

Abstract

Drug discovery building blocks available commercially or within an internal inventory cover a diverse range of chemical space and yet describe only a tiny fraction of all chemically feasible reagents. Vendors will eagerly provide tools to search the former; there is no straightforward method of mining the latter. We describe a procedure and use case in assembling chemical structures not available for purchase but that could likely be synthesized in one robust chemical transformation starting from readily available building blocks. Accessing this vast virtual chemical space dramatically increases our curated collection of reagents available for medicinal chemistry exploration and novel hit generation, almost tripling the number of those with 10 or fewer atoms.
Keywords: Machine learning, synthesis prediction, ultrahigh-throughput virtual screening
Medicinal chemistry structure–activity relationship (SAR) investigation covers a broad variety of goals and activities which can be generalized into two categories: early stage exploratory research where SAR is largely undiscovered and later-stage focused augmentation of the chemical space earlier identified as promising. In the exploratory phase, the chemist is usually less focused on synthesizing building blocks, preferring instead to order building blocks from a chemical vendor or internal inventory and then devoting time and effort to the rapid evaluation of diverse SAR. Reagent price1 and chemical diversity2 can impact this stage, yet speed is often a primary goal. Exceptions can be found in the area of novel hit generation,3,4 but here also the amount of hit-like chemical space that can be generated from orderable reagents is vast.5
In the latter stage of more focused medicinal chemistry, after readily available building blocks have been exhausted, SAR may point toward chemical space that cannot be purchased. When beginning to explore this space, a chemist utilizes both creativity and ideas of drug-likeness,6−8 aided by cheminformatics tools and, more recently, automated models and processes.9−13 The use of readily available building blocks to augment hit- or lead-like chemical space has long been well-established, but a systematic method to explore unavailable building block space that balances chemical novelty and synthetic accessibility would be of great value to medicinal chemists.
Both the exploratory and focused stages of SAR investigation could benefit from a larger set of available reagents, but we see a greater need in the latter, more-focused, stage. To demonstrate the potential utility of larger reagent sets, we conducted an experiment in the course of an unpublished therapeutic project in the lead-optimization (LO) stage. The lead series contained an ether moiety prepared from the corresponding alcohols, and we constructed a virtual library with the aim of prioritizing and selecting alcohols for incorporation into the LO chemotype. Every computational tool used was fast enough to process millions of compounds, so we included not only readily available building blocks but also alcohols found in PubChem14 and GDB-13.15,16 A virtual library of the fully enumerated molecules was prepared and scored. The modeling identified reagents 1,172, and 3 (Figure 1) as high-scoring hits; i.e. when attached to the rest of the LO molecule (after enumerating stereoisomers), they were predicted to have favorable properties.
Figure 1.

Three high-scoring hits from a virtual library exercise, along with a possible (in two cases) synthetic precursor reported to be commercially available.
Identifying 1–3 as moieties of interest to the project team is a key step in the decision for synthesis, but there are other considerations. There is little doubt they could be prepared given unlimited resources; however, a medicinal chemistry program must prioritize limited resources among competing activities. Compound 4(18) is structurally related and reported19 to be commercially available, and it was identified using a substructure search of commercially available reagents with a focus on structures shared by 1–3. Examination of these four suggests 4 likely could serve as a precursor to desired reagents 1 and 2, but not 3. The result of this process is that the chemistry team selected 1 and 2 as synthetic targets for incorporation into the LO molecule. Although 4 shares a pharmacophore similar to that of 1–3 and when connected to the rest of the LO molecule would be expected to have similar binding potency, known SAR suggests the presence of an additional hydrogen bond donor would likely give the LO molecule poor ADME properties. If the virtual library had contained only available 4 and not virtual 1–3, this moiety would not have been selected as a synthetic target.
This simple synthetic evaluation (useful as a proof of concept) was conducted using manual techniques not well-suited to much larger applications. Inspired by this example, we envision automating this process using a cheminformatics method with an awareness of all orderable reagents (from an internal inventory or commercially available) and knowledge of all other reagents that could be derived from them via one robust chemical transformation, such as the N-alkylation required for 1 and 2, and unlike the selective C-alkylation needed for 3.
Our goal is to identify large numbers of drug discovery building blocks neither commercially available nor present in our internal inventory but that could be prepared with one chemical transformation from a readily available precursor. With these ground rules in place, we conclude the compounds shown in Figure 1 would not make a suitable test case; compound 4 turns out to be unavailable.20 The first step in the process is to identify all orderable reagents, using both internal proprietary and also commercial building blocks; an easily overlooked but necessary component is the exclusion of nonorderable compounds.
