

Abstract
Chromosome copy number variations are a hallmark of cancers. Among them, the prevalent chromosome 17p deletions are associated with poor prognosis and can promote tumorigenesis more than TP53 loss. Here we utilize multiple functional genetic strategies and identify a new 17p tumor suppressor gene, PHD finger protein 23 (PHF23). Its deficiency impairs B cell differentiation and promotes immature B lymphoblastic malignancy. Mechanistically, we demonstrate that PHF23, an H3K4me3 reader, directly binds the SIN3-HDAC complex through its N-terminus and represses its deacetylation activity on H3K27ac. Thus, the PHF23-SIN3-HDAC (PSH) complex coordinates these two major active histone markers for the activation of downstream TSGs and differentiation-related genes. Further, dysregulation of the PSH complex is essential for the development and maintenance of PHF23 deficient and 17p deleted tumors. Hence, our study reveals a novel epigenetic regulatory mechanism that contributes to the pathology of 17p-deleted cancers and suggests novel susceptibility to this disease.
Keywords: del(17p), PHF23, the PHF23-SIN3-HDAC complex
Introduction
Chromosome copy number variations (CNVs), which generally consist of hundreds or thousands of genes, are a common feature of human cancers(1,2). Accumulating evidence indicate that specific CNVs are recurrent in many types of cancer, and some of them can occur at the early stages of tumorigenesis, suggesting that at least some cancer-associated CNVs may function as the drivers of human cancers(3,4). For example, chromosome 3p deletions are detected in more than 90% of lung cancers, while chromosome 8q is frequently amplified in hepatocellular carcinoma(5,6). Among them, heterozygous chromosome 17p deletions (del(17p)) are the most frequent chromosome loss in both solid cancers and hematopoietic malignancies, including lymphomas and leukemias(2–4,7–10). It has been proposed that TSGs are enriched in cancer-associated chromosome deletion regions while oncogenes are enriched in chromosome amplicons(11,12). However, given that a large number of genes are involved in each CNV, it has been challenging to functionally identify potential TSGs and oncogenes for better mechanistic insights of CNVs in cancer. It had long been assumed that the only function of del(17p) is for TP53 loss of heterozygosity until recently our previous work(8). Utilizing a conditional chromosome 11B3 (syntenic to human chromosome 17p13, a commonly deleted region in human cancers) deletion mouse model, we demonstrated that del(17p) could promote cancer development through both TP53-dependent and -independent mechanisms. However, the additional TSGs in this region remain to be identified.
There are about 500 genes on chromosome 17p and a large amount of effort has been made to identify new TSGs in this region(8,13). According to the “two-hit” theory, most previous studies focused on searching for candidate TSGs with mutations/focal deletions in one allele and complete loss of the other allele through del(17p), similar to TP53(14). Unfortunately, limited success has been achieved with this strategy(15,16). The majority of 17p genes involved in del(17p) maintain intact in the other allele and are expressed in 17p deleted cancers, although generally at reduced levels. Recently, it has been reported that EIF5A and ALOX15B, both located on chromosome 17p13.1, can prevent tumorigenesis by promoting apoptosis of pre-malignant B cells(8,13). Interestingly, both EIF5A and ALOX15B are haploinsufficient TSGs and neither of them has the second hit on the other allele. These studies imply that potential TSGs involved in del(17p) have been largely underestimated and emphasize functional studies for further identifying TSGs on chromosome deletions.
In this study, we utilized our previous in vivo functional shRNA screening results(8) and analyzed the potential roles of PHD finger protein 23 (PHF23), located on chromosome 17p13.1, in immature B lymphoblastic malignancy. As a putative epigenetic reader, the PHD domain of PHF23 specifically binds to tri-methylated lysine 4 of histone H3 (H3K4me3) (17,18). Interestingly, PHF23 is reported to be also involved in recurrent chromosomal translocations, forming an oncofusion gene NUP98-PHF23 in human acute myeloid leukemia (AML) (17–20). NUP98-PHF23 promotes leukemogenesis through its dysregulated chromatin-modifying activity. In contrast, in del(17p) cancer, one allele of PHF23 is lost while without recurrent mutations on the second allele, suggesting that it might be loss of function in these cancers. It would be interesting to investigate whether and how an epigenetic reader involved in del(17p) plays a role in cancer biology.
