

Abstract

Immunomodulatory drugs (IMiDs) thalidomide, lenalidomide, and pomalidomide engage cereblon and mediate a protein interface with neosubstrates such as zinc finger transcription factors promoting their polyubiquitination and degradation. The IMiDs have garnered considerable excitement in drug discovery, leading to exploration of targeted protein degradation strategies. Although the molecular modes-of-action of the IMiDs and related degraders have been the subject of intense research, their pharmacokinetics and disposition have been relatively understudied. Here, we assess the effects of physicochemistry of the IMiDs, the phthalimide EM-12, and the candidate drug CC-220 (iberdomide) on lipophilicity, solubility, metabolism, permeability, intracellular bioavailability, and cell-based potency. The insights yielded in this study will enable the rational property-based design and development of targeted protein degraders in the future.
Keywords: physicochemistry, pharmacokinetics, molecular glue, targeted protein degradation, intracellular bioavailability
The molecular mode of action of the immunomodulatory drugs (IMiDs) thalidomide, lenalidomide, and pomalidomide was elucidated in the past decade following affinity isolation of their common binding protein cereblon.1 Following engagement of cereblon, a component of an E3 ubiquitin ligase complex, the IMiDs mediate new interactions with “neosubstrates” such as the zinc finger transcription factor Ikaros (IKZF1), resulting in polyubiquitination and proteasomal degradation of the target.2 IMiDs have also been chemically linked to protein ligands to create heterobifunctional molecules, called proteolysis-targeting chimeras (PROTACs), that hijack the ubiquitination machinery to mediate degradation of target proteins.3
IMiD drugs, often referred to as “molecular glues”, possess a critical glutarimide motif that binds in a tritryptophan cage in the cereblon pocket, but their aromatic ring structures (phthalimide or isoindolinone) differ subtly (Figure 1). Additionally, lenalidomide and pomalidomide contain a polar amino group that has been shown to bind through a water molecule to residues in cereblon and IKZF1.4 Despite minor variations in structure, we hypothesized that the physicochemical features of the phthalimide, that possesses an extended bicyclic delocalized π-electron system, may diverge substantially from the isoindolinone (the carbon atom of the −CH2– group, which is formally sp3 hybridized, hinders π-conjugation). Here, we detail these differences and, for completeness, explore the properties of the related derivative EM-12 to be able to perform a pairwise analysis. We also profiled the elaborated and structurally differentiated molecular glue CC-220 (iberdomide) that is currently in clinical trials for multiple myeloma, non-Hodgkin lymphoma, and systemic lupus erythematosus.5
Figure 1.
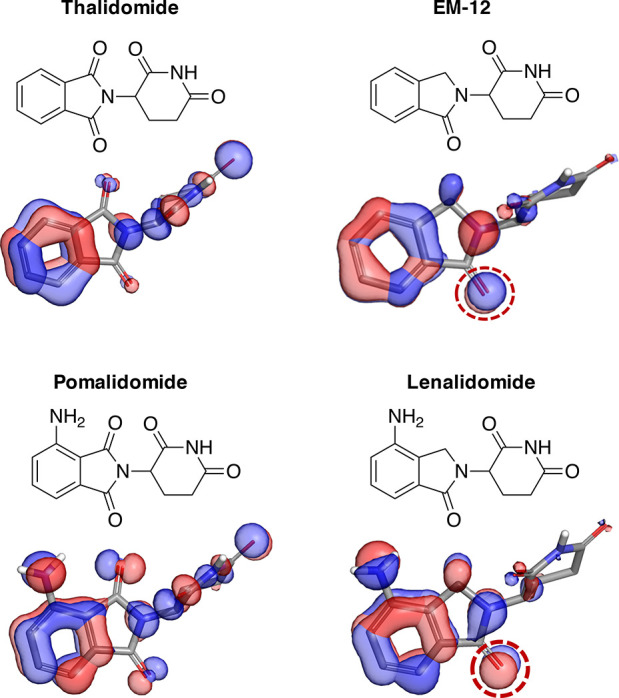
Electronic structure calculations for thalidomide, EM-12, pomalidomide, and lenalidomide using Jaguar (Schrödinger release 2021-1).17 The three highest occupied molecular orbitals are shown as isosurfaces (depicted at the level of −0.5) for each molecule (color indicates phase).
