

Abstract
Human β-nerve growth factor (β-NGF) and its associated receptor, human tropomyosin receptor kinase A (hTrkA), have been demonstrated to be key factors in the perception of pain. However, efficacious small molecule therapies targeting the intracellularly located hTrkA kinase have not been explored thoroughly for pain management. Herein, we report the pharmacological properties of a selective hTrkA allosteric inhibitor, 1. 1 was shown to be active against the full length hTrkA, showing preferential binding for the inactive kinase, and was confirmed through the X-ray of hTrkA···1 bound complex. 1 was also found to inhibit β-NGF induced neurite outgrowth in rat PC12 cells. Daily oral administration of 1 improved the joint compression threshold of rats injected intra-articularly with monoiodoacetate over a 14-day period. The efficacy of 1 in a relevant chronic pain model of osteoarthritis coupled with in vitro confirmation of target mediation makes allosteric hTrkA inhibitors potential candidates for modulating pain.
Keywords: β-NGF, osteoarthritis pain, hTrkA, allosteric, kinase
Despite tremendous efforts over the past few decades, uncovering novel mechanistic replacements for nonsteroidal anti-inflammatory drugs (NSAIDs) and opioids for the treatment of chronic pain has proved exceedingly difficult. In addition, chronic pain may or may not be associated with ongoing inflammation. New analgesics are in great demand for the myriad chronic pain conditions for which adequate therapies do not exist, making long-term pain management a significant unmet medical need.1
Levi-Montalcini discovered nerve growth factor (NGF) as an essential protein for the prenatal growth of sensory and sympathetic nerves.2−4 Subsequently, it was discovered that in addition to NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5) were other factors involved in nerve growth and collectively designated as “neurotrophic factors”. The discovery of the receptors of each of these factors followed, with tropomyosin receptor kinase A (TrkA) for NGF displaying high sequence homology to tropomyosin receptor kinase B (TrkB) that binds BDNF and NT-4/5 and tropomyosin receptor kinase C (TrkC) which partners with NT-3.5,6
Advances in molecular biology eventually led to the discovery of a genetic mutation in TrkA as the root cause of a congenital insensitivity to pain.7 Also, it was found that when NGF actions are continuously suppressed by anti-NGF antibodies from the prenatal through the neonatal period, growth of sensory and sympathetic nerves is completely inhibited. However, if NGF is suppressed postnatally, only partial inhibition of nerve growth occurs.8 In addition, it was discovered that NGF administered to adult rats induces hyperalgesia, confirming NGF as one of the chemical substances that induces pain in adults.9,10
Anti-NGF antibody therapies have shown efficacy in preclinical models of pain and in the human clinical setting. However, research into efficacious small molecule therapies has progressed more slowly.1 The binding of small molecules to the kinase domain of the TrkA receptor can fall into three main categories. Type 1 inhibitors occupy the catalytic adenosine triphosphate (ATP) binding site of an active state conformation of the kinase domain and are competitive with ATP. Type 2 inhibitors occupy the same site but are noncompetitive with respect to ATP, arising from an inactive state kinase conformation that restricts access to ATP. Allosteric inhibitors bind to the kinase domain but distal to the ATP site and can effectively bind either kinase conformation.11 Manipulating the protein conformational versatility and the non-ATP binding pockets can be instrumental in achieving small molecule inhibitor selectivity against Trk subtypes and across the kinome.
Although numerous examples of preclinically efficacious small molecules are known, many are pan-Trk inhibitors of the intracellularly located Trk kinase.12−15 Due to potential hyperphagia associated with inhibiting TrkB16−18 and propioception-related adverse events with inhibiting TrkC,19,20 finding inhibitors that exhibit not only kinase selectivity but also selectivity for the Trk isoforms is of potential value for chronically administered analgesics. Subtype-selective, allosteric inhibitors of hTrkA kinase are emerging in the literature21−26 and could serve as potential alternatives to orthosteric inhibitors with improved safety over pan-Trk inhibitors.27 Although the allosteric inhibitors described in the literature exhibit good potency for TrkA, little has been published on the pharmacology of these inhibitors.