Results
PHF23 is a new 17p TSG
In our unbiased in vivo tumorigenesis screening with an shRNA library targeting genes on mouse chromosome 11B3, syntenic to human chromosome 17p13, the common deletion region of chromosome 17p, multiple shRNAs targeting PHD finger protein 23 (Phf23) were enriched in the resulting lymphoma/leukemias (8) (Fig. 1A). By analyzing the TCGA diffused large B cell lymphoma (DLBCL) cohorts, we found that PHF23 expressions were significantly reduced in DLBCL with del(17p) compared to their counterparts, consistently with its DNA copy number loss (Fig. 1B). Therefore, we hypothesized that PHF23 might be a new TSG on chromosome 17p.
Figure 1 |. Phf23 deficiencies promoted lymphomagenesis.
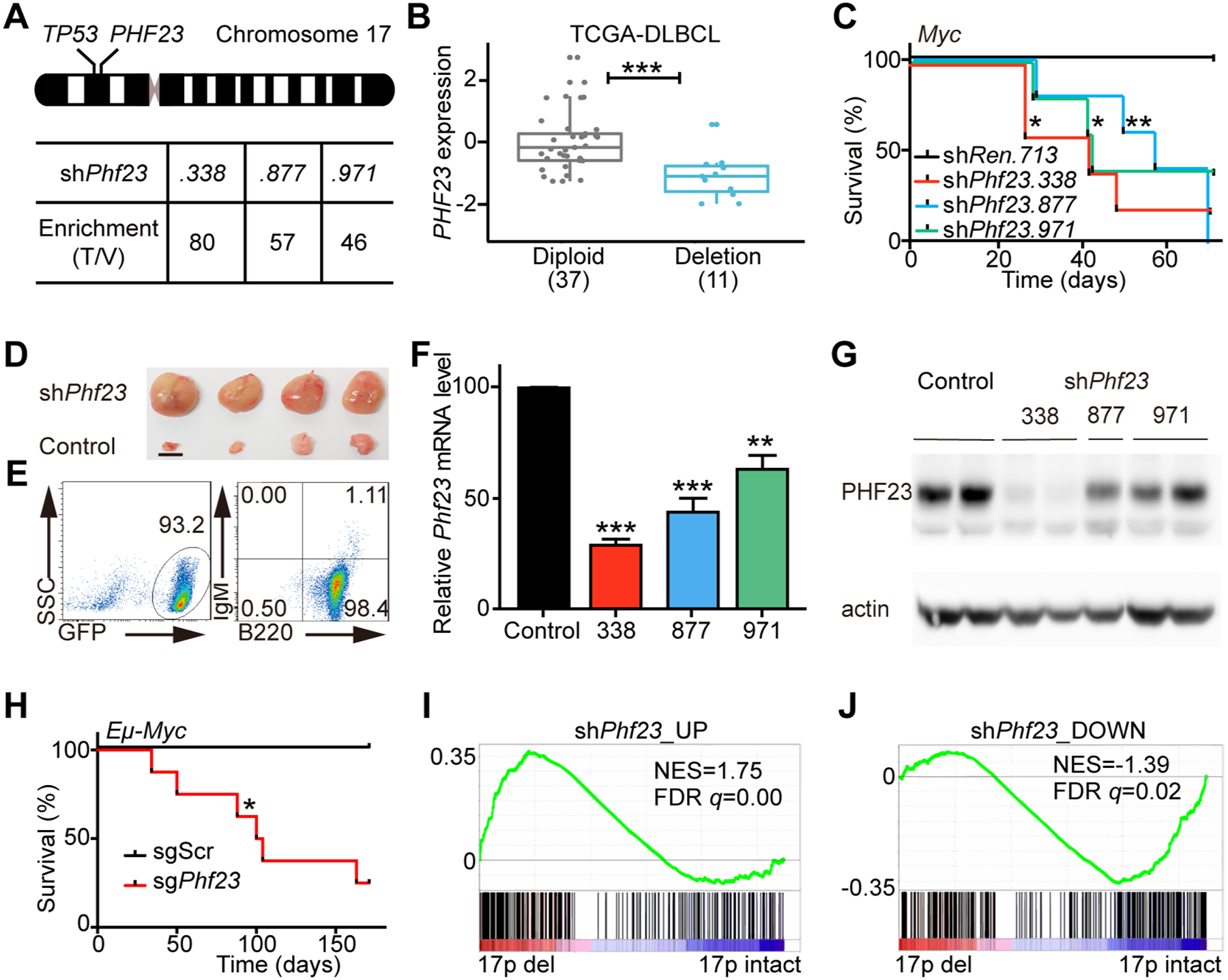
A, Top, a schematic diagram of human chromosome 17, showing the relative positions of TP53 and PHF23 on 17p. Bottom, the enrichment folds of Phf23 shRNAs in the shRNA library screening of mouse chromosome 11B3 genes in Myc-driven lymphoma/leukemia in mice. The enrichment folds were calculated by dividing the percentages of each shRNA reads in tumors by those in the library of plasmids. B, The expression levels of PHF23 in DLBCL patients with (Deletion) or without (Diploid) chromosome 17p deletions. RNA-seq data with RSEM were analyzed from TCGA. *** padj < 0.001, Wilcoxon signed-rank test. C, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with pre-B cells infected with Myc cDNA and indicated shRNAs. n=5 for each group. *p<0.05, **p<0.01 (log-rank test). D, Representative picture showing enlarged lymph nodes in recipient mice with shPhf23. Scale bar, 5mm. E, Representative flow plots showing the expressions of GFP, B220 and IgM of shPhf23 lymphoma/leukemia cells. F, Relative expression levels of Phf23 in shPhf23 lymphoma/leukemia cells, measured by qPCR. Control, shTrp53;Myc lymphoma/leukemia cells. G, Representative Western blotting picture showing the protein levels of PHF23 