Our building blocks come from two sources: one for purchased reagents and one for proprietary synthesized intermediates. From the intermediates database, we included only those molecules reported to have at least 50 mg available. Commercial reagent structures were obtained from a selected set of trusted vendors and some specialty catalogs. We requested only available compounds, excluding compounds requiring synthesis upon ordering.21,22 From all sources, we removed a few undesirable molecules (e.g., inorganics, isotopes). Details on the chemical filters are in the Supporting Information, along with a list of reagent vendors.
We looked through this curated building block set for a molecule conceptually similar to 4, identifying the structural isomer 5(23) (Figure 2). Ignoring stereochemistry, there are eight methylated analogues of 5, none of which are commercially available. As a test case for the informatics system we envision, we would expect predictions of compounds 6 and 7 as easily synthesizable, in contrast to compounds 8–13.
Figure 2.
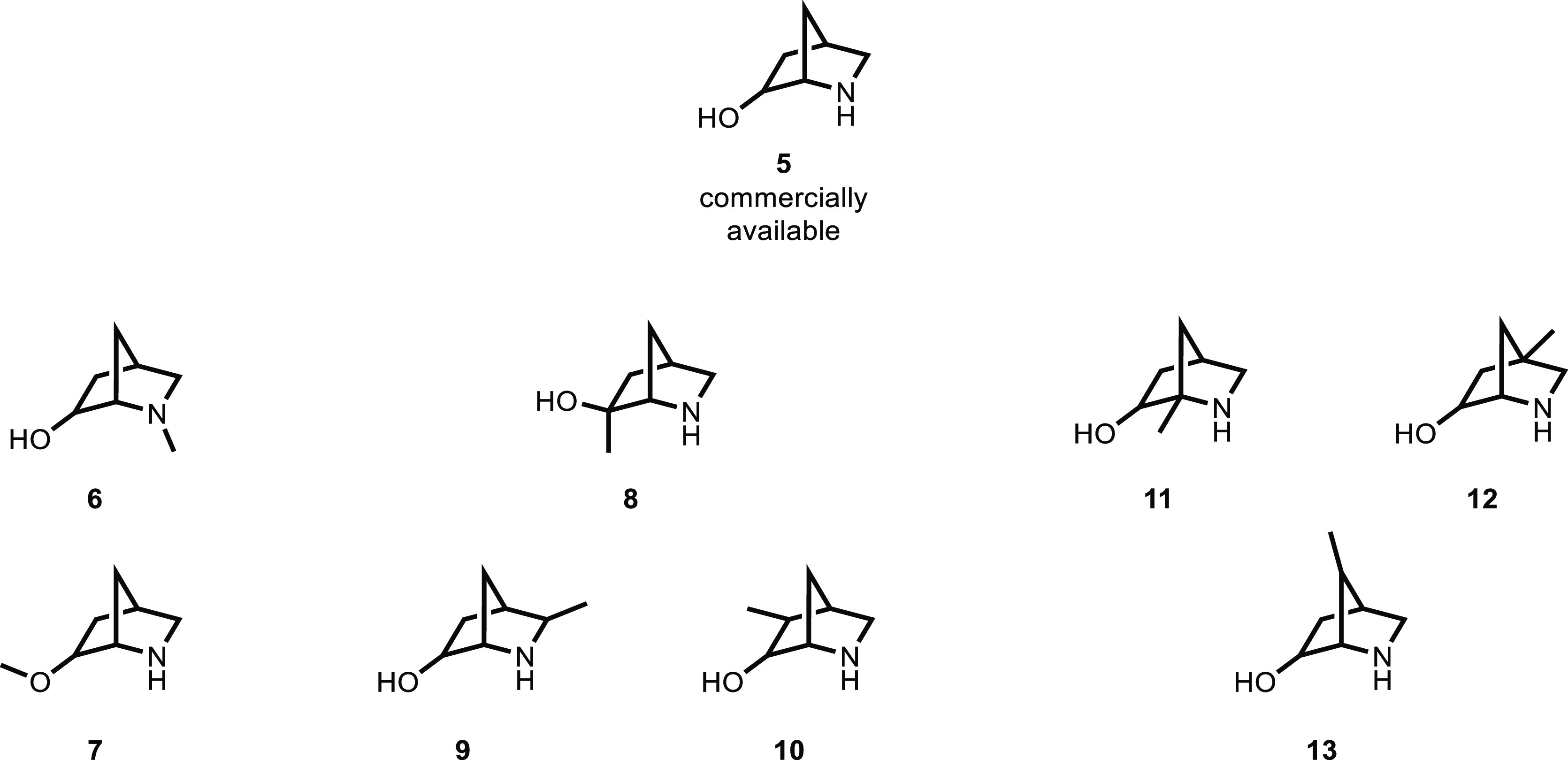
Orderable compound 5 and all eight methylated analogues, with some (e.g., 6, 7) more easily prepared from 5 than others (e.g., 11, 12).