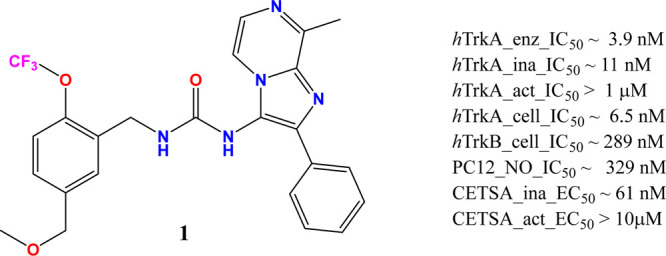
This paper describes the pharmacological profile of a novel benzylic urea lead, 1 [1-(5-(methoxymethyl)-2-(trifluoromethoxy)benzyl)-3-(8-methyl-2-phenylimidazo[1,2-a]pyrazin-3-yl)urea], that emerged from the medicinal chemistry lead optimization efforts (Figure 1) at Zoetis. The effect of 1 on neurite outgrowth (NO) from PC12 cells and its nociceptive effects in a chronic rodent model of osteoarthritis (OA) positions the lead for further evaluation in the discovery and development process.
Figure 1.
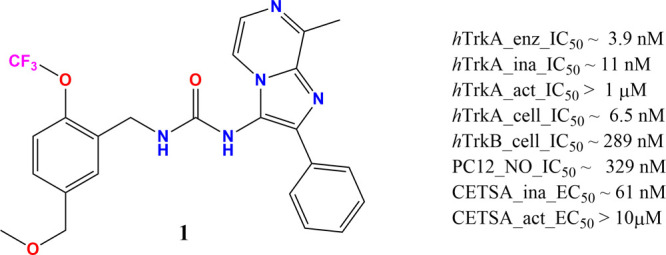
Structure and in vitro profile of 1.
Results
In Vitro Profile of 1
The chemical series to which 1 belongs was initially identified and evaluated using a high throughput TR-FRET kinase assay that utilizes the inactive form of hTrkA enzyme. 1 inhibited hTrkA enzymatic phosphorylation with a mean (±SD) IC50 of 3.9 (±4.9) nM. To evaluate phosphorylation-state-dependent binding, caliper technology was used to separate and directly quantify substrate phosphorylation for both inactive and active states of hTrkA enzyme. The IC50 for 1 using the inactive hTrkA enzyme was 10.97 (±4.71) nM and showed no activity for the active enzyme up to a maximum of 1 μM screening concentration. Clearly, 1 showed evidence of phosphorylation state binding. In the cell based PathHunter assay, 1 displayed 6.5 (±6.6) nM and 289 (±174) nM IC50 values for hTrkA and hTrkB, respectively, indicating potential selectivity gains against the subfamily member. 1 also exhibited prolonged binding to the inactive state of hTrkA kinase relative to AZ-23,28 a known type-1 pan-Trk inhibitor, displaying residence times of 78.25 and 8.05 min, respectively. Conversely, 1 did not bind to the active form of hTrkA kinase, displaying a residence time of 1.68 min relative to 111.86 min for AZ-23. Finally, 1 produced a dose-dependent inhibition of rat β-NGF-induced neurite outgrowth in PC12 cells29 with an IC50 of 329.4 (±1.8) nM. Additionally, 1 was evaluated via a label-free CETSA assay using PC12 cells30 and biasing the kinase conformational state by altering the temperature. This assay also revealed 1 to possess EC50 ∼ 61 nM when screened at 40 °C (inactive state) and EC50 > 10 μM at 52 °C (active state) providing support for the target engagement using a full length hTrkA with β-NGF stimulation
X-ray Structure of the Kinase Domain hTrkA···1 Complex
Crystals of the juxtamembrane (JM) domain including hTrkA kinase domain spanning residues 485–795 in complex with 1 were prepared using reported protocols.30 The hTrkA ligand bound complex structure was subsequently solved and refined to 2.06 Å final resolution (PDB code 6PL4). The crystal asymmetric unit contains only one TrkA monomer with the electron density unambiguously defining the ligand orientation in an allosteric binding pocket away from the orthosteric ATP binding site (Figure 2). The overall structure has two short loops missing between residues 491–499 and 609–611 due to the poor electron density. The protein folding integrity assessed using the Ramachandran plot reveals 93.3% of the amino acids to be in the most favored regions and 6.3% in the additional allowed regions. The completeness of the model, however, was achieved by building the missing loop structures and assigning side chains to residues that could not be clearly defined by the experimental electron density as part of the protein preparation module within the MAESTRO application suite from Schrödinger. This modeled structure was used for all analyses and comparisons.
