

SUMMARY
Mechanistic insights into the role of the human microbiome in the predisposition to and treatment of disease are limited by the lack of methods to precisely add or remove microbial strains or genes from complex communities. Here, we demonstrate that engineered bacteriophage M13 can be used to deliver DNA to Escherichia coli within the mouse gastrointestinal (GI) tract. Delivery of a programmable exogenous CRISPR-Cas9 system enables the strain-specific depletion of fluorescently marked isogenic strains during competitive colonization and genomic deletions that encompass the target gene in mice colonized with a single strain. Multiple mechanisms allow E. coli to escape targeting, including loss of the CRISPR array or even the entire CRISPR-Cas9 system. These results provide a robust and experimentally tractable platform for microbiome editing, a foundation for the refinement of this approach to increase targeting efficiency, and a proof of concept for the extension to other phage-bacterial pairs of interest.
Graphical Abstract
In brief
Lam et al. show that filamentous bacteriophage can be harnessed as agents of gene delivery to bacteria colonizing the gastrointestinal tract. Using M13 to deliver CRISPR-Cas9, they demonstrate sequence-specific targeting of GFP-marked E. coli in the gut and show that CRISPR-Cas9 can induce genomic deletions at the target site.
INTRODUCTION
Current strategies for manipulating the microbiome either lack species- or strain-level precision (Basolo et al., 2020; Smillie et al., 2018) or require the introduction of an exogenous bacterium into the host (Isabella et al., 2018; Shepherd et al., 2018). Pioneering studies of pathogenic bacteria that colonize the skin (Bikard et al., 2014; Citorik et al., 2014; Park et al., 2017) and gut (Hsu et al., 2020a; Selle et al., 2020) support the potential for the use of engineered bacteriophage carrying an exogenous CRISPR-Cas system that could be directed to any target of interest; however, these methods have yet to be broadly applied to the human or mouse gut microbiome. Recent progress has been made using lysogenic phage and transcriptional repression (Hsu et al., 2020b), but more work is needed to enable stable manipulation of microbial community structure and gene content without the need to integrate a viral genome. Given the tremendous diversity within both the bacterial (Human Microbiome Project Consortium, 2012) and viral (Camarillo-Guerrero et al., 2021) components of the human gut microbiota, we sought to establish a tripartite model system that builds upon tools for the genetic manipulation of a bacteriophage and its bacterial target coupled to an experimentally tractable mammalian host.
We focused on M13, a single-stranded DNA (ssDNA) filamentous inovirus (Ackermann, 2009; Hofschneider, 1963) able to replicate and release virions without causing cell lysis (Salivar et al., 1964). M13 infects E. coli and related Enterobacteriaceae carrying the F sex factor necessary to form the conjugative F pilus (Guiney, 1982; Lee and Ames, 1984). M13 phagemid vectors combine the advantages of plasmid DNA manipulation with the ability to easily package recombinant DNA into virions (Zinder and Boeke, 1982). M13 has been used to target E. coli (Lu and Collins, 2009; Westwater et al., 2003) and Helicobacter pylori (Cao et al., 2000), including engineered M13 carrying CRISPR-Cas9 in an insect model of bacterial infection (Citorik et al., 2014). However, the use of M13 to deliver genetic constructs (including CRISPR-Cas systems) to bacteria within the mammalian gastrointestinal (GI) tract had not been previously demonstrated. We also leveraged the streptomycin (Sm)-treated mouse model for E. coli colonization within the mouse gut microbiota (Myhal et al., 1982), providing a robust and accessible model that could be used by any group with access to a mouse colony.
RESULTS
Bacteriophage M13 enables the delivery of DNA to the gut microbiome
We utilized phagemid pBluescript II (Alting-Mees and Short, 1989) carrying the bla (β-lactamase) gene and a β-lactam antibiotic in the drinking water to select for successfully infected E. coli. pBluescript II conferred in vitro resistance to ampicillin and the semi-synthetic analog carbenicillin at concentrations exceeding 1 mg/mL (Figure S1). We used Sm-resistant (SmR) E. coli to colonize the GI tract of Sm-treated mice. As expected, Sm altered gut microbial community structure while decreasing diversity and overall colonization level (Figures S2A–S2D). SmR E. coli colonized at a high proportion (median 18% of the gut microbiota; range 1.4%–43%) 4 days after gavage (Figure S2E). We introduced a SmR E. coli population that was a mixture of 99.9% AmpS (no plasmid) and 0.1% AmpR cells (pBluescript II), split the mice into 2 groups with access to water containing only Sm or both Sm and ampicillin, and tracked both total E. coli and AmpR E. coli in mouse feces. At 6 h post-E. coli introduction, the percentage of AmpR E. coli in the feces of all mice was at or close to 0.1%, consistent with the gavaged mixture transiting through the GI tract. Within 1 to 2 days, mice on water containing ampicillin exhibited an increase in the percentage of AmpR E. coli by 3 orders of magnitude, reaching complete or near complete colonization (Figure 1A). In contrast, the AmpR subpopulation was lost in mice on water without ampicillin. These results demonstrate that β-lactam antibiotics can be used to select for resistant E. coli in mice.
Figure 1. M13 bacteriophage can deliver a plasmid-borne antibiotic resistance gene to E. coli in the mouse gut.
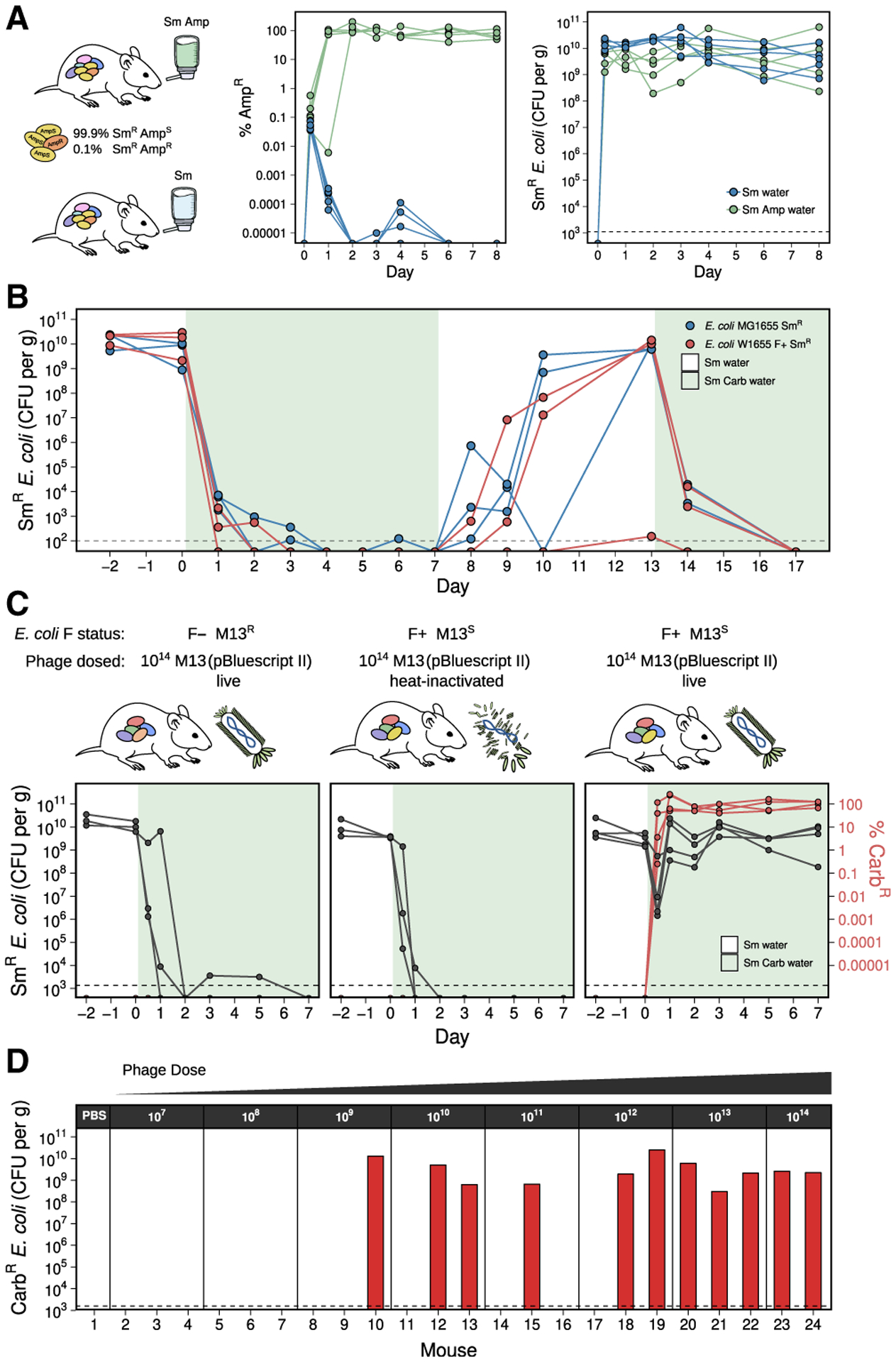
(A) A resistant subpopulation of E. coli can be selected in the gut when a β-lactam antibiotic is provided in the water. Streptomycin (Sm)-treated mice were gavaged with SmR MG1655 containing 99.9% ampicillin-sensitive (AmpS) and 0.1% ampicillin-resistant (AmpR) cells and provided water containing Sm (n = 5) or Sm+Amp (n = 6).
(B) A sensitive E. coli population is unable to maintain colonization in the gut when carbenicillin (Carb) is provided in the water. Mice were colonized with either SmR MG1655 or SmR W1655 F+ (n = 3 each) and Carb was provided in the water (shaded time points).
(C) M13(pBluescript II) can infect F+ E. coli in the gut. Mice were split into 3 groups based on colonization and phage treatment: (1) SmR W1655 F− and live phage (n = 3); (2) SmR W1655 F+ and heat-inactivated phage (n = 3); and (3) SmR W1655 F+ and live phage (n = 4). 1014 phage were dosed, and Carb was provided in the water. Total E. coli (black) and percentage of Carb-resistant (CarbR) colonies (red) in mouse fecal pellets are shown.
(D) M13-based delivery of an antibiotic resistance gene is dose-dependent. Varying doses (107–1014) of M13(pBluescript II) were given tomice (n = 2–3/dose), and Carb was provided in the water. After 2 days, CarbR CFU/gram feces was determined. Dashed line indicates our limit of detection.
See also Figures S1, S2, and S3.
Antibiotics were capable of eradicating a sensitive population of E. coli that had established stable colonization in the mouse gut. We colonized Sm-treated mice with SmR E. coli MG1655 or W1655 F+ and tracked colonization levels during treatment with the β-lactam antibiotic carbenicillin. Carbenicillin decreased the median E. coli colonization level from 9.6 × 109 to 2.0 × 103 colony-forming units (CFU)/gram feces in the first day, and levels decreased to below our limit of detection (~102 CFU/g) in all mice over the course of treatment (Figure 1B). When selection was lifted on day 7, recolonization was observed for 5 out of 6 mice. When carbenicillin was reintroduced on day 13, colonization again dropped below our limit of detection. The low background of E. coli in the gut during carbenicillin treatment, as well as the lack of spontaneous resistant cells able to expand, supports the utility of this model for assessing the phage-mediated delivery of a resistance gene.
Next, we tested our ability to deliver an antibiotic resistance gene to E. coli within the gut. We colonized Sm-treated mice with either SmR E. coli W1655 F+ (M13S) or W1655 F− (M13R as a control) and dosed each animal with either live or heat-inactivated M13 carrying pBluescript II (Figure 1C). After dosing the mice with 1 × 1014 M13(pBluescript II), we immediately transferred them to water containing carbenicillin and tracked both the total E. coli and carbenicillin-resistant (CarbR) E. coli in the feces. E. coli colonization fell rapidly and stayed near or below the limit of detection in control mice that were either colonized with F− and given live phage or colonized with F+ but given heat-inactivated phage. In contrast, mice colonized with F+ and dosed with live phage had a transient drop in colonization on the first day, during which the rise of CarbR cells occurred, and colonization was re-established within 1 day by an E. coli population resistant to carbenicillin (Figure 1C). These results suggest that orally dosed M13 phage were able to infect E. coli in the gut and deliver a plasmid-conferring resistance to carbenicillin.
We replicated M13-mediated pBluescript II delivery to E. coli in the gut in an independent animal experiment. Sm-treated mice were colonized with SmR E. coli W1655 F+ and orally dosed with 10-fold serial dilutions of M13(pBluescript II). Colonization by CarbR E. coli was consistent at high doses but variable at lower doses, representing a significantly higher probability of successful colonization with increasing phage dose (Figure 1D; p = 0.009; odds ratio = 2.5; logistic regression). Plasmid DNA of the expected size was detected in fecal CarbR E. coli isolates from all 11 mice that were successfully colonized (Figure S3A). Genome sequencing confirmed the presence of pBluescript II in these 11 isolates, which was undetectable in the parent strain (Figure S3B). These results indicate that plasmid DNA was transferred from M13 phage into recipient E. coli colonizing the GI tract.
Finally, we repeated this experiment in the absence of carbenicillin selection. We colonized mice with SmR E. coli, gavaged each mouse with M13(pBluescript II), and tracked both infected (CarbR) and total (SmR) E. coli in feces. The fraction of phage-infected CarbR E. coli was low, reaching a maximum of 0.1% of the total population (Figures 2A and 2B), potentially indicative of poor phage survival during GI transit. We gavaged mice with M13(pBluescript II) and assayed for viable phage in the feces. The median output of viable M13(pBluescript II) from feces over a 24 h period was 1 × 106 relative to an input of 6 × 1013 (Figure 2C). M13(pBluescript II) is acid-tolerant in vitro (Figure 2D), suggesting that additional factors may be responsible for the low in vivo viability and emphasizing the benefits of pairing gene delivery with antibiotic selection.
Figure 2. Low frequency of CarbR E. coli following treatment with M13(pBluescript II) in the absence of Carb selection may be due to low survival of M13 in the gut despite high acid tolerance in vitro.