For the next step, we needed a mechanism to assess whether a desired building block can be readily synthesized. For this purpose, we investigated several modules available to us within the ASKCOS v0.3.124 suite of retrosynthetic tools: SCScore, One-Step Retrosynthesis Fast Filter Score, and One-Step Retrosynthesis Score. SCScore is a single numerical estimation of a molecule’s synthetic complexity, not an assessment of any particular reaction or synthetic path;25 it has recently been used to analyze and improve the synthesizability of compounds proposed by generative models.26 The One-Step Retrosynthesis Fast Filter score provides a likelihood that conditions exist for which the reactants will form the desired product. The One-Step Retrosynthesis Score is an assessment of whether the specific forward reaction will proceed as expected.27,28
We evaluated available compound 5 and virtual compounds 6–13 with SCScore and the Fast Filter and One-Step Retrosynthesis scores from the highest ranked reaction produced by the One-Step Retrosynthesis module of ASKCOS (Table 1). The SCScore module provided roughly similar predictions for all compounds. As has been previously noted, SCScore is a measure of overall molecular complexity rather than an assessment of synthesizability given a certain reaction and set of starting materials. Our results confirm SCScore is not an appropriate metric for ease of practical synthesis. Similar to SCScore, the One-Step Retrosynthesis Fast Filter score does not distinguish 6 and 7 from the others; interestingly, both SCScore and Fast Filter predict 10 to be the most significant synthetic challenge within this set. We were pleased to see the desired result from the Score prediction of the One-Step Retrosynthesis module, with compounds 6 and 7 having significantly higher scores than all other methylated analogues. Based on this and other manual examinations, we propose a preliminary rule-of-thumb: a Score of −15 or higher indicates the compound can be prepared in a single robust chemical transformation from a readily available reagent, a Score of −100 or lower indicates an inaccessible compound, and a (relatively rare) Score between −15 and −100 requires further examination (additional discussion/examples are in the Supporting Information).
Table 1. ASKCOS Module Results for Compounds 5–13.
| compd | SCScore | One-Step Retrosynthesis: Fast Filter Score | One-Step Retrosynthesis: Score |
|---|---|---|---|
| 5 | 2.27 | 1.000 | –0.02 |
| 6 | 2.55 | 0.996 | –0.04 |
| 7 | 2.70 | 0.960 | –4.48 |
| 8 | 2.77 | 0.998 | –686 |
| 9 | 2.82 | 0.990 | –350 |
| 10 | 3.44 | 0.775 | –1386 |
| 11 | 2.70 | 0.984 | –1068 |
| 12 | 2.21 | 0.998 | –946 |
| 13 | 2.73 | 0.992 | –574 |
Virtual Library Design and Synthetic Target Selection
With this manual exploration showing promise, we next moved to a larger and realistic case study. For the same unpublished therapeutic project that was the subject of Figure 1, we collected from GDB-13 all examples of alcohols with no more than 10 heavy atoms and exactly 1 hydroxyl group. As before, these alcohols would be attached to the LO molecule as the corresponding ether. Project design parameters required a neutral substituent at this position; using pKa predictions from Pipeline Pilot,29 we removed any structure with an acidic or basic moiety, leaving 223 163 candidate alcohols. Knowing this mature project had explored chemical space of available commercial and internal reagents, we removed the 1437 alcohols in this set from GBD-13 that also corresponded to available compounds (see the Supporting Information). The remaining 221 726 alcohols were filtered by performing a One-Step Retrosynthesis with ASKCOS30 and removing compounds scoring less than −100, leaving 15 681 for further consideration.31
At this point, the selection process became analogous to any virtual library exercise with available reagents. Several thousand reagents predicted to be readily accessible by synthesis were eliminated because they contained chemical functionality not desired in the final LO molecule, although useful for other purposes (e.g., aldehydes); details of this filtering can be found in the Supporting Information. The remaining 765 alcohols were virtually attached to a key template to allow the calculation of ADME-relevant properties. We excluded any reagent that led to a final compound with a cLogP32 of less than 2 or greater than 4 or with a topological polar surface area of less than 85 or greater than 125. From the remaining 338 alcohols, we used interactive cheminformatics tools33 to select 12 for synthesis. We next examined these 12 in the graphical web-based version of the ASKCOS retrosynthesis tool; as shown in Figure 3, the proposed reactions cover a variety of robust chemical transformations.