in shPhf23 lymphoma/leukemia cells. Control, shTrp53;Myc lymphoma/leukemia cells. H, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with Eμ-Myc fetal liver cells (FLCs) with CRISPR/Cas9 targeting Phf23 or scramble sequence. n=5, sgScr; n=8, sgPhf23. *p<0.05 (log-rank test). I, Gene Set Enrichment Analysis (GSEA) showing the positive enrichment of the shPhf23 lymphoma/leukemia upregulated gene set in 17p deleted TCGA DLBCL, comparing to 17p intact ones (NES=1.75; FDR q=0.00). J, GSEA showing the negative enrichment of the shPhf23 lymphoma/leukemia downregulated gene set in 17p deleted TCGA DLBCL, comparing to 17p intact ones (NES=−1.39; FDR q=0.02).
To validate the role of PHF23 in B cell malignancies, multiple independent Phf23 shRNAs (shPhf23.338, shPhf23.877 and shPhf23.971) or control Renilla shRNA (shRen) were co-introduced with GFP and Myc into pre-B cells isolated from C57BL/6 mouse bone marrow (BM). These shPhf23 or shRen cells were then transplanted into sub-lethally irradiated congenic recipient mice(21) (Supplementary Fig. S1). The recipient mice transplanted with shPhf23 cells developed tumors with enlarged lymph nodes while none of the recipient mice with shRen did in the observation period (Fig. 1C–D). The median tumor onset times were 40 days for shPhf23.338, 40 days for shPhf23.971 and 53 days for shPhf23.877, respectively. Flow cytometry analysis showed that all of the tumor cells were GFP positive, indicating originating from donor cells with shPhf23 (Fig. 1E). The expression of B cell marker B220 and hematoxylin and eosin (H/E) staining results indicated that these mouse tumors with shPhf23 represented the characteristics of high grade lymphoblastic lymphoma/leukemia (Fig. 1E and Supplementary Fig. S2A). The knockdown efficiencies of Phf23 shRNAs in the resulting immature B lymphoblastic malignant cells from enlarged lymph nodes of recipients were confirmed by qPCR and Western blotting (Fig. 1F–G).