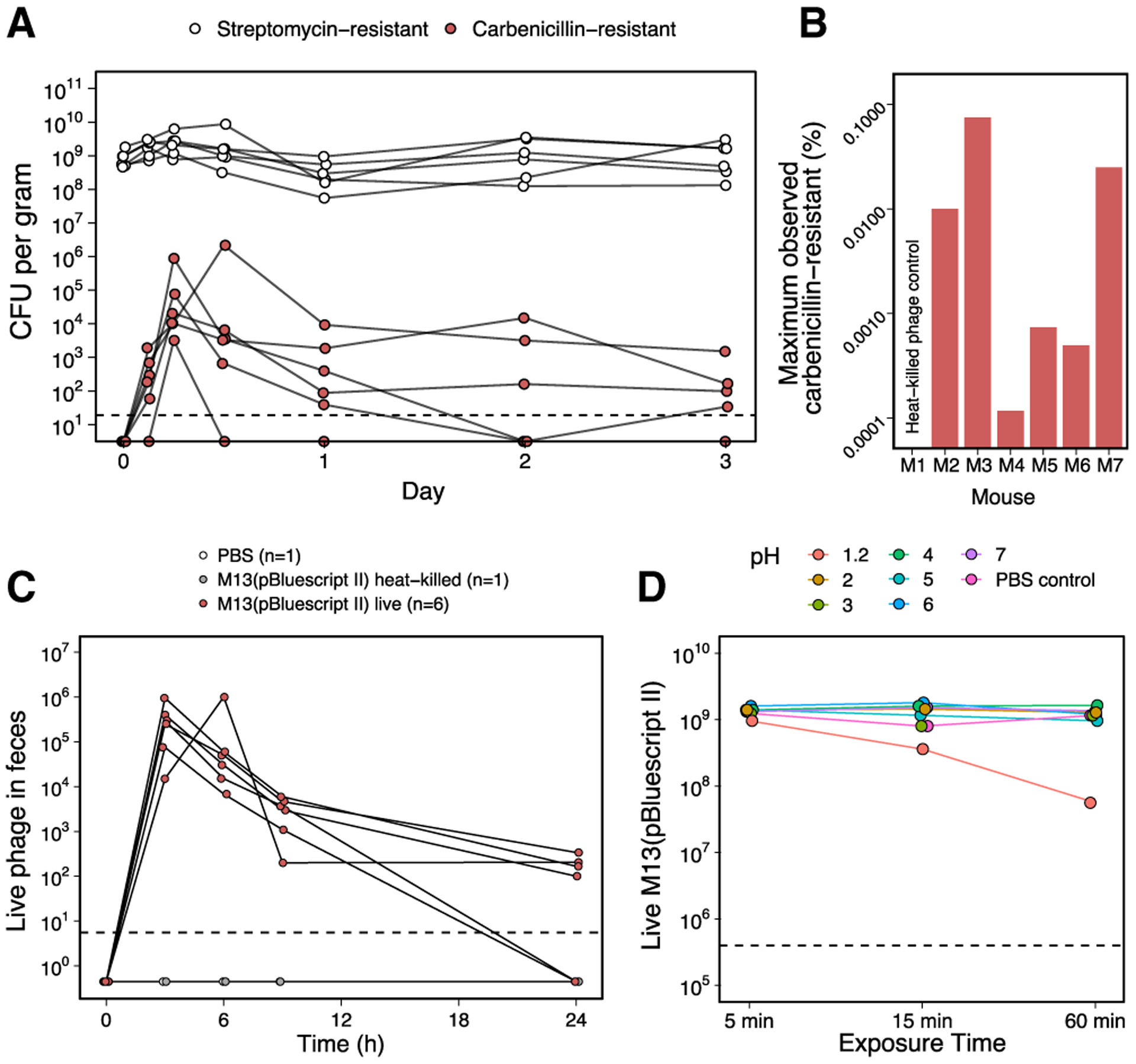
(A) SmR E. coli W1655 F+ was introduced and maintained in Sm-treated mice (n = 6). Oral gavage of 1013 M13(pBluescript II) was performed on day 0, but contrary to Figure 1C, Carb was not added to the drinking water to select for phagemid delivery. SmR and CarbR CFUs were enumerated up to 3 days post-gavage. SmR CFUs indicative of total E. coli remained steady, while CarbR CFUs indicative of phage-infected cells were much lower in number, peaked between 6 and 12 h after gavage, and decreased over time. Dashed line indicates limit of detection.
(B) The maximum observed percentage of CarbR CFUs (phage-infected) over SmR CFUs (total) was approximately 0.1%. For each of the 6 mice in (A), the maximum percentage of CarbR/SmR was calculated, with values ranging over 3 orders of magnitude between ~0.0001% and ~0.1%. Time-series fecal samples from a mouse orally gavaged with heat-killed phage were also assayed as a negative control.
(C) Enumeration of phage M13(pBluescript II) in feces of conventionally raised mice after oral gavage. Mice were treated with 1013 M13(pBluescript II) (n = 6) or as negative controls, heat-killed M13(pBluescript II), or PBS. Live phage in fecal samples were assayed using indicator strain XL1-Blue MRF’ at t = 0, 3, 6, 9, and 24 h post-gavage. Using the median live phage output in the feces at each time point for the 6 mice gavaged with live M13(pBluescript II), the area under the curve from 0 to 24 h is 1 × 106.
(D) Phage M13 displays resistance to acidic conditions as extreme as pH 2. 109 M13(pBluescript II) were incubated in pH solutions 1.2 to 7 and sampled over the course of 60 min to assay for viability. Dashed line indicates limit of detection.
M13 carrying CRISPR-Cas9 can target E. coli in vitro
We generated 2 fluorescently marked isogenic derivatives of SmR W1655 F+ using the mcherry (red fluorescence) or the sfgfp (green fluorescence) marker gene. Next, we constructed M13-compatible non-targeting (NT) and GFP-targeting (GFPT) CRISPR-Cas9 vectors by cloning the spacers sequences, bla gene, and f1 origin of replication into the previously described low-copy vector pCas9 (Jiang et al., 2013), generating pCas9-NT-f1A/B and pCas9-GFPT-f1A/B (Figure S4A). The bla and f1 ori were cloned as a fragment from pBluescript II in both possible orientations (A or B) to make possible M13 ssDNA packaging of either strand of vector DNA. We packaged these phagemids into M13 using a helper strain and called the resulting phage NT-M13 or GFPTM13. The 2 phages were used to infect the GFP+ or mCherry+ strains, and cells were diluted and spotted on solid media containing carbenicillin to select for the transferred phagemid. GFP+ E. coli infected with GFPT-M13 exhibited impaired colony growth relative to the NT-M13 control (Figure 3A; Figure S4B). Total CFUs were not markedly affected, indicating that cells can recover from M13-delivered CRISPR-Cas9 targeting.
Figure 3. M13-mediated delivery of CRISPR-Cas9 to E. coli in vitro causes impaired colony growth and can induce chromosomal deletions that encompass the targeted gene.
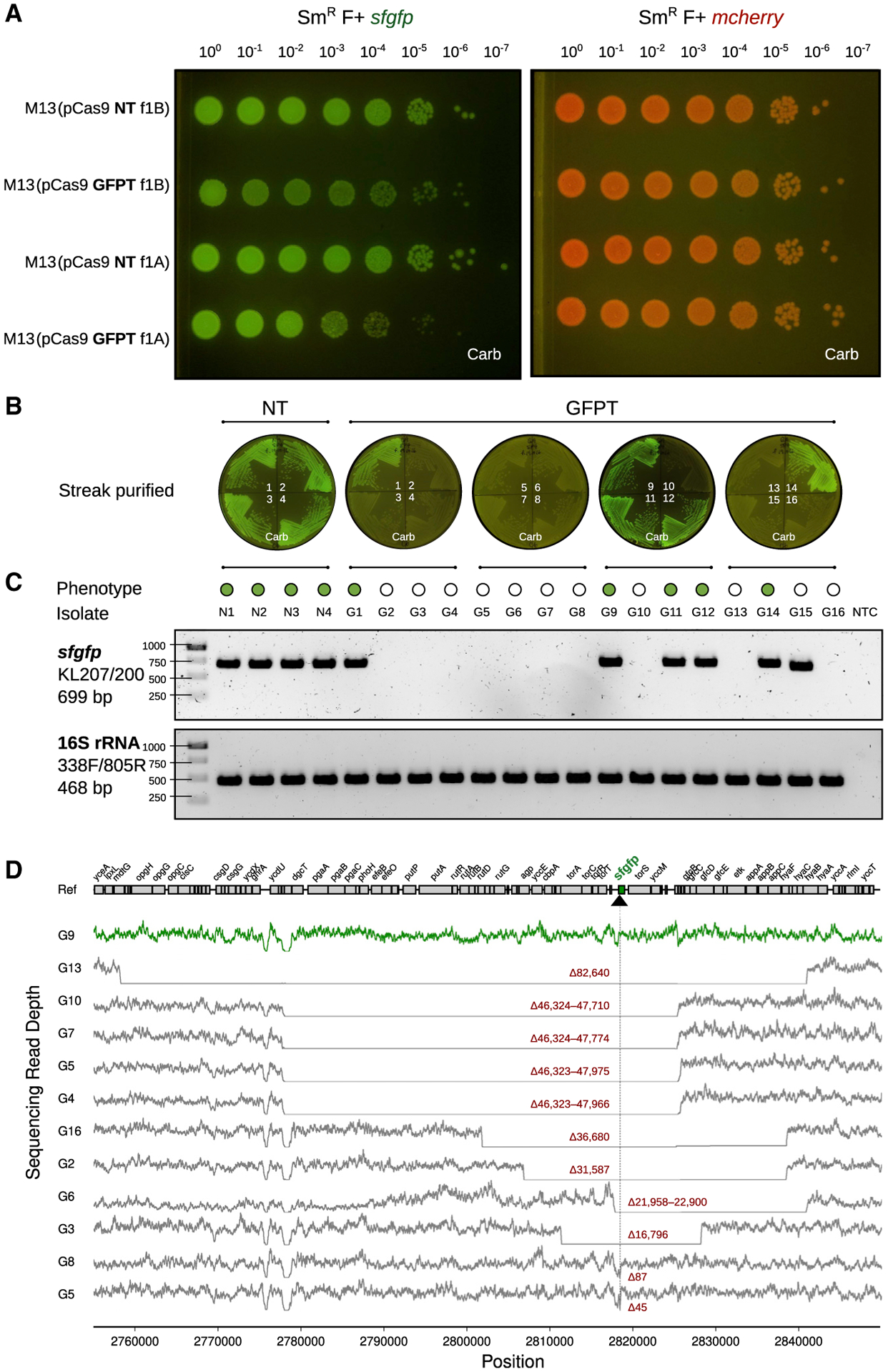
(A)GFP+E. coliexhibit a sick colony morphologyafter infection with M13 phage carrying GFP-targeting (GFPT) CRISPR-Cas9. NT (non-targeting) or GFPT M13 were used to infect SmR W1655 F+ sfgfp or SmR W1655 F+ mcherry as a control. Cells were infected, diluted, and spotted onto media with selection for the vector; f1A or f1B indicates vector version.
(B) CRISPR-Cas9 targeting the sfgfp gene can induce loss of fluorescence. Colonies arising from infection with NT-M13 or GFPT-M13 were subjected to several rounds of streak purification on selective media to ensure phenotypic homogeneity and clonality. The majority (11/16) of GFPT clones exhibited a loss of fluorescence.
(C) Clones exhibiting loss of fluorescence either lack an sfgfp PCR amplicon or exhibit an amplicon of decreased size. Genomic DNA was isolated from streak-purified clones, and PCR was used to determine whether the sfgfp gene was present. PCR for the 16S rRNA gene was performed as a positive control.
(D) Genome-sequencing results confirm that non-fluorescent clones have chromosomal deletions encompassing the targeted gene. Read depth surrounding sfgfp locus for a fluorescent control clone G9 (green line) and all non-fluorescent clones (gray lines). Deletion size is indicated in red; range indicates a deletion flanked by repetitive sequences. Black arrow and vertical line denote position of targeting. See also Figure S4.
Analysis of the surviving cells provided mechanistic insights. Colonies arising from infection with NT-M13 or GFPT-M13 were streak purified, allowing us to pick a mixture of bright and dim colonies. Of 16 GFPT clones analyzed, 11 were non-fluores-cent (Figure 3B). PCR amplification of sfgfp confirmed the intact gene in 4 NT controls and all 5 GFPT clones that retained fluorescence (Figure 3C). Sanger sequencing revealed that 1 GFPT clone had a point mutation in the sfgfp target (Figure S4C), while the 4 others had lost the spacer in the CRISPR-Cas9 phagemid that leads to targeting (Figure S4D). All 11 non-fluorescent GFPT clones retained the spacer (Figure S4D) and had chromosomal deletions at the target locus: 10 were PCR-negative (Figure 3C), and 1 had a small deletion within sfgfp (Figure S4C). Finally, we used whole-genome sequencing to define the size of each deletion, which ranged from 45 bp to 82.6 kb (Figure 3D), consistent with prior work demonstrating that E. coli can repair Cas9-induced double-stranded breaks through homologous recombination (Cui and Bikard, 2016).
These results led us to hypothesize that targeted cells would be less able to recover during competitive growth. We co-cultured GFP+ and mCherry+ E. coli, adding either NT-M13 or GFPT-M13 followed by carbenicillin to select for DNA delivery. GFPT-M13 decreased the frequency of GFP+ colonies by 4 h, relative to the NT-M13 control (Figure 4A). At later time points (16–24 h), healthy GFP+ colonies increased in abundance, consistent with low levels of carbenicillin after 4 h in cultures expressing the β-lactamase resistance gene (Figure 4B). We confirmed the loss of selection for the phagemid by analysis on selective media (Figure S5A). Next, we used flow cytometry to better quantify the 2 strains in an independent experiment. Compared to the NT-M13 control, GFPT-M13 co-cultures exhibited fewer GFP+ events (Figure 4C) and a bimodal distribution of fluorescence (Figure 4D). Counts of GFP+ cells were higher by flow cytometry than on solid media for the same co-cultures (Figure 4D, inset), consistent with an impaired growth of these cells. GFP+ events further decreased at 24 h in the GFPT-M13 group (Figure S5B). Taken together, these results suggest that competitive growth can increase the efficiency of targeting a strain for depletion due to the resulting growth impairments in the targeted strain.