Figure 3.

Selected alcohol reagents predicted to be easily synthesizable from readily available precursors.
Proposed Synthesis via Nucleophilic Addition to Ketone
As shown in Scheme 1, alcohol 14 was predicted to be synthesizable from commercially available ketone 26 by addition of a nucelophilic methyl group. The best scoring ASKCOS route suggested a Grignard reagent (the Supporting Information has details for all ASKCOS predictions). Our first attempt at synthesis used methylmagnesium bromide in THF and was unsuccessful, producing a complex mixture by NMR with no diagnostic methyl peak as anticipated near 1.2 ppm.34 An equivalent route using a different nucleophilic methyl source was successful (Scheme 1); ketone 26 was added to a preformed complex of methyllithium and TiCl4, providing a usable amount of 14 in 26% yield (as a 2.5:1 mixture of diastereomers).35 Methyllithium was also suggested by the ASKCOS tool, but with a lower score (−211). For the synthesis of 14 and subsequent alcohols, when the initial conditions did not produce the desired product the ASKCOS conditions were not exhaustively explored. Rather, alternate synthetically equivalent reagents were substituted to efficiently reach the target structures.
Scheme 1. Synthesis of Compound 14.

Reagents and conditions: (a) MeMgBr, THF, 0 °C, (b) MeLi, TiCl4, toluene, −5 °C.
Proposed Syntheses Using Amide Coupling
Scheme 2 shows proposed retrosyntheses for the three compounds (15–17) to be derived from amide couplings. Although 15 and 16 both contain the same hydroxyacetate moiety, the ASKCOS tool suggested different precursors for each. An initial attempt to prepare 16 using carboxylic acid 30, amine 31, and N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDAC) as a coupling reagent did not engage in productive coupling; however, the final route for both 15 and 16 used acetyl-protected acid chloride 34 to provide the common synthon.
Scheme 2. Synthesis of Compounds 15–17.

Reagents and conditions: (a) DIPEA, DCM, rt; (b) K2CO3, methanol, rt; (c) NaH, THF, rt.
The route used to prepare 15 is shown in Scheme 2. The proposed retrosynthesis called for aziridine (29), but due to the high level of acute toxicity and DNA reactivity, and its classification as a mutagen,36 we elected to use 2-chloroethylamine hydrochloride 35. Acylation of 35 with 34 proceeded smoothly to provide 36; after unmasking of the acetate-protected alcohol with K2CO3, ring closure to provide aziridine 15 was accomplished using NaH. Alcohol 16 was also prepared via 34, which upon coupling to commercially available amine 31 (as the HCl salt) followed by deprotection with K2CO3 provided the desired building block.
Alcohol 17 is the first example where preparation was unsuccessful. Synthesis of 17 was attempted using a variety of starting materials, bases/additives, solvents and temperatures. To screen various solvents, the ethyl ester of 33 was combined with 32 (as the HCl salt) and triethylamine then dissolved in solvent and heated to 150 °C under microwave. The solvents screened were DMSO, DMA, DMF, dioxane, toluene, and ethanol (the last was also screened at 80 °C). Other bases and additives tested include triethylamine/EDC (DCM, 25 °C) and sodium methoxide (methanol, 50 °C). Variations of the starting materials were also utilized. The free base 32 was combined with the ethyl ester of 33, triethylamine, and ethanol and then stirred at 80 °C. Other attempts were made with the acid and acid chloride versions of 33. We do not doubt 17 is synthesizable, but it was not readily synthesizable as predicted by the ASKCOS tool.
Proposed Syntheses via SN2
Four alcohols (18–21) were thought to be accessible via SN2 displacement. Scheme 3 shows the proposed retrosyntheses, with alcohol 18 arising from epoxide opening with commercially available 37 and the other three from SN2 displacement of alkyl halides. The synthesis of 19 is shown in Scheme 3 and, in this case, exactly matched the retrosynthesis.37 Unprotected amine 39 was selectively alkylated by chloride 40 to provide over 50 mg of 19. Likewise, the alkylation to prepare 20 proceeded smoothly, albeit through the TBDPS ether of 41. An attempt to prepare alcohol 21 via alkylation of sultam 43 with bromide 44 was not successful; the sultam was consumed, and none of the desired alcohol was isolated, potentially due to the tendency of β-sultams to open under basic conditions.38
Scheme 3. Synthesis of Compounds 18–21.

