

Abstract

Prostate cancer is among the leading causes of cancer related death of men in the United States. The ERG gene fusion leading to overexpression of near full-length ERG transcript and protein represents most prevalent (50–65%) prostate cancer driver gene alterations. The ERG oncoprotein overexpression persists in approximately 35% of metastatic castration resistant prostate cancers. Due to the emergence of eventual refractoriness to second- and third-generation androgen axis-based inhibitors, there remains a pressing need to develop drugs targeting other validated prostate cancer drivers such as ERG. Here we report the new and more potent ERG inhibitor ERGi-USU-6 developed by structure–activity studies from the parental ERGi-USU. We have developed an improved procedure for the synthesis of ERGi-USU-6 and identified a salt formulation that further improves its activity in biological assays for selective targeting of ERG harboring prostate cancer cells.
Keywords: ERG, Oncogene, TMPRSS2-ERG, RIOK2, Inhibitor, Small molecule, Prostate cancer, Metastatic castration resistant prostate cancer, SAR, Precision medicine
The 2021 projection of prostate cancer (PCa) prevalence predicts alarming increases that have not been seen in the past two decades. PCa is one of the most prevalent noncutaneous malignancy and second leading cause of cancer deaths in men in the United States.1 PCa is a global public health issue, exceeding one million people succumbing to the disease each year.2 It is estimated that globally one in eight men will be diagnosed with PCa worldwide over his lifetime. Surgery, radiation therapy, and active surveillance are the few choices for the management of localized disease. The main line of treatment for advanced PCa is androgen deprivation therapy (ADT), which is widely used for treating PCa that has escaped the primary treatment regimens.3
It is remarkable that approximately half of primary PCas harbor gene fusions (prevalent fusion of the TMPRSS2 gene promoter with the protein coding region of the ETS Related Gene [ERG]) which drive overexpression of the ERG fusion transcripts and ERG oncoprotein.4,5 In the gene fusion context ERG expression is driven by the androgen receptor (AR) due to the male hormone activated TMPRSS2 promoter. The ERG was originally discovered as a retroviral oncogene.6 In castration resistant prostate cancer (CRPC) and metastatic CRPC the promiscuous activation of the AR can keep ERG levels high in a large number of cases. The global assessment of ERG in PCa even after accounting for racial/ethnic differences7 indicated that ERG is proposed as one of the most prevalent oncologic targets known in cancer. Oncogenic activation of ERG is not restricted to PCa, aggressive forms of acute myeloid leukemia (AML) and Kaposi sarcoma-endotheliomas are also driven by this oncogene.8 In the context of malignancies, ERG activates cell invasion, abrogates normal differentiation, and facilitates a broad range of cancer cell survival mechanisms.
The realization of ERG fusion and overexpression in PCa has led to a significant effort in developing potential treatment by targeting the ERG oncoprotein.9−12 The ETS transcription factors including ERG are emerging drug targets in cancer.13 Initial small molecule screening for inhibition of ERG oncoprotein expression using mouse monoclonal anti-ERG antibody (9FY) developed in our lab14,15 resulted in the identification of ERGi-USU ((E)1-[2-thiazolylazo]-2-naphthol) that selectively inhibited the growth of ERG positive cancer cells without affecting normal cells in vitro and in vivo.12 Following up on the intriguing hypothesis that an upstream regulator kinase is responsible for the cancer selective ERG inhibition, we subsequently identified an atypical kinase, RIOK212−17 as the direct binding partner of ERGi-USU. This was accomplished by a kinome screen of a broad kinase panel in a substrate competition assay and by the tryptophan fluorescence quenching assay. The compound ERGi-USU (1-[2-thiazolylazo]-2-naphthol (NSC139021) has been tested for its chemopreventive properties in the context of cancer.18,19
The objective of the current study was to develop new derivatives of ERGi-USU utilizing a structure–activity design approach, which show enhanced biological activity.20−24 Therefore, we designed new analogs around ERGi-USU and identified a more effective inhibitor, ERGi-USU-6. This led to development of salt formulations of the ERGi-USU-6. Approximately half of all therapeutic molecules are available in salt forms, as they provide benefits such as solubility, dissolution rate, permeability, and potency over the corresponding parent molecules.25 Salt selection is a lucid and well-known process, where they are evaluated for their precise characteristics that consist of crystallinity, hygroscopicity, and polymorphism.26−28 First, we developed a new facile synthetic procedure that can be applicable for the synthesis of the ERGi-USU-6 in high yields. Subsequently, a set of pharmaceutically acceptable ERGi-USU-6 salts were prepared and evaluated for their efficacies for inhibition of growth of the ERG positive prostate cancer cell line, ERG protein, and RIOK2 protein read-out as an indicator associated with ERG inhibition. Herein, we report the results of our studies, describing the synthesis and pharmacological inhibition of ERG in prostate cancer experimental models.
