

Abstract

Comprehensive synthetic strategies afforded a diverse set of structurally unique bicyclic proline-containing arginase inhibitors with a high degree of three-dimensionality. The analogs that favored the Cγ-exo conformation of the proline improved the arginase potency over the initial lead. The novel synthetic strategies reported here not only enable access to previously unknown stereochemically complex proline derivatives but also provide a foundation for the future synthesis of bicyclic proline analogs, which incorporate inherent three-dimensional character into building blocks, medicine, and catalysts and could have a profound impact on the conformation of proline-containing peptides and macrocycles.
Keywords: Bicyclic proline, arginase inhibitor, conformation control
l-Proline (proline, symbol Pro or P) is an essential amino acid that is used in the biosynthesis of proteins. Proline is the only proteinogenic amino acid that bears a secondary amine moiety, as its α-amino group is constrained within a pyrrolidine ring. The distinctive cyclic structure of proline’s side chain gives proline an exceptional conformational rigidity compared with other amino acids, which affects the secondary structure of proteins/peptide near a proline residue.1 Proline acts as a structural disruptor in the middle of regular secondary structure elements such as α helices and β strands. Proline is also commonly found in turns (another kind of secondary structure) and aids in the formation of β turns.2 Crystallographic studies3 have indicated that proline generally adopts two predominant conformations: Cγ-exo and Cγ-endo (Figure 1). The energy difference between the endo and exo puckers for proline is ∼0.5 kcal/mol, and the two forms rapidly interconvert at room temperature.4
Figure 1.

Conformational analysis of proline.
Arginase is an enzyme that metabolizes l-arginine to l-ornithine and urea. In addition to its fundamental role in the hepatic urea cycle and its enablement of nitric oxide biosynthesis,5 arginase also influences T-cell activation and proliferation in humans.6 Arginase expression and l-arginine depletion are hypothesized to be an immune-suppressive pathway in mammals. Myeloid-derived suppressor cells (MDSCs) exploit arginase activity within the tumor microenvironment to suppress T-cell activation and proliferation. The depletion of l-arginine renders cytotoxic T-cells unable to proliferate and therefore unable to effectively mount an antitumor attack.7 The analysis of clinical samples derived from patients undergoing anti-PD-1 checkpoint blockade suggested that arginase expression correlates strongly with poor patient survival.8 Thus small-molecule arginase inhibitors are promising therapeutics for the treatment of several diseases, including cardiovascular diseases (atherosclerosis and hypertension), cancer, and induced or spontaneous immune disorders.9−12 Recently, we discovered proline-containing compound 1 to be a potent arginase inhibitor.13−15 The crystal structure of 1 in human arginase 1 (hArg1) showed that the boronic acid side chain occupies a narrow channel, with the boronic acid in its hydrate form interacting with the Mn2+ ions (Figure 2). The proline moiety occupies space toward the solvent front, with the NH engaging in a salt-bridge interaction with Asp183 and the CO2H in a hydrogen-bonding interaction with Ser137.
Figure 2.

X-ray cocrystal structures of boronic acid inhibitor 1 bound to hArg1 (PDB code 7K4G).
Given that the proline in compound 1 adopts an exo conformation and is solvent-exposed, we envisioned rigidifying the proline ring through the fusion of a small bicyclic ring to probe the impact of the proline conformation on the potency (Figure 3). Specifically, cyclization from Cα to Cβ of proline in compound 1 will lead to compounds like 2 and 3 (Figure 3a). Cyclization from Cα to Cγ will lead to bridged compounds like 4 and 5 (Figure 3b). Cyclization from Cβ to Cγ will lead to bicyclic compounds like 6 and 7 (Figure 3c). Building a ring on Cγ to Cδ will lead to bicyclic compounds like 8 (Figure 3d). These bicyclic analogs significantly impact and constrain the conformation of the proline ring. For example, in the model of compound 2, proline adopts a Cγ-endo conformation (Figure 4, left panel).16 On the contrary, the conformation of proline is locked into a Cγ-exo conformation in compound 5 (Figure 4, right panel).
Figure 3.

Structure of proline-containing arginase inhibitor 1 and examples of constrained bicyclic proline arginase inhibitors.
Figure 4.

Model of compound 2 (yellow, left) overlaid with the crystal structure of 1 (green), suggesting that the proline adopted a Cγ-endo conformation. Model of compound 5 (magenta, right) overlaid with the crystal structure of 1 (green), suggesting that it locked the proline into the Cγ-exo conformation.
On the contrary, even though bicyclic prolines are very important for proline conformational control and have been utilized in building blocks,17 medicine,18 and catalysis,19,20 their synthesis is under-represented in the literature compared with that of monocyclic proline derivatives. There are very limited synthetic strategies for bicyclic prolines, in general,21−23 and there are no synthetic methods for some bicyclic proline analogs. For example, there are over 32 000 hits associated with l-proline from a SciFinder substructure search using absolute stereo match (Figure 5). There is only one hit associated with the bicyclic core of 3, and no synthetic method is reported. There is not no relevant hit that could be associated with the bicyclic cores of 4 and 6. Thus there is a significant need for synthetic innovation to access such bicyclic proline analogs. Here we report our comprehensive synthetic strategies for constructing a series of bicyclic proline-containing arginase inhibitors. More broadly, the work herein describes synthetic strategies to access novel, constrained, and highly stereochemically complex proline analogs that will have broad utility in future synthetic, biological, and biomedical explorations.
Figure 5.

SciFinder substructure search hits using absolute stereo match criteria and synthetic precedence.
Synthetic Strategy A
For the cyclization from Cα to Cβ (Figure 3a), we envisioned utilizing the key enamine intermediate 9 to undergo 2 + 1, 2 + 2, and 2 + 3 cyclizations to efficiently construct fused bicyclic rings [3,1,0] 10, [3,2,0] 2, and [3,3,0] 3 correspondingly (Figure 6).
Figure 6.