Reagents and conditions: (a) K2CO3, THF, rt.
Regarding alcohol 18, our initial attempt to effect the transformation proposed by the ASKCOS tool (retrosynthesis shown in Scheme 3) was unsuccessful; upon treatment with NaH in THF, 37 and 38 did not provide any of the desired product (Scheme 4). However, a modified route using the same available reagent was successful at attaching moiety 18 to the LO molecule to provide 47. As shown in Scheme 4, the epoxide opening was performed by LO precursor 45 to afford 46 after tosylation. Displacement of the tosylate with 37 was at first unsuccessful using either NaH or LHMDS in THF but was accomplished with NaH in DMA.
Scheme 4. Synthesis of Compound 47,

Reagents and conditions: (a) NaH, THF; (b) Cs2CO3, DMF, 60 °C, 1 h; (c) TsCl, DMAP, DCM, rt; (d) 37, NaH, DMA, rt, overnight.
R = synthetic precursor of the (unpublished) LO molecule.
Proposed Syntheses of Ester or Ketone Reduction to Alcohol
Retrosyntheses of the final alcohols are shown in Scheme 5. The two secondary alcohols (22 and 23) were predicted to be available via reduction of the corresponding orderable ketones, and the primary alcohols (24 and 25) from esters. Alcohol 22 is the third and final example from these 12 where attempted synthesis was unsuccessful. Although 48 is commercially available, it is expensive and we purchased only 100 mg ($1192); a single attempt at reduction using NaBH4 in ethanol produced a complex mixture not containing 22 as a major product.
Scheme 5. Retrosynthesis of Alcohols Derived from Ketone/Ester Reduction.

The attempted preparation of 23 is noteworthy: of the 12 alcohols we hoped to synthesize, this is the only example where the necessary starting material could not be easily ordered. Our curated set of reagents included ketone 49, and at the time of our order the vendor claimed this was available.39 Upon further inquiry it was found to be unavailable and also not available from any other vendor except for a willingness to attempt delivery within 3 months, a certainty and timeline not consistent with our concept of “readily available”.
Much more straightforward was the synthesis of alcohols 24 and 25 from ester reduction (Scheme 6). The first attempt at reducing commercially available ester 50 was with NaBH4 in methanol, which gave no reaction after 2 h at room temperature. Only decomposition was seen with LAH/THF at 0 °C. The first sign of desired alcohol 24 came from an overnight room temperature reaction with NaBH4 in ethanol with added CaCl2; partial optimization of these conditions provided 24 as part of a crude mixture. In the case of 51, reduction with NaBH4 in methanol provided enough material to allow coupling of 25 to the LO molecule.
Scheme 6. Synthesis of Compounds 24 and 25.

Reagents and conditions: (a) NaBH4, ethanol, rt, overnight; (b) NaBH4, methanol, rt, 2 h.
There are several potential reasons for the remaining four compounds not being readily accessible despite the apparently robust synthetic methods ASKCOS suggested for their synthesis. Low molecular weight building blocks can be particularly difficult to detect, purify, and isolate due to the potential volatility, lack of UV absorption, and weak ionizability of the starting materials and products. These challenges may contribute to the lack of commercial vendors for these building blocks.
Future Direction
We envision a comprehensive set of theoretical reagent structures, precalculated to identify those easily synthesizable from orderable building blocks. Although currently beyond the scope of our algorithms and hardware, for a step in this direction, we scored all the GDB-13 drug-like reagent structures with 10 or fewer heavy atoms. Figure 4 shows a comparison between this chemical space of 2.2 million and the corresponding (10 or fewer heavy atoms) Janssen curated building blocks, currently numbering 74 000. For this analysis, we have very loosely defined a drug-like reagent as any compound with at least one carbon and one noncarbon heavy atom (excluding hydrocarbons and inorganics from GDB-13 and the building block set, respectively).
Figure 4.

Collection of reagent structures among GDB-13 and the curated Janssen building block collection, including only compounds with 10 or fewer heavy atoms containing at least 1 carbon and 1 noncarbon atom. An “available” structure is one in the curated building block collection, otherwise “novel” or “not available”. A structure is “synthesizable” if the ASKCOS score is > –100. Pie charts show the distribution for compounds with 4 (A), 6 (B), 8 (C), and 10 (D) heavy atoms.
