

Important Compound Classes
Title
KRAS G12D Inhibitors
Patent Publication Number
WO 2021/041671 A1
Publication Date
March 04, 2021
Priority Application
62/893,604; 63/052,840; 63/058,188 US
Priority Date
August 29, 2019; July 16, 2020; July 29, 2020
Inventors
Wang, X.; Burns, A. C.; Christensen, J. G.; Ketcham, J. M.; Lawson, J. D.; Marx, M. A.; Smith, C. R.; Allen, S.; Blake, J. F.; Chicarelli, M. J.; Dahlke, J. R.; Dai, D.; Fell, J. B.; Fischer, J. P.; Mejia, M. J.; Newhouse, B.; Nguyen, P.; O’Leary, J. M.; Pajk, S.; Rodriguez, M. E.; Savechenkov, P.; Tang, T. P.; Vigers, G. P. A.; Zhao, Q.
Assignee Company.
Mirati Therapeutics, Inc. [US/US]; 9393 Towne Center Dr., Suite 200, San Diego, CA 92121 (US). Array Biopharma Inc. [US/US]; 3200 Walnut Street, Boulder, CO 803301 (US).
Disease Area
Cancer
Biological Target.
KRAS G12D Mutant
Summary
This Patent Highlight showcases exemplary compounds to treat cancer in patients with KRAS G12D-associated disease or disorder. RAS genes were first identified as viral genes, and RAS proteins were found to be a member of a large superfamily of small GTPases, including Ras, Arf, Rab, Rho, and Ran families. In general, the RAS superfamily proteins function as GDP/GTP-regulated binary on–off switches. RAS proteins (KRAS4A, KRAS4B, NRAS, and HRAS) control the cytoplasmic signaling networks and regulate diverse normal cellular functions, and mutations occur in approximately a third of all human solid tumors. Consequently, intensive efforts on RAS structure, biochemistry, and biology have been made during the last 3 decades for therapeutic intervention in RAS mutations. The six proposed strategies for targeting RAS signaling are the direct target of RAS proteins, exploiting synthetic lethal partners of RAS mutant, disrupting RAS membrane association, disrupting the metabolic habit of RAS mutant cells, targeting RAS downstream pathways, and harnessing the immune response.
Kirsten Rat Sarcoma 2 Viral Oncogene Homologue (KRAS) is a small GTPase and a member of the RAS family of oncogenes. KRAS serves as a molecular switch that cycles between the inactive (GDP-bound) and active (GTP-bound) states. This helps transduce upstream cellular signals received from multiple tyrosine kinases to downstream effectors that regulate a wide variety of processes, including cellular proliferation. Multiple cancers have altered metabolic processes, and oncogenic KRAS has been shown to be a key player in promoting metabolic modulation. It has been demonstrated that mutant KRAS G12D is responsible for metabolic phenotype of pancreatic cancer cells, partly through its role in reprogramming anabolic glucose metabolism. However, the specific actions on metabolic regulation may differ depending on tumor types and genetic context, including differences in mutant KRAS protein. For instance, in vivo evidence of metabolic rewiring during lung cancer malignant progression have demonstrated that mutant KRAS G12D homozygous cells exhibited an increase in glucose metabolism toward the tricarboxylic acid cycle and glutathione synthesis, which leads to an enhanced glutathione-mediated detoxification. Single nucleotide substitutions that result in missense mutations at codons 12 and 13 of the KRAS primary amino acid sequence comprise approximately 40% of these KRAS driver mutations in lung adenocarcinoma. Recent pancreatic ductal adenocarcinoma (PDAC) patients sequencing studies show that activating mutations affected codon 12 in 93% of all patients (G12D, 38%; G12 V, 38%; G12R, 14%; G12C, 2%; G12S, 1%), codon 61 in 6% of the patients (Q61H. 3.5%; Q61R. 1.5%; Q61K, 1%), and codon 13 or codon 146 in 0.5% of all patients.
PDAC is the fourth leading cause of cancer death in the Western world and is predicted to become the second leading cause of cancer death by 2030, just after lung cancer. Pancreatitis is an inflammatory disease of the pancreas, which enables and accelerates the transformation of pancreatic cells when the KRAS oncogene is activated. The pathogenesis of pancreatic adenocarcinoma has been widely studied and is determined to be caused by genetic alterations such as oncogenic mutations in the KRAS gene, which have been frequently detected to be more than 90% of cases. Activation of the oncogene KRAS signals pancreatic cells to undergo acinar-to-ductal metaplasia (ADM), the main origin of pancreatic preneoplastic lesions that eventually develop into pancreatic ductal adenocarcinoma (PDA) is an essential step in the formation of premalignant lesions, in addition to the inactivation of tumor suppressor genes, such as TP53, CDKN2A, and SMAD4, allow the progression to invasive cancer.