To further confirm its tumor suppressor function, we designed sgRNAs targeting the genomic DNA sequence encoding the PHD domain of PHF23, which recognizes and binds H3K4me3 (20). Fetal liver cells (FLCs) enriched of hematopoietic stem and progenitor cells from E13.5 Eμ-Myc mouse embryos(22) were transduced with Cas9 and sgRNAs for Phf23 (sgPhf23) or scramble sequence (sgScr) and then transplanted into sub-lethally irradiated congenic recipient mice (Supplementary Fig. S1). While none of the sgScr recipients developed tumors during the observation period, 6 of 8 sgPhf23 recipient mice developed lymphoma/leukemia with median tumor-free survival of 102 days (Fig. 1H and Supplementary Fig. S2B). sgPhf23 generated loss-of-function mutations in the resulting lymphoma/leukemias, as shown by T7E1 assay, Sanger sequencing of clonal lymphoma/leukemia and Western blotting (Supplementary Fig. S2C–E).
To test whether our mouse lymphoma/leukemia model resembles human cancers with 17p deletions, we analyzed RNA-seq (RNA-sequencing) data of mouse lymphoma/leukemia cells with or without Phf23 loss. Gene set enrichment analyses (GSEA) showed that genes upregulated in shPhf23 lymphoma/leukemia cells were significantly positively enriched in 17p deleted human DLBCLs, comparing to those without 17p deletions (NES=1.75, FDR q=0.00), while the downregulated genes in mouse shPhf23 lymphoma/leukemia cells were significantly negatively enriched in human DLBCLs with 17p deletions (NES=−1.39, FDR q=0.02) (Fig 1I–J). These correlations suggested that our mouse lymphoma/leukemia with Phf23 loss partially resembled human DLBCLs with 17p deletions and PHF23 was a key TSG on 17p. Thus, with both shRNA and CRISPR/Cas9-mediated genome editing experiments, we identified PHF23 as a new TSG on chromosome 17p.
PHF23 is a haploinsufficient TSG and cooperates with TP53
Chromosome 17p deletions usually occur as a heterozygous loss(3,4,23). TSGs on chromosome deletions presumably need loss of heterozygosity through various mechanisms, such as mutations, focal deletions and epigenetic silencing during tumorigenesis(24). However, mutations on PHF23 with del(17p) were rare in human cancers (Supplementary Fig. S2F) and the expressions of PHF23 in del(17p) DLBCL patients were significantly reduced compared to those with intact chromosome 17p (Fig. 1B). Similarly, some other genes involved in large chromosomal deletions, such as MLL3 on chromosome 7q, have been shown to be haploinsufficient TSGs (12,25,26). Hence, we hypothesized that PHF23 might also be a haploinsufficient TSG on chromosome 17p. Consistently, although the expression levels of Phf23 were significantly reduced, there were remarkable remaining Phf23 mRNA and protein in shPhf23 lymphoma/leukemia (Fig. 1F–G). Heterozygous mutations of Phf23 were also detected in sgPhf23 lymphoma/leukemia (Supplementary Fig. S2D).
To test this hypothesis, we generated Phf23 knockout mice with CRISPR/Cas9 editing, which resulted in a 14-bp insertion on exon 5 and a presumable premature stop codon (Fig. 2A–B). Phf23−/− mice were embryonically lethal (E15.5–16.5, unpublished data). Western blotting showed that the expression levels of PHF23 in FLCs from Phf23+/− embryos were precisely reduced to 50% of that in wildtype, while it was completely lost in Phf23−/− cells (Fig. 2C). Phf23 wildtype, heterozygous and homozygous knockout FLCs were transduced with Myc cDNA, followed by transplantation into sub-lethally irradiated congenic recipient mice (Supplementary Fig. S1). Consistent with the aforementioned tumorigenesis experiments with shRNA and CRISPR/Cas9 (Fig. 1C and 1H), recipients transplanted with Phf23−/− FLCs transduced with Myc had significantly shorter tumor onset times than those with wildtype FLCs with Myc overexpression (Fig. 2D). Surprisingly, Phf23+/− FLCs also gave rise to tumors with a similar latency as Phf23−/− cells. Interestingly, Myc-overexpressing Phf23+/+ FLCs led to typical myeloid leukemia, indicated by expression of myeloid lineage markers CD11b and Gr-1 but not B220, consistent with the previous report(27). In contrast, recipient mice with both Phf23−/− and Phf23+/− cells had enlarged lymph nodes and their tumor cells were B220+CD11b−Gr-1− (Fig. 2E–F). Therefore, Phf23 heterozygosity, similar to Phf23 null, promoted Myc-driven lymphoma/leukemia. To rule out the possibility of loss-of-heterozygosity of Phf23 during Phf23+/− lymphomagenesis, we measured the protein levels of PHF23 in the resulting lymphoma/leukemia by Western blotting. The results showed that the protein levels of PHF23 in Phf23+/− lymphoma/leukemia remained at about half of that in Phf23+/+ cells, indicating that the second allele of Phf23 remained intact during lymphomagenesis (Fig. 2G). Moreover, the WT Phf23 allele was confirmed to remain in the Phf23+/− tumor cells (data not shown). Altogether, these results strongly suggested that PHF23 was a haploinsufficient TSG.