At least two results depicted in Figure 4 are not surprising. First, the total number of building blocks increases rapidly as the number of atoms increases (especially true for molecules from GDB-13). Second, the value of this method is greatest for larger reagents. Among the 295 building blocks with 4 atoms (Figure 4a), only 5 are novel reagents from GDB-13 predicted to be easily synthesizable. The set of reagents with 10 atoms (Figure 4d) totals almost 2 million, with the vast majority not (easily) synthesizable. Although a small percentage, there are over 90 000 molecules not readily available and yet can be prepared in one step from orderable precursors. This analysis identified almost 124 000 novel, yet synthesizable, building blocks.
Future improvements could include expanding the prediction to encompass two or more chemical transformations. Alternatively, and partly accomplishing this effect through different means, the curated set of available building blocks could be augmented by virtually generating molecules derived from common protecting/deprotecting steps. As an example, although compounds 1 and 2 are not available in one chemical transformation from a readily available precursor, compound 4 may be available via deprotection of the available N-Boc analogue.40 Such a synthesis prediction process could recognize that 1 and 2 were accessible from an orderable reagent, although using a slightly more complicated route. Whether protecting group manipulations ought to be considered trivial or not, including them in this process would be valuable.
Another valuable improvement would be more precision in the machine learning-predicted synthesis. In all cases the algorithm provided enough information for a skilled organic chemist to attempt the transformation. Yet, for the 11 alcohols where synthesis was attempted, none saw a successful preparation of the desired alcohol on the first attempt. In many cases, the ASKCOS tool suggests only a transformation; even in cases where a specific organic reagent was suggested, the predicted best route was not always successful. For example, the preparation of alcohol 14 from ketone 26 (Scheme 1) was predicted to be best accomplished using Grignard reagent 27, with methyllithium a low-scoring afterthought. The lab-based reality in this case was the opposite. The difference in money, time, and effort of a skilled chemist between “ordering a reagent” and “preparing a reagent in one attempt” is significant; so too is the difference between one attempt and many. Advances in the predictive power of this method would also be a helpful precursor for an expansion to include multiple transformations; any failure rate in one step becomes much more significant when applied to multiple steps. Likewise, there is room for improvement in chemical sophistication (e.g., stereochemistry awareness).
Finally, more chemical structures could be scored with this method, including those larger than 10 atoms and also structures from other sources. Our analysis of GDB-13 members with 10 or fewer atoms identified 124 000 synthesizable compounds not available from our internal inventories or commercial suppliers, yet GDB-13 is not designed to be a fully comprehensive representation of this chemical space. GDB-13 uses filters and rules to avoid inclusion of unstable or otherwise unrealistic molecules. This removes numerous unsuitable compounds but also removes a small number that could be useful as reagents. For example, 1-methyl-1H-pyrazol-3-amine (PubChem CID 137254) is not in GDB-13 because of an element ratio filter. GDB-13 also does not contain every element that could be in a useful reagent (e.g., some chlorines are present but not fluorine). Any file or generative model is a potential source of structures for this analysis, including collections that do not distinguish between compounds synthesized in one step versus those resulting from a thesis project (e.g., PubChem).
There are times when a medicinal chemist can adequately explore chemical space using readily available reagents. For other times, the history of medicinal chemistry has been a balance between the imagined (and thought useful) on the one hand and the synthesizable on the other. In the age of big data and ultrahigh-throughput virtual screening, a multitude of new options is emerging. The central questions (what should we make? what can we make?) are not changing, but new tools are providing new answers.
We describe a method to augment our curated collection of orderable building blocks for drug discovery. Starting with a set of theoretical organic molecules, we used a machine-learning method to identify those which could be synthesized in a single chemical transformation of an available precursor. We show our use of this process with an internal therapeutic project, designing a virtual library from novel (but easily prepared) alcohol reagents and selecting for synthesis 12 predicted to be suitable for the project. We prepared 8 of the 12, although in one case (18) we made not the alcohol itself but the fully elaborated ether. We will report more details on the LO project in due course, but among these reagents it was 24 that led to the most potent fully elaborated molecule. This moiety occupies a water-filled hydrophobic pocket when bound to the protein, and the unique arrangement of the rigid and polar ether and nitrile likely contributed to potency. We searched our database of available/purchasable reagents for this combination of ether/nitrile; the most similar alcohols are shown in Figure 5. All contain the same pharmacophoric features as 24, yet none place those features in the same positions in the binding pocket. Thus, the identification of alcohol 24 as a synthetically accessible building block provided access to valuable SAR not available from commercial reagents.