Physicochemistry plays a central role in drug design because it determines important characteristics that influence candidate development, including lipophilicity, solubility, membrane permeability, nonspecific binding, metabolism, and chemical stability, which in turn affect in vivo pharmacokinetics, disposition, efficacy, and safety.6 In this study, side-by-side in vitro profiling of the IMiDs revealed key differences in these important characteristics, yielding useful insights that will impact the future design, property-based optimization, and development of cereblon-modulating drugs and chemical probes.
To understand the differences in electronic structures between the IMiDs, a simple calculation of their molecular orbitals was performed, as shown in Figure 1 (further described in the Supporting Information). The extended delocalization of the phthalimide system enhances the resonance of the π-electron ring density. Replacement of an imide carbonyl with the sp3 hybridized −CH2– present in the isoindolinones hinders π-conjugation into the five-membered ring and localizes electron density to the remaining carbonyl oxygen of the cyclic benzamide group (dotted ring, Figure 1). These electronic features significantly influence the physicochemical properties, pharmacokinetics, and disposition of the IMiDs, as demonstrated in Table 1 and described in more detail below.
Table 1. In Vitro Profiling of Cereblon-Modulating Drugsa.
| thalidomide | EM-12 | pomalidomide | lenalidomide | CC-220 | |
|---|---|---|---|---|---|
| molecular weight | 258 | 244 | 273 | 259 | 449 |
| cLogP | 0.3 | 0.2 | 0.2 | –0.5 | 1.1 |
| Chrom LogD (pH 7.4) | 0.900 ± 0.001 | 0.498 ± 0.001 | 0.787 ± 0.001 | –0.058 ± 0.001 | 1.900 ± 0.001 |
| Caco-2 (10–6 cm/s)b | 35.6 ± 1.6 (0.9) | 22.2 ± 0.9 (0.99) | 24.9 ± 2.0 (1.1) | 1.45 ± 0.05 (1.7) | 5.6 ± 2.5 (5.1) |
| kinetic solubility (μM) | 345 ± 47 | 451 ± 48 | 78 ± 17 | 461 ± 47 | 380 ± 28 |
| HLM T1/2 (min) | >186 | >186 | >186 | 124 | 115 |
| human plasma T1/2 (min) | 22 | 119 | 159 | 284 | >746 |
| fu,cell | 0.34 ± 0.2 | 1.0c | 0.90 ± 0.1 | 1.0c | 0.06 ± 0.006 |
| Kp | 8.9 ± 3.4 | 2.2 ± 0.1 | 4.5 ± 0.4 | 5.0 ± 0.4 | 9.3 ± 1.8 |
| Fic | 3.0 ± 0.68 | 2.2 ± 0.1 | 4.1 ± 0.04 | 5.0 ± 0.4 | 0.56 ± 0.01 |
| IKZF1 DC50 (nM) | >10 000 | 192.7 ± 52.5 | 26.7 ± 3.8 | 90 ± 15.2 | 2.4 ± 0.13 |
Experimental details are provided in the Supporting Information.
Efflux ratios in parentheses.
Intracellular unbound fractions reported as 1.0 due to homogenate unbound fractions marginally lower (<3%) than buffer (see Supporting Information). HLM = human liver microsomes. Chrom LogD = chromatographic assessment of the logarithm of the distribution coefficient (LogD).21fu,cell = intracellular unbound fraction (4 h incubation, MOLT4). Kp = intracellular drug accumulation (4 h incubation, MOLT4). Fic = intracellular bioavailability = fu,cell × Kp. DC50 = concentration of drug required to reduce protein signal in the HiBiT assay by 50% (details in the Supporting Information).
The extended π-delocalization of the phthalimide ring in pomalidomide, in conjunction with the electron-donating amino group, creates a push–pull electronic system, not dissimilar to that of benzoxadiazole dyes,7 that renders the drug fluorescent. The intrinsic fluorescence of pomalidomide has been used as a clinical biomarker of drug levels in ex vivo human plasma samples.8 Fluorescent amino phthalimides were described previously,9−11 but surprisingly, the photophysical properties of molecular glues and PROTACs containing this motif have not been reported. The fluorescent nature of degraders possessing the amino phthalimide structure should be considered as a potential complicating factor when screening such compounds in fluorescence-based assays.