We have previously identified (E)1-[2-thiazolylazo]-2-naphthol (ERGi-USU, Figure 1) as a selective inhibitor of ERG positive cancer cells from a library of 2407 compounds from the National Cancer Institute diversity set.12 The demonstration of the nontoxic effect of the parental compound ERGi-USU in primary normal endothelial HUVEC cells and in in vivo experiments led to further development of ERGi-USU derivative compounds. As described before, ERGi-USU binds to the RIOK2 atypical kinase and affects the ERG levels presumably through ERG upstream mechanisms selective for the context of cancer cells.12
Figure 1.

Identification of lead compound ERGi-USU-6 from ERGi-USU and its optimization to identify a salt derivative 7b.
Since, RIOK2 protein levels are consistently decreased in response to ERGi-USU, we also measured the RIOK2 protein levels as indicators throughout our experiments in both ERG positive cancer cells and in ERG positive normal endothelial HUVEC cells. In order to improve the efficacy of ERGi-USU, structural activity relationship studies defined 134 compounds that were synthesized and tested for ERG protein inhibition levels and cell growth inhibition in the ERG positive prostate cancer cell line (VCaP), ERG negative prostate cancer cell line (LNCaP), and ERG positive normal primary endothelial cells (HUVEC). For high throughput screening, we utilized a highly specific anti-ERG mouse monoclonal antibody, 9FY, in an In-Cell Western assay developed by our group.12
Evaluation of 57 (see the supporting document for the structures) prioritized ERGi-USU analogs by In-Cell Western, Western blot, and cell growth assay resulted in the identification of 6 compounds with similar or better activity in relation to the parental ERGi-USU. Among these, ERGi-USU-6 (Figures 1 and S1) was designated as the lead compound based on its favorable characteristics and biological activity. In the next step, we focused on preparing pharmaceutically acceptable ERGi-USU-6 salts to further improve efficacy. Though the ERGi-USU compounds show similarities to chemical structures of pan-assay interference compounds (PAINS)29 complexing metal ions with pleiotropic activities, we observed that ERGi-USU-6 selectively inhibits the growth of ERG positive cancer cells with no detectable effect on ERG positive normal cells or ERG negative cancer cells, removing any concern of its pleiotropic biological activity. In the future, we also plan to investigate the possibility of the involvement of metal ion complexing mechanisms in the context of ERG positive cancer cells.
Acquiring ERGi-USU-6 in large quantities was found to be a major limitation for preparation of a variety of ERGi-USU-6 salts, as the reported procedures did not afford the desired compound in good yield and were inconsistent with previous literature reports.30−32 In addition, the method described by Grudpan and Taylor33 required harsh reaction conditions and laborious workup procedure to obtain the desired products. Moreover, the yields obtained in this method were very low and may not be applicable to synthesize ERGi-USU-6 in large quantities. Therefore, we first modified the literature procedure and developed a facile route to synthesize ERGi-USU-6 in large quantities (Scheme 1), that was also used to synthesize similar analogs.34
Scheme 1. Synthesis of ERGi-USU-6.

Conditions (a) m-CPBA, acetone, room temperature; (b) NaNO2, HBF4, −10 to −5 °C; (c) 3-(dimethylamino)phenol, methanol, 10–15 °C; (d) MoO2[(C2H5)2NCS2]2, Ph3P, acetone, room temperature.
The synthetic path utilizes 2-aminopyridine 1 as the starting material, which was then reacted with m-CPBA in acetone to afford 2-aminopyridine-1-oxide 2. Treating 2 with sodium nitrite in 40% HBF4 solution resulted the key intermediate, pyridin-2-diazonium 1-oxide tetrafluoroborate 3, which usually exists in mesomeric form. Isolation of 3 is crucial in order to significantly improve the yields of the final compound. Since the compound 3 is hygroscopic, upon completion of the reaction, it was collected under a nitrogen atmosphere by filtration. Compound 5 was prepared by a diazotization reaction of 3 with 3-(dimethylamino) phenol 4. Treating 5 with a freshly prepared Mo(VI) complex,35 MoO2[(C2H5)2NCS2]2 and PPh3 afforded the final compound. 1H and 13C NMR and LC-MS data confirmed the identity of compound 6. A highly deshielded 1H NMR signal at δH 15.86 of 6 (Supporting Information) was observed, demonstrating that the H in phenolic OH may participate in an intramolecular hydrogen-bonding with the adjacent N in 6.