Figure 5.

Orderable alcohols most similar to 24, none with the same shape/electrostatic profile.
The workflow we describe uses informatics to profile a virtual chemical space several orders of magnitude larger than what can be perused manually and provides the expectation that most of the reagents selected from this huge chemical space can be readily synthesized by a skilled medicinal chemist.
Acknowledgments
We are grateful to Prof. Scott E. Denmark for insightful discussions and Dr. Vladimir Chupakhin and Charlotte Pooley Deckhut for technical assistance.
Glossary
Abbreviations
- GDB
generated database
- LO
lead optimization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00340.
Our curated list of reagent vendors and information about prefiltering, example with specific vendor of virtual building blocks, other examples of ASKCOS scoring, criteria used to remove virtual library compounds, comprehensive high-scoring results from ASKCOS for all 12 alcohols, and experimental details (PDF)
Molecular formula strings (CSV)
Author Present Address
§ Cullgen Inc., San Diego, California 92130, United States
Author Present Address
∥ Ferring Research Institute, San Diego, California 92121, United States
The authors declare no competing financial interest.
Supplementary Material
References
- Kalliokoski T. Price-focused analysis of commercially available building blocks for combinatorial library synthesis. ACS Comb. Sci. 2015, 17, 600–607. 10.1021/acscombsci.5b00063. [DOI] [PubMed] [Google Scholar]
- Brown D. G.; Gagnon M. M.; Boström J. Understanding our love affair with p-chlorophenyl: present day implications from historical biases of reagent selection. J. Med. Chem. 2015, 58, 2390–2405. 10.1021/jm501894t. [DOI] [PubMed] [Google Scholar]
- Gerry C. J.; Schreiber S. L. Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discovery 2018, 17, 333–352. 10.1038/nrd.2018.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T. E.; Schreiber S. L. Towards the optimal screening collection: a synthesis strategy. Angew. Chem., Int. Ed. 2008, 47, 48–56. 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters W. P. Virtual chemical libraries. J. Med. Chem. 2019, 62, 1116–1124. 10.1021/acs.jmedchem.8b01048. [DOI] [PubMed] [Google Scholar]
- Murcko M. A. What makes a great medicinal chemist? A personal perspective. J. Med. Chem. 2018, 61, 7419–7424. 10.1021/acs.jmedchem.7b01445. [DOI] [PubMed] [Google Scholar]
- Campos K. R.; Coleman P. J.; Alvarez J. C.; Dreher S. D.; Garbaccio R. M.; Terrett N. K.; Tillyer R. D.; Truppo M. D.; Parmee E. R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363, eaat0805. 10.1126/science.aat0805. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Green J.; Weiss J. R.; Murcko M. A. What do medicinal chemists actually make? A 50-year retrospective. J. Med. Chem. 2011, 54, 6405–6416. 10.1021/jm200504p. [DOI] [PubMed] [Google Scholar]
- Schneider G. Automating drug discovery. Nat. Rev. Drug Discovery 2018, 17, 97–113. 10.1038/nrd.2017.232. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Baumgartner M. P. Creating a virtual assistant for medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 1051–1055. 10.1021/acsmedchemlett.9b00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N.; Ertl P.; Lewis R.; Luksch T.; Reker D.; Schneider N. Artificial intelligence in chemistry and drug design. J. Comput.-Aided Mol. Des. 2020, 34, 709–715. 10.1007/s10822-020-00317-x. [DOI] [PubMed] [Google Scholar]
- Wilbraham L.; Mehr S. H. M.; Cronin L. Digitizing chemistry using the chemical processing unit: from synthesis to discovery. Acc. Chem. Res. 2021, 54, 253–262. 10.1021/acs.accounts.0c00674. [DOI] [PubMed] [Google Scholar]
- Makara G. M.; Kovács L.; Szabó I.; Pőcze G. Derivatization design of synthetically accessible space for optimization: in silico synthesis vs deep generative design. ACS Med. Chem. Lett. 2021, 12, 185–194. 10.1021/acsmedchemlett.0c00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum L. C.; Reymond J.-L. 970 Million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- https://pubchem.ncbi.nlm.nih.gov/compound/20583540 (accessed 2020-06-11).
- https://pubchem.ncbi.nlm.nih.gov/compound/18541295 (accessed 2020-06-11).