Figure 2 |. PHF23 was a haploinsufficient TSG and cooperated with p53.
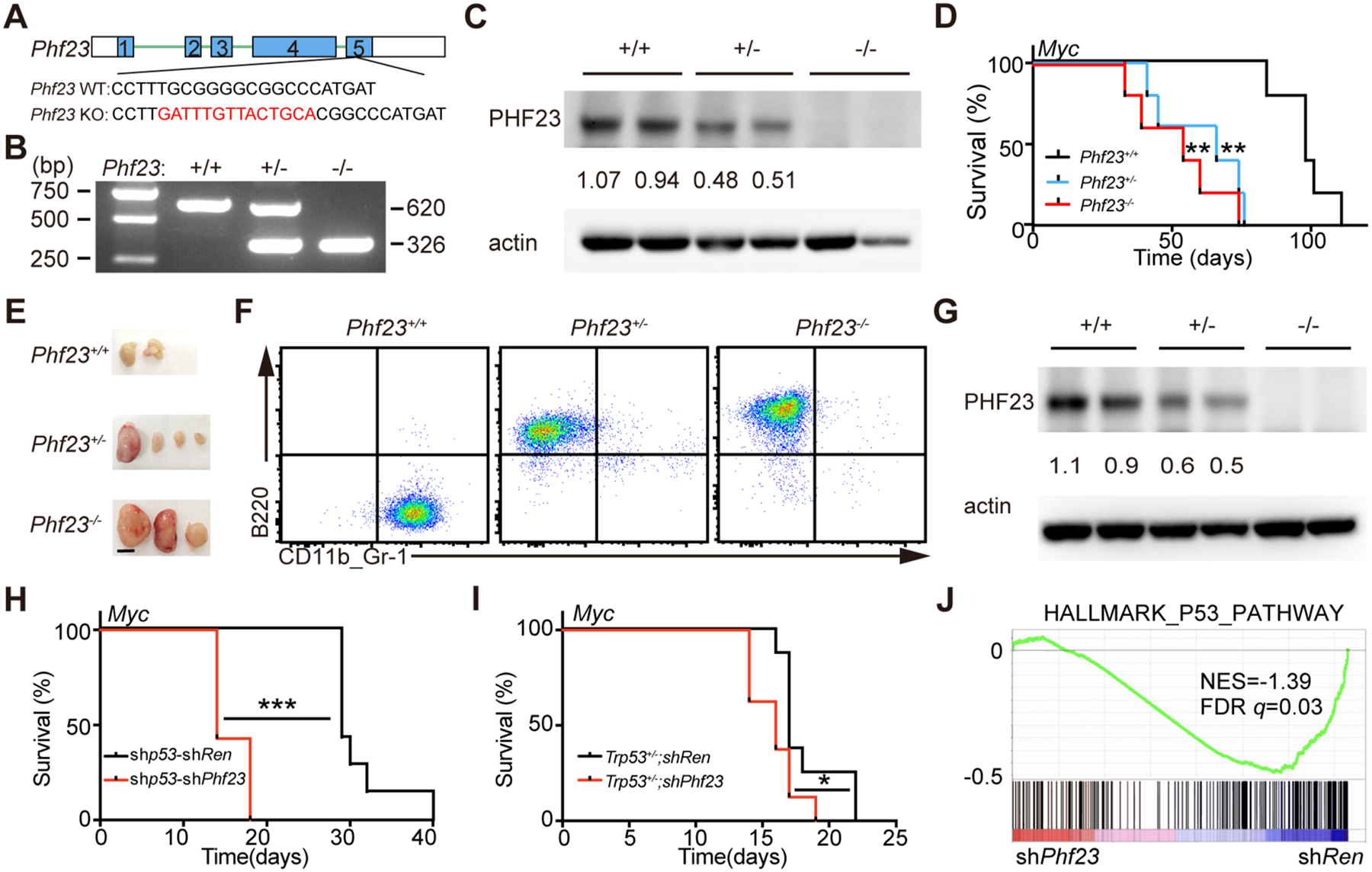
A, Diagram showing Phf23 mutation with a 14bp insertion on one allele introduced by CRISPR/Cas9 in exon 5 in mice. B, Representative picture showing genotyping of Phf23+/+, Phf23+/− and Phf23−/− mice. C, Representative Western blotting picture showing the protein levels of PHF23 in Phf23+/+, Phf23+/− and Phf23−/− E14.5 FLCs. D, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with Myc transduced E14.5 FLCs derived from Phf23+/+, Phf23+/− and Phf23−/− embryos. n=5 for each group. **p<0.01 (log-rank test). E, Representative pictures showing enlarged lymph nodes in recipient mice with Myc transduced Phf23+/− or Phf23−/− FLCs, but not Phf23+/+ FLCs in Fig. 2D. Scale bar, 5mm. F, Representative flow plots showing the expressions of B220 and CD11b/Gr-1 in Phf23+/+, Phf23+/− and Phf23−/− tumor cells from recipient mice in Fig. 2D. G, Western blotting of PHF23 in Phf23+/+;Myc, Phf23+/−;Myc and Phf23−/−;Myc lymphoma/leukemia cells from recipient mice in Fig. 2D. (Note: Phf23+/+;Myc lymphoma/leukemia cells had Trp53 mutations by CRISPR/Cas9). H, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with pre-B cells infected with Myc-linked tandem shRNAs. n=7, ***p<0.001 (log-rank test). I, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with