A general scheme to synthesize salts of ERGi-USU-6 is outlined in Scheme 2. Reacting ERGi-USU-6 (free base) with an appropriate solvent and the counterion precursor (Table 1) resulted in a variety of pharmaceutically acceptable organic and inorganic salts of the parent compound.
Scheme 2. Synthesis of ERGi-USU-6 Salts.

Conditions: (a) counter ion B, solvent, room temperature.
Table 1. Details of Counterions, Solvents, and Methods for the Preparation of ERGi-USU-6 Salts and Their Inhibition Concentrations.

In order to enhance the solubility while maintaining the selectivity, we synthesized formulations of 21 salt derivatives (Table 1). Among these, 7b was found to be the most effective salt derivative which exhibited increased solubility, inhibition of ERG and RIOK2 protein levels in ERG positive prostate cancer cells (VCaP) in three independent experiments (Figure 2).
Figure 2.

ERGi-USU-6 salt derivative 7b showing improved activity. (A) 7b inhibits the growth of ERG positive VCaP cells. (B) 7b is a potent inhibitor of ERG and RIOK2 proteins. (C–E) 7b showed improved cell growth, ERG protein, and RIOK2 protein inhibition with IC50 values of 0.089, 0.17, and 0.13 μM, respectively.
We then assessed whether 7b has any adverse effect on ERG positive normal endothelial cells (HUVEC). To this end, HUVEC were treated with same dose concentration of 7b as were VCaP cells. It was observed that HUVEC were not affected by 7b salt derivatives at its highest doses (Figure 3).
Figure 3.

Cell growth, ERG protein, and RIOK2 protein levels are not affected in ERG positive normal endothelial HUVEC cells in response to ERGi-USU-6 salt formula 7b treatment. (A) Cell growth inhibition response of HUVEC was tested in the 0–0.25 μmol/L concentration range selected within the IC50 (0.089 μM) of VCaP prostate cancer cells. (B) No significant decrease of endogenous ERG protein and RIOK2 protein inhibition was observed in normal endothelial HUVEC cells.
We next examined the maximum tolerated dose (MTD) of ERGi-USU-6 in vivo. The MTD was estimated based on the maximum at which all animals survived. Athymic nude mice were used to evaluate the MTD of ERGi-USU-6 in parallel comparison with ERGi-USU. Based on the dosages, the animals were divided into five groups. Group 1 designated as control/vehicle (90% PEG + 10% DMSO). Group 2 (50 mg/kg in control vehicle), group 3 (100 mg/kg in control vehicle), group 4 (150 mg/kg in control vehicle), and group 5 (200 mg/kg in control vehicle). Dosing and survival details are summarized in Figure 4. All mice receiving doses up 100 mg/kg survived, but mortality occurred in 2 mice out of 5 at 150 mg/kg and 4 out of 5 at 200 mg/kg in the ERGi-USU-6 treatment group. To further define MTD, the overall toxicity as revealed by body weights was monitored throughout the study. The mice receiving the highest dose of 200 mg/kg showed significant weight loss (Figure 4C and D). Gross examination of major organs also revealed damage in tissues and vasculature as a result of the compound administration at the highest dose. In some events, localized inflammation at the site of the injection was observed at the highest dose raising solubility concerns. We therefore conclude that the MTD for ERGi-USU-6 is 100 mg/kg when compared with a 150 mg/kg dose of the parental compound ERGi-USU. During the observation period, no deterioration in health was observed in mice treated with the 100 mg/kg dose. These results point out that ERGi-USU-6 at a dose range of 100 mg/kg is safe and support the use of ERGI-USU-6 for further tumor xenograft experiments.
Figure 4.