Figure 2.
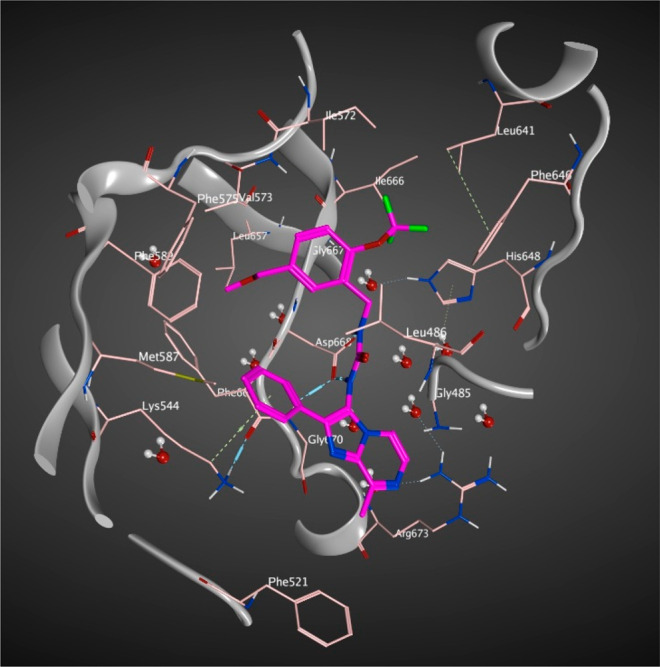
Binding mode of 1 (magenta) bound to extended hTrkA kinase domain (PDB code 6PL4).
The DFG residues (668–670) side chain orientation and the activation loop (A-loop) unequivocally reveal the kinase to adapt a DFG-out and α C-helix in conformation, typical for the kinase inactive state.31 This A-loop conformation partially occludes the orthosteric site, preventing ATP from binding and thereby rendering the enzyme to be catalytically ineffective. In addition, the glycine-rich loop (516–524) is pushed further upward when compared to kinase active state structures. The highly unstructured JM domain and the A-loop residues carve out the allosteric binding site that allows small molecule allosteric inhibitors to bind in an ATP noncompetitive way.24 The catalytic lysine, K544-E560 salt bridge break-off typically encountered in the enzymatically competent kinase active state also facilitates an additional binding volume used for the ligand occupancy. Superposition of the current hTrkA-ligand complex with the reported structure of ATP-competitive inhibitor AZ-23 (PDB code 4AOJ)28 bound to hTrkA clearly reveals the macromolecular motions of the glycine-rich and A-loop that defines the two unique conformational states of the kinase and removes any ambiguity in defining the kinase conformational state observed via X-ray.
The X-ray diffraction data further reveal a clear binding mode for 1 (Figure 2) with the appropriate binding orientation and unconstrained ligand conformation that maximizes the surrounding residues’ interaction in the binding pocket and showcases the contributions of the JM region residues like L486 to the allosteric modulation. It is worth noting that the JM region is crucial as it serves as the SHC32 phosphotyrosine binding site for protein/protein interaction and the lower sequence identity in this region may also help achieve inhibitor selectivity across the TrkA, -B, and -C subfamily members. On the basis of the standard binding site cutoff distances, the urea NH groups of the ligand effectively H-bond with the carboxylate side chain of D668 (Scheme 1). Apart from this anchoring interaction, the ligand is covered by several hydrophobic residues that strengthen the protein–ligand interaction as reflected by the high binding affinity for hTrkA. The binding site also includes several solvent water molecules (Figure 2) captured from the X-ray that seem to be responsible for keeping the protein and binding site intact. However, none of these water molecules are involved in any solvent mediated H-bond between the ligand and the protein.
Scheme 1. . hTrkA···1 Binding Interaction Derived from the Solved X-ray Complex.
