motifs with scores less than this were discarded. TFs with < 12,000 peaks not used. Hits inside ChIP-Seq peaks were considered true positives.
Figure S4. RT-seq yields similar RT profiles across varied cell types, related to Figure 3
RT for all cell types shown on Figure 2. RT is expressed as the log2 enrichment over genomic median coverage for all panels except for Myoblast and LCL data. These data were obtained in processed form from the Replication Domain database. All panels depict chromosome 12. Sample names are explained briefly in Figure 3 and in detail in Tables S1, S2 and S3.
Figure S5. Experimentally derived RT varies as a function of the starting population, related to Figure 4
(A) Four sub-populations of meiotic S-phase were isolated from early S (T1) through late S (T4). RT was inferred from WGS (see STAR Methods). The earliest and latest replicating regions exhibit substantial differences in RT across these four populations. (B-G) Properties of DNA replication inferred from best-fitting models for Meiotic (pink) and ESCs (gray). (B) The cumulative amount of replicated DNA at each simulation cycle is shown. We infer S-phase duration from the time at which 90% of the genome is replicated (magenta line). The rate of replication is reduced in the final 10%–15% of the genome. (C) The final 10%–15% of the genome is slow to replicate because the number of active forks decreases rapidly. (D) The total number of replication origins fired per simulated S-phase is shown. Each simulation models replication of a single haploid genome. (E) Replication tract length is longer in meiotic cells compared to ESCs. Note that meiotic cells use fewer origins. (F) Model estimates of the per S-phase frequency of origin firing. (G) The rate at which new origins fire throughout simulation cycles.
Figure S6. Modeling recapitulates experimental RT genome-wide, related to Figure 4
Experimental RT is shown as filled green (early replicating) and violet (late replicating) regions. The best-fitting RT-Sim predicted RT is shown as a solid black line. Per-chromosome Pearson correlation coefficients are shown. Both (A) Meiotic and (B) ESC RT can be accurately modeled in silico. Both experimental and predicted RT are normalized by mean and standard deviation (s.d.); (RTnorm = (RT-mean(RT)) / s.d.(RT)).
Figure S7. Meiotic S-phase duration is unaffected in Spo11−/− male mice, related to Figure 4
Meiotic S-phase nuclei, B-type spermatogonia and undifferentiated spermatogonia were isolated from a Spo11−/− male mouse testis (SPO11 in sample names) and a wild-type littermate (wtLM in sample names). Details for each sample are in Tables S1, S2, and S3. (A) 2–4C nuclei were sorted and (B) gated by DMRT1 and STRA8 immunofluorescence signal. Gates were defined while sorting nuclei from the Spo11−/− mouse and retained, unchanged for sorting nuclei from the littermate. (C) Summary statistics for best-fitting RT-Sim models for all samples. (D) Comparison of S-phase duration across cell types. The median S-phase duration of good models for each cell-type is compared to meiotic S-phase. (E) The ‘‘fit’’ of RT-Sim models for Spo11−/− samples is comparable to the fit in other cell types. (F) The duration of meiotic S-phase is unaffected in Spo11−/− mice relative to the wild-type littermate control. Notably, both mice exhibit slightly shorter S-phase duration compared to the three wild-type replicates. This is likely because for the Spo11−/− and wild-type littermate, the sorting gates were defined in the Spo11−/− mouse and left unchanged when sorting nuclei from the littermate. Cellularity differences from the loss of Spo11 may result in the capture of a slightly different population than when sorting gates are defined using wild-type. (G) The firing rate is similarly unaffected.
Data Availability Statement
Sequencing data are deposited at the GEO (GSE148327). Replication origin locations, replication timing data, and associated data used to generate figures are available in Data S1 (mouse) and Data S2 (human). Custom code is available at Zenodo:
| Analytic pipeline | https://zenodo.org/record/4634002 |
| RT-Seq pipeline | https://zenodo.org/record/4634128 |
| RT-sim | https://zenodo.org/record/4634118 |