Figure 4. M13-delivered CRISPR-Cas9 for sequence-specific targeting of E. coli during the in vitro co-culture of fluorescently marked isogenic strains.
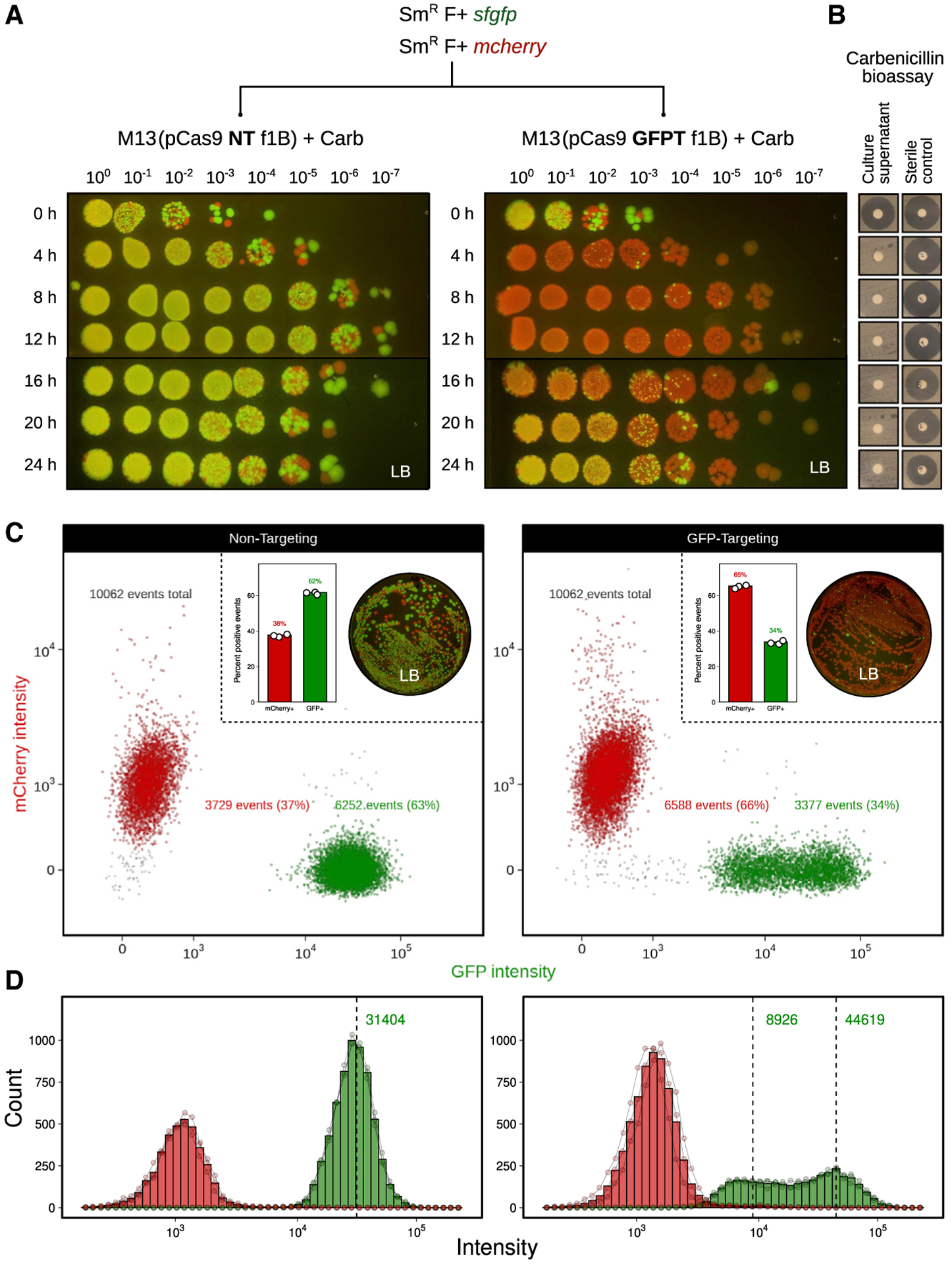
(A) M13-delivered GFPT CRISPR-Cas9 leads to reduced competitive fitness of the GFP-marked strain. A co-culture of SmR F+ sfgfp and SmR F+ mcherry was incubated with NT-M13 or GFPT-M13 at a starting MOI of ~500. Carb was added to a final concentration of 100 μg/mL to select for phage infection. Co-cultures were sampled every 4 h over 24 h; cells were washed, serially diluted, and spotted onto non-selective media to assess targeting of the GFP-marked strain.
(B) Carb in culture supernatants was not detectable within 4 h of growth using a Carb bioassay against indicator strain Bacillus subtilis 168; bioassay detection limit approximately 2.5 μg/mL.
(C) Flow cytometry of co-cultures 8 h following the addition of phage and Carb show reduced GFP+ events in the GFPT versus NT condition. Representative flow plots show data from 1 of 3 biological replicates. Inset: bar graph quantifying percentage of GFP+ and mCherry+ events for 3 replicates (left); plating results for a single replicate on non-selective media (right).
(D) GFPT CRISPR-Cas9 changes the shape of the distribution of GFP+ population. Histogram of mCherry+ and GFP+ events by intensity shows that a proportion of GFP+ cells in the GFPT condition have shifted to a state of lower fluorescence. Bars indicate the mean of 3 biological replicates; connected points are individual replicates.
See also Figure S5.
Sequence-specific depletion of E. coli within the mouse gut microbiota
We co-colonized Sm-treated mice with both SmR F+ sfgfp and SmR F+ mcherry strains, orally dosed them with either 1011 NT-M13 or GFPT-M13 and added carbenicillin in the water to select for DNA delivery. After 1 week, carbenicillin was removed from the water, and mice were followed for an additional week to determine whether phagemid-induced changes would persist in the absence of maintaining selection (Figure 5A). Flow cytometry on mouse stool samples revealed that the GFP+ strain outcompeted the mCherry+ strain in the NT-M13 group (Figures 5B and 5C; Figures S6A and S6B). In contrast, GFP+ events in the GFPT-M13 group exhibited a sharp decrease on day 2, followed by a recovery on days 7 and 14 to levels below the NT-M13 group (Figures 5B and 5C). Culturing from mouse stool confirmed the decreased GFP+ events on day 2 (Figures S6C–S6E). In 4 out of 10 mice that received GFPT-M13, the mCherry+ strain fixed in the population (GFP+ events were below background), an outcome that was not observed for any mouse in the NT-M13 group (Figure 5D). Despite lifting the carbenicillin selection for 1 week, endpoint GFP+ events remained significantly lower in the GFPT-M13 group relative to NT-M13 controls (Figure 5E; p = 0.0002; Mann-Whitney test). These data support the utility of M13-delivered CRISPR-Cas9 for sequence-specific depletion of an otherwise isogenic bacterial strain in the mouse gut.
Figure 5. M13-delivered CRISPR-Cas9 for sequence-specific depletion of E. coli in the gut of mice colonized by competing fluorescently marked isogenic strains.
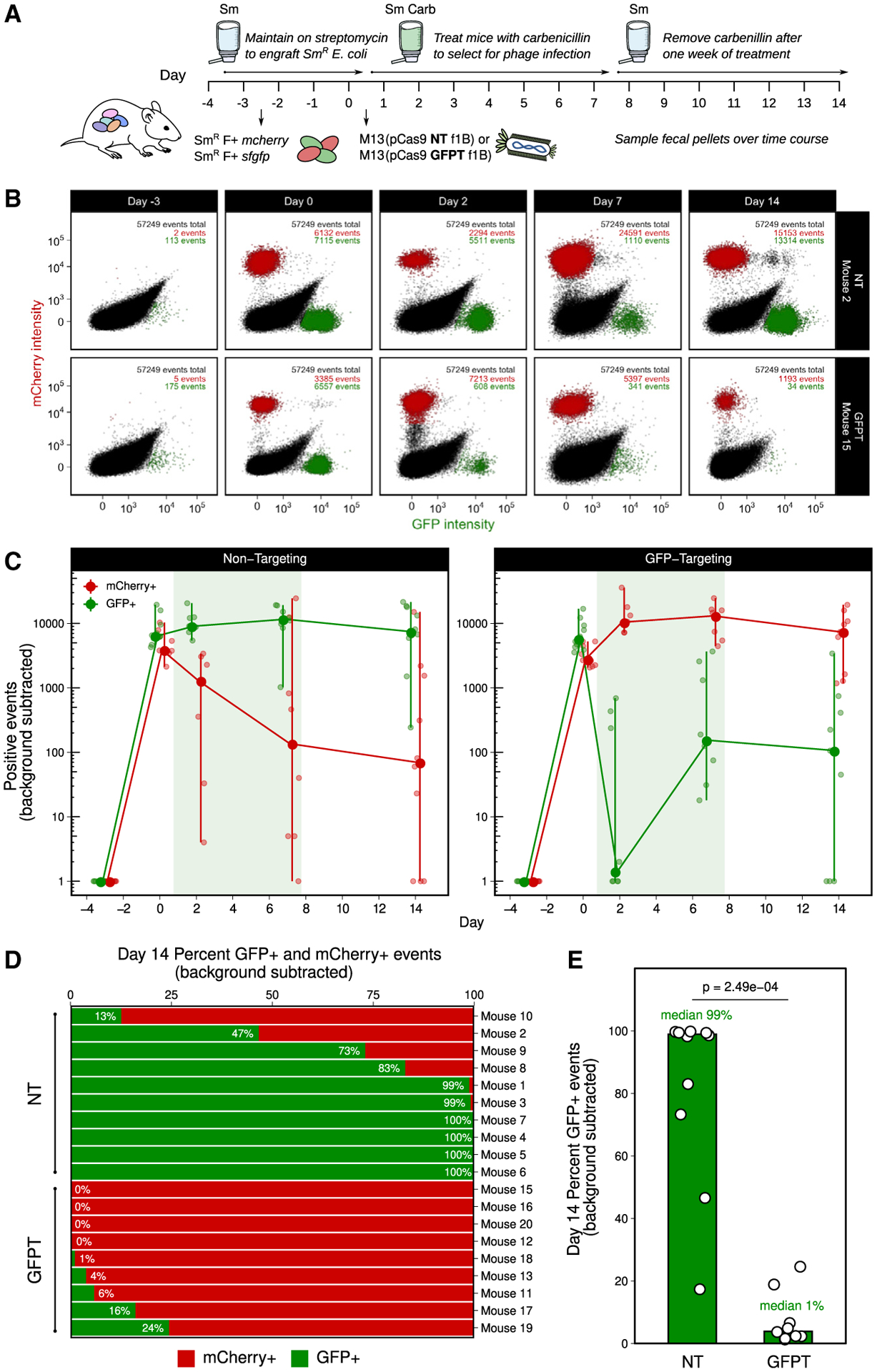
(A) Timeline: day −3, colonize with 50/50 mixture of SmR F+ sfgfp and SmR F+ mcherry; day 0, dose with 1011 NT-M13 or GFPT-M13 (n = 10/group), and provide Carb in the water; day 7, remove Carb.
(B) GFPT-M13 can lead to loss of the GFP-marked strain. Time series flow plots of fecal samples for 1 mouse from each of NT and GFPT groups. Top right: number of total, mCherry+, and GFP+ events.
(C) Mice in GFPT group exhibited a decrease in number of fecal GFP+ events over time compared to the NT group; time points were excluded if both GFP+ and mCherry+ events were below background thresholds. Line graph: large points indicate median; vertical lines, range.
(D) Mice in GFPT group exhibited depletion or loss of the GFP-marked strain. Percentage of GFP+ and mCherry+ events for each mouse on day 14. Mice were excluded if both GFP+ and mCherry+ events were both below background thresholds (final n = 9 GFPT and n = 10 NT).
(E) A significant difference was observed in the percentage of GFP+ events in fecal samples at day 14 in the GFPT group compared to NT. Bars are medians; p value, Mann-Whitney test.
See also Figure S6.
M13-delivered CRISPR-Cas9 induces chromosomal deletions in the gut microbiome
We constructed a double-marked SmR F+ sfgfp mcherry strain to quantify the efficiency of gene deletion. We introduced this strain into Sm-treated mice, orally dosed each mouse with either 1011 NT-M13 or GFPT-M13, and added carbenicillin in the water. After 1 week, we removed carbenicillin and followed the mice for a final week (Figure 6A). GFP− mCherry+ events were detectable in GFPT-M13 but not NT-M13 mice, indicative of successful CRISPR-Cas9 delivery and gene loss (Figure 6B; Figure S7A). By the final time point, GFP− mCherry+ events were detected in 3 out of 8 mice (Figure 6C; Figure S7B). The relative abundance of GFP− mCherry+ cells varied from 12% to 96% (Figure 6C). Culturing on solid media confirmed the presence of viable red fluorescent colonies in proportions consistent with flow cytometry results (Figure 6D; Figure S7C).
Figure 6. M13-delivered CRISPR-Cas9 can induce chromosomal deletions encompassing the targeted gene in E. coli colonizing the mouse gut.