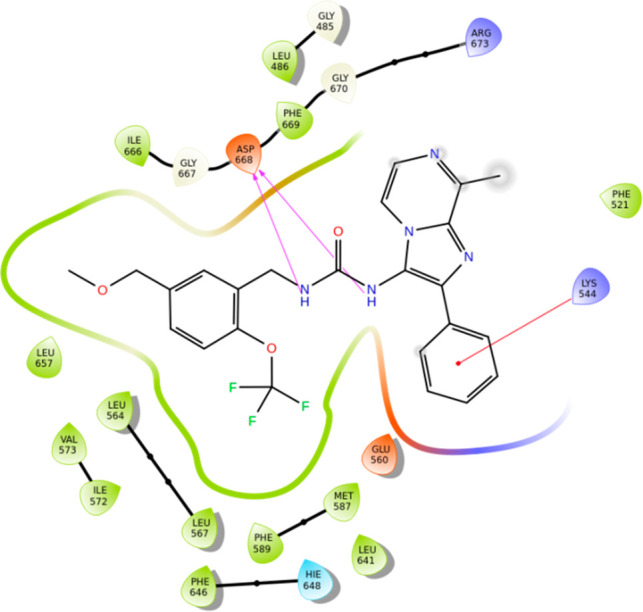
PDB accession code is 6PL4.
Reduction of Pain in Chronic Osteoarthritis Model
On day −14, MIA was injected into the left intra-articular space, producing OA-induced pain, as demonstrated by significant decreases in JCTs in the ipsilateral joints compared to the contralateral joints of the vehicle-treated animals. On day 0, 1 dosed qd produced a significant increase in JCT at 2 h postdose. On day 7, both qd and BID treatments of 1 produced a significant increase in JCTs 5 h postdose. On day 14, qd treatment with 1 produced a significant effect 2 h postdose, and BID treatment produced significant effects at 5 and 8 h postdose (Figure 3). Both treatment groups for 1 showed an increase in pretreatment baselines across the entire study that were significantly different from zero, but vehicle treatment did not produce a similar effect. None of the treatments had any statistically significant effect on the contralateral limb, suggesting any effect was a direct result of altering the pain response induced by intra-articular MIA.
Figure 3.

Joint compression threshold (mean ± SEM) over time for qd and BID administration of 1 in a rat MIA model (N = 10 rats/treatment). Animals were injected with MIA on day −14, and treatment with either vehicle or 1 began on day 0 continuing through day 15. Baseline (BL) recordings occurred immediately prior to the AM dose administration. JCTs measured on day 16 occurred 24 h after the last administration of vehicle or 1. Significance of effects of 1 treatment (qd or BID administration) compared to vehicle-treated animals was calculated at each time point using a two-way repeated-measures ANOVA followed by Tukey’s multiple comparisons (*P < 0.05; **P < 0.01).
Discussion
In this Letter, we report on the pharmacological characterization of a hTrkA allosteric kinase inhibitor, 1. The in vitro and in vivo pharmacological profiles of 1 were evaluated. These studies characterize 1 as a potent, inhibitor of TrkA with selectivity over TrkB.
The clinical administration of Trk inhibitors has been associated with potential liabilities that have limited their use. It has been noted that pan-Trk inhibition leads to ataxia and hyperphagia.17,20 Isoform selectivity was measured through in vitro functional assay against TrkB and kinase conformation selectivity through binding kinetics using SPR. With a preference for binding the inactive state of TrkA kinase as evidenced from the biophysical and label-free CETSA assays, coupled with the observed selectivity in the functional assay, 1 was suggested to be a novel, allosteric inhibitor of hTrkA. The hTrkA···1 extended kinase domain X-ray complex confirmed the inactive state kinase conformation and binding to the distal non-ATP site.