The extended π-electron system of thalidomide and pomalidomide also affords higher lipophilicity (higher LogD) and lower solubility compared to the isoindolinone congeners EM-12 and lenalidomide (Table 1). The calculated solvation energies (Supporting Information) are also more favorable for lenalidomide (−28.0 kcal/mol) and EM-12 (−23.7 kcal/mol) compared with their counterparts pomalidomide (−25.8 kcal/mol) and thalidomide (−22.4 kcal/mol), consistent with higher polarity. Additionally, the intramolecular hydrogen bonding of the amino group with the phthalimide ring of pomalidomide further increases relative lipophilicity, cf. ΔLogD (thalidomide/EM-12) = 0.4 versus ΔLogD (pomalidomide/lenalidomide) = 0.85 (Table 1). The calculated LogPs appear to capture the differences in lipophilicity resulting from intramolecular hydrogen-bonding but not π-delocalization (Table 1).
For these same reasons, lenalidomide has the highest hydrophilicity of the IMiDs that imparts high aqueous solubility and remarkably low permeability in Caco-2 cells due to a high desolvation penalty in water (Table 1). The low permeability of lenalidomide translates to impaired CNS penetration, and the IMiD thus lacks the neurological side effects (sedation and neuropathy) of thalidomide.12 Due to enhanced permeability resulting from higher lipophilicity and intramolecular hydrogen-bonding, pomalidomide appears to possess improved CNS penetration over lenalidomide.13
Lenalidomide undergoes rapid urinary excretion resulting in a short half-life of 3 h in humans, in concordance with its physicochemistry.12,14 Indeed, lenalidomide is a very weak substrate for CYP P450 enzymes and phase II conjugative metabolism. Therefore, an important feature of the drug is that renal impairment can affect drug clearance, resulting in patient variability.15 Although the IMiDs possess excellent stability in human liver microsomes (HLM), somewhat unexpectedly, lenalidomide appears to be the least stable (Table 1), which may be a reflection of the higher electron density of the aniline ring being more prone to oxidation in this in vitro assay. Although the differences in HLM stability do not directly mirror in vivo human PK observations, a small amount (<5%) of 5-hydroxy-lenalidomide was identified in human plasma following oral administration revealing a minor propensity for oxidative metabolism.16
It is also noticeable that lenalidomide is the most stable IMiD derivative in human plasma, while thalidomide is the least stable (Table 1). The propensity for glutarimide hydrolysis is similar for all of the drugs, but the phthalimide ring, essentially an imide derivative of phthalic anhydride, appears to be more prone to nonenzymatic hydrolysis compared to the isoindolinone.18 In humans, thalidomide undergoes phthalimide and glutarimide hydrolysis, and these polar products are renally excreted with little to no metabolism.19 Although EM-12 is more stable than thalidomide, hydrolysis products are also formed and renally cleared in marmoset monkeys.20
CC-220 (iberdomide, Figure 2a) was developed recently as a potent cereblon modulator that degrades IKZF1 (and the related target IKZF3) more effectively than the IMiDs.22 CC-220 possesses a lipophilic tail that binds in a hydrophobic region of the cereblon pocket (PDB code 5V3O, Figure 2b). The changes in molecular weight and lipophilicity compared to the IMiDs (Table 1) leads to an oxidative mode of metabolism in humans, mediated by CYP3A4/5 (cf. lower HLM stability in Table 1), and a lower apparent total clearance than the IMiDs.12,23 CC-220 also contains a basic center that increases the volume of distribution compared to the neutral IMiDs, and together with the relatively low clearance, this yields a 9–13 h half-life in humans.24
Figure 2.