General strategy for cyclization A.
The synthesis of the common intermediate 9 started from the commercially available bromopyrrole 11 (Scheme 1). Stille coupling of 11 with allyltributylstannane gave olefin 12. Hydroboration of 12 with 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (HBpin) afforded 13,24 which was hydrogenated to give racemic 14. To obtain a more acid-stable protecting group and additionally provide a chromophore for LC-MS analysis, the Boc group on 14 was exchanged with Cbz to give compound 15. Inspired by Kublitskii’s original work, we deprotonated compound 15 with LiHMDS at −20 °C to form lithium enolate 16 followed by the addition of neat bromine at −90 °C; the dehydrohalogenation of intermediate bromide 17 gave the desired key enamine 9.25 Whereas we were delighted that Kublitskii’s protocol worked on substrate 15 and that the Bpin moiety was tolerated, the yield of this reaction often varied from 25 to 75%. To yield a more robust reaction and enable the gram-scale synthesis of enamine 9 for future explorations, we sought to further optimize this transformation. Gratifyingly, we found that the addition of LiHMDS to a solution of 15 in tetrahydrofuran (THF) at 0 °C cleanly afforded enolate 16 without side reactions. The original conditions required bromine at −90 °C to avoid side reactions; however, we found that the slow addition of bromine at −35 °C directly afforded the desired product 9 in 84% yield on a 76 g scale without the intermediacy of bromide 17. The conversion of this reaction was carefully monitored via ReactIR, as shown in Figure 7, which illustrates the clean profile of these new conditions.26
Scheme 1. Synthesis of Key Intermediate 9.

Figure 7.

Deprotonation of 15 with LiHMDS and the addition of bromine were monitored with ReactIR.
With ample enamine 9 in hand, we explored two 2 + 1 cyclization strategies. The first was a Rh-catalyzed carbene addition generated from ethyl diazoacetate (Scheme 2).27 The slow addition of ethyl diazoacetate to the mixture of 9 and (Rh(OAc)2)2 in 1,2-dichloroethane (DCE) gave the major isomer 18 in 5:1 dr. Taking advantage of the steric hindrance of the tertiary methyl ester, the exposed secondary ethyl ester was selectively hydrolyzed and then transformed to a mixed anhydride and reduced to alcohol 19 with NaBH4.28
Scheme 2. 2 + 1 Cyclization of Enamine 9 with Ethyl Diazoacetate.

Converting the primary alcohol in 19 with phthalimide through Mitsunobu condition gave 20.29 Four protecting groups including two different protecting groups for nitrogen, one boronic ester, and one carboxylic ester needed to be removed to obtain the final product 21. After significant reaction optimization, we were delighted to find that global deprotection under acidic conditions afforded the final racemic boronic acid 21 in modest yield. It is noteworthy that the cyclopropyl group survived under such harsh conditions (2:1:1 mixture of 12 N aq. HCl and acetic acid in water under microwave heating at 130 °C for 90 min).
To obtain the unsubstituted fused cyclopropane analog 10, we employed a second cyclopropanation strategy utilizing the Simmons–Smith reaction (Scheme 3).30 The steric hindrance of tetra-substituted enamine 9 prevented the cycloaddition from occurring under conventional conditions. It was not until modified reaction conditions using trifluoroacetic acid (TFA)31 were employed that the desired product 22 could be obtained in low yield after reprotection of the boronic acid formed during the reaction. Global deprotection of 22 afforded racemic 10.
Scheme 3. 2 + 1 Cyclization of Enamine 9 with Modified Simmons–Smith Reaction.

Our next cyclization strategy for enamine 9 was a 2 + 2 reaction (Scheme 4). Even though electron-deficient olefins32 have been reported to react with ethylene gas, little was known about the reactivity of an electron-rich enamine like compound 9.33 We discovered that after bubbling a toluene solution of 9 and benzophenone with ethylene gas followed by irradiation of the resulting solution with a Na/Hg lamp at room temperature (rt) for 3 h, the desired cyclization product 23 could be obtained in 13% yield. Global deprotection of 23 afforded racemic 2.
Scheme 4. 2 + 2 Cyclization of Enamine 9 with Ethylene Gas under UV Light.

Our 2 + 3 strategy utilized a 1,3-dipolar cyclization of azomethine ylide 25 (derived from N-benzyl-N-(methoxymethyl)trimethylsilyl methyl-amine 24, Scheme 5) with olefin 9. Williams and Fegley reported that azomethine ylide 25 could be generated with a catalytic amount of TFA (condition a) and reacted with a 1,1-disubstituted olefin substrate.34 Unfortunately, in our case, the desired product racemic 26 was formed in only <5% yield under Williams’ conditions. Increasing the equivalents (equiv) of TFA or increasing the temperature did not improve the yield. This suggested the tetrasubstituted olefin in 9 was too sterically hindered and much less reactive compared with the 1,1-disubstituted olefin substrate reported by Williams. Then, we turned our attention to Chalyk and colleagues’ report on using LiF to generate the azomethine ylide 25 (condition b).35 No reaction was observed under the original conditions at 60 °C, but we were encouraged that compound 9 stayed intact. Considering the low reactivity of the electron-rich and sterically hindered 9, we further optimized the reaction temperature and solvent. Not until the reaction was heated in dioxane to 165 °C in a microwave reactor could the desired product 26 be obtained in an appreciable 10% yield from 1 g of 9 (condition c). Because the size of the our microwave vessel (20 mL) limited the scale of the reaction, we changed the solvent from dioxane to dimethylformamide (DMF) to permit conventional heating at 165 °C in an oil bath, and the use of larger conventional flasks was part of our plan to improve the scale of the reaction. Given that azomethine ylide 25 could be formed at 60 °C, as reported by Chalyk,35 and due to the relative inertness of 9, we rationalized that the rate-limiting step was the 1,3-dipolar cyclization. To prevent the decomposition of azomethine ylide 25 at such a high temperature, using a syringe pump, we slowly added 24 to the hot mixture of LiF and a solution of 9 in DMF. These optimizations indeed improved the yield of the reaction to 38% on a 12 g scale of compound 9. Purification by chiral supercritical fluid chromatography (SFC) of compound 26 provided the enantiomerically pure 27. Debenzylation using hydrogenation gave amine 28, which was further deprotected to give the [3,3,0] bicyclic proline analog 3.
Scheme 5. 1,3-Dipolar Cyclization of Enamine 9 with Azomethine Ylide 25.