In addition to the binding and selectivity characteristics, 1 proved to be effective at inhibiting NO in rat PC12 cells. It has been hypothesized that growing and damaged peripheral nerves display sensitization and are associated with increased pain sensation.33,34 In OA, articular cartilage becomes vascularized and these new vessels may be associated with new sensory nerves.35−37 Likewise, patients with chronic lower back pain due to degenerative disks have demonstrated that the damaged disks release a combination of factors, including NGF, leading to neurite sprouting and increased calcitonin gene related-peptide (CGRP) expression, which is strongly associated with pain.38 Therefore, it is thought that effectively arresting neurite outgrowth via inhibition of the action of NGF during OA is a possible mechanism for the relief of perceived pain.39,40
The combination of in vivo methods and in vitro biochemical and biophysical techniques in engineered cells used to characterize 1 represents the most thorough characterization of an allosteric TrkA inhibitor reported to date for pain indication. Preliminary immunohistochemistry of the DRGs and spinal cord explants performed to comprehend the expression and internalization of the TrkA receptor in the rat MIA model also provides evidence for receptor internalization in the efficacy model. Interestingly, 1 appeared to display some degree of analgesia in rats beyond the dosing period of 15 days, continuing into the final 16th day time point. Although not validated experimentally, one possible mechanism for this action is through the modulation of CGRP release. Synthesis and release of substance P (SP) and CGRP are known to occur as a result of NGF stimulation, and this excessive release may lead to neurogenic inflammation. CGRP potentiates SP release at the spinal cord level and delays its degradation, thus augmenting the response of nociceptive dorsal horn neurons to repetitive type IV/C fiber stimulation.41 Others have reported elevated levels of CGRP as well as activated peptidergic afferent C fibers in the spinal cord of rats in MIA models of OA pain.42
It is also worth noting that 1 produced statistically significant reduction in JCT on day 0 at the 8 h time point. This is interesting, as it is generally accepted that there is an NSAID-sensitive and NSAID-insensitive phase of the MIA model. The NSAID-sensitive phase occurs early after MIA injection (within 14 days) and is responsive to treatment with NSAIDs, most likely due to an inflammatory component of the model. The fact that 1 produced a statistically significant effect during this NSAID-sensitive phase suggests that this effect may be due to an anti-inflammatory effect of 1.23,43,44
In conclusion, 1 is a potent and selective hTrkA inhibitor. The selectivity of this molecule is likely to afford a much wider margin of safety over known pan-Trk inhibitors. 1 was able to effectively inhibit neurite outgrowth in rat PC12 cells which has been demonstrated to have high translatability to human dorsal root ganglion.45 Robust efficacy in a rat MIA model of OA has been shown to be translatable to field efficacy for this pathway.46 The combined efficacy in a translatable rodent model and potency in the NO assay make 1 a potential candidate for human use in the relief of pain associated with OA.
Acknowledgments
The authors thank Nicole Roush, Kevin Esch, Phyllis Malpas, Catherine Rugg, Jim Messamore, Jennifer Paquette, Robert Sturm, Leslie Kuiper, Roger Martin, Ann Janssen, and Graeme Bainbridge for technical support. G.S. also thanks Stefan Steinbacher from Proteros Biostructures GmbH, Germany, for structural biology, and Arthur Wittwer and Jeffrey Hirsch from Confluence Discovery Technologies for the caliper assays.
Glossary
Abbreviations
- qd
quarter in die
- BID
bis in die
- JCT
joint compression threshold
- SPR
surface plasmon resonance
- CETSA
cellular thermal shift assay
- DRG
dorsal root ganglion
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00483.
Complete experimental information for the chemistry, in vitro biochemical and biophysical assays, and in vivo pharmacological assay, along with the table for the X-ray data (PDF)
Author Present Address
‡ Medtronic, 7000 Central Avenue NE, Minneapolis, MN55432, United States
Author Contributions
† G.S. and B.D. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Authors are past or present employees of Zoetis and have financial interest in the company.
Supplementary Material
References
- Norman B. H.; McDermott J. S. Targeting the nerve growth factor (NGF) pathway in drug discovery. Potential Applications to new therapies for chronic pain. J. Med. Chem. 2017, 60, 66–88. 10.1021/acs.jmedchem.6b00964. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. Effects of mouse tumor transplantation on the nervous system. Ann. N. Y. Acad. Sci. 1952, 55, 330–344. 10.1111/j.1749-6632.1952.tb26548.x. [DOI] [PubMed] [Google Scholar]