- https://pubchem.ncbi.nlm.nih.gov/substance/343147461 and https://pubchem.ncbi.nlm.nih.gov/substance/316495598 (accessed 2020-06-11).
- Unpublished results: a chemist attempted to purchase 4 and synthesize 1 and 2; 4 was not orderable.
- Although the request for only readily available compounds is emphasized, sometimes follow up is required.
- See the Supporting Information for related discussion.
- https://www.keyorganics.net/2-azabicyclo221heptan-6-olhcl-c6h12clno.html (accessed 2020-06-11).
- Table 1 numbers are from the version specific to Janssen, which includes our curated set of available reagents.
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. SCScore: Synthetic complexity learned from a reaction corpus. J. Chem. Inf. Model. 2018, 58, 252–261. 10.1021/acs.jcim.7b00622. [DOI] [PubMed] [Google Scholar]
- Gao W.; Coley C. W. The synthesizability of molecules proposed by generative models. J. Chem. Inf. Model. 2020, 60, 5714–5723. 10.1021/acs.jcim.0c00174. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Thomas D. A. III; Lummiss J. A. M.; Jaworski J. N.; Breen C. P.; Schultz V.; Hart T.; Fishman J. S.; Rogers L.; Gao H.; Hicklin R. W.; Plehiers P. P.; Byington J.; Piotti J. S.; Green W. H.; Hart A. J.; Jamison T. F.; Jensen K. F. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, eaax1566. 10.1126/science.aax1566. [DOI] [PubMed] [Google Scholar]
- Segler M. H. S.; Preuss M.; Waller M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Dassault Systèmes BIOVIA . BIOVIA Pipeline Pilot, release 2020; Dassault Systèmes: San Diego, 2020.
- The Janssen installation includes a URL-based API called Pipeline Pilot. Input is the molecule as SMILES; text-based output is returned. Running with four parallel processes, typical throughput is 100–200K per day.
- Of 15 681 compounds, 3308 have a score < −15.
- BioByte Corp., Claremont, CA.
- Agrafiotis D. K.; Alex S.; Dai H.; Derkinderen A.; Farnum M.; Gates P.; Izrailev S.; Jaeger E. P.; Konstant P.; Leung A.; Lobanov V. S.; Marichal P.; Martin D.; Rassokhin D. N.; Shemanarev M.; Skalkin A.; Stong J.; Tabruyn T.; Vermeiren M.; Wan J.; Xu X. Y.; Yao X. Advanced Biological and Chemical Discovery (ABCD): centralizing discovery knowledge in an inherently decentralized world. J. Chem. Inf. Model. 2007, 47, 1999–2014. 10.1021/ci700267w. [DOI] [PubMed] [Google Scholar]
- Kurouchi H.; Singleton D. A. Labelling and determination of the energy in reactive intermediates in solution enabled by energy-dependent reaction selectivity. Nat. Chem. 2018, 10, 237–241. 10.1038/nchem.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel M. E.; Chen P.; Carrera D. E.; Zhang H.; Gosselin F. Highly diastereoselective α-arylation of cyclic nitriles. Org. Lett. 2017, 19, 3446–3449. 10.1021/acs.orglett.7b01421. [DOI] [PubMed] [Google Scholar]
- Musser S. M.; Pan S.-S.; Egorin M. J.; Kyle D. J.; Callery P. S. Alkylation of DNA with aziridine produced during the hydrolysis of N,N′,N″-triethylenethiophosphoramide. Chem. Res. Toxicol. 1992, 5, 95–99. 10.1021/tx00025a016. [DOI] [PubMed] [Google Scholar]
- Desroy N.; Joncour A. M.; Peixoto C.; Temal-Laib T.; Tirera A.; Bucher D.; Amantini D.; De Vos S. I. J.; Brys R. C. X.. Novel Compounds and Pharmaceutical Compositions Thereof for the Treatment of Diseases. WO2019/238424A1, December 19, 2019.
- Koller W.; Linkies A.; Rehling H.; Reuschling D. Synthesis and properties of β-sultams. Tetrahedron Lett. 1983, 24, 2131–2134. 10.1016/S0040-4039(00)81862-6. [DOI] [Google Scholar]
- http://www.combi-blocks.com/cgi-bin/find.cgi?QM-7935 (accessed 2020-09-30).
- https://www.keyorganics.net/tert-butyl7-hydroxy-2-azabicyclo221heptane-2-carboxylate-mfcd18792130-1221818-31-0-c11h19no3.html (accessed 2020-06-11).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.