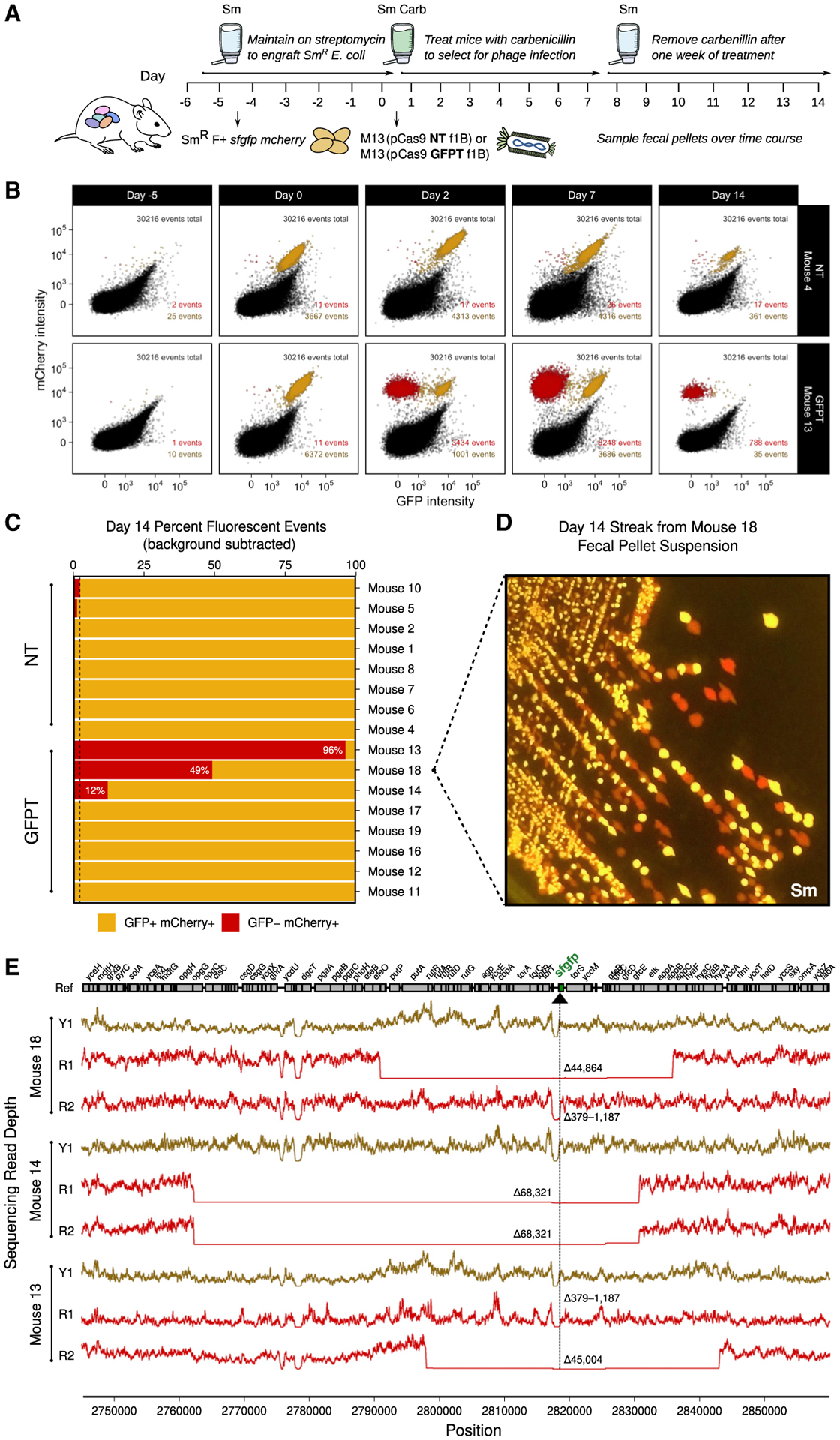
(A) Timeline: day −5, colonize with double-marked SmR F+ sfgfp mcherry; day 0, dose with 1011 NT-M13 or GFPT-M13 (n = 10/group), and provide Carb in the water; day 7, remove Carb.
(B) GFPT-M13 can cause loss of GFP fluorescence in double-marked E. coli. Time series flow plots of fecal samples for select mice, 1 from each of NT and GFPT groups. Top right: total number of events; bottom right: GFP− mCherry+ events and GFP+ mCherry+ events.
(C) Day 14 fecal samples of 3 mice in GFPT group exhibited mCherry-only fluorescence. Percentage of GFP+ mCherry+ and GFP− mCherry+ events for each mouse; mice were excluded if both populations were below the background threshold (final n = 8/group). Dashed line indicates maximum mCherry fluorescence for NT group.
(D) Colonies arising from culture of mouse 18 day 14 fecal sample confirmed presence of red-only fluorescence.
(E) Genome-sequencing results confirm red fluorescent isolates from mouse 13, 14, and 18 have chromosomal deletions encompassing the targeted gene. Read depth surrounding sfgfp locus for GFP+ mCherry+ (Y1; yellow lines) and GFP− mCherry+ fluorescent (R1 or R2; red lines) isolates from day 2 fecal samples. Deletion size indicated in red; range indicates a deletion flanked by repetitive sequences. Black arrow and vertical line denote position of targeting.
See also Figure S7.
To more definitively assess the presence or absence of the targeted genomic locus and the CRISPR-Cas system, we isolated GFP− mCherry+ as well as GFP+ mCherry+ E. coli from day-2 mouse stool. All of the GFP+ mCherry+ isolates from the NT-M13 group and the GFP− mCherry+ isolates from the GFPT-M13 group had an intact spacer sequence (Figure 7A). In contrast, 4 out of 5 GFP+ mCherry+ isolates from the GFPT-M13 group had lost the spacer (Figure 7A). Of note, the remaining isolate lost a larger fragment of the phagemid corresponding to cas9 and parts of the CRISPR array and tracrRNA (Figures 7B and 7C). Whole-genome sequencing was used to confirm putative chromosomal deletions and to quantify their size. 2 representative colonies were analyzed from each of the 3 mice with detectable GFP− mCherry+ cells, revealing a wide range in deletion sizes that were not observed in a control GFP+ mCherry+ isolate from each animal (Figure 6E). These results indicate that while it is possible for CRISPR-Cas9-induced genomic deletion events to occur in vivo, resultant deletion strains may or may not outcompete the parent strain due to the potential to evade targeting through loss of some or all of the exogenous CRISPR-Cas system.
Figure 7. Mechanisms enabling GFP+ mCherry+ E. coli to escape CRISPR-Cas9 targeting.
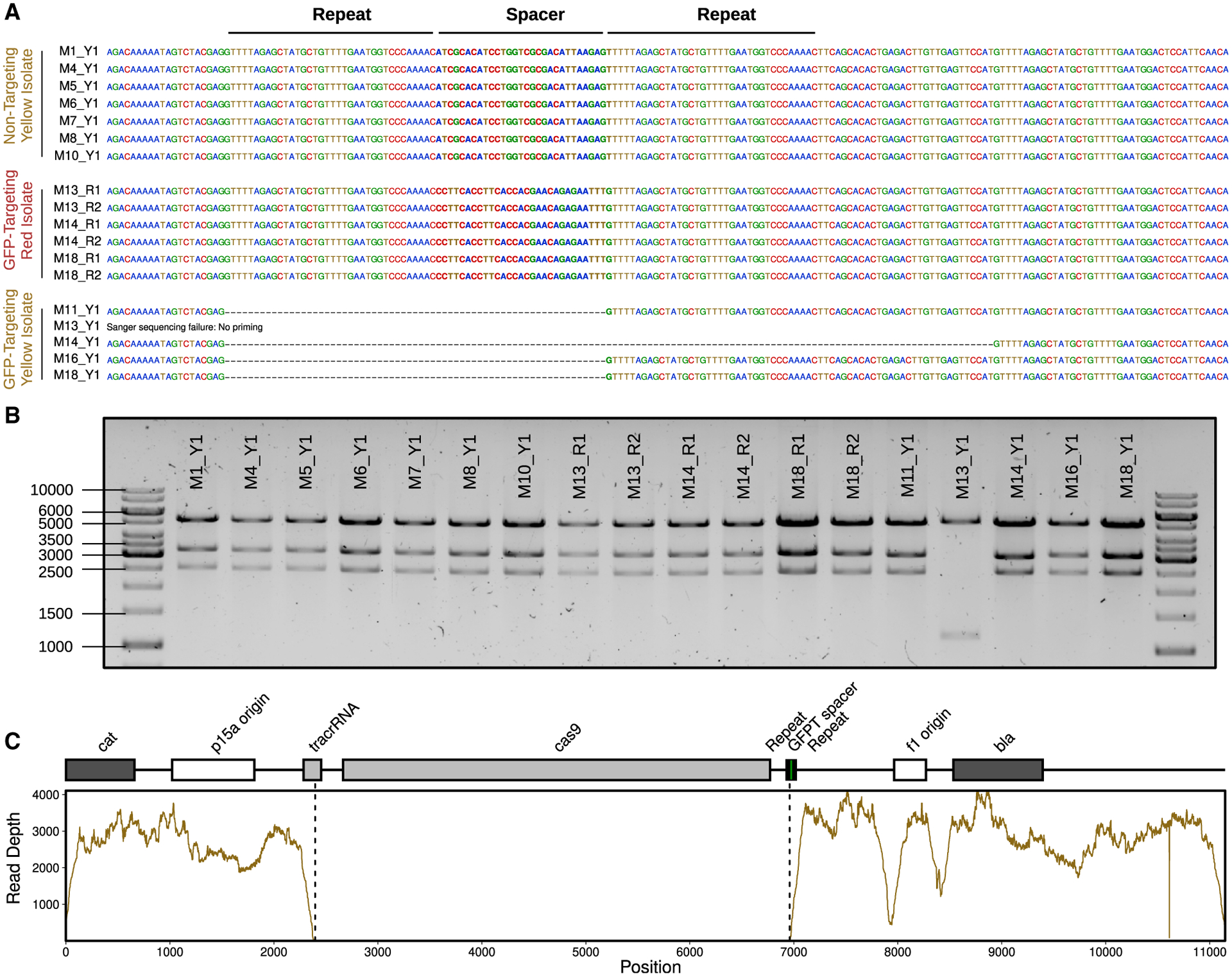
(A) Sanger-sequencing results confirm the expected spacer present in phagemid DNA extracted from fluorescent yellow isolates (Y1) colonizing NT mice (M1, M4, M5, M6, M7, M8, and M10) and fluorescent red isolates (R1 and R2) colonizing GFPT mice (M13, M14, and M18). In contrast, 4 out of 5 fluorescent yellow isolates colonizing GFPT mice (M11, M13, M14, M16, and M18) were confirmed to have lost the spacer. No Sanger-sequence data were obtained for the last isolate (M13) with the report for failing being “no priming,” suggesting loss of a larger fragment from the phagemid.
(B) Diagnostic digest of CRISPR-Cas9 phagemid DNA indicates loss of a portion of phagemid DNA for the phagemid extracted from M13 Y1. Expected fragment sizes from KpnI-XbaI double digest: 5,289, 3,285, and 2,573 bp.
(C) Genome-sequencing data for M13 Y1 confirms loss of DNA from phagemid. Sequencing coverage across the GFPT phagemid reveals lack of reads corresponding to the cas9 gene and parts of the CRISPR array and tracrRNA.
DISCUSSION
Our results emphasize that foundational, reductionist, and highly controlled studies will be necessary to assess the feasibility, utility, and limitations of phage-based gene delivery as a tool for microbiome editing. While our results provide a proof-of-principle for strain-specific targeting within the GI tract, the full eradication of the targeted strain was difficult to achieve due to the ability of bacterial cells to survive Cas9-induced double-stranded breaks by homologous recombination (Dillingham and Kowalczykowski, 2008). We propose that CRISPR-Cas9 may be better suited to induce targeted genomic deletions, leveraging the conserved DNA repair pathways present in bacteria. An advantage of this approach is that the deletion of a single genomic locus is unlikely to have as large an impact on the rest of the gut microbiota than if the strain were to be removed entirely. Remarkably, we detected a wide range of deletion sizes (379–68,321 bp), highlighting the ability of bacteria to survive large deletions and opening up the potential for the in vivo removal of entire biosynthetic gene clusters or pathogenicity islands. In turn, our data suggest that it may also be feasible to deliver more complex genetic circuits to E. coli, with the goal of boosting metabolic pathways beneficial to its mammalian host or altering immune function. Furthermore, the size of resulting chromosomal deletions could be controlled by providing a DNA-repair template alongside CRISPR-Cas9 (Jiang et al., 2013).
This work provides a valuable step toward establishing a modular toolkit for microbiome editing. The extension of these approaches to enable the genetic manipulation of a more diverse panel of bacteria found within the human microbiota will require a renewed effort to isolate and characterize bacteriophages that target strains of interest (Soto-Perez et al., 2019). Robust in vitro methods to study and genetically modify novel bacteriophage are also needed, given that most of their host bacteria remain genetically intractable (Bisanz et al., 2020). Finally, our work emphasizes the value of model systems for understanding the rules of engagement, complementing ongoing efforts to advance candidate phage-based therapies into the clinic.
Limitations of the study
There are several limitations in our current approach that could be refined through iterative “design-build-test” cycles. Perhaps most importantly, we identified multiple mechanisms through which cells can escape CRISPR-Cas9 targeting during in vitro growth and/or within the GI tract, including the loss of the spacer in the CRISPR array, target site mutations, and even one case in which the entire CRISPR-Cas9 system was deleted. Spacer loss could be reduced through the use of single guide RNAs (sgRNAs) (Guo et al., 2019), mutation of the repeat sequences that flank each spacer (Csörgő et al., 2020), or by incorporating multiple spacers. Target-site mutation could be countered by targeting multiple sites simultaneously or by prioritizing conserved regions of the target genes essential for activity. Our system provides a tool for the robust design and validation of putative spacers and sgRNAs in vivo.
Another important caveat is that antibiotics were used to select for successful gene delivery, which is not ideal due to potential disruptions to the gut microbiota (Basolo et al., 2020) and/or selection for antibiotic resistance. Removing this selection resulted in a low penetrance of cells carrying the delivered cargo, similar to that observed for delivery to bacteria in a soil community in the absence of selection (Rubin et al., 2020). Low rates of gene delivery may be due to the lower adsorption rate of M13 compared to other phages with a greater number of host binding sites (Kasman et al., 2002) but are likely driven by the observed loss of viable M13 bacteriophage during transit through the GI tract. We confirmed that M13 is acid tolerant; however, it is sensitive to artificial gastric juice (Tóthová et al., 2012), suggesting that gastric proteases may be responsible for low phage survival. The aggregative behavior of filamentous phage in response to microbial and/or host polymers (Secor et al., 2015) may also be important to consider in addition to potential differences in bacterial physiology that could disrupt targeting (Porter et al., 2020). Encapsulating phage for oral transit may be able to circumvent these limitations or alternatively, it may be possible to use non-antibiotic selection strategies such as dietary supplementation of an exclusive substrate (Shepherd et al., 2018) that can only be used by phagemid recipients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Turnbaugh (peter.turnbaugh@ucsf.edu).