- Abbott A. One hundred years of Rita. Nature 2009, 458, 564–567. 10.1038/458564a. [DOI] [PubMed] [Google Scholar]
- Hirose M.; Kuroda Y.; Murata E. NGF/TrkA signaling as a therapeutic target for pain. Pain Pract. 2016, 16, 175–182. 10.1111/papr.12342. [DOI] [PubMed] [Google Scholar]
- Landreth G. E.; Shooter E. M. Nerve growth factor receptors on PC12 cells: ligand- induced conversion from low- to high-affinity states. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 4751–4755. 10.1073/pnas.77.8.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton D. L.; Sutherland J.; Gripp J.; Camerato T.; Armanini M. P.; Phillips H. S.; Carroll K.; Spencer S. D.; Levinson A. D. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain adhesions. J. Neurosci. 1995, 15, 477–491. 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y.; Tsuruta M.; Hayashida Y.; Karim M. A.; Ohta K.; Kawano T.; Mitsubuchi H.; Tonoki H.; Awaya Y.; Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhydrosis. Nat. Genet. 1996, 13, 485–488. 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M. The changing sensitivity in the life of the nociceptor. Pain 1999, 82, S93–S102. 10.1016/S0304-3959(99)00142-6. [DOI] [PubMed] [Google Scholar]
- Kromer L. F. Nerve growth factor treatment after brain injury prevents neuronal death. Science 1987, 235, 214–216. 10.1126/science.3798108. [DOI] [PubMed] [Google Scholar]
- Lewin G. R.; Ritter A. M.; Mendell L. M. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J. Neurosci. 1993, 13, 2136–2148. 10.1523/JNEUROSCI.13-05-02136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Smaill J. B.; Ding K. New promise and opportunities for allosteric kinase inhibitors. Angew. Chem., Int. Ed. 2020, 59, 13764–13776. 10.1002/anie.201914525. [DOI] [PubMed] [Google Scholar]
- Stachel S. J.; Sanders J. M.; Henze D. A.; Rudd M. T.; Su H. P.; Li Y.; Nanda K. K.; Egbertson M. S.; Manley P. J.; Jones K. L.; Brnardic E. J.; Green A.; Grobler J. A.; Hanney B.; Leitl M.; Lai M. T.; Munshi V.; Murphy D.; Rickert K.; Riley D.; Krasowska-Zoladek A.; Daley C.; Zuck P.; Kane S. A.; Bilodeau M. T. Maximizing diversity from a kinase screen: identification of novel and selective pan-Trk inhibitors for chronic pain. J. Med. Chem. 2014, 57, 5800–5816. 10.1021/jm5006429. [DOI] [PubMed] [Google Scholar]
- Skerratt S. E.; Andrews M.; Bagal S. K.; Bilsland J.; Brown D.; Bungay P. J.; Cole S.; Gibson K. R.; Jones R.; Morao I.; Nedderman A.; Omoto K.; Robinson C.; Ryckmans T.; Skinner K.; Stupple P.; Waldron G. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem. 2016, 59, 10084–10099. 10.1021/acs.jmedchem.6b00850. [DOI] [PubMed] [Google Scholar]
- Bailey J. J.; Schirrmacher R.; Farrell K.; Bernard-Gauthier V. Tropomyosin receptor kinase inhibitors: an updated patent review for 2010-2017 – Part II. Expert Opin. Ther. Pat. 2017, 27, 831–849. 10.1080/13543776.2017.1297797. [DOI] [PubMed] [Google Scholar]
- Yan W.; Lakkaniga N. R.; Carlomagno F.; Santoro M.; McDonald N. Q.; lv R.; Gunaganti N.; Frett B.; Li H. Y. Insights into current tropomyosin receptor kinase (TRK) inhibitors: Development and clinical application. J. Med. Chem. 2019, 62, 1731–1760. 10.1021/acs.jmedchem.8b01092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. C.; Tsao D.; Barras P.; Bastarrachea R. A.; Boyd B.; Chou J.; Rosete R.; Long H.; Forgie A.; Abdiche Y.; Dilley J.; Stratton J.; Garcia C.; Sloane D. L.; Comuzzie A. G.; Rosenthal A. Appetite enhancement and weight gain by peripheral administration of TrkB agonists in non-human primates. PLoS One 2008, 3, e1900. 10.1371/journal.pone.0001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.; Tse M. C. L.; Liu X.; Zhang S.; Schmidt R.; Otten R.; Liu L.; Ye K. Activation of muscular TrkB by its small molecular agonist 7,8-dihydroxyflavone sex-dependently regulates energy metabolism in diet-induced obese mice. Chem. Biol. 2015, 22, 355–368. 10.1016/j.chembiol.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B.; Xie X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016, 17, 282–292. 10.1038/nrn.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R.; Silos-Santiago I.; Smeyne R. J.; Lira S. A.; Brambilla R.; Bryant S.; Zhang L.; Snider W. D.; Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 1994, 368, 249–251. 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- Genç B.; Özdinler P. H.; Mendoza A. E.; Erzurumlu R. S. A chemoattractant role for NT-3 in proprioceptive axon guidance. PLoS Biol. 2004, 2, e403. 10.1371/journal.pbio.0020403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwosu L. N.; Mapp P. I.; Chapman V.; Walsh D. A. Blocking the tropomyosin receptor kinase A (TrkA) receptor inhibits pain behaviors in two rat models of osteoarthritis. Ann. Rheum. Dis. 2016, 75, 1246–1254. 10.1136/annrheumdis-2014-207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S.; Andrews S. W.; Baer B.; Crane Z.; Liu W.; Watson D. J.. 1-((3S,4R)-4-(3-Fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1h-pyrazol-5-yl)urea as a TrkA kinase inhibitor. WO2015/175788, 2015.