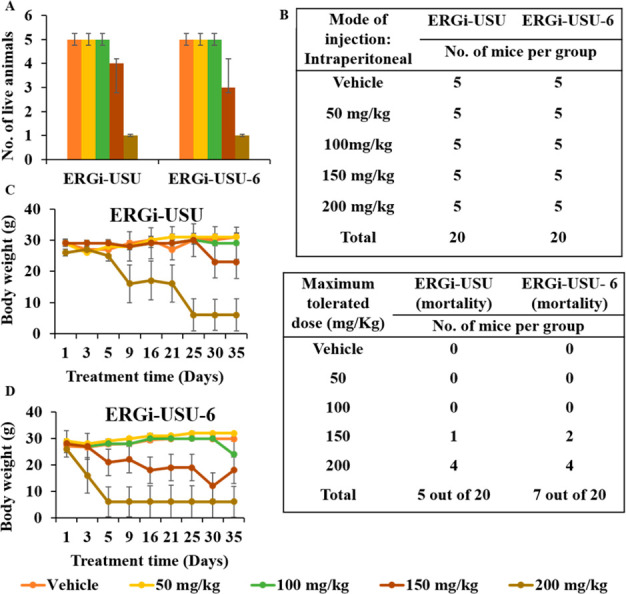
Maximum tolerated dose of ERGi-USU-6 is 100 mg/kg in athymic nude mice. (A, B) Assessment of maximum tolerated dose (MTD) of ERGi-USU and ERGi-USU-6. The table and bar diagram summarize the survival and mortality in each treatment group. (C and D) Body weight at different time points of the experiment. At the highest doses of ERGi-USU-6, body weight losses were observed when compared to the parental in ERGi-USU.
The TMPRSS2-ERG fusion driven activation of the ERG oncogene is one of the main cancer driver gene alterations that occurs in early stages of PCa and is retained in a subset of metastatic CRPCs. Early studies including reports from our laboratory have demonstrated that knockdown of ERG by siRNAs resulted in growth inhibition of TMPRSS2-ERG positive CaP cells in cell culture and xenograft models.12 Along these lines emerging data from in vitro and in vivo models continue to underscore biological roles of ERG in PCa initiation and progression.13
These studies and the unprecedented prevalence and emerging PCa mortality reports provide a strong rationale for the development of ERG targeted therapies. Our goal has been to develop ERG inhibitors with tolerable systemic toxic effects. During this endeavor we consistently monitor the selectivity of ERG inhibitors for cancer cells harboring ERG overexpression and likely driven by oncogenic addiction and lack of inhibitory effect ERG inhibitors on normal endothelial cells, where endogenous regulated ERG expression plays essential function in cell renewal.36 In other contexts, ERG is known to be expressed during development, restricted to precartilage and hematopoietic tissues including megakaryocytes.37,38
Our previous and current in vitro and in vivo work and SAR studies on ERGi-USU led to the discovery of improved derivatives. Among these ERGi-USU-6 was found to be more effective and chosen for further studies. Drug molecules administrated through salt form offer many potential benefits, as it can improve the solubility, dissolution rate, permeability, and efficacy of the drug. Salt selection is a rational and stepwise process in which salts are analyzed with regards to particular properties that include crystalline, hygroscopicity, and polymorphism assessment. Our primary objective was to identify an ERG inhibitor which can encounter pharmacological, technological conditions, possibilities and restrictions during development, and manufacturing and storage of the dosage forms. For this goal, selecting an optimal salt form of ERG USU-6 with an appropriate dosage form was crucial. From these studies we identified the most suitable new salt formula 7b through SAR studies.
While there is no evidence for the direct binding of ERGi-USUs to ERG protein itself, there has been recurring themes on similar inhibitory profiles for various ERGi-USU derivatives for ERG and RIOK2 proteins in a given ERG positive cancer cell line. Although the mechanistic connection between RIOK2 and ERG is currently not known, based on our observations on direct binding of ERGi-USU to RIOK2, it is conceivable that a high-resolution crystal structure of ERGi-USU and RIOK2 will advance our knowledge by defining a new direct inhibitor of this atypical kinase. This is significant as pioneering research endeavors already point to the role of RIOK2 in key cancer driver pathways.16,17,39,40
Previously, we reported the discovery of ERGi-USU as a selective inhibitor of ERG positive cancer cells.12 Subsequent, development of its analogs and medicinal chemistry campaign has led to the development of ERGi-USU-6 through a new and more efficient chemical synthesis method for the large-scale production. Analysis of new ERGi-USU analogs and various salts resulted in a compound that inhibits the growth of ERG positive PCa cells at 0.089 μM IC50. To the best of our knowledge, this is the best small molecule inhibitor of ERG protein and merits further development for therapeutic applications.13 Encouraged from this data, our future studies will include the pharmacokinetics of this lead compound determination of favorable administration route, evaluations of its effect in immune-competent mice
Glossary
Abbreviations
- PCa
prostate cancer
- CRPC
castration resistant prostate cancer
- ERG
ETS related gene
- ADT
androgen deprivation therapy
- TMPRSS2
transmembrane protease, serine 2
- AR
androgen receptor
- AML
acute myeloid leukemia
- RIOK2
RIO kinase 2
- VCaP
vertebral cancer of the prostate
- HUVEC
human umbilical vein endothelial cells
- MTD
maximum tolerated dose
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00308.