Materials availability
Plasmids or strains generated in this study will be made available upon request.
Data and code availability
Sequence data have been deposited at the NCBI Sequence Read Archive under NCBI BioProject: PRJNA642411 and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. Additional supporting data is publicly available at https://github.com/turnbaughlab/2021_Lam_M13_CRISPRCas9.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| MG1655 (Derivative of K-12) | ATCC | ATCC 700926 |
| DH5α (Routine cloning; phage propagation with helper HP4_M13) | Thermo Fisher | Thermo Fisher 18265017 |
| NEB 5-alpha (Routine cloning) | NEB | NEB C2987H |
| XL1-Blue MRF’ (Phage propagation with helper M13KO7; TcR) | Agilent | Agilent 212208 |
| MG1655 rpsL-SmR (Spontaneous rpsL-SmR (Lys42Arg) derivative of MG1655) | This study | KL52 |
| W1655 F− (Derivative of K-12; M13R) | ATCC | ATCC 23737 |
| W1655 F+ (Derivative of K-12; M13S) | ATCC | ATCC 23590; KL68 |
| W1655 F− rpsL-SmR (Recombineered rpsL-SmR (Lys42Arg) derivative of W1655 F−) | This study | KL89 |
| W1655 F+ rpsL-SmR (Recombineered rpsL-SmR (Lys42Arg) derivative of W1655 F+) | This study | KL90 |
| AV01::pAV01 (MG1655 with constitutive sfgfp clonetegrated at HK022 att site; KmR) | Vigouroux et al., 2018 | AV01::pAV01; KL106 |
| AV01::pAV02 (MG1655 with constitutive mcherry clonetegrated at lambda att site; KmR) | Vigouroux et al., 2018 | AV01::pAV02; KL107 |
| W1655 F+ rpsL-SmR sfgfp (W1655 F+ rpsL-SmR with sfgfp transduced from AV01::pAV01; KmS) | This study | KL114 |
| W1655 F+ rpsL-SmR mcherry (W1655 F+ rpsL-SmR with mcherry transduced from AV01::pAV02; KmS) | This study | KL115 |
| W1655 F+ rpsL-SmR sfgfp mcherry (W1655 F+ rpsL-SmR sfgfp with mcherry transduced from AV01::pAV02; KmS) | This study | KL204 |
| Bacteriophage: M13KO7 (helper phage, KmR) | NEB | NEB N0315S |
| Bacteriophage: VCSM13 (helper phage, KmR) | Agilent | Agilent 200251 |
| Bacteriophage: P1 (transducing phage) | ATCC | ATCC 25404-B1 |
| Chemicals, peptides, and recombinant proteins | ||
| MacConkey Agar | Thermo Fisher | Cat#212123 |
| USP-grade streptomycin sulfate | VWR | Cat#0382 |
| USP-grade ampicillin sodium salt | Teknova | Cat#A9510 |
| USP-grade carbenicillin disodium salt | Teknova | Cat#C2110 |
| Critical commercial assays | ||
| ZymoBIOMICS 96 MagBead DNA Kit | Zymo | Cat#D4302 |
| SequelPrep Normalization Plate Kit | Life Tech | Cat#A10510-01 |
| KAPA Library Quantification Kit | KAPA | Cat#KK4824 |
| Ligation Sequencing Kit | Oxford Nanopore | Cat#SQK-LSK109 |
| Native Barcoding Kit | Oxford Nanopore | Cat#EXP-NBD114 |
| Deposited data | ||
| Sequencing data repository | This paper | NCBI BioProject: PRJNA642411 |
| 16S rRNA gene sequencing data | This paper | NCBI SRA: SRR12118792-SRR12118959 |
| Isolates from mouse fecal samples after delivery of pBluescript II | This paper | NCBI SRA: SRR14278062-SRR14278073 |
| Illumina and Nanopore data for the hybrid assembly of reference strain KL68 (W1655 F+ or ATCC 23590) as well as SmR fluorescent derivatives KL114 (sfgfp) and KL204 (sfgfp mcherry) | This paper | NCBI SRA: SRR14296642-SRR14296644; SRR14297452-SRR14297454 |
| Sequencing of isolates after targeting with phage-delivered CRISPR-Cas9 | This paper | NCBI SRA: SRR14289086-SRR14289109 |
| Experimental models: Organisms/strains | ||
| BALB/c mice | Taconic | Taconic BALB-F MPF |
| Oligonucleotides | ||
| See Table S1 | See Table S1 | See Table S1 |
| Recombinant DNA | ||
| Plasmid: pBluescript II KS(−) (Commercial phagemid; CarbR) | Alting-Mees and Short, 1989 | Agilent 212208 |
| Plasmid: pSIJ8 (Temperature-sensitive; lambda Red recombineering; CarbR) | Jensen et al., 2015 | RRID: Addgene_68122 |
| Plasmid: pE-FLP (Temperature sensitive; constitutive flippase expression; CarbR) | St-Pierre et al., 2013 | RRID: Addgene_45978 |
| Plasmid: pCas9 (Low-copy vector carrying cas9, tracrRNA, and CRISPR array; CmR) | Jiang et al., 2013 | RRID: Addgene_42876 |
| Plasmid: HP4_M13 (helper plasmid, KmR) | Praetorius et al., 2017 | RRID: Addgene_120340 |
| Plasmid: pCas9-GFPT-f1A (pCas9 with GFP-targeting spacer; f1-bla in orientation A; CmR CarbR) | This study | pCas9-GFPT-f1A; pKL100 |
| Plasmid: pCas9-GFPT-f1B (pCas9 with GFP-targeting spacer; f1-bla in orientation B; CmR CarbR) | This study | pCas9-GFPT-f1B; pKL101 |
| Plasmid: pCas9-NT-f1A (pCas9 with Non-targeting spacer; f1-bla in orientation A; CmR CarbR) | This study | pCas9-NT-f1A; pKL102 |
| Plasmid: pCas9-NT-f1B (pCas9 with Non-targeting spacer; f1-bla in orientation B; CmR CarbR) | This study | pCas9-NT-f1B; pKL103 |
| Software and algorithms | ||
| QIIME2 | Bolyen et al., 2019 | https://qiime2.org/ |
| DADA2 | Callahan et al., 2016 | https://benjjneb.github.io/dada2/ |
| phyloseq | McMurdie and Holmes, 2013 | https://github.com/joey711/phyloseq |
| phylosmith | Smith, 2019 | https://github.com/schuyler-smith/phylosmith |
| flowCore | Hahne et al., 2009 | https://github.com/RGLab/flowCore |
| Phenoflow | Props et al., 2016 | https://github.com/rprops/PhenoFlow |
| ggcyto | Van et al., 2018 | https://github.com/RGLab/ggcyto |
| Guppy | Oxford Nanopore Technologies | https://nanoporetech.com/community |
| qcat | Oxford Nanopore Technologies | https://github.com/nanoporetech/qcat |
| porechop | Wick et al., 2017a | https://github.com/rrwick/Porechop |
| NanoFilt | De Coster et al., 2018 | https://github.com/wdecoster/nanofilt |
| fastp | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| Unicycler | Wick et al., 2017b | https://github.com/rrwick/Unicycler |
| bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| breseq | Deatherage and Barrick, 2014 | https://github.com/barricklab/breseq |
| samtools | Li et al., 2009 | https://github.com/samtools/samtools |
| Other | ||
| Resource website for supporting data | This paper | https://dx.doi.org/10.5281/zenodo.5517961 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animal procedures were approved by the University of California, San Francisco (UCSF) Institutional Animal Care and Use Committee (IACUC), and animal experiments performed were in compliance with ethical regulations. Specific-pathogen-free 6–10 week old female BALB/c mice from the vendor Taconic were used for all mouse experiments. Mice that arrived co-housed were distributed equally across experimental groups. Mice were orally gavaged with known doses of phage and provided food and antibiotic water ad libitum. All phage experiments were carried out using singly-housed mice to reduce the risk of cross-contamination of modified bacterial strains.
Microorganisms
E. coli strains, plasmids, and phage used in this study, including descriptions of relevant characteristics, are provided in the Key Resources Table (TcR, tetracycline-resistant; SmR, streptomycin-resistant; KmR, kanamycin-resistant; KmS, kanamycin-sensitive; M13R, M13-resistant; M13S, M13-sensitive; CarbR, carbenicillin-resistant; CmR, chloramphenicol-resistant). E. coli strains were routinely cultured in liquid using lysogeny broth (LB) or terrific broth (TB) and on solid media using LB or MacConkey agar at 37°C; strains harboring a temperature-sensitive plasmid were cultured at 30°C. M13 phage carrying phagemid DNA were generated using helper phage M13KO7 or VCSM13, or helper plasmid HP4_M13, and were stored in PBS at −80°C with added glycerol as cryoprotectant. P1 phage lysates for transduction were generated using plate lysates and stored at 4°C.
METHOD DETAILS
Oligonucleotides
Oligonucleotides used in this study are listed in Table S1.
Minimum inhibitory concentration (MIC) assay
Cells were prepared by standardizing an overnight culture to an OD600 of 0.1 using saline (0.85% NaCl), and further diluted ten-fold in saline then ten-fold in LB. The drug was prepared by dissolving the antibiotic in vehicle (sterile distilled water) and filter-sterilizing, then serially diluting two-fold in vehicle to prepare 100 × stock solutions, and finally diluting ten-fold in LB for 10 × stock. To wells of a 96-well plate, 60 μl of LB, 15 μl of drug, and 75 μl of cells were added and mixed well. Final drug concentrations ranged between 0.002 μg/ml to 1000 μg/ml for ampicillin and 0.24 μg/ml to 2000 μg/ml for carbenicillin. The plate was incubated overnight at 37°C without shaking and OD600 was measured the following morning after agitation.
16S rRNA gene sequencing
Mouse fecal pellets were stored at −80°C. DNA was extracted from single pellets using a ZymoBIOMICS 96 MagBead DNA Kit (Zymo D4302) and 16S rRNA gene sequencing was performed using a dual indexing strategy (Gohl et al., 2016). Briefly, a 22-cycle primary PCR was performed using KAPA HiFi Hot Start DNA polymerase (KAPA KK2502) and V4 515F/806R Nextera primers. The reaction was diluted in UltraPure DNase/RNase-free water (Life Tech 0977–023) and used as template for a 10-cycle secondary (indexing) PCR using sample-specific dual indexing primers. The reactions were normalized using a SequelPrep Normalization plate (Life Tech A10510–01) and the DNA was eluted and pooled. To purify and concentrate the DNA, 5 volumes of PB Buffer (QIAGEN 28004) were added, mixed, and purified using a QIAquick PCR Purification Kit (QIAGEN 28106). The DNA was gel extracted using a MinElute Gel Extraction Kit (QIAGEN 28604), quantified by qPCR using a KAPA Library Quantification Kit for Illumina Platforms (KAPA KK4824), and paired-end sequenced on the Illumina MiSeq platform. Data were processed using a 16S rRNA gene analysis pipeline (https://github.com/turnbaughlab/AmpliconSeq) based on QIIME2 (Bolyen et al., 2019) incorporating DADA2 (Callahan et al., 2016), and analyzed using R packages qiime2R (v0.99.23; https://github.com/jbisanz/qiime2R), phyloseq (v1.33.0) (McMurdie and Holmes, 2013), and phylosmith (v1.0.4) (Smith, 2019).
Construction of streptomycin-resistant E. coli strains
Strains resistant to the antibiotic streptomycin were generated by either selection for spontaneous resistance or by lambda Red recombineering (Datsenko and Wanner, 2000; Jensen et al., 2015). Spontaneous resistant mutants were selected by plating overnight cultures on LB supplemented with 500 μg/ml streptomycin. Lambda Red recombineering was later used to introduce a specific allele for genetic consistency between strains as different mutations in the rpsL gene can confer resistance to streptomycin (Timms et al., 1992). Briefly, cells were transformed with the CarbR temperature-sensitive plasmid pSIJ8 (Jensen et al., 2015), and electrocompe-tent cells were prepared from cells grown in LB carbenicillin at 30°C to early exponential phase and lambda Red recombinase genes were induced by addition of L-arabinose to 7.5 mM. Cells were electroporated with an rpsL-SmR PCR product amplified from a spontaneous streptomycin-resistant mutant of MG1655 using primers PS-rpsL1 and PS-rpsL2, and recombinants were selected on LB supplemented with 500 μg/ml streptomycin. The pSIJ8 plasmid was cured by culturing in liquid at 37°C in the absence of carbenicillin, plating for single colonies, and confirming CarbS. The rpsL gene of SmR strains was confirmed by Sanger sequencing.
Construction of fluorescently marked E. coli strains
P1 lysates were generated of AV01∷pAV01 and AV01∷pAV02 carrying “clonetegrated” sfgfp and mcherry, respectively (Vigouroux et al., 2018). Briefly, 150 μl of overnight culture in LB supplemented with 12.5 μg/ml kanamycin was mixed with 1 μl to 25 μl P1 phage (initially propagated from ATCC on MG1655). The mixture was incubated for 10 min at 30°C to aid adsorption, added to 4 mL LB 0.7% agar, and overlaid on pre-warmed LB agar supplemented with 25 μg/ml kanamycin 10 mM MgSO4. Plates were incubated overnight at 30°C, and phage were harvested by adding 5 mL SM buffer, incubating at room temperature for 10 min, and breaking and scraping off the top agar into a conical tube. Phage suspensions were centrifuged to pellet agar; the supernatant was passed through a 100 μm cell strainer, then through a 0.45 μm syringe filter, and lysates were stored at 4°C. For transduction, 1–2 mL of recipient overnight culture was pelleted and resuspended in 1/3 volume LB 10 mM MgSO4 5 mM CaCl2. 100 μl of cells was mixed with 1 μl to 10 μl P1 lysate and incubated at 30°C for 60 minutes. To minimize secondary infections, 200 μl 1 M sodium citrate was added, followed by 1 mL of LB. The mixture was incubated at 30°C for 2 h, then plated on LB 10 mM sodium citrate 25 μg/ml kanamycin to select for transductants. For excision of the vector backbone including the kanamycin resistance gene and heat-inducible integrase, cells were electroporated with pE-FLP (St-Pierre et al., 2013); transformants were selected on carbenicillin and confirmed for KmS. pE-FLP was cured by culturing in liquid at 37°C in the absence of carbenicillin, plating for single colonies, and confirming CarbS. Strains were subsequently grown routinely at 37°C. For imaging fluorescent strains on agar, plates were typically incubated at 37°C overnight, transferred to room temperature to allow fluorescence intensity to increase, and then imaged.