The exact mechanism on how pancreatitis promotes the development of PDAC is a fundamental question in the field of pancreatology that has not been fully answered. However, the systematic activation of autophagy (removal of damaged organelles in order to maintain cellular homeostasis) during pancreatitis helps protect the pancreatic cells, decreases disease progression, and aids in the recovery phase. The overexpression of the vacuole membrane protein 1 (VMP1) gene triggers autophagy in numerous types of cells. The complexity of Ras proteins in different tumor tissues and the less reproducibility of identified KRAS synthetic lethal interactors provide the opportunity for improvement by taking into account KRAS specific mutations, cell models, data validation, cancer subclassification, and elucidation mechanism. Furthermore, a major challenge with current cancer treatments is drug resistance, which necessitate the need for combination therapies that simultaneously target multiple cancer-associated pathways for efficacious anti-RAS treatments. Nonetheless, recent studies of small-molecule approaches to directly inhibit oncogenic KRAS G12C have raised the possibility of drugging Ras that has been long considered “undruggable”, which has led to the approval of Lumakras (AMG-510).
Definitions
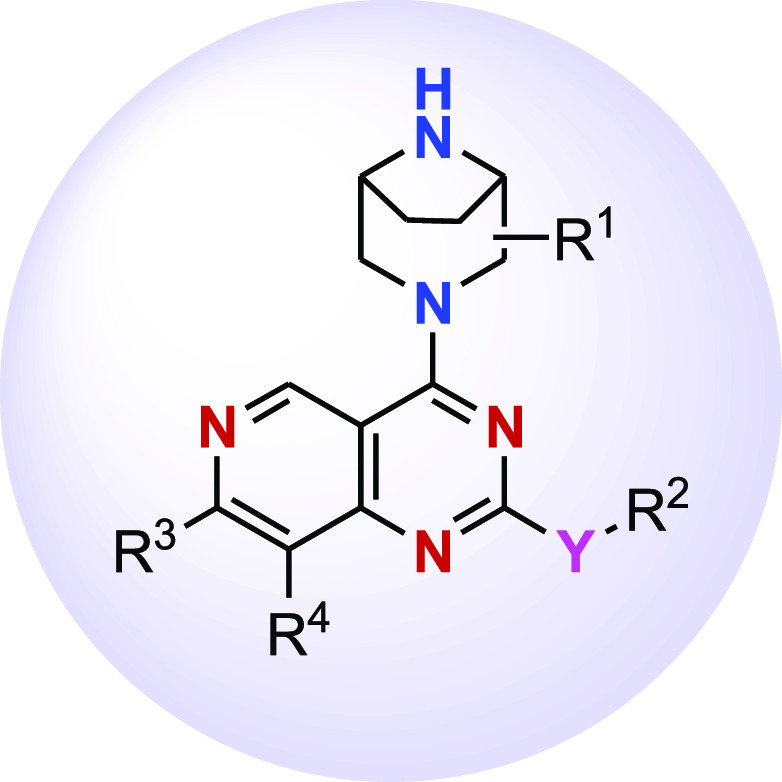
R1 = Hydrogen, hydroxy, halogen, C1–C3 alkyl, C1–C3 cyanoalkyl, C1–C3 hydroxyalkyl, HC(=O)–;
Y = O or NR5
R2 = hydrogen, −N(R5)2, heterocyclyl, C1–C6 alkyl, −L–COOH, −L–OR5;
L = C1–C4 alkylene, hydroxyalkyl, or heteroaryl
R3 = aryl or heteroaryl;
R4 = hydrogen, halogen, or C1–C3 alkyl.
Key Structures

Biological Assay
KRAS G12D surface plasmon resonance (SPR) binding assay.
Biological Data
The table below
shows the binding of
exemplary compounds to KRAS G12D (IC50).
Recent Review Articles
-
1.
Ward R. A.; Fawell S.; Floc’h N.; Flemington V.; McKerrecher D.; Smith P. D.. Chem. Rev. 2021, 121, 3297.
-
2.
Chen K.; Zhang Y.; Qian L.; Wang P.. J. Hematol. Oncol. 2021, 14, 116.
-
3.
Merz V.; Gaule M.; Zecchetto C.; Cavaliere A.; Casalino S.; Pesoni C.; Contarelli S.; Sabbadini F.; Bertolini M.; Mangiameli D.. et al. Front. Oncol. 2021, 11, 638360.
-
4.
Tani T.; Kitajima S.; Conway E. B.; Knelson E. H.; Barbie D. A.. Expert Opin. Ther. Targets 2021, 25, 167.
-
5.
Malapelle U.; Pepe F.; Pisapia P.; Passiglia F.. Eur. J. Cancer 2021, 146, 74.
-
6.
Chen H.; Smaill J. B.; Liu T.; Ding K.; Lu X.. J. Med. Chem. 2020, 63, 14424.
The author declares no competing financial interest.