Synthetic Strategy B
For the ring closure from Cα to Cγ, we investigated several synthetic strategies. Eventually, Grygorenko’s intramolecular cyclization between the anion at Cα and the bromide at Cγ (intermediate 29) turned out to be general for both the [3,2,1] bridged compound 4 and the [2,2,1] bridged compound 5 (Figure 8).36 Expectedly, the reaction was stereospecific, and the stereochemistry at Cβ and Cγ in 29 determined the stereochemistry in compound 30.
Figure 8.

General strategy B.
For the synthesis of compound 4, the key was to prepare the enantiomerically pure 37 as a precursor to the ring closure, which contained three contiguous chiral centers and had the correct chirality at Cβ and Cγ (Scheme 6). The known ketone 31 was used to prepared 37.37 Ketone 31 existed as a 2:1 mixture of trans/cis isomers at Cβ. The reduction of 31 with NaBH4 directly afforded a complex mixture of alcohol isomers predominantly consisting of the Cα,Cβ-trans isomer due to the rapid epimerization at Cβ under the reduction conditions.37 The Cβ of 31 was first deprotonated with potassium bis(trimethylsilyl)amide (KHMDS), then trapped with the bulky proton source (1S)-10-camphorsulfonic acid to enrich the Cα,Cβ-cis ketone to prepare the cis isomer 32. The corresponding ketone was reduced in situ with NaBH4 to afford cis alcohol 32 in 44% yield. The next required transformation was the stereospecific conversion the Cγ–OH of compound 32 to Cγ-NH2 in 37. Because Cl-methylsulfonate was reported to be a better leaving group and less prone to elimination compared with the mesylate,3832 was reacted with chloromethanesulfonyl chloride to afford 33. The displacement of Cl-methylsulfonate in 33 with Bu4NN3 and the reduction of the corresponding azide with the polymer-supported Ph3P (PB-Ph3P) afforded amine 34. The mono reductive amination of 34 afforded benzylamine 35. To attach the two-carbon linker bromides, we used triflate 36 in the alkylation of 35 to afford the key intermediate 37.39 Cα deprotonation of 37 with KHMDS at −78 °C followed by warming up to −20 °C gave the desired bridged compound 38 in excellent yield (82%). Hydroboration of 38 gave compound 39. Debenzylation and global deprotection of 40 gave the [3,2,1] bridged proline 4.
Scheme 6. Synthesis of Compound 4.

For target 5, the key was the preparation of the ring-closure precursor 48, which contained the correct configuration at Cβ and Cγ and an undesired configuration at Cα (Scheme 7). It is noteworthy that the undesired configuration at Cα was abolished during the deprotonation, and the desired configuration was established and directed by the stereochemistry at Cγ during the cyclization. The key was that the undesired configuration at Cα in starting material 41 was used to set the desired configuration at Cβ and Cγ.40 Wittig–Horner olefination afforded the desired thermodynamically stable trans isomer compound 43 in 37% yield as the major isomer after chiral SFC separation. For the hydroboration of 43, given that pinacol boronate is prone to hydrolysis to the corresponding boronic acid during the purification on silica gel and reverse-phase high-performance liquid chromatography (HPLC), (+)-pinanediolborane 44 was used to prepare boronate 45 with improved stability to facilitate sequential purifications.41 The saturation of the olefin and debenzylation of 45 under hydrogenation gave acid 46. Acid 46 was transformed to the mixed anhydride and reduced to alcohol 47 with NaBH4. The bromination of 47 with CBr4/PPh3 afforded the cyclization precursor 48. The cyclization of 48 utilizing KHMDS worked smoothly as designed to afford the bridged compound 49 in 51% yield. Global deprotection gave the desired [2,2,1] bicyclic proline 5.
Scheme 7. Synthesis of Compound 5.

Synthetic Strategy C
Next, we focused our attention on developing a strategy toward the introduction of a variety of heterocycles via cyclization between the oxygen-containing functional groups at Cβ and Cγ in the common intermediate 50 (Figure 9). Cyclization with one carbon linker (n = 1) would lead to [3,2,0] compounds 6 and 51. Cyclization with two (n = 2) and three (n = 3) carbons linkers would afford [3,3,0] 52 and [4,3,0] 53 correspondingly. Additionally, one carbon extension on Cγ intermediate 54 could provide access to [3,3,0] 7.
Figure 9.

General strategy C.
For bicyclic compounds 6 and 51, the common key step was a double displacement of the key bis-sulfonate 57 with corresponding sulfur nucleophiles42 and nitrogen nucleophiles, respectively (Scheme 8).43 The synthesis of 57 started with ketone 31; catalysis by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), deprotonation at the Cβ of ketone 31, and alkylation with formaldehyde afforded ketone 55 as the major isomer with the Cβ hydroxymethyl trans to the Cα ester. The reduction of ketone 55 with NaBH4 gave diol 56 as the major isomer with the desired stereochemistry at Cγ. Diol 56 was then reacted with chloromethanesulfonyl chloride to afford bis-sulfonate 57. Double displacement of the bis-Cl-methylsulfonate in 57 with Na2S proceeded with the inversion of stereochemistry at Cγ affording thietane 60, which was converted to [3,2,0] bicyclic proline 6 through hydroboration and global deprotection. The double displacement of 57 with benzylamine afforded azetidine 61, which was then converted to [3,2,0] bicyclic proline 51 through sequential hydroboration, debenzylation, and global deprotection. It is worth noting that the Cl-methylsulfonate in 57 was the key to the success of the double displacement reaction. The reaction of benzyl amine with either methylsulfonate 58 or p-tolylsulfonate 59 afforded only a trace amount of the desired product 61 (1% yield by LC-MS). Similarly, [4,3,0] bicyclic proline 53 was also prepared from the analogous displacement reaction with the corresponding three-carbon linker bis-Cl-methylsulfonate.44
Scheme 8. Synthesis of Compounds 6 and 51.