Mouse experiments with E. coli colonization, antibiotic water, and phage treatment
Streptomycin water was prepared by dissolving USP grade streptomycin sulfate (VWR 0382) in autoclaved tap water to a final concentration of 5 mg/ml and passing through 0.45 μm filtration units. Mice were provided streptomycin water for 1 day, followed by oral gavage of 0.2 mL containing approximately 109 CFU of streptomycin-resistant E. coli. Mice were kept on streptomycin water thereafter to maintain colonization. For selection with β-lactam antibiotics, USP grade ampicillin sodium salt (Teknova A9510) or USP grade carbenicillin disodium salt (Teknova C2110) was also dissolved in the water to a final concentration of 1 mg/ml; carbenicillin was preferred for its increased stability over ampicillin (Bobrowski and Borowski, 1971). Drinking water containing streptomycin was prepared fresh weekly; with the addition of a β-lactam antibiotic, it was prepared fresh every 3–4 days. For phage treatment, filtered phage solutions stored at −80°C were thawed and used directly for oral gavage. Unfiltered phage solutions were precipitated by diluting approximately 5-fold in PBS, adding 0.2 volumes phage precipitation solution (20% PEG-8000, 2.5 M NaCl), incubating for 15 min on ice, pelleting at 15,000–21,000 g for 15 min at 4°C, resuspending in PBS, centrifuging to pellet insoluble matter, and filtering through 0.45 μm. Heat-inactivated phage were prepared by incubating 1 mL aliquots at 95°C in a water bath for 30 min. Streptomycin-treated mice colonized with SmR E. coli were orally gavaged with 0.2 mL of phage and placed on drinking water containing both streptomycin and carbenicillin.
Enumeration and culture of E. coli from mouse feces
Fecal pellets were collected from individual mice and CFU counts were performed on the same day to determine CFU per gram feces. Briefly, fecal samples (typically 10–40 mg) were weighed on an analytical balance and 250 μl to 500 μl PBS or saline was added. Samples were incubated for 5 min at room temperature and suspended by manual mixing and vortexing. Large particulate matter was pelleted by centrifuging at 100 g, ten-fold serial dilutions were made in PBS, and 5 μl of each dilution was spotted on Difco MacConkey agar (Thermo Fisher 212123) supplemented with the appropriate antibiotics, i.e., streptomycin (100 μg/ml) or carbenicillin (50 μg/ml). For qualitative assessment of the fluorescent strains in feces, samples were spotted onto LB supplemented with the appropriate antibiotics. For isolating E. coli from fecal samples for genomic or plasmid DNA analysis, the fecal suspension was streaked on agar, and single colonies were further streak-purified.
Construction of CRISPR-Cas9 phagemid vectors
Cultures were grown in LB or TB media supplemented with the appropriate antibiotics. Plasmid DNA was prepared by QIAprep Spin Miniprep Kit (QIAGEN 27106), eluted in TE buffer, and incubated at 60°C for 10 min. Samples were quantified using a NanoDrop One spectrophotometer. The vector pCas9 (Jiang et al., 2013) was digested with BsaI (NEB R0535) and gel extracted with a QIAquick Gel Extraction Kit (QIAGEN 28706). Spacers were generated by annealing and phosphorylating the two oligos (PSP116 and PSP117 for GFPT; PSP120 and PSP121 for NT (Vigouroux et al., 2018)) at 10 μM each in T4 ligation buffer (NEB B0202S) with T4 polynucleotide kinase (NEB M0201S) by incubating at 37°C for 2 h, 95°C for 5 min, and ramping down to 20°C at 5°C/min. The annealed product was diluted 1 in 200 in sterile distilled water and used for directional cloning by ligating (Thermo Scientific FEREL0011) to 60 ng of BsaI-digested, gel extracted pCas9 overnight at room temperature. Ligations were used to transform NEB 5-alpha competent cells (NEB C2987H) and the cloned spacer was verified by Sanger sequencing using primer PSP108. The trailing repeat was later confirmed to lack the starting 5′G, which did not interfere with GFP-targeting function. The 1.8-kb fragment carrying the f1 origin of replication and β-lactamase gene (f1-bla) was amplified from pBluescript II with SalI adapters using primers KL215 and KL216 and KOD Hot Start DNA polymerase (Millipore 71842–3). The PCR product was purified using a QIAquick PCR Purification Kit (QIAGEN 28104), digested with SalI (Thermo Fisher FD0644), gel extracted, and used to ligate to SalI-digested, FastAP-dephosphorylated (Thermo Fisher FEREF0651) vector. Ligations were used to transform DH5α and clones were screened by restriction digest for both possible insert orientations (A or B) using XbaI (Thermo Scientific FD0684) and one of each orientation was saved for both the GFPT and NT phagemids, generating pCas9-GFPT-f1A, pCas9-GFPT-f1B, pCas9-NT-f1A, and pCas9-NT-f1B.
Preparation of M13 carrying pBluescript II
This protocol was adapted from those to generate phage display libraries(Tonikian et al., 2007). XL1-Blue MRF’ was transformed with pBluescript II (Agilent 212208). An overnight culture of this strain was prepared in 5 mL LB supplemented with tetracycline (5 μg/ml) and carbenicillin (50 μg/ml) and subcultured the following day 1-in-100 into 5 mL 2YT supplemented with the same antibiotics. At an OD600 of 0.8, cells were infected with helper phage M13KO7 (NEB N0315S) or VCSM13 (Agilent 200251) at a multiplicity of infection of approximately 10-to-1 for 1 h at 37°C The infected cells were used to seed 2YT supplemented with carbenicillin (100 μg/ml) and kanamycin (25 μg/ml) at 1-in-100, and the culture was grown overnight to produce phage. Cells were pelleted at 10,000 g for 15 min, and the supernatant containing phage was transferred. Phage were precipitated by adding 0.2 volumes phage precipitation solution, inverting to mix well, and incubating for 30 min on ice. Phage were pelleted at 15,000 g for 15 min at 4°C and the supernatant was discarded. The phage pellet was resuspended in PBS at 1%–4% of the culture volume. The resuspension was centrifuged to pellet insoluble material and transferred to a new tube. Glycerol was added to a final concentration of 10%–15%. Phage preparations were aliquoted into cryovials and stored at −80°C.
Preparation of M13 carrying CRISPR-Cas9 phagemids
DH5α(HP4_M13) (Praetorius et al., 2017) was transformed with the GFPT phagemid (pCas9-GFPT-f1A or pCas9-GFPT-f1B) or the NT phagemid (pCas9-NT-f1A or pCas9-NT-f1B) and plated on LB media containing carbenicillin and kanamycin. Transformants were inoculated into 5 mL 2YT supplemented with 100 μg/ml carbenicillin and 25 μg/ml kanamycin, incubated overnight, used 1-in-100 to seed a large volume of the same media, and incubated overnight. Cells were pelleted at 10,000 g for 15 min, and the supernatant containing phage was transferred. Phage were precipitated by adding 0.2 volumes phage precipitation solution, inverting to mix well, and incubating for 30 min on ice. Phage were pelleted at 20,000 g for 20 min at 4°C with slow deceleration. The supernatant was completely removed, phage were resuspended in PBS at 1% of the culture volume, and glycerol was added to a final concentration of 10%–15%. The phage solution was centrifuged at 21,000 g to pellet insoluble matter, filtered through 0.45 μm, and stored at −80°C.
Titration of M13 phage carrying phagemid DNA
Phage titer was determined using indicator strain XL1-Blue MRF’ or SmR W1655 F+. An overnight culture of the indicator strain in LB supplemented with the appropriate antibiotics was subcultured 1-in-100 or 1-in-200 into fresh media and grown to an OD600 of 0.8. To estimate titer, serial ten-fold dilutions of the phage preparation were made in PBS, and 10 μl of each dilution was used to infect 90 μl of cells. After incubating at 37°C for 30 min with shaking, 10 μl of the infection mix was spotted onto LB supplemented with carbenicillin. For more accurate titration, 100 μl of phage dilutions were mixed with 900 μl cells in culture tubes, incubated at 37°C for 30 min with shaking, and 100 μl was plated on LB carbenicillin.
Enumeration of viable M13 from mouse feces
Mice were orally gavaged with 6 × 1013 M13(pBluescript II) or as negative controls, heat-inactivated phage or PBS. Approximately 100 mg of feces were collected at 0, 3, 6, 9, and 24 h post-gavage, and samples at each time point were processed immediately. 500 μl PBS was added, samples were incubated for 5 min at room temperature, then suspended by manual mixing and vortexing. Samples were centrifuged at 21,000 g for 1 min, the supernatant was transferred to a new tube, and phage titer was determined against indicator strain XL1-Blue MRF’ by diluting samples in PBS, incubating with cells, and plating on LB supplemented with carbenicillin. For all dilutions and the undiluted suspension, 10 μl was used to infect 90 μl cells; additionally, for the undiluted suspension, 100 μl was used to infect 900 μl cells to maximize the limit of detection.
Assay for acid survival
Phage M13(pBluescript II) stored in PBS was diluted 1-in-100 in saline. Solutions varying in pH (1.2, 2, 3, 4, 5, 6, and 7) were prepared by mixing different ratios of 0.2 M sodium phosphate dibasic and 0.1 M citric acid and adjusting with concentrated HCl. 200 μl of each pH solution was transferred to the wells of a microtiter plate, and 10 μl of phage was added containing 1 × 109 M13(pBluescript II). Phage were incubated in the solution, and 10 μl was sampled at 5, 15, and 60 min. Samples were diluted 1-in-100 in PBS to make acidic samples neutral and phage titer was determined against indicator strain XL1-Blue MRF’ by plating on LB supplemented with carbenicillin. Solution-only controls were assayed simultaneously and cells were plated on LB to confirm viability of the indicator strain in the presence of samples originating from an acidic pH.
Targeting experiments in vitro with M13 CRISPR-Cas9
Overnight cultures of fluorescently marked SmR W1655 F+ sfgfp and mcherry were prepared in LB supplemented with streptomycin, subcultured 1 in 200 into fresh media, and grown to an OD600 of 0.8. 900 μl cells (approximately 1 × 109) was transferred to a culture tube, 100 μl phage (approximately 1 × 1010 for f1A vectors and approximately 5 × 1010 for f1B vectors) was added, and the tube was incubated at 37°C for 30 min. The infection culture was transferred to a microfuge tube, cells were pelleted at 21,000 g for 1 min, and the supernatant was removed. Cells were washed twice by adding 1 mL PBS, vortexing, pelleting cells, and removing supernatant. Cells were resuspended in 1 mL PBS, and ten-fold serially diluted in PBS. 10 μl of each dilution was spotted onto LB supplemented with carbenicillin and 100 μl was plated on larger plates for isolating single colonies for analysis. Colonies were picked and streak-purified four times to ensure phenotypic homogeneity and clonality.
Co-culture experiments of strains infected with M13 CRISPR-Cas9
Overnight cultures of fluorescently marked SmR W1655 F+ sfgfp and mcherry were prepared in LB supplemented with streptomycin. For each culture, three serial ten-fold dilutions were made in PBS, followed by a fourth ten-fold dilution into LB. Equal volumes of each were combined and 5 mL aliquots were transferred to culture tubes. Using a CFU assay, the input was determined to be 6 × 106 CFU of each strain or 1 × 107 CFU total. 10 μl (5 × 109) M13 carrying CRISPR-Cas9 was added, the co-culture was incubated at 37°C for 30 min, and carbenicillin was added to a final concentration of 100 μg/ml. The co-culture was sampled for the t = 0 time point and then incubated for 24 h with further sampling every 4 h. At each time point, 200 μl was taken; 100 μl was used to assay carbenicillin in the media (see section: Carbenicillin bioassay) and the remaining 100 μl was used for plating as follows. To the 100 μl sample of culture, 900 μl was added and cells were washed by vortexing. Cells were pelleted by centrifuging at 21,000 g for 1 min, and 900 μl of the supernatant was removed. To remove residual phage and antibiotic, the wash was repeated once more by adding 900 μl PBS, vortexing, pelleting cells, and removing 900 μl. Cells were resuspended in the remaining 100 μl. Serial ten-fold dilutions were made in PBS and 10 μl of each dilution was spotted onto LB or LB carbenicillin.
Carbenicillin bioassay
Cultures were sampled over time, cells were pelleted at 21,000 g for 1 min, and the supernatant was transferred to a new tube and frozen at −20°C until all time points were collected. The supernatants were thawed and assayed using a Kirby-Bauer disk diffusion test. An overnight culture of the indicator organism (Bacillus subtilis 168) was diluted in saline to an OD600 of 0.1. A cotton swab was dipped into this dilution and spread across LB agar, antibiotic sensitivity disks (Fisher Scientific S70150A) were overlaid using tweezers, and 20 μl of the supernatant was applied to the disk. At the same time, carbenicillin standards were prepared from 1 μg/ml to 100 μg/ml and also applied to discs. Plates were incubated overnight at 37°C and imaged the following morning.