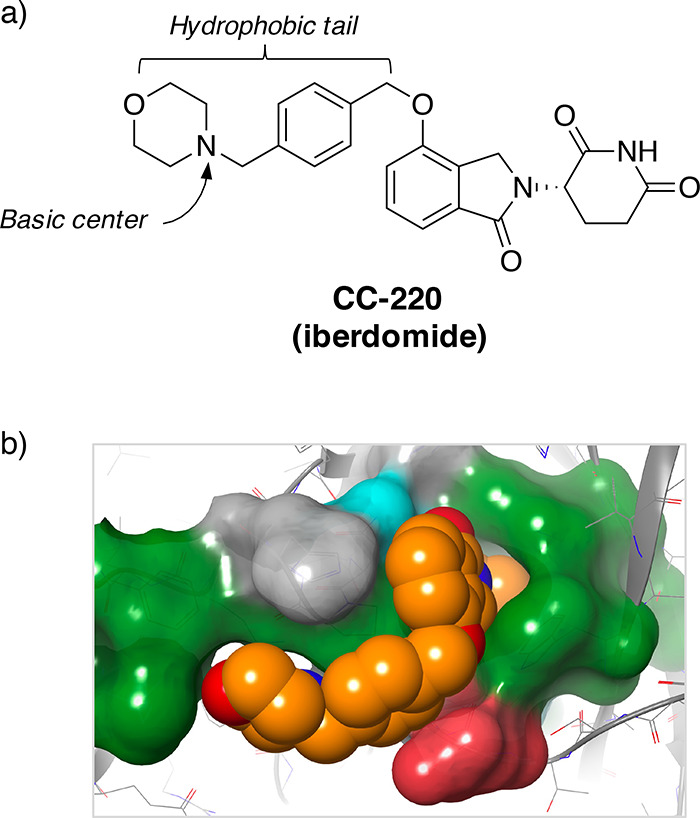
(a) Structure of the cereblon-modulating molecular glue drug candidate CC-220 (iberdomide). (b) CC-220/cereblon crystal structure (PDB 5V3O) highlighting binding surface (green = hydrophobic residues, drawn using Maestro, Schrödinger).
The “free drug hypothesis” states that unbound drug levels are the pharmacologically relevant concentrations in cells and in vivo.25 It is therefore important to measure free drug concentrations to understand in vitro–in vivo correlations, pharmacokinetic–pharmacodynamic (PKPD), and structure–activity relationships (SARs) of derivatives with the same mode of action, as for the cereblon modulators described here. We were particularly interested in the effects of physicochemistry on the levels of free drug in cells, suspecting that the higher lipophilicity and ionizable basic center of CC-220 would reduce the unbound fraction relative to the IMiDs. The protocols used to measure unbound fractions and cellular accumulation are provided in the Supporting Information.26,27
EM-12, pomalidomide, and lenalidomide achieved high unbound levels (fu,cell ∼ 1) in MOLT4 cells due to their physicochemistry (low molecular weight, polar and nonionizable at physiological pH). Thalidomide instability, as described above, yielded a large error in the results obtained for this IMiD in the assay (Supporting Information, Figure S1). Interestingly, pomalidomide and lenalidomide accumulate in MOLT4 cells and the intracellular bioavailability Fic is greater than 1 which is indicative of potential cellular uptake processes (Table 1).26 Due to the significantly higher lipophilicity of CC-220, and the presence of an ionizable basic center, an increase in nonspecific binding to proteins and membranes would be expected, and this was reflected in the lower observed fu,cell. However, intracellular accumulation Kp is elevated compared to pomalidomide and lenalidomide, and although unconfirmed, CC-220 may accumulate to a certain degree in the acidic lysosome (pH ∼ 5) due to protonation of the morpholine nitrogen basic center, which is a common lysosome-targeting motif used in fluorescent imaging probes.28
To explore the implications of these effects on cell-based pharmacology, an IKZF1 HiBiT assay was developed in the same cell line (MOLT4) which uses CRISPR-mediated tagging of the endogenous protein with a luminescent peptide to enable quantitative measurement of IKZF1 degradation by the cereblon-modulating molecular glues (Supporting Information, Figure S2).29 MOLT4 is a human T-cell line which was originally chosen for this study due to the involvement of IKZF1 in immune cell differentiation, and the line serves as a workhorse screening model in our center.30 The IKZF1 DC50 (half degradation concentration) of CC-220 was found to be 2.4 nM (in agreement with published potency data in an ePL fusion chemiluminescence assay)22 yielding an intracellular unbound DC50 of 1.3 nM, compared to 111 and 450 nM for pomalidomide and lenalidomide, respectively. These data thus enable a direct comparison of the cell-based activity of different drugs that possess distinct intracellular bioavailabilities. The considerably higher potency of CC-220 translates to antimultiple myeloma activity greater than that of pomalidomide and lenalidomide, particularly in IMiD resistant cells that have low cereblon expression.31
Immunomodulatory drugs represent an exciting therapeutic modality of targeted protein degraders that have garnered considerable R&D investment in recent years. Despite numerous pharmacological studies of the IMiDs and related degraders, there has been little investigation into their fundamental physicochemical properties. Property-based design, which describes the optimization of absorption, metabolic and chemical stability, distribution, and biopharmaceutical properties, requires a deep understanding of physicochemical parameters.32 Here, we show that the phthalimide ring is less polar than the isoindolinone motif due to extended π-delocalization over the heterobicyclic system. Further analysis highlighted physicochemical differences among the IMiDs that govern their pharmacokinetics and disposition.