- Ashraf S.; Bouhana K. S.; Pheneger J.; Andrews S. W.; Walsh D. A. Selective inhibition of tropomyosin receptor kinase A (TrkA) reduces pain and joint damage in two rat models of inflammatory arthritis. Arthritis Res. Ther. 2016, 18, 97. 10.1186/s13075-016-0996-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H. P.; Rickert K.; Burlein C.; Narayan K.; Bukhtiyarova M.; Hurzy D. M.; Stump C. A.; Zhang X.; Reid J.; Krasowska-Zoladek A.; Tummala S.; Shipman J. M.; Kornienko M.; Lemaire P. A.; Krosky D.; Heller A.; Achab A.; Chamberlin C.; Saradjian P.; Sauvagnat B.; Yang X.; Ziebell M. R.; Nickbarg E.; Sanders J. M.; Bilodeau M. T.; Carroll S. S.; Lumb K. J.; Soisson S. M.; Henze D. A.; Cooke A. J. Structural characterization of nonactive site, TrkA-selective kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E297–306. 10.1073/pnas.1611577114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya N.; Momose T.; Katsuno K.; Fushimi N.; Muranaka H.; Handa C.; Ozawa T.; Kinoshita T. The juxtamembrane region of TrkA kinase is critical for inhibitor selectivity. Bioorg. Med. Chem. Lett. 2017, 27, 1233–1236. 10.1016/j.bmcl.2017.01.056. [DOI] [PubMed] [Google Scholar]
- Bagal S. K.; Omoto K.; Blakemore D. C.; Bungay P. J.; Bilsland J. G.; Clarke P. J.; Corbett M. S.; Cronin C. N.; Cui J. J.; Dias R.; Flanagan N. J.; Greasley S. E.; Grimley R.; Johnson E.; Fengas D.; Kitching L.; Kraus M. L.; McAlpine I.; Nagata A.; Waldron G. J.; Warmus J. S. Discovery of allosteric, potent, subtype selective, and peripherally restricted TrkA kinase inhibitors. J. Med. Chem. 2019, 62, 247–265. 10.1021/acs.jmedchem.8b00280. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Yu P.; Dong L.; Wang W.; Duan S.; Wang B.; Gong X.; Ye L.; Wang H.; Tian J. Discovery of the next-generation pan-TRK kinase inhibitors for the treatment of cancer. J. Med. Chem. 2021, 64, 10286–10296. 10.1021/acs.jmedchem.1c00712. [DOI] [PubMed] [Google Scholar]
- Wang T.; Lamb M. L.; Scott D. A.; Wang H.; Block M. H.; Lyne P. D.; Lee J. W.; Davies A. M.; Zhang H. J.; Zhu Y.; Gu F.; Han Y.; Wang B.; Mohr P. J.; Kaus R. J.; Josey J. A.; Hoffmann E.; Thress K.; Macintyre T.; Wang H.; Omer C. A.; Yu D. Identification of 4-aminopyrazolylpyrimidines as potent inhibitors of Trk kinases. J. Med. Chem. 2008, 51, 4672–4684. 10.1021/jm800343j. [DOI] [PubMed] [Google Scholar]
- Greene L. A.; Tischler A. S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 2424–2428. 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian G.; Johnson P. D.; Zachary T.; Roush N.; Zhu Y.; Bowen S. J.; Janssen A.; Duclos B. A.; Williams T.; Javens C.; Dekki-Shalaly N.; Martinez-Molina D.; Wittwer A. J.; Hirsch J. L. Deciphering the Allosteric Binding Mechanism of the Human Tropomyosin Receptor Kinase A (hTrkA) Inhibitors. ACS Chem. Biol. 2019, 14, 1205–1216. 10.1021/acschembio.9b00126. [DOI] [PubMed] [Google Scholar]
- van Linden O. P. J.; Kooistra A. J.; Leurs R.; de Esch I. J. P.; de Graaf C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- Stephens R. M.; Loeb D. M.; Copeland T. D.; Pawson T.; Greene L. A.; Kaplan D. R. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron 1994, 12, 691–705. 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Mayer D. J.; Mao J.; Holt J.; Price D. D. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 7731–7736. 