Flow cytometry
For turbid in vitro cultures, samples were diluted 1-in-10,000 in PBS. For mouse fecal pellets, samples were used fresh or thawed from −80°C, and suspended in 500 μl PBS by manual mixing and vortexing. Fecal suspensions were incubated aerobically at 4°C overnight to improve fluorescence signal. Samples were vortexed to mix, large particulate matter was pelleted by centrifuging at 100 g for 30 s, and the sample was diluted 1-in-100 in PBS. Samples were run on a BD LSRFortessa flow cytometer using a 530/30 nm filter for GFP fluorescence and 610/20 nm for mCherry fluorescence, with the following voltages: 750 V for FSC, 400 V for SSC, 700 V for mCherry, and 700–800 V (in vivo) or 650 V (in vitro) for GFP. Flow cytometry data were analyzed in R using packages flowCore (v1.52.1) (Hahne et al., 2009), Phenoflow (v1.1.2) (Props et al., 2016), and ggcyto (v1.14.0) (Van et al., 2018). Typically, between 10,000 and 100,000 events were collected per sample, and data were rarefied after gating on FSC and SSC. Background events were accounted for on a per-mouse basis. For co-colonization with the sfgfp-marked and mcherry-marked strains, GFP+ and mCherry+ events from Day −3 (pre-E. coli) were used to subtract background at subsequent time points. For colonization with the double-marked strain, GFP+ mCherry+ events from Day −5 (pre-E. coli) were used to subtract background of double fluorescence at subsequent time points, and GFP− mCherry+ events from Day 0 (pre-phage) were used to subtract background of red fluorescence at subsequent time points. For exclusion of time points due to lack of colonization, the background threshold was calculated as the maximum background observed for that population across all time points multiplied by a factor of three. See Code Availability for more information.
Quick extraction and PCR analysis of genomic DNA from in vitro or in vivo isolates
Genomic DNA was extracted crudely to use as template for PCR. Briefly, 1.5 mL to 3 mL of culture was transferred to a microfuge tube, cells were pelleted by centrifuging, and the supernatant was discarded. The pellet was frozen, allowed to thaw on ice, resuspended in 100 μl TE, and incubated at 100°C for 15 min in an Eppendorf ThermoMixer. Samples were cooled on ice, cell debris was pelleted by centrifuging at 21,000 g for 1 min, the supernatant was transferred to a new tube, and diluted 1-in-100 in TE to use as template DNA. PCR was performed using KOD Hot Start DNA polymerase (Millipore 71842–3) using primers KL207/KL200 for the sfgfp gene and primers BAC338F/BAC805R for the 16S rRNA gene (Yu et al., 2005).
Extraction of DNA for hybrid assembly
E. coli strains KL68 (W1655 F+ or ATCC 23590), KL114 (W1655 F+ rpsL-SmR sfgfp), and KL204 (W1655 F+ rpsL-SmR sfgfp mcherry) were cultured in 50 mL LB supplemented with streptomycin. Cells were collected by centrifuging at 6,000 g for 10 min at room temperature, washed in 10 mL 10 mM Tris 25 mM EDTA (pH 8.0), and resuspended in 4 mL of the same buffer. 12.5 mg lysozyme (Sigma-Aldrich L6876), 100 μl 5 M NaCl, and 50 μl 10 mg/ml RNase A (Thermo-Fisher EN0531) were added and the mixture was incubated at 37°C for 15 min. To lyse cells, 350 μl 5 M NaCl, 20 μl 20 mg/ml Proteinase K (Ambion AM2546), and 500 μl 10% SDS were added, and the mixture was incubated at 60°C for 1 h with gentle inversions. 2.75 mL of 7.5 M ammonium acetate was added, and the mixture was incubated on ice 20 min to precipitate proteins. Debris was removed by centrifuging 20,000 g for 10 min and the supernatant was transferred to a new tube. To extract, an equal volume of chloroform was added and mixed; phases were separated by centrifuging at 2,000 g for 10 min, and the aqueous phase was transferred to a new tube. To precipitate the DNA, 1 volume of isopropanol was added, and the tube was inverted until a white precipitate formed. The DNA was pelleted by centrifuging at 2,000 g for 10 min and the supernatant was removed. The pellet was washed with 500 μl ice-cold 70% ethanol, allowed to dry, 1 mL TE was added, and the pellet allowed to dissolve overnight at 4°C. To further remove RNA, 250 μl of the genomic prep was transferred to a new tube, 12.5 μl 10 mg/ml RNase A was added, and the mixture was incubated at 37°C for 2 h with mixing every 30 min. To precipitate the DNA, 0.1 volume of 3 M sodium acetate was added followed by 3 volumes of 100% ethanol, and the mixture was inverted until a white precipitate formed. DNA was pelleted by centrifuging at 2,000 g for 10 min, the supernatant was removed, the pellet washed with 100 μl 70% ethanol, allowed to dry, and resuspended in 100 μl TE. Samples were quantified by Qubit dsDNA BR Assay and DNA integrity was confirmed by 0.4% agarose gel electrophoresis using GeneRuler High Range DNA Ladder (Thermo-Fisher FERSM1353). DNA was used for both Oxford Nanopore sequencing and Illumina sequencing.
Illumina whole genome sequencing
DNA concentration was quantified using PicoGreen (Thermo Fisher). Genomic DNA was normalized to 0.18 ng/μl for library preparation. Nextera XT libraries were constructed in 384-well plates using a custom, miniaturized version of the standard Nextera XT protocol. Small volume liquid handlers such as the Mosquito HTS (TTP LabTech) and Mantis (Formulatrix) were used to aliquot precise reagent volumes of < 1.2 μl to generate a total of 4 μl per library. Libraries were normalized and 1.2 μl of each normalized library was pooled and sequenced on the Illumina NextSeq or MiSeq platform using 2×146 bp configurations. 12 bp unique dual indices were used to avoid index hopping, a phenomenon known to occur on ExAmp based Illumina technologies. See Data Availability for more information.
Oxford Nanopore sequencing and hybrid Nanopore/Illumina assembly
PCR-free long read libraries were prepared using the Ligation Sequencing Kit (SQK-LSK109), multiplexed using the Native Barcoding Kit (EXP-NBD114), and sequenced on the MinION platform using flow cell version MIN106 (Oxford Nanopore Technologies). Base-calling of MinION raw signals was done using Guppy (v2.2.2, Oxford Nanopore Technologies). Reads were demultiplexed with qcat (v1.1.0, Oxford Nanopore Technologies). Quality control was achieved using porechop (v0.2.3 seqan2.1.1) (Wick et al., 2017a) using the discard middle option. Reads were filtered using NanoFilt (v2.6.0) (De Coster et al., 2018) with the following parameters: minimum average read quality score of 10 (−q 10) and minimum read length of 100 (−l 100). Illumina reads were quality filtered using fastp (v0.20.1) (Chen et al., 2018) with the following parameters: cut front, cut tail, cut window size 4, cut mean quality 20, length required 60. Filtered MinION and Illumina reads were then provided to Unicycler (v0.4.8) (Wick et al., 2017b) for hybrid assembly; default parameters were used unless otherwise noted.
Analysis of isolates after in vitro or in vivo M13-mediated delivery of phagemid
Isolates from in vitro GFP-targeting experiments were streak purified 4 times to ensure clonality. Isolates from in vivo experiments were obtained by streaking fecal suspensions followed by streak purification of single colonies. For DNA extraction, colonies were inoculated into LB or TB supplemented with the appropriate antibiotics. For analysis of the phagemid, plasmid DNA was extracted using a QIAprep Spin Miniprep Kit (QIAGEN 27106), eluted in TE buffer, and incubated at 60°C for 10 min. DNA was quantified using a NanoDrop One spectrophotometer and 200–600 ng was digested with FastDigest restriction enzymes (KpnI, Thermo Scientific FD0524; XbaI, Thermo Scientific FD0684) for 10 min at 37°C followed by gel electrophoresis. Spacer sequences on phagemids were confirmed by Sanger sequencing using primer PSP108. For genome sequencing, genomic DNA was either extracted using a DNeasy Blood & Tissue Kit (QIAGEN 69506) or an in-house protocol. Briefly, isolates were cultured in 3 mL TB supplemented with streptomycin and carbenicillin. Cells were pelleted and resuspended in 460 ul of freshly prepared buffer [per sample: 400 ul 10 mM Tris (pH 8.0) 25 mM ETDA, 50 μl 5 M NaCl, and 10 μl 10 mg/ml RNase A (Thermo-Fisher EN0531)]. 50 μl 10% SDS was added, mixed, and samples were incubated at 60°C for 1 h with periodic inversions. 260 μl of 7.5 M ammonium acetate was added, and the mixture was incubated on ice for 20 min to precipitate proteins. Precipitate was removed by centrifuging 21,000 g for 5 min and the supernatant was transferred to a new tube. An equal volume of chloroform was added and mixed; phases were separated by centrifuging at 21,000 g for 2.5 min. The aqueous phase was transferred to a new tube, centrifuged at 21,000 g for 2.5 min, and 500 ul was transferred to a new tube. To precipitate the DNA, 500 μl isopropanol was added, and the tube was inverted until a white precipitate formed. Using a pipette tip, the clump was transferred to a new tube, washed with 100 μl cold 70% ethanol, and allowed to dry. 50 μl TE was added and the pellet was allowed to dissolve at 4°C overnight. DNA integrity was confirmed by gel electrophoresis and used for Illumina whole genome sequencing (see section: Illumina whole genome sequencing). Sequence reads were quality filtered using fastp (v0.20.1) (Chen et al., 2018) and reads were aligned using bowtie2 (v2.3.5.1) (Langmead and Salzberg, 2012) to reference genomes and phagemid sequences; complete reference genomes were generated using hybrid assembly (see section: Oxford Nanopore sequencing and hybrid Nanopore/Illumina assembly). For CarbR isolates obtained after delivery of the pBluescript II phagemid, reads were simultaneously aligned to the genome of strain KL68 (W1655 F+) and the pBluescript II sequence (NCBI accession X52329.1). For CarbR isolates obtained after delivery of CRISPR-Cas9 phagemids, reads were simultaneously aligned to the genome of the strain used for in vitro (KL114; sfgfp) or in vivo (KL204; sfgfp mcherry) experiments and the sequence of the delivered phagemid. For isolates from GFP-targeting experiments, breseq (v0.35.4) (Deatherage and Barrick, 2014) was used to assess deletion size; additionally, deletions were visualized by using samtools (v1.9) (Li et al., 2009) to filter multi-mapping and low-quality read alignments with MAPQ < 2 (view −q 2), and depth was calculated using a sliding window of 20.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of biological and/or technical replicates is specified in each figure legend, along with the definition of center and variance used. Statistical significance was evaluated using R (version 3.6.2) unpaired Mann-Whitney tests (Figure 5E, wilcox.test, alternative = “less”; Figure S2, wilcox.test, alternative = “two.sided”) and a logistic regression (Figure 1D, glm, family = “binomial”).
Supplementary Material
Highlights.
Filamentous phage M13 can deliver DNA to E. coli cells colonizing the mouse gut
Engineered M13 carrying CRISPR-Cas9 can specifically target a strain in the gut
M13-delivered CRISPR-Cas9 can induce chromosomal deletions in vitro and in vivo
ACKNOWLEDGMENTS
We thank the UCSF Gnotobiotics Core staff (Jessie Turnbaugh, Kimberly Ly, and Jolie Ma) for animal care support and assistance; Daryll Gempis, Bernarda Lopez, and Ernesto Valencia (G.W. Hooper Foundation) for laboratory and administrative support; Katja Engel (University of Waterloo) for translating the original publication on the isolation of M13 from German; Antoine Vigouroux (Institut Pasteur) for sharing MG1655 derivatives carrying sfgfp and mcherry marker genes; the Chan Zuckerberg Biohub and the Stanford Microbiome Therapies Initiative for sequencing support; and Joseph Bondy-Denomy and Oren Rosenberg (UCSF) for constructive criticism of the manuscript. The graphic of a laboratory mouse was adapted from a Wikimedia Commons graphic distributed under CC-BY-SA-4.0 by Gwilz. Salary support was provided by the Canadian Institutes of Health Research (CIHR; K.N.L. and P.S.); the Natural Sciences and Engineering Research Council of Canada (NSERC; J.E.B.); the National Institutes of Health (F32AI147456, M.A.; K08AR073930, R.R.N.); and the UCSF Discovery Fellows Program (P.S.-P.). Research support was provided by the National Institutes of Health (R01HL122593 and R01AT011117, P.J.T.). P.J.T. held an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, was a Chan Zuckerberg Biohub investigator, and was a Nadia’s Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation (DRR-42-16) and the Searle Scholars Program (SSP-2016-1352).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109930.