p53+/− pre-B cells infected with Myc-linked shRen or shPhf23. *p<0.05 (log-rank test). J, GSEA showing the negative enrichment of the HALLMARK_P53_PATHWAY gene set in shPhf23 lymphoma/leukemia cells, comparing to shRen cells (NES=−1.39; FDR q=0.03).
Co-deleted TSGs through large chromosomal deletions have been suggested to be able to cooperate during tumorigenesis(8,13). Here, we wondered whether Phf23 deficiency would cooperate with Trp53 loss, arguably the most important TSG on chromosome 17p, to promote lymphomagenesis. While Trp53 knockdown promoted Myc-driven immature B lymphoblastic malignancy, co-knockdown of both Trp53 and Phf23 by shRNA (shp53-shPhf23) significantly shortened the latency from 29 days to 14 days (Fig. 2H). A similar cooperative effect between Trp53 and Phf23 deficiencies was observed with Trp53+/− pre-B cells (Fig. 2I). To explore the potential molecular mechanisms of the synergy of Phf23 and Trp53, RNA-seq analyses of shRen and shPhf23 B lymphoma /leukemia cells were performed. GSEA showed that the genes in p53 pathway were significantly negatively enriched in Phf23-deficient tumor cells, compared to shRen cells (Fig. 2J). Hence, loss of PHF23 and TP53 through 17p deletions may cooperate to drive tumorigenesis through syngenic repressions of the p53 pathway.