These findings are of significance to the design and development of future degraders, including PROTACs, because they provide insights that will impact the optimization of physicochemical parameters needed to achieve desirable PKPD profiles. For example, the metabolic stability of a lipophilic phthalimide/pomalidomide-based degrader could be improved by switching to the more polar isoindolinone/lenalidomide scaffold. Conversely, the low permeability of a polar lenalidomide analogue could be addressed through conversion to the phthalimide congener.
Determination of free drug levels in cells enabled the assessment of intracellular unbound DC50 values, which confirmed the exceptional functional potency of the clinical molecular glue degrader CC-220. Therefore, these assays may supplement an experimental screening platform to help advance the development of targeted degraders.33 Such analyses could also become important for elucidating complex degrader cell-based SARs because off-target competitive recruitment of cereblon determines on-target activities due to a limiting pool of the E3 ligase.34
Acknowledgments
We thank Domainex for measuring unbound fractions, cellular accumulation, and Chrom LogD and BioDuro for measuring Caco-2 permeability, plasma stability, kinetic solubility, and HLM stability.
Glossary
Abbreviations
- Chrom LogD
chromatographic assessment of the logarithm of the distribution coefficient (LogD)
- CRISPR
clustered regularly interspaced short palindromic repeats
- DC50
concentration of drug required to reduce protein signal by 50%
- Fic
intracellular bioavailability
- fu,cell
intracellular unbound fraction
- HLM
human liver microsomes
- IMiD
immunomodulatory drug
- Kp
intracellular drug accumulation
- PKPD
pharmacokinetic–pharmacodynamic
- PROTAC
proteolysis-targeting chimera
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00475.
Experimental section containing computational calculations of molecular orbitals and desolvation energies and methods for determining Chrom LogD, kinetic solubility, plasma stability, human liver microsome stability, Caco-2 permeability, drug accumulation in cells, intracellular unbound fraction, and IKZF1 HiBiT potency (including dose–response curves) (PDF)
The authors declare the following competing financial interest(s): All authors receive research funding from Deerfield. L.H.J. serves on the scientific advisory board for, and holds equity in, Interline Therapeutics, holds equity in Jnana Therapeutics, and consults for Carnot Pharma and EoCys. J.C. is a consultant for Soltego, Jengu, Allorion and EoCys and holds equity in Soltego, Allorion, EoCys, and M3 Bioinformatics & Technology Inc.