DECLARATION OF INTERESTS
K.N.L., P.S., and P.J.T. are listed inventors on a US provisional patent application related to this work (33167/55262P1). P.J.T. is on the scientific advisory boards for Kaleido, Pendulum, and SNIPRbiome. All other authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Ackermann H-W (2009). Phage classification and characterization. Methods Mol. Biol 501, 127–140. [DOI] [PubMed] [Google Scholar]
- Alting-Mees MA, and Short JM (1989). pBluescript II: gene mapping vectors. Nucleic Acids Res. 17, 9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basolo A, Hohenadel M, Ang QY, Piaggi P, Heinitz S, Walter M, Walter P, Parrington S, Trinidad DD, von Schwartzenberg RJ, et al. (2020). Effects of underfeeding and oral vancomycin on gut microbiome and nutrient absorption in humans. Nat. Med 26, 589–598. [DOI] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, and Marraffini LA (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol 32, 1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz JE, Soto-Perez P, Noecker C, Aksenov AA, Lam KN, Kenney GE, Bess EN, Haiser HJ, Kyaw TS, Yu FB, et al. (2020). A Genomic Toolkit for the Mechanistic Dissection of Intractable Human Gut Bacteria. Cell Host Microbe 27, 1001–1013.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrowski M, and Borowski E (1971). Interaction between carbenicillin and β-lactamases from Gram-negative bacteria. J. Gen. Microbiol 68, 263–272. [DOI] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD, and Lawley TD (2021). Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Sun Y, Berglindh T, Mellgård B, Li Z, Mårdh B, and Mårdh S (2000). Helicobacter pylori-antigen-binding fragments expressed on the filamentous M13 phage prevent bacterial growth. Biochim. Biophys. Acta 1474, 107–113. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citorik RJ, Mimee M, and Lu TK (2014). Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol 32, 1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csörgő B, León LM, Chau-Ly IJ, Vasquez-Rifo A, Berry JD, Mahendra C, Crawford ED, Lewis JD, and Bondy-Denomy J (2020). A compact Cascade-Cas3 system for targeted genome engineering. Nat. Methods 17, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, and Bikard D (2016). Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 44, 4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, D’Hert S, Schultz DT, Cruts M, and Van Broeckhoven C (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, and Barrick JE (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol 1151, 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, and Kowalczykowski SC (2008). RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev 72, 642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R, et al. (2016). Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol 34, 942–949. [DOI] [PubMed] [Google Scholar]
- Guiney DG (1982). Host range of conjugation and replication functions of the Escherichia coli sex plasmid Flac. Comparison with the broad host-range plasmid RK2. J. Mol. Biol 162, 699–703. [DOI] [PubMed] [Google Scholar]
- Guo C-J, Allen BM, Hiam KJ, Dodd D, Van Treuren W, Higginbottom S, Nagashima K, Fischer CR, Sonnenburg JL, Spitzer MH, and Fischbach MA (2019). Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 366, eaav1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, and Gentleman R (2009). flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 10, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofschneider PH (1963). Untersuchungen über „kleine“ E. coli K 12 Bakteriophagen. Zeitschrift Für Naturforschung B 18, 203–210. [PubMed] [Google Scholar]
- Hsu BB, Way JC, and Silver PA (2020a). Stable Neutralization of a Virulence Factor in Bacteria Using Temperate Phage in the Mammalian Gut. mSystems 5, e00013–e00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu BB, Plant IN, Lyon L, Anastassacos FM, Way JC, and Silver PA (2020b). In situ reprogramming of gut bacteria by oral delivery. Nat. Commun 11, 5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, et al. (2018). Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol 36, 857–864. [DOI] [PubMed] [Google Scholar]
- Jensen SI, Lennen RM, Herrgård MJ, and Nielsen AT (2015). Seven gene deletions in seven days: Fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Sci. Rep 5, 17874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, and Marraffini LA (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol 31, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasman LM, Kasman A, Westwater C, Dolan J, Schmidt MG, and Norris JS (2002). Overcoming the phage replication threshold: a mathematical model with implications for phage therapy. J. Virol 76, 5557–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, and Ames GF (1984). Analysis of promoter mutations in the histidine transport operon of Salmonella typhimurium: use of hybrid M13 bacteriophages for cloning, transformation, and sequencing. J. Bacteriol 159, 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TK, and Collins JJ (2009). Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 106, 4629–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhal ML, Laux DC, and Cohen PS (1982). Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol 1, 186–192. [DOI] [PubMed] [Google Scholar]
- Park JY, Moon BY, Park JW, Thornton JA, Park YH, and Seo KS (2017). Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci. Rep 7, 44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NT, Hryckowian AJ, Merrill BD, Fuentes JJ, Gardner JO, Glowacki RWP, Singh S, Crawford RD, Snitkin ES, Sonnenburg JL, and Martens EC (2020). Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol 5, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius F, Kick B, Behler KL, Honemann MN, Weuster-Botz D, and Dietz H (2017). Biotechnological mass production of DNA origami. Nature 552, 84–87. [DOI] [PubMed] [Google Scholar]
- Props R, Monsieurs P, Mysara M, Clement L, and Boon N (2016). Measuring the biodiversity of microbial communities by flow cytometry. Methods Ecol. Evol 7, 1376–1385. [Google Scholar]
- Rubin BE, Diamond S, Cress BF, Crits-Christoph A, He C, Xu M, Zhou Z, Smock DC, Tang K, Owens TK, et al. (2020). Targeted Genome Editing of Bacteria Within Microbial Communities. bioRxiv. 10.1101/2020.07.17.209189. [DOI] [Google Scholar]
- Salivar WO, Tzagoloff H, and Pratt D (1964). Some Physical-Chemical and Biological Properties of the Rod-Shaped Coliphage M13. Virology 24, 359–371. [DOI] [PubMed] [Google Scholar]
- Secor PR, Sweere JM, Michaels LA, Malkovskiy AV, Lazzareschi D, Katznelson E, Rajadas J, Birnbaum ME, Arrigoni A, Braun KR, et al. (2015). Filamentous Bacteriophage Promote Biofilm Assembly and Function. Cell Host Microbe 18, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selle K, Fletcher JR, Tuson H, Schmitt DS, McMillan L, Vridhambal GS, Rivera AJ, Montgomery SA, Fortier L-C, Barrangou R, et al. (2020). In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. MBio 11, 00019–00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Sauk J, Gevers D, Friedman J, Sung J, Youngster I, Hohmann EL, Staley C, Khoruts A, Sadowsky MJ, et al. (2018). Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 23, 229–240.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SD (2019). phylosmith: an R-package for reproducible and efficient microbiome analysis with phyloseq-objects. J. Open Source Softw 4, 1442. [Google Scholar]
- Soto-Perez P, Bisanz JE, Berry JD, Lam KN, Bondy-Denomy J, and Turnbaugh PJ (2019). CRISPR-Cas System of a Prevalent Human Gut Bacterium Reveals Hyper-targeting against Phages in a Human Virome Catalog. Cell Host Microbe 26, 325–335.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre F, Cui L, Priest DG, Endy D, Dodd IB, and Shearwin KE (2013). One-step cloning and chromosomal integration of DNA. ACS Synth. Biol 2, 537–541. [DOI] [PubMed] [Google Scholar]
- Timms AR, Steingrimsdottir H, Lehmann AR, and Bridges BA (1992). Mutant sequences in the rpsL gene of Escherichia coli B/r: mechanistic implications for spontaneous and ultraviolet light mutagenesis. Mol. Gen. Genet 232, 89–96. [DOI] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Boone C, and Sidhu SS (2007). Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc 2, 1368–1386. [DOI] [PubMed] [Google Scholar]
- Tóthová L, Bábícková J, and Celec P (2012). Phage survival: the biodegradability of M13 phage display library in vitro. Biotechnol. Appl. Biochem 59, 490–494. [DOI] [PubMed] [Google Scholar]
- Van P, Jiang W, Gottardo R, and Finak G (2018). ggCyto: next generation open-source visualization software for cytometry. Bioinformatics 34, 3951–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux A, Oldewurtel E, Cui L, Bikard D, and van Teeffelen S (2018). Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol 14, e7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwater C, Kasman LM, Schofield DA, Werner PA, Dolan JW, Schmidt MG, and Norris JS (2003). Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob. Agents Chemother 47, 1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, and Holt KE (2017a). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom 3, e000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, and Holt KE (2017b). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Lee C, Kim J, and Hwang S (2005). Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng 89, 670–679. [DOI] [PubMed] [Google Scholar]
- Zinder ND, and Boeke JD (1982). The filamentous phage (Ff) as vectors for recombinant DNA–a review. Gene 19, 1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data have been deposited at the NCBI Sequence Read Archive under NCBI BioProject: PRJNA642411 and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. Additional supporting data is publicly available at https://github.com/turnbaughlab/2021_Lam_M13_CRISPRCas9.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| MG1655 (Derivative of K-12) | ATCC | ATCC 700926 |
| DH5α (Routine cloning; phage propagation with helper HP4_M13) | Thermo Fisher | Thermo Fisher 18265017 |
| NEB 5-alpha (Routine cloning) | NEB | NEB C2987H |
| XL1-Blue MRF’ (Phage propagation with helper M13KO7; TcR) | Agilent | Agilent 212208 |
| MG1655 rpsL-SmR (Spontaneous rpsL-SmR (Lys42Arg) derivative of MG1655) | This study | KL52 |
| W1655 F− (Derivative of K-12; M13R) | ATCC | ATCC 23737 |
| W1655 F+ (Derivative of K-12; M13S) | ATCC | ATCC 23590; KL68 |
| W1655 F− rpsL-SmR (Recombineered rpsL-SmR (Lys42Arg) derivative of W1655 F−) | This study | KL89 |
| W1655 F+ rpsL-SmR (Recombineered rpsL-SmR (Lys42Arg) derivative of W1655 F+) | This study | KL90 |
| AV01::pAV01 (MG1655 with constitutive sfgfp clonetegrated at HK022 att site; KmR) | Vigouroux et al., 2018 | AV01::pAV01; KL106 |
| AV01::pAV02 (MG1655 with constitutive mcherry clonetegrated at lambda att site; KmR) | Vigouroux et al., 2018 | AV01::pAV02; KL107 |
| W1655 F+ rpsL-SmR sfgfp (W1655 F+ rpsL-SmR with sfgfp transduced from AV01::pAV01; KmS) | This study | KL114 |
| W1655 F+ rpsL-SmR mcherry (W1655 F+ rpsL-SmR with mcherry transduced from AV01::pAV02; KmS) | This study | KL115 |
| W1655 F+ rpsL-SmR sfgfp mcherry (W1655 F+ rpsL-SmR sfgfp with mcherry transduced from AV01::pAV02; KmS) | This study | KL204 |
| Bacteriophage: M13KO7 (helper phage, KmR) | NEB | NEB N0315S |
| Bacteriophage: VCSM13 (helper phage, KmR) | Agilent | Agilent 200251 |
| Bacteriophage: P1 (transducing phage) | ATCC | ATCC 25404-B1 |
| Chemicals, peptides, and recombinant proteins | ||
| MacConkey Agar | Thermo Fisher | Cat#212123 |
| USP-grade streptomycin sulfate | VWR | Cat#0382 |
| USP-grade ampicillin sodium salt | Teknova | Cat#A9510 |
| USP-grade carbenicillin disodium salt | Teknova | Cat#C2110 |
| Critical commercial assays | ||
| ZymoBIOMICS 96 MagBead DNA Kit | Zymo | Cat#D4302 |
| SequelPrep Normalization Plate Kit | Life Tech | Cat#A10510-01 |
| KAPA Library Quantification Kit | KAPA | Cat#KK4824 |
| Ligation Sequencing Kit | Oxford Nanopore | Cat#SQK-LSK109 |
| Native Barcoding Kit | Oxford Nanopore | Cat#EXP-NBD114 |
| Deposited data | ||
| Sequencing data repository | This paper | NCBI BioProject: PRJNA642411 |
| 16S rRNA gene sequencing data | This paper | NCBI SRA: SRR12118792-SRR12118959 |
| Isolates from mouse fecal samples after delivery of pBluescript II | This paper | NCBI SRA: SRR14278062-SRR14278073 |
| Illumina and Nanopore data for the hybrid assembly of reference strain KL68 (W1655 F+ or ATCC 23590) as well as SmR fluorescent derivatives KL114 (sfgfp) and KL204 (sfgfp mcherry) | This paper | NCBI SRA: SRR14296642-SRR14296644; SRR14297452-SRR14297454 |
| Sequencing of isolates after targeting with phage-delivered CRISPR-Cas9 | This paper | NCBI SRA: SRR14289086-SRR14289109 |
| Experimental models: Organisms/strains | ||
| BALB/c mice | Taconic | Taconic BALB-F MPF |
| Oligonucleotides | ||
| See Table S1 | See Table S1 | See Table S1 |
| Recombinant DNA | ||
| Plasmid: pBluescript II KS(−) (Commercial phagemid; CarbR) | Alting-Mees and Short, 1989 | Agilent 212208 |
| Plasmid: pSIJ8 (Temperature-sensitive; lambda Red recombineering; CarbR) | Jensen et al., 2015 | RRID: Addgene_68122 |
| Plasmid: pE-FLP (Temperature sensitive; constitutive flippase expression; CarbR) | St-Pierre et al., 2013 | RRID: Addgene_45978 |
| Plasmid: pCas9 (Low-copy vector carrying cas9, tracrRNA, and CRISPR array; CmR) | Jiang et al., 2013 | RRID: Addgene_42876 |
| Plasmid: HP4_M13 (helper plasmid, KmR) | Praetorius et al., 2017 | RRID: Addgene_120340 |
| Plasmid: pCas9-GFPT-f1A (pCas9 with GFP-targeting spacer; f1-bla in orientation A; CmR CarbR) | This study | pCas9-GFPT-f1A; pKL100 |
| Plasmid: pCas9-GFPT-f1B (pCas9 with GFP-targeting spacer; f1-bla in orientation B; CmR CarbR) | This study | pCas9-GFPT-f1B; pKL101 |
| Plasmid: pCas9-NT-f1A (pCas9 with Non-targeting spacer; f1-bla in orientation A; CmR CarbR) | This study | pCas9-NT-f1A; pKL102 |
| Plasmid: pCas9-NT-f1B (pCas9 with Non-targeting spacer; f1-bla in orientation B; CmR CarbR) | This study | pCas9-NT-f1B; pKL103 |
| Software and algorithms | ||
| QIIME2 | Bolyen et al., 2019 | https://qiime2.org/ |
| DADA2 | Callahan et al., 2016 | https://benjjneb.github.io/dada2/ |
| phyloseq | McMurdie and Holmes, 2013 | https://github.com/joey711/phyloseq |
| phylosmith | Smith, 2019 | https://github.com/schuyler-smith/phylosmith |
| flowCore | Hahne et al., 2009 | https://github.com/RGLab/flowCore |
| Phenoflow | Props et al., 2016 | https://github.com/rprops/PhenoFlow |
| ggcyto | Van et al., 2018 | https://github.com/RGLab/ggcyto |
| Guppy | Oxford Nanopore Technologies | https://nanoporetech.com/community |
| qcat | Oxford Nanopore Technologies | https://github.com/nanoporetech/qcat |
| porechop | Wick et al., 2017a | https://github.com/rrwick/Porechop |
| NanoFilt | De Coster et al., 2018 | https://github.com/wdecoster/nanofilt |
| fastp | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| Unicycler | Wick et al., 2017b | https://github.com/rrwick/Unicycler |
| bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| breseq | Deatherage and Barrick, 2014 | https://github.com/barricklab/breseq |
| samtools | Li et al., 2009 | https://github.com/samtools/samtools |
| Other | ||
| Resource website for supporting data | This paper | https://dx.doi.org/10.5281/zenodo.5517961 |