Cell lines, reagents, general protocol for the protein inhibition studies, cell growth inhibition studies, maximum tolerated dose assay, statistical analysis, and synthesis of ERG-USU-6 and its derivatives (PDF)
Author Present Address
○ Department of Biochemistry and Molecular & Cell Biology, Georgetown University School of Medicine, Washington, DC, 20057
Author Contributions
⊥ B.E. and M.P. contributed equally to this work
This research was supported by the CPDR-USU program HU0001-20-2-0032, the USU & HJF Office of Technology Transfer FY2015 IP Development Award to S.S. and A.D. and the John P. Murtha Cancer Center Translational Research Fellowship Award to C.X. under the mentorship of S.S. and S.V.M.
The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions, or policies of the Uniformed Services University of the Health Sciences (USUHS), the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the Department of Defense (DoD), or the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Fuchs H. E.; Jemal A. Cancer Statistics, 2021. Ca-Cancer J. Clin. 2021, 71 (1), 7–33. 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- Taitt H. E. Global trends and prostate cancer: a review of incidence, detection, and mortality as influenced by race, ethnicity, and geographic location. Am. J Men's Health 2018, 12 (6), 1807–1823. 10.1177/1557988318798279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. J.; Rumble R. B.; Basch E.; Hotte S. J.; Loblaw A.; Rathkopf D.; Celano P.; Bangs R.; Milowsky M. I. Optimizing anticancer therapy in metastatic non-castrate prostate cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36 (15), 1521–1539. 10.1200/JCO.2018.78.0619. [DOI] [PubMed] [Google Scholar]
- Tomlins S. A.; Rhodes D. R.; Perner S.; Dhanasekaran S. M.; Mehra R.; Sun X.-W.; Varambally S.; Cao X.; Tchinda J.; Kuefer R. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310 (5748), 644–648. 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Petrovics G.; Liu A.; Shaheduzzaman S.; Furasato B.; Sun C.; Chen Y.; Nau M.; Ravindranath L.; Chen Y.; Dobi A.; et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene 2005, 24 (23), 3847–3852. 10.1038/sj.onc.1208518. [DOI] [PubMed] [Google Scholar]
- Reddy E.; Rao V. N.; Papas T. S. The erg gene: a human gene related to the ets oncogene. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (17), 6131–6135. 10.1073/pnas.84.17.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarsky J.; Degon M.; Srivastava S.; Dobi A. Ethnicity and ERG frequency in prostate cancer. Nat. Rev. Urol. 2018, 15 (2), 125–131. 10.1038/nrurol.2017.140. [DOI] [PubMed] [Google Scholar]
- Miettinen M.; Wang Z.-F.; Paetau A.; Tan S.-H.; Dobi A.; Srivastava S.; Sesterhenn I. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. American journal of surgical pathology 2011, 35 (3), 432. 10.1097/PAS.0b013e318206b67b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins S. A.; Laxman B.; Varambally S.; Cao X.; Yu J.; Helgeson B. E.; Cao Q.; Prensner J. R.; Rubin M. A.; Shah R. B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10 (2), 177–IN9. 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.; Dobi A.; Mohamed A.; Li H.; Thangapazham R.; Furusato B.; Shaheduzzaman S.; Tan S.; Vaidyanathan G.; Whitman E.; et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008, 27 (40), 5348–5353. 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A. A.; Tan S.-H.; Xavier C. P.; Katta S.; Huang W.; Ravindranath L.; Jamal M.; Li H.; Srivastava M.; Srivatsan E. S.; et al. Synergistic activity with NOTCH inhibition and androgen ablation in ERG-positive prostate cancer cells. Mol. Cancer Res. 2017, 15 (10), 1308–1317. 