For the synthesis of [3,3,0] bicyclic proline 52, the two-carbon linker ketone 62 was prepared as the major isomer from 31 (Scheme 9). Reduction of the ketone followed by deprotection of the tert-butyldimethylsilyl (TBS) ester and Swern oxidation of the corresponding diol gave ketone 63. The cyclization of 63 with benzylamine under reductive amination conditions led to the [3,3,0] 64.45 Hydroboration of 64 with (+)-pinanediolborane afforded boronate 65. Debenzylation of 65 and global deprotection with aq. HCl gave [3,3,0] bicyclic proline 52.
Scheme 9. Synthesis of Compound 52.

The synthesis of [3,3,0] bicyclic proline 7 started from 66 (Scheme 10).46 Similar to the preparation of compound 55, the alkylation of ketone 66 with formaldehyde catalyzed by DBU afforded ketone 67 as the major isomer. TBS ether protection of the primary hydroxy group in 67 gave ketone 68, which was reacted with dimethyl (1-diazo-2-oxopropyl)phosphonate 69 to afford vinyl ether 70 with one carbon extension at Cγ.47 Aqueous HCl promoted the deprotection of the TBS and methyl ether in one step and directly afforded the cyclized thermodynamically stable cis-cyclic acetal 71. The reduction of acetal 71 under the reductive esterification conditions afforded cyclic ether 72 in 77% yield.48 The hydroboration of 72 and global deprotection utilizing BBr3 afforded bicyclic proline 7.
Scheme 10. Synthesis of Compound 7.

Synthetic Strategy D
For the cyclization from Cγ to Cδ, a strategic olefin intermediate 73 was envisioned to undergo 2 + 1 and 2 + 2 cyclization reactions to efficiently construct fused bicyclic rings, including [3,1,0] bicyclic proline 74 and [3,2,0] bicyclic proline 8 (Figure 10).
Figure 10.

General strategy D.
The synthesis of olefin 73 started with 66 (Scheme 11). Cα deprotonation followed by the addition of MeI afforded the desired methylated product with a moderate diastereoselectivity (dr 3:1). The mixture was separated by chiral SFC to provide the major product ketone 75 (53% yield) in an enantiomerically pure form. The reduction of ketone 75 gave alcohol 76 as a mixture of diastereoisomers. 76 was then converted to Cl-methanesulfonate 77, which was treated with DBU in MeCN at reflux to give 73 in 53% yield over two steps.49
Scheme 11. Synthesis of Compound 73.

For the synthesis of 74, the terminal olefin in 73 was selectively hydroborated with (+)-pinanediolborane 44 to give 78 (Scheme 12). The modified Simmons–Smith cyclopropanation of 78 provided cyclopropane 79 in 52% yield.31 After saponification, the removal of Cbz via hydrogenation, and acidic hydrolysis, 79 was converted to the [3,1,0] bicyclic proline 74. Interestingly, 2D nuclear magnetic resonance (NMR) studies revealed that the cyclopropane formed on the more sterically hindered face of the olefin, cis to the adjacent ester. This unique stereochemical outcome was consistent with the report by Wang’s group and was presumably due to the ester group chelation of the putative “IZnCH2I” species to direct the cyclopropanation on the same face of the ester.50
Scheme 12. Synthesis of Compound 74.

For the synthesis of [3,2,0] bicyclic proline 8, the key step was a [2 + 2] cycloaddition reaction (Scheme 13). The ketene generated in situ from 2,2-dichloroacetyl chloride and TEA gratifyingly only reacted with the electron-rich enamine olefin in 73 and did not touch the terminal olefin to construct the [3,2,0] bicyclic proline 80 in good yield.51 The reductive dechlorination of 80 with zinc afforded ketone 81. The reduction of ketone 81 gave an inconsequential diastereomeric mixture of alcohol 82, which was directly used in the successive Barton deoxygenation reaction through 83 to give 84.52 The hydroboration of 84 followed by the global deprotection of 85 using BBr3 afforded [3,2,0] bicyclic proline 8.
Scheme 13. Synthesis of Compound 8.

Exquisitely Potent Bicyclic Proline Arginase Inhibitors
With the comprehensive synthetic strategies and innovations reported herein, novel, constrained, and highly stereochemically complex proline analogs were obtained, and their Arg1 potency was assessed using an in vitro fluorimetric assay measuring the production of thioornithine from thioarginine.15
For Cα to Cβ cyclized analogs, surprisingly, [3,1,0] analogs 10 and 21 showed only very weak Arg1 potency (IC50 > 10 000 and 8784 nM respectively), presumably due to the large conformational change in the proline ring (Figure 11). This was supported as the less constrained [3,2,0] analog 2 and [3,3,0] analog 3 had significantly improved Arg1 potency (370 and 46 nM respectively) compared with that of 10. Even though the prediction of compound 3 favored the Cγ-exo conformation, it was still less potent compared with the initial lead compound 1 (16 nM). This indicated that besides the Cγ conformation, other factors could also contribute to the observed Arg1 potency.
Figure 11.