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W.; Strong J. A.; Li H.; Zhang J. M. Sympathetic sprouting near sensory neurons after nerve injury occurs preferentially on spontaneously active cells and is reduced by early nerve block. J. Neurophysiol. 2007, 97, 492–502. 10.1152/jn.00899.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens R. W.; Ghosh P.; Taylor T. K. The pathogenesis of osteoarthrosis. Med. Hypotheses 1979, 5, 809–816. 10.1016/0306-9877(79)90041-0. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade J.; Mantyh P. W. Sensory and sympathetic nerve fibers undergo sprouting and neuroma formation in the painful arthritic joint of geriatric mice. Arthritis Res. Ther. 2012, 14, R101–116. 10.1186/ar3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grässel S.; Muschter D. Peripheral nerve fibers and their neurotransmitters in osteoarthritis pathology. Int. J. Mol. Sci. 2017, 18, 931–954. 10.3390/ijms18050931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock E.; Rosenzweig D. H.; Chabot-Dore A. J.; Jarzem P.; Weber M. H.; Ouellet J. A.; Stone L. S.; Haglund L. Painful, degenerating intervertebral discs up-regulate neurite sprouting and CGRP through nociceptive factors. J. Cell. Mol. Med. 2014, 18, 1213–1225. 10.1111/jcmm.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet C. S.; Walsh D. A. Osteoarthritis, angiogenesis and inflammation. Rheum. 2005, 44, 7–16. 10.1093/rheumatology/keh344. [DOI] [PubMed] [Google Scholar]
- Chen Y. J.; Chang W. A.; Wu L. Y.; Hsu Y. L.; Chen C. H.; Kuo P. L. Systematic analysis of transcriptomic profile of chondrocytes in osteoarthritic knee using next-generation sequencing and bioinformatics. J. Clin. Med. 2018, 7, 535–559. 10.3390/jcm7120535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konttinen Y. T.; Kemppinen P.; Segerberg M.; Hukkanen M.; Rees R.; Santavirta S.; Sorsa T.; Pertovaara A.; Polak J. M. Peripheral and spinal neural mechanisms in arthritis, with particular reference to treatment of inflammation and pain. Arthritis Rheum. 1994, 37, 965–982. 10.1002/art.1780370701. [DOI] [PubMed] [Google Scholar]
- Otis C.; Guillot M.; Moreau M.; Martel-Pelletier J.; Pelletier J. P.; Beaudry F.; Troncy E. Spinal neuropeptide modulation, functional assessment and cartilage lesions in a monosodium iodoacetate rat model of osteoarthritis. Neuropeptides 2017, 65, 56–62. 10.1016/j.npep.2017.04.009. [DOI] [PubMed] [Google Scholar]
- Bove S. E.; Calcaterra S. L.; Brooker R. M.; Huber C. M.; Guzman R. E.; Juneau L.; Schrier D. J.; Kilgore K. S. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis and Cartilage 2003, 11, 821–830. 10.1016/S1063-4584(03)00163-8. [DOI] [PubMed] [Google Scholar]
- Pomonis J. D.; Boulet J. M.; Gottshall S. M.; Phillips S.; Sellers R.; Bunton T.; Walker K. Development and pharmacological characterization of a rat model of osteoarthritis pain. Pain 2005, 114, 339–346. 10.1016/j.pain.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Schwaid A. G.; Krasowka-Zoladek A.; Chi A.; Cornella-Taracido I. Comparison of the rat and human dorsal root ganglion proteome. Sci. Rep. 2018, 8, 13469–13478. 10.1038/s41598-018-31189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa G.; Koya Y.; Tanaka H.; Nagakura Y. Long-term analgesic effect of a single dose of anti-NGF antibody on pain during motion without notable suppression of joint edema and lesion in a rat model of osteoarthritis. Osteoarthritis and Cartilage 2015, 23, 925–932. 10.1016/j.joca.2015.02.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.