10.1158/1541-7786.MCR-17-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A. A.; Xavier C. P.; Sukumar G.; Tan S.-H.; Ravindranath L.; Seraj N.; Kumar V.; Sreenath T.; McLeod D. G.; Petrovics G.; Rosner I. L.; Srivastava M.; Strovel J.; Malhotra S. V.; LaRonde N. A.; Dobi A.; Dalgard C. L.; Srivastava S. Identification of a small molecule that selectively inhibits ERG-positive cancer cell growth. Cancer Res. 2018, 78 (13), 3659–3671. 10.1158/0008-5472.CAN-17-2949. [DOI] [PubMed] [Google Scholar]
- Hsing M.; Wang Y.; Rennie P. S.; Cox M. E.; Cherkasov A. ETS transcription factors as emerging drug targets in cancer. Med. Res. Rev. 2020, 40 (1), 413–430. 10.1002/med.21575. [DOI] [PubMed] [Google Scholar]
- Furusato B.; Tan S.; Young D.; Dobi A.; Sun C.; Mohamed A.; Thangapazham R.; Chen Y.; McMaster G.; Sreenath T.; et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis. 2010, 13 (3), 228–237. 10.1038/pcan.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A. A.; Tan S.-H.; Mikhalkevich N.; Ponniah S.; Vasioukhin V.; Bieberich C. J.; Sesterhenn I. A.; Dobi A.; Srivastava S.; Sreenath T. L. Ets family protein, erg expression in developing and adult mouse tissues by a highly specific monoclonal antibody. J. Cancer 2010, 1, 197. 10.7150/jca.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameismeier M.; Cheng J.; Berninghausen O.; Beckmann R. Visualizing late states of human 40S ribosomal subunit maturation. Nature 2018, 558 (7709), 249–253. 10.1038/s41586-018-0193-0. [DOI] [PubMed] [Google Scholar]
- Ferreira-Cerca S.; Sagar V.; Schäfer T.; Diop M.; Wesseling A.-M.; Lu H.; Chai E.; Hurt E.; LaRonde-LeBlanc N. ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat. Struct. Mol. Biol. 2012, 19 (12), 1316. 10.1038/nsmb.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talalay P.; De Long M. J.; Prochaska H. J. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 1988, 85 (21), 8261–8265. 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks J. D.; Goldberg M. F.; Nelson L. A.; Wu D.; Nelson W. G. Identification of potential prostate cancer preventive agents through induction of quinone reductase in vitro. Cancer Epidemiology and Prevention Biomarkers 2002, 11 (9), 868–875. [PubMed] [Google Scholar]
- Kumar V.; LaJevic M.; Pandrala M.; Jacobo S. A.; Malhotra S. V.; Zabel B. A. Novel CMKLR1 Inhibitors for application in demyelinating disease. Sci. Rep. 2019, 9 (1), 1–13. 10.1038/s41598-019-43428-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoli M.; Persico M.; Kumar A.; Orteca N.; Kumar V.; Pepe A.; Mahalingam S.; Alegria A. E.; Petrella L.; Sevciunaite L.; et al. Identification of the first inhibitor of the GBP1: PIM1 interaction. Implications for the development of a new class of anticancer agents against paclitaxel resistant cancer cells. J. Med. Chem. 2014, 57 (19), 7916–7932. 10.1021/jm5009902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Kumar R.; Pandrala M.; Kaur P.; Gupta S.; Tailor D.; Malhotra S. V.; Salunke D. B. Facile synthesis of C6-substituted benz [4, 5] imidazo [1, 2-a] quinoxaline derivatives and their anticancer evaluation. Arch. Pharm. 2021, 354, e2000393 10.1002/ardp.202000393. [DOI] [PubMed] [Google Scholar]
- Ferroni C.; Pepe A.; Kim Y. S.; Lee S.; Guerrini A.; Parenti M. D.; Tesei A.; Zamagni A.; Cortesi M.; Zaffaroni N.; et al. 1,4-Substituted triazoles as nonsteroidal anti-androgens for prostate cancer treatment. J. Med. Chem. 2017, 60 (7), 3082–3093. 10.1021/acs.jmedchem.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses M. A.; Kim Y. S.; Rivera-Marquez G. M.; Oshima N.; Watson M. J.; Beebe K. E.; Wells C.; Lee S.; Zuehlke A. D.; Shao H.; et al. Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res. 2018, 78 (14), 4022–4035. 10.1158/0008-5472.CAN-17-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl P. H.; Wermuth C. G.. Pharmaceutical salts: Properties, selection and use; John wiley & sons, 2002. [Google Scholar]