Structures and Arg1 potencies of bicyclic proline Arg1 inhibitors. The details of the calculation of Cγ-endo/exo ratios can be found in the Supporting Information.
Gratifyingly, Cα to Cγ cyclized [3,2,1] analog 4 (2.2 nM) and [2,2,1] analog 5 (3.9 nM) significantly improved the Arg1 potency compared with compound 1. As we hypothesized, the bridged ring in 4 and 5 locked the proline into the Cγ-exo conformation to lower the entropic penalty, thus improving their inhibitory activity. This was supported by the quantum mechanical (QM) calculations in which the Cγ-exo conformation was greatly favored and the Cγ-endo conformation could not be generated for compounds 4 and 5. The Cγ-exo conformation of compound 4 was further confirmed by the crystal structure of 4 with Arg1 protein (Figure 12). The crystal structure of 4 (blue) clearly demonstrated that the proline was rigidified into the Cγ-exo conformation and matched perfectly with the crystal structure of compound 1 (green). In addition to the salt-bridge interaction of NH with Asp183 and the hydrogen-bond interaction of CO2H with Ser 137, the crystal structure revealed that the NH at Cγ engaged a new salt-bridge interaction with Asp181.
Figure 12.
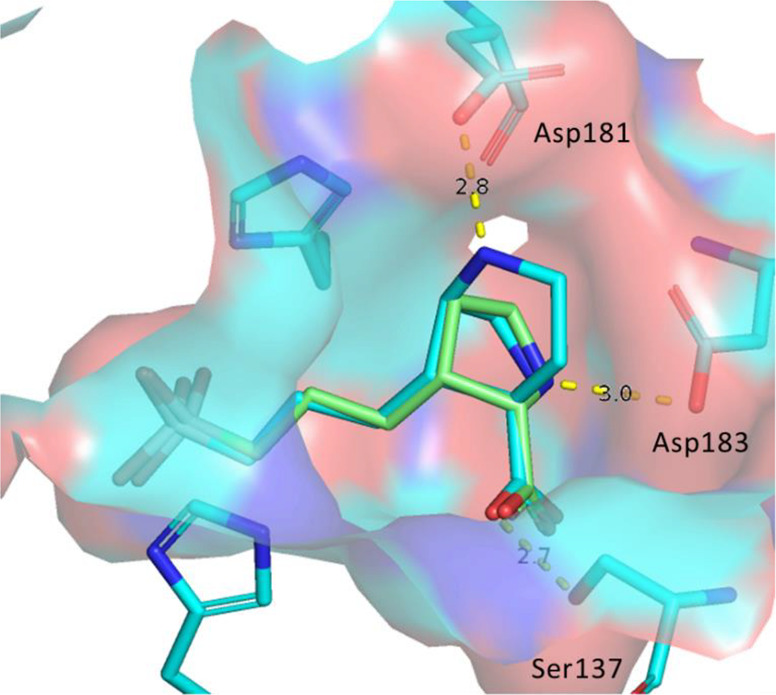
Overlay of compound 1 (green) with the cocrystal structure of 4 (blue, PDB code 7K4H) bound to hArg1.
For the Cβ to Cγ cyclized analogs, [3,2,0] analog 51 (104 nM) was less potent than [3,2,0] analog 6 (25 nM). The crystal structures of 51 (Figure 13) and 6 (Figure 14) showed very similar binding interactions, with a salt-bridge interaction of NH with Asp183 in 51 and a sulfur–carbonyl oxygen interaction in 6.53 The crystal structures supported two hypotheses on why 51 was less potent. One was that the distance between the NH and Asp181 was 3.2 Å in 51, which is slightly longer than the typical distance (2.7 to 3.0 Å).54 Another hypothesis was that the larger sulfur atom reduced the strain of the bicyclic ring to allow the proline to adopt a more bioactive conformation and contributed to the improved potency of 6. Indeed, the QM calculation of compound 6 suggested that the Cγ-exo conformation was greatly favored over the Cγ-endo conformation (>99/1).
Figure 13.

X-ray cocrystal structure of 51 bound to hArg1 (PDB code 7K4J).
Figure 14.