Supplementary Material
References
- Ito T.; Ando H.; Suzuki T.; Ogura T.; Hotta K.; Imamura Y.; Yamaguchi Y.; Handa H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–50. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Chamberlain P. P.; Hamann L. G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers Q. L.; Petzold G.; Bunker R. D.; Renneville A.; Słabicki M.; Liddicoat B. J.; Abdulrahman W.; Mikkelsen T.; Ebert B. L.; Thomä N. H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, 362. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer P. H.; Ye Y.; Wu L.; Kosek J.; Ringheim G.; Yang Z.; Liu L.; Thomas M.; Palmisano M.; Chopra R. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1516–1523. 10.1136/annrheumdis-2017-212916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinworth C. P.; Young R. J. Facts, Patterns, and Principles in Drug Discovery: Appraising the Rule of 5 with Measured Physicochemical Data. J. Med. Chem. 2020, 63, 10091–10108. 10.1021/acs.jmedchem.9b01596. [DOI] [PubMed] [Google Scholar]
- Thooft A. M.; Cassaidy K.; VanVeller B. A Small Push-Pull Fluorophore for Turn-on Fluorescence. J. Org. Chem. 2017, 82, 8842–8847. 10.1021/acs.joc.7b00939. [DOI] [PubMed] [Google Scholar]
- Shahbazi S.; Peer C. J.; Polizzotto M. N.; Uldrick T. S.; Roth J.; Wyvill K. M.; Aleman K.; Zeldis J. B.; Yarchoan R.; Figg W. D. A sensitive and robust HPLC assay with fluorescence detection for the quantification of pomalidomide in human plasma for pharmacokinetic analyses. J. Pharm. Biomed. Anal. 2014, 92, 63–8. 10.1016/j.jpba.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Ruiz R.; Fichtler R.; Diaz Miara Y.; Nicoul M.; Schaniel D.; Neumann H.; Beller M.; Blunk D.; Griesbeck A. G.; Jacobi von Wangelin A. On the photophysical properties of new luminol derivatives and their synthetic phthalimide precursors. J. Fluoresc. 2010, 20, 657–64. 10.1007/s10895-010-0598-0. [DOI] [PubMed] [Google Scholar]
- Eugenio Vazquez M.; Rothman D. M.; Imperiali B. A new environment-sensitive fluorescent amino acid for Fmoc-based solid phase peptide synthesis. Org. Biomol. Chem. 2004, 2, 1965–6. 10.1039/B408001G. [DOI] [PubMed] [Google Scholar]
- Wörner S.; Rönicke F.; Ulrich A. S.; Wagenknecht H. A. 4-Aminophthalimide Amino Acids as Small and Environment-Sensitive Fluorescent Probes for Transmembrane Peptides. ChemBioChem 2020, 21, 618–622. 10.1002/cbic.201900520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.; Zhou S.; Palmisano M. Clinical Pharmacokinetics and Pharmacodynamics of Lenalidomide. Clin. Pharmacokinet. 2017, 56, 139–152. 10.1007/s40262-016-0432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Qiu Y.; Personett D.; Huang P.; Edenfield B.; Katz J.; Babusis D.; Tang Y.; Shirely M. A.; Moghaddam M. F.; Copland J. A.; Tun H. W. Pomalidomide shows significant therapeutic activity against CNS lymphoma with a major impact on the tumor microenvironment in murine models. PLoS One 2013, 8, e71754 10.1371/journal.pone.0071754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma M. V.; Feng B.; Obach R. S.; Troutman M. D.; Chupka J.; Miller H. R.; El-Kattan A. Physicochemical determinants of human renal clearance. J. Med. Chem. 2009, 52, 4844–52. 10.1021/jm900403j. [DOI] [PubMed] [Google Scholar]
- Chen N.; Lau H.; Kong L.; Kumar G.; Zeldis J. B.; Knight R.; Laskin O. L. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J. Clin. Pharmacol. 2007, 47, 1466–75. 10.1177/0091270007309563. [DOI] [PubMed] [Google Scholar]
- Chen N.; Wen L.; Lau H.; Surapaneni S.; Kumar G. Pharmacokinetics, metabolism and excretion of [(14)C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother. Pharmacol. 2012, 69, 789–97. 10.1007/s00280-011-1760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
- Lepper E. R.; Smith N. F.; Cox M. C.; Scripture C. D.; Figg W. D. Thalidomide metabolism and hydrolysis: mechanisms and implications. Curr. Drug Metab. 2006, 7, 677–85. 10.2174/138920006778017777. [DOI] [PubMed] [Google Scholar]