- Berge S. M.; Bighley L. D.; Monkhouse D. C. Pharmaceutical salts. J. Pharm. Sci. 1977, 66 (1), 1–19. 10.1002/jps.2600660104. [DOI] [PubMed] [Google Scholar]
- Farag Badawy S. I Effect of salt form on chemical stability of an ester prodrug of a glycoprotein IIb/IIIa receptor antagonist in solid dosage forms. Int. J. Pharm. 2001, 223 (1–2), 81–87. 10.1016/S0378-5173(01)00726-8. [DOI] [PubMed] [Google Scholar]
- Makary P. Principles of salt formation. UK J. Pharm. Biosci. 2016, 2, 1–4. 10.20510/ukjpb/2/i4/91101. [DOI] [Google Scholar]
- Jasial S.; Hu Y.; Bajorath J. How frequently are pan-assay interference compounds active? Large-scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J. Med. Chem. 2017, 60, 3879–3886. 10.1021/acs.jmedchem.7b00154. [DOI] [PubMed] [Google Scholar]
- Brown E. V. Mass spectra of some phenylazopyridines and quinolines. J. Heterocycl. Chem. 1969, 6 (4), 571–573. 10.1002/jhet.5570060419. [DOI] [Google Scholar]
- Martí A.; Costero A. M.; Gaviña P.; Gil S.; Parra M.; Brotons-Gisbert M.; Sánchez-Royo J. F. Functionalized gold nanoparticles as an approach to the direct colorimetric detection of DCNP nerve agent simulant. Eur. J. Org. Chem. 2013, 2013 (22), 4770–4779. 10.1002/ejoc.201300339. [DOI] [Google Scholar]
- Brown E. V.; Granneman G. R. Cis-trans isomerism in the pyridyl analogs of azobenzene. Kinetic and molecular orbital analysis. J. Am. Chem. Soc. 1975, 97 (3), 621–627. 10.1021/ja00836a025. [DOI] [Google Scholar]
- Grudpan K.; Taylor C. G. Some azo-dye reagents for the spectrophotometric determination of cadmium. Talanta 1989, 36 (10), 1005–1009. 10.1016/0039-9140(89)80183-3. [DOI] [PubMed] [Google Scholar]
- Kreicberga J.; Jecs E.; Kampars V. Synthesis of 5-nitropyridin-2-ylazo push-pull derivatives. Chem. Heterocycl. Compd. 2008, 44 (1), 29–34. 10.1007/s10593-008-0013-9. [DOI] [Google Scholar]
- Moore F. W.; Larson M. L. Dialkyldithiocarbamate complexes of molybdenum (V) and molybdenum (VI). Inorg. Chem. 1967, 6 (5), 998–1003. 10.1021/ic50051a031. [DOI] [Google Scholar]
- Baltzinger M.; Mager-Heckel A. M.; Remy P. Xl erg: expression pattern and overexpression during development plead for a role in endothelial cell differentiation. Dev. Dyn. 1999, 216 (4–5), 420–433. . [DOI] [PubMed] [Google Scholar]
- Iwamoto M.; Tamamura Y.; Koyama E.; Komori T.; Takeshita N.; Williams J. A.; Nakamura T.; Enomoto-Iwamoto M.; Pacifici M. Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev. Biol. 2007, 305 (1), 40–51. 10.1016/j.ydbio.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainis L.; Toki T.; Pimanda J. E.; Rosenthal E.; Machol K.; Strehl S.; Göttgens B.; Ito E.; Izraeli S. The proto-oncogene ERG in megakaryoblastic leukemias. Cancer Res. 2005, 65 (17), 7596–7602. 10.1158/0008-5472.CAN-05-0147. [DOI] [PubMed] [Google Scholar]
- Asquith C. R.; East M. P.; Zuercher W. J. RIOK2: straddling the kinase/ATPase line. Nat. Rev. Drug Discovery 2019, 18, 574. 10.1038/d41573-019-00107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S.; Sha Z.; Zhou J.; Wu Y.; Song Y.; Li C.; Liu X.; Zhang T.; Yu R. BYSL contributes to tumor growth by cooperating with the mTORC2 complex in gliomas. Cancer Biol. Med. 2021, 18 (1), 88. 10.20892/j.issn.2095-3941.2020.0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.