X-ray cocrystal structure of 6 bound to hArg1 (PDB code 7K4I).
Larger ring [3,3,0] analogs 52 and 7 were also designed. It is very interesting to note that the QM calculation suggested that compound 52 favored the Cγ-endo conformation and compound 7 favored the Cγ-exo conformation. This could be rationalized by the fact that compound 7 has an oxygen that is very close to the NH for an attractive intramolecular interaction. On the contrary, compound 52 has two NHs (possibly charged) facing each other that would be repulsive. (See the Supporting Information for details.) Both compounds 52 and 7 had excellent Arg1 potency. The crystal structure of 52 with Arg1 protein confirmed that the NH at Cγ engaged in an optimal salt-bridge interaction with Asp181 (2.8 Å, Figure 15), which significantly contributed to its Arg1 potency. On the contrary, the crystal structure suggested that the proline ring in 52 adopted the Cγ-exo conformation, which is most likely the bioactive conformer of 52. The larger ring size [4,3,0] analog 53 was still a very potent Arg1 inhibitor (3.2 nM).
Figure 15.
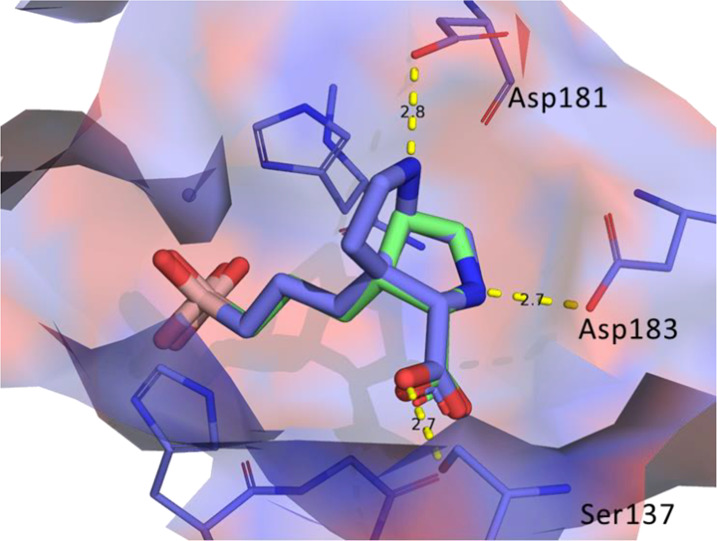
Overlay of compound 1 (green) with the cocrystal structure of 52 (blue, PDB code 7K4K) bound to hArg1.
For Cγ to Cδ cyclized analogs, the [3,1,0] analog 74 expectedly showed only very weak Arg1 potency (5742 nM), as the cyclopropane ring fusion resided on the undesired face of the proline ring. On the contrary, [3,2,0] analog 8 showed good Arg1 inhibitory activity (67 nM), with the Cγ-exo conformation favored over the Cγ-endo conformation (>99/1) by QM calculations.
4
Our comprehensive synthetic strategies afforded a diverse set of structurally unique bicyclic proline-containing Arg1 inhibitors with a high degree of stereochemical complexity and three-dimensionality. Compounds , 5, and 53 locked in the Cγ-exo conformation of the proline ring and thus improved the Arg1 potency compared with our initial lead 1. Importantly, the synthetic strategies we outlined in this Letter also provide a foundation for the future synthesis and application of bicyclic compounds featuring densely functionalized and stereochemically complex proline derivatives with high Csp3 character. Such compounds are currently underrepresented as a concept in building blocks, medicine, and catalysts. The synthetic routes we outline could also have a profound impact on the access to novel building blocks to further explore the conformation of proline-containing peptides. This work complements the seminal work by Prof. Rapoport on the use of proline as a starting point for the synthesis of therapeutics.55 Synthesis is one the fundamental pillars of medicinal chemistry and drug discovery.56,57 What we reported here further expands the significant impact of synthesis on medicinal chemistry.
Acknowledgments
Brian Hall and Angie Sun are acknowledged for provided purified Arg1 protein for crystallography studies. Wilfredo Pinto is acknowledged for the high-resolution mnass analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00258.
This work was funded entirely by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
The authors declare no competing financial interest.
Author Status
○ P.S. is deceased (April 15, 2019).
Supplementary Material
References
- MacArthur M. W.; Thornton J. M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991, 218, 397–412. 10.1016/0022-2836(91)90721-H. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui Y.; Tsuboi M.; Iitaka Y. The crystal structure of dl-proline hydrochloride. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1969, B25, 2182–2192. 10.1107/S0567740869005449. [DOI] [Google Scholar]
- DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. Collagen stability: insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J. Am. Chem. Soc. 2002, 124, 2497–2505. 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- Pudlo M.; Demougeot C.; Girard-Thernier C. Arginase inhibitors: a rational approach over one century. Med. Res. Rev. 2017, 37, 475–513. 10.1002/med.21419. [DOI] [PubMed] [Google Scholar]
- Bronte V.; Serafini P.; Mazzoni A.; Segal D. M.; Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003, 24, 301–305. 10.1016/S1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yu Y.; Yang S.; Zeng B.; Zhang Z.; Jiao G.; Zhang Y.; Cai L.; Yang R. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2009, 58, 687–697. 10.1007/s00262-008-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steggerda S. M.; Bennett M. K.; Chen J.; Emberley E.; Huang T.; Janes J. R.; Li W.; MacKinnon A. L.; Makkouk A.; Marguier G.; Murray P. J.; Neou S.; Pan A.; Parlati F.; Rodriguez M. L. M.; Van de Velde L. A.; Wang T.; Works M.; Zhang J.; Zhang W.; Gross M. I. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101–119. 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilies M.; Di Costanzo L.; Dowling D. P.; Thorn K. J.; Christianson D. W. Binding of alpha,alpha-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 2011, 54, 5432–5443. 10.1021/jm200443b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zandt M. C.; Whitehouse D. L.; Golebiowski A.; Ji M. K.; Zhang M.; Beckett R. P.; Jagdmann G. E.; Ryder T. R.; Sheeler R.; Andreoli M.; Conway B.; Mahboubi K.; D’Angelo G.; Mitschler A.; Cousido-Siah A.; Ruiz F. X.; Howard E. I.; Podjarny A. D.; Schroeter H. Discovery of (R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J. Med. Chem. 2013, 56, 2568–2580. 10.1021/jm400014c. [DOI] [PubMed] [Google Scholar]
- Van Zandt M. C.; Jagdmann G. E.; Whitehouse D. L.; Ji M.; Savoy J.; Potapova O.; Cousido-Siah A.; Mitschler A.; Howard E.; Pyle A. M.; Podjarny A. D. Discovery of N-substituted 3-amino-4-(3-boronopropyl)pyrrolidine-3-carboxylic acids as highly potent thirdgeneration inhibitors of human arginase I and II. J. Med. Chem. 2019, 62, 8164–8177. 10.1021/acs.jmedchem.9b00931. [DOI] [PubMed] [Google Scholar]