- Chung F.; Lu J.; Palmer B. D.; Kestell P.; Browett P.; Baguley B. C.; Tingle M.; Ching L. M. Thalidomide pharmacokinetics and metabolite formation in mice, rabbits, and multiple myeloma patients. Clin. Cancer Res. 2004, 10, 5949–56. 10.1158/1078-0432.CCR-04-0421. [DOI] [PubMed] [Google Scholar]
- Schmahl H. J.; Heger W.; Nau H. The enantiomers of the teratogenic thalidomide analogue EM 12. 2. Chemical stability, stereoselectivity of metabolism and renal excretion in the marmoset monkey. Toxicol. Lett. 1989, 45, 23–33. 10.1016/0378-4274(89)90155-0. [DOI] [PubMed] [Google Scholar]
- Young R. J.; Green D. V.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–30. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Matyskiela M. E.; Zhang W.; Man H. W.; Muller G.; Khambatta G.; Baculi F.; Hickman M.; LeBrun L.; Pagarigan B.; Carmel G.; Lu C. C.; Lu G.; Riley M.; Satoh Y.; Schafer P.; Daniel T. O.; Carmichael J.; Cathers B. E.; Chamberlain P. P. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- Gaudy A.; Atsriku C.; Ye Y.; MacGorman K.; Liu L.; Xue Y.; Surapaneni S.; Palmisano M. Evaluation of iberdomide and cytochrome p450 drug-drug interaction potential in vitro and in a phase 1 study in healthy subjects. Eur. J. Clin. Pharmacol. 2021, 77, 223–231. 10.1007/s00228-020-03004-w. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Gaudy A.; Schafer P.; Thomas M.; Weiss D.; Chen N.; Liu L.; Xue Y.; Carayannopoulos L.; Palmisano M. First-in-Human, Single- and Multiple-Ascending-Dose Studies in Healthy Subjects to Assess Pharmacokinetics, Pharmacodynamics, and Safety/Tolerability of Iberdomide, a Novel Cereblon E3 Ligase Modulator. Clin. Pharmacol. Drug Dev. 2021, 10, 471–485. 10.1002/cpdd.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–39. 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Mateus A.; Treyer A.; Wegler C.; Karlgren M.; Matsson P.; Artursson P. Intracellular drug bioavailability: a new predictor of system dependent drug disposition. Sci. Rep. 2017, 7, 43047. 10.1038/srep43047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus A.; Matsson P.; Artursson P. Rapid measurement of intracellular unbound drug concentrations. Mol. Pharmaceutics 2013, 10, 2467–78. 10.1021/mp4000822. [DOI] [PubMed] [Google Scholar]
- Gao P.; Pan W.; Li N.; Tang B. Fluorescent probes for organelle-targeted bioactive species imaging. Chem. Sci. 2019, 10, 6035–6071. 10.1039/C9SC01652J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinn M. K.; Machleidt T.; Zimmerman K.; Eggers C. T.; Dixon A. S.; Hurst R.; Hall M. P.; Encell L. P.; Binkowski B. F.; Wood K. V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13, 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- Huang C. C.; Hou Y.; Woods L. K.; Moore G. E.; Minowada J. Cytogenetic study of human lymphoid T-cell lines derived from lymphocytic leukemia. J. Natl. Cancer Inst 1974, 53, 655–60. 10.1093/jnci/53.3.655. [DOI] [PubMed] [Google Scholar]
- Bjorklund C. C.; Kang J.; Amatangelo M.; Polonskaia A.; Katz M.; Chiu H.; Couto S.; Wang M.; Ren Y.; Ortiz M.; Towfic F.; Flynt J. E.; Pierceall W.; Thakurta A. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. 10.1038/s41375-019-0620-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van De Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–33. 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- Thompson J. W.; Clayton T.; Khambatta G.; Bateman L. A.; Carroll C. W.; Chamberlain P. P.; Matyskiela M. E. Profiling CELMoD-Mediated Degradation of Cereblon Neosubstrates. Methods Mol. Biol. 2021, 2365, 283–300. 10.1007/978-1-0716-1665-9_15. [DOI] [PubMed] [Google Scholar]
- Sperling A. S.; Burgess M.; Keshishian H.; Gasser J. A.; Bhatt S.; Jan M.; Słabicki M.; Sellar R. S.; Fink E. C.; Miller P. G.; Liddicoat B. J.; Sievers Q. L.; Sharma R.; Adams D. N.; Olesinski E. A.; Fulciniti M.; Udeshi N. D.; Kuhn E.; Letai A.; Munshi N. C.; Carr S. A.; Ebert B. L. Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 2019, 134, 160–170. 10.1182/blood.2019000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.