- Mitcheltree M. J.; Li D.; Achab A.; Beard A.; Chakravarthy K.; Cheng M.; Cho H.; Eangoor P.; Fan P.; Gathiaka S.; Kim H. Y.; Lesburg C. A.; Lyons T. W.; Martinot T. A.; Miller J. R.; McMinn S.; O’Neil J.; Palani A.; Palte R. L.; Sauri J.; Sloman D. L.; Zhang H.; Cumming J. N.; Fischer C. Discovery and Optimization of Rationally Designed Bicyclic Inhibitors of Human Arginase to Enhance Cancer Immunotherapy. ACS Med. Chem. Lett. 2020, 11, 582–588. 10.1021/acsmedchemlett.0c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Zhang H.; Li D.; Childers M.; Pu Q.; Palte R. L.; Gathiaka S.; Lyons T. W.; Palani A.; Fan P. W.; Spacciapoli P.; Miller J. R.; Cho H.; Cheng M.; Chakravarthy K.; O’Neil J.; Eangoor P.; Beard A.; Kim H.-Y.; Sauri J.; Gunaydin H.; Sloman D. L.; Siliphaivanh P.; Cumming J.; Fischer C. Structure-Based Discovery of Proline-Derived Arginase Inhibitors with Improved Oral Bioavailability for Immuno-Oncology. ACS Med. Chem. Lett. 2021, 12, 1380–1388. 10.1021/acsmedchemlett.1c00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons T. W.; Martinot T. A.; He C. Q. X.; Qi J.; Shao G. X. Development of a Zinc-Mediated Approach to a 2,3-cis-Pyrrolidine Arginase Inhibitor. Org. Process Res. Dev. 2020, 24, 1457–1466. 10.1021/acs.oprd.0c00171. [DOI] [Google Scholar]
- Arg1 assay conditions:Han S.; Viola R. E. A spectrophotometric assay of arginase. Anal. Biochem. 2001, 295, 117–119. 10.1006/abio.2001.5189. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for docking methods.
- Cox B.; Duffy J.; Zdorichenko V.; Bellanger C.; Hurcum J.; Laleu B.; Booker-milburn K. I.; Elliott L. D.; Robertson-ralph M.; Swain C. J.; Bishop S. J.; Hallyburton I.; Anderson M. Escaping from flatland: Antimalarial Activity of Sp 3 - Rich Bridged Pyrrolidine Derivatives. ACS Med. Chem. Lett. 2020, 11, 2497–2503. 10.1021/acsmedchemlett.0c00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A. Y. M.; Venkatraman S. The discovery and development of boceprevir: A novel, first-generation inhibitor of the hepatitis C virus NS3/4A serine protease. J. Clin. and Transl. Hepatol. 2013, 1, 22–32. 10.14218/JCTH.2013.002XX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Example of bicyclic proline analogues as organocatalysts:Shinisha C. B.; Sunoj R. B. Bicyclic proline analogues as organocatalysts for stereoselective aldol reactions: an in silico DFT study. Org. Biomol. Chem. 2007, 5, 1287–1294. 10.1039/b701688c. [DOI] [PubMed] [Google Scholar]
- Example of chiral, bicyclic proline derivatives and their application as ligands:Södergren M. J.; Andersson P. G. Chiral, bicyclic proline derivatives and their application as ligands for copper in the catalytic asymmetric allylic oxidation of olefins. Tetrahedron Lett. 1996, 37, 7577–7580. 10.1016/0040-4039(96)01664-4. [DOI] [Google Scholar]
- Calaza M. I.; Sayago F. J.; Laborda P.; Cativiela C. Diastereoselective direct amidation/aza-Michael cascade reaction to synthesize cis-1, 3-disubstituted isoindolines. Eur. J. Org. Chem. 2015, 2015, 1633–1658. 10.1002/ejoc.201403121. [DOI] [Google Scholar]
- Koep S.; Gais H.-J.; Raabe G. Asymmetric Synthesis of Unsaturated, Fused Bicyclic Proline Analogues through Amino Alkylation of Cyclic Bis(allylsulfoximine)titanium Complexes and Migratory Cyclization of δ-Amino Alkenyl Aminosulfoxonium Salts. J. Am. Chem. Soc. 2003, 125, 13243–13251. 10.1021/ja030324y. [DOI] [PubMed] [Google Scholar]
- Belov D. S.; Ratmanova N. K.; Andreev I. A.; Kurkin A. V. Synthesis of Bicyclic Proline Derivatives by the Aza-Cope–Mannich Reaction: Formal Synthesis of (±)-Acetylaranotin. Chem. - Eur. J. 2015, 21, 4141–4147. 10.1002/chem.201405811. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y.; Fujikawa R.; Umemoto T.; Miyaura N. Iridium-catalyzed hydroboration of alkenes with pinacolborane. Tetrahedron 2004, 60, 10695–10700. 10.1016/j.tet.2004.09.014. [DOI] [Google Scholar]
- Kublitskii V. S.; Stepanov A. E.; Trukhan V. M. A New Method of Synthesis of Methyl N-Boc-2,3-dehydropyrrolidine- and piperidine-2-carboxylates. Russ. J. Org. Chem. 2008, 44, 933–934. Because Br2 reacts violently with THF and other organic solvents, do not mix Br2 with other organic solvents, including THF. 10.1134/S1070428008060274. [DOI] [Google Scholar]
- See the Supporting Information for the details of the monitoring reaction with ReatIR.
- Doyle M. P.; Griffin J. H.; Bagheri V.; Dorow R. L. Correlations between catalytic reactions of diazo compounds and stoichiometric reactions of transition-metal carbenes with alkenes. Mechanism of the cyclopropanation reaction. Organometallics 1984, 3, 53–61. 10.1021/om00079a011. [DOI] [Google Scholar]
- Fadel A.; Arzel P. Asymmetric construction of benzylic quaternary carbons from chiral malonates: Formal synthesis of natural (−) aphanorphine. Tetrahedron: Asymmetry 1995, 6, 893–900. 10.1016/0957-4166(95)00097-9. [DOI] [Google Scholar]
- Sen S. E.; Roach S. L. A convenient two-step procedure for the synthesis of substituted allylic amines from allylic alcohols. Synthesis 1995, 7, 756–758. 10.1055/s-1995-4012. [DOI] [Google Scholar]
- Simmons H. E.; Cairns T. L.; Vladuchick S. A.; Hoiness C. M. Cyclopropanes from unsaturated compounds, methylene iodide, and zinc-copper couple. Org. React. 2011, 1. 10.1002/0471264180.or020.01. [DOI] [Google Scholar]
- Lorenz J. C.; Long J.; Yang Z.; Xue S.; Xie X.; Shi Y. A Novel Class of Tunable Zinc Reagents (RXZnCH2Y) for Efficient Cyclopropanation of Olefins. J. Org. Chem. 2004, 69, 327–334. 10.1021/jo030312v. [DOI] [PubMed] [Google Scholar]
- Williams J. D.; Nakano M.; Gerardy R.; Rincon J. A.; de Frutos O.; Mateos C.; Monbaliu J.-C. M.; Kappe C. O. Finding the perfect match: a combined computational and experimental study toward efficient and scalable photosensitized [2+ 2] cycloadditions in flow. Org. Process Res. Dev. 2019, 23, 78–87. 10.1021/acs.oprd.8b00375. [DOI] [Google Scholar]
- Example of intramolecular [2 + 2]:Zhu M.; Zheng C.; Zhang X.; You S.-L. Synthesis of cyclobutane-fused angular tetracyclic spiroindolines via visible-light-promoted intramolecular dearomatization of indole derivatives. J. Am. Chem. Soc. 2019, 141, 2636–2644. 10.1021/jacs.8b12965. [DOI] [PubMed] [Google Scholar]
- Williams R. M.; Fegley G. J. Asymmetric synthesis of S-(−)-cucurbitine. Tetrahedron Lett. 1992, 33, 6755–6758. 10.1016/S0040-4039(00)61768-9. [DOI] [Google Scholar]
- Chalyk B. A.; Butko M. V.; Yanshyna O. O.; Gavrilenko K. S.; Druzhenko T. V.; Mykhailiuk P. K. Synthesis of spirocyclic pyrrolidines: advanced building blocks for drug discovery. Chem. - Eur. J. 2017, 23, 16782–16786. 10.1002/chem.201702362. [DOI] [PubMed] [Google Scholar]
- Grygorenko O. O.; Komarov I. V.; Cativiela C. A novel approach to 2, 4-ethanoproline. Tetrahedron: Asymmetry 2009, 20, 1433–1436. 10.1016/j.tetasy.2009.05.003. [DOI] [Google Scholar]
- Holladay M. W.; Lin C. W.; May C. S.; Garvey D. S.; Witte D. G.; Miller T. R.; Wolfram C. A. W.; Nadzan A. M. trans-3-n-propyl-L-proline is a highly favorable, conformationally restricted replacement for methionine in the C-terminal tetrapeptide of cholecystokinin. Stereoselective synthesis of 3-allyl- and 3-n-propyl-L-proline derivatives from 4-hydroxy-L-proline. J. Med. Chem. 1991, 34, 455–457. 10.1021/jm00105a068. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Hiranuma S.; Nakata T. Efficient method for inversion of secondary alcohols by reaction of chloromethanesulfonates with cesium acetate. Tetrahedron Lett. 1996, 37, 6145–6148. 10.1016/0040-4039(96)01333-0. [DOI] [Google Scholar]
- Rolla G. A.; Tei L.; Fekete M.; Arena F.; Gianolio E.; Botta M. Responsive Mn (II) complexes for potential applications in diagnostic magnetic resonance imaging. Bioorg. Med. Chem. 2011, 19, 1115–1122. 10.1016/j.bmc.2010.07.064. [DOI] [PubMed] [Google Scholar]
- Compound 41 was prepared according to the synthesis of compound 31 using 1-(tert-butyl) 2-methyl (R)-4-oxopyrrolidine-1,2-dicarboxylate as the starting material.
- Matteson D. S.; Jesthi P. K.; Sadhu K. M. Synthesis and properties of pinanediol. alpha.-amido boronic esters. Organometallics 1984, 3, 1284–1288. 10.1021/om00086a024. [DOI] [Google Scholar]
- Sulphur nucleophile:Choo H.; Chen X.; Yadav V.; Wang J.; Schinazi R. F.; Chu C. K. Synthesis and Anti-HIV Activity of d- and l-Thietanose Nucleosides. J. Med. Chem. 2006, 49, 1635–1647. 10.1021/jm050912h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitrogen nucleophile:Juaristi E.; Madrigal D. Use of hexamethylphosphoramide chmpa in the alkylation of aromatic amines: synthesis of azetidines, pyrrolidines, piperidines and hexahydroazepines. Tetrahedron 1989, 45, 629–636. 10.1016/0040-4020(89)80091-2. [DOI] [Google Scholar]
- The synthesis of compound 53 can be found in the Supporting Information.
- Goli N.; Kallepu S.; Mainkar P. S.; Lakshmi J. K.; Chegondi R.; Chandrasekhar S. Synthetic Strategy toward the Pentacyclic Core of Melodinus Alkaloids. J. Org. Chem. 2018, 83, 2244–2249. 10.1021/acs.joc.7b03138. [DOI] [PubMed] [Google Scholar]
- Compound 66 was prepared according to the synthesis of compound 31 using 1-benzyl 2-methyl (S)-4-oxopyrrolidine-1,2-dicarboxylate as the starting material.
- Yoneyama H.; Numata M.; Uemura K.; Usami Y.; Harusawa S. Transformation of carbonyl compounds into homologous alkynes under neutral conditions: fragmentation of tetrazoles derived from cyanophosphates. J. Org. Chem. 2017, 82, 5538–5556. 10.1021/acs.joc.7b00346. [DOI] [PubMed] [Google Scholar]
- Athawale P. R.; Kumari N.; Dandawate M. R.; Kashinath K.; Srinivasa Reddy D. Synthesis of Chiral Tetrahydrofuran Building Blocks from Pantolactones: Application in the Synthesis of Empagliflozin and Amprenavir Analogs. Eur. J. Org. Chem. 2019, 30, 4805–4810. 10.1002/ejoc.201900718. [DOI] [Google Scholar]
- Underwood B. S.; Tanuwidjaja J.; Ng S.-S.; Jamison T. F. Total syntheses of the squalene-derived halogenated polyethers ent-dioxepandehydrothyrsiferol and armatol A via bromonium- and Lewis acid-initiated epoxide-opening cascades. Tetrahedron 2013, 69, 5205–5220. 10.1016/j.tet.2013.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; James C. A.; Meanwell N. A.; Hamann L. G.; Belema M. A scalable synthesis of (1R,3S,5R)-2-(tert-butoxycarbonyl)-2-azabicyclo[3.1.0]hexane-3-carboxylic acid. Tetrahedron Lett. 2013, 54, 6722–6724. 10.1016/j.tetlet.2013.09.114. [DOI] [Google Scholar]
- De Faria A. R.; Matos C. R. R.; Correia C. R. D. [2 + 2] Cycloaddition reaction of cyclic enecarbamates and enamides with ketenes. A short and efficient synthesis of the Geissman-Waiss lactone. Tetrahedron Lett. 1993, 34, 27–30. 10.1016/S0040-4039(00)60049-7. [DOI] [Google Scholar]
- Barton D. H. R.; McCombie S. W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc., Perkin Trans. 1 1975, 16, 1574–1585. 10.1039/p19750001574. [DOI] [PubMed] [Google Scholar]
- Brameld K. A.; Kuhn B.; Reuter D. C.; Stahl M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 2008, 48, 1–24. 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardina F. J.; Rapoport H. Enantiospecific synthesis of heterocycles from α-amino acids. Chem. Rev. 1996, 96, 1825–1872. 10.1021/cr9300348. [DOI] [PubMed] [Google Scholar]
- Blakemore D. C.; Castro L.; Churcher I.; Rees D. C.; Thomas A. W.; Wilson D. M.; Wood A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394. 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]
- Campos K. R.; Coleman P. J.; Alvarez J. C.; Dreher S. D.; Garbaccio R. M.; Terrett N. K.; Tillyer R. D.; Truppo M. D.; Parmee E. R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363, eaat0805. 10.1126/science.aat0805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.