PHF23 co-located with H3K4me3 on gene promoters and is required for B cell differentiation
PHF23 contains a PHD domain at its C-terminus, which has been shown to specifically bind H3K4me3(20). In NUP98-PHF23 fusion-driven AML, NUP98-PHF23 binds a specific subset of H3K4me3 and regulates the expressions of multiple AML-associated genes(18,28). Here, we studied the functions of wildtype PHF23 in Ba/F3 cells, a mouse pro/pre-B cell line(29), by chromatin immunoprecipitation-sequencing (ChIP-seq) assay. We found that the majority (>70%) of PHF23-binding sites were located in gene promoters with a similar pattern as H3K4me3 peaks (Fig. 3A and Supplementary Fig. S3A). More than 90% of PHF23-binding peaks were overlapped with H3K4me3 peaks (Fig. 3B). H3K4me3 is commonly enriched on active promoters(30), and consistently the average expression levels of H3K4me3 binding genes were significantly higher than those without binding in Ba/F3 cells (31) (Fig. 3C). Interestingly, PHF23 binding was associated with further mildly but significantly increased expressions of genes (average log2 TPM of genes with both PHF23 and H3K4me3 binding vs. with only H3K4me3 binding: 3.46 vs 3.05, padj=3.90e-17; and those with only PHF23 binding vs. without binding: 2.21 vs. −3.28, padj=5.70e-61) (Fig. 3C). A similar gene expression pattern was also observed in freshly isolated pre-B cells (Supplementary Fig. S3B). Thus, PHF23 co-located with H3K4me3 and regulated gene activation.
Figure 3 |. PHF23 co-located with H3K4me3 in pre-B cells and was required for B cell differentiation.
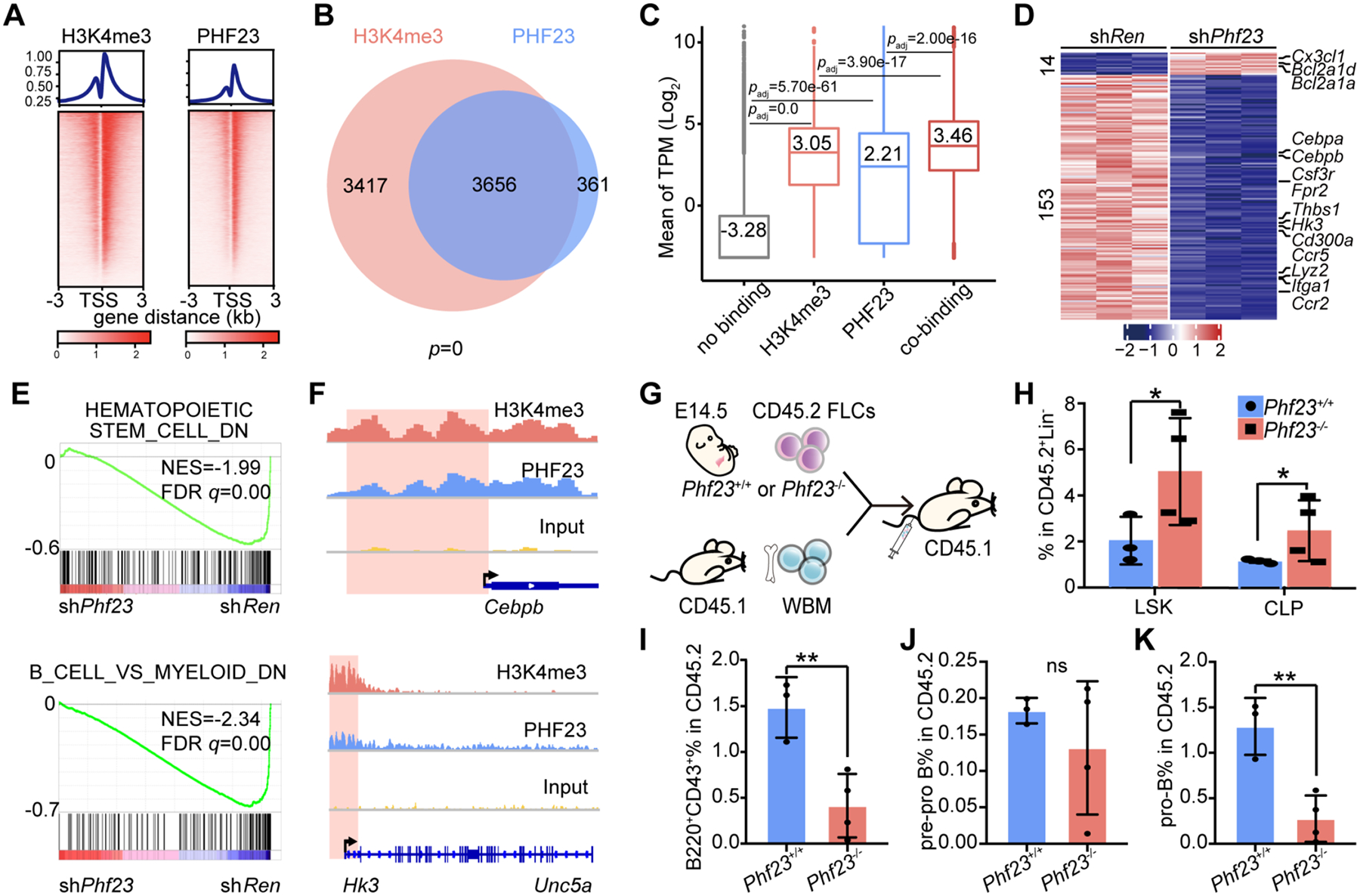
A, Top, profiles showing average H3K4me3 (left) and PHF23 (right) bound peak densities in TSS regions by ChIP-seq analyses. Bottom, heatmaps showing H3K4me3 and PHF23 bound peaks in TSS regions. B, Venn diagram showing overlapping of PHF23 and H3K4me3 bound genes; p, Fisher’s exact test. C, Mean log2 TPM (Transcripts Per Kilobase Million) of genes with no binding (without H3K4me3 and PHF23 binding), H3K4me3 only, PHF23 only or co-binding (both have H3K4me3 and PHF23 binding) in Ba/F3 cells. padj, Wilcoxon signed-rank test. D, Heatmap showing the differentially expressed genes (DEGs) (padj<0.05, log2 Fold change>1 or <−1) in shPhf23 pre-B cells compared to those with shRen. E, GSEA showing the negative enrichments of the HEMATOPOIETIC_STEM_CELL_DOWN (NES=−1.99; FDR q=0.00) and B_CELL_VS_MYELOID_DOWN gene sets (NES=−2.34; FDR q=0.00) in shPhf23 pre-B cells, comparing to shRen cells. F, Integrative Genomics Viewer (IGV) showing binding peaks of H3K4me3 and PHF23 on Cebpb (top) and Hk3 (bottom). G, Schematic diagram of the hematopoiesis analyses of Phf23+/+ and Phf23−/− FLCs. H, Percentages of donor-type LSK (Lin−ScaI+cKit+) and CLP (Lin−Sca1lowcKitlow) populations in the bone marrow of recipient mice 4 months after BMT. I, Percentages of donor-type B220+CD43+ populations in the bone marrow of recipient mice. J, Percentages of donor-type B220+CD43+CD24− (pre-pro B) populations in the bone marrow of recipient mice. K, Percentages of donor-type B220+CD43+ CD24+ (pro-B) populations in the bone marrow of recipient mice. H-K, *p<0.05, **p<0.01, ns, not significant (t-test).
Accordingly, there were more significantly downregulated genes than upregulated ones (153 vs. 14) in Phf23 knockdown pre-B cells, comparing to those in shRen cells (Fig. 3D and Supplementary Table S1). Consistent with the tumor suppression function of Phf23, upregulated genes in shPhf23 cells were enriched in pathways of cell survival and proliferation, such as negative regulation of apoptosis signaling pathway and positive regulation of ERK1 and ERK2 cascade, while the downregulated genes were enriched in pathways related to leukocyte differentiation and functions, suggesting potential roles of PHF23 in hematopoiesis (Supplementary Fig. S3C–D). GSEA showed that the HEMATOPOIETIC_STEM_CELL_DOWN gene set and B_CELL_VS_MYELOID_DOWN gene set were significantly negatively enriched in shPhf23 cells (NES=−1.99 and −2.34; FDR q=0.00 and 0.00, respectively) (Fig. 3E). Among the most downregulated genes, Cebpb and Hk3 had been shown to be important for B cell development and function (32,33) (Fig. 3F). Interestingly, the genes bound by PHF23 were significantly overlapped with those by NUP98-PHF23 (34) (Supplementary Fig. S3E). The overlapped genes included well-known tumor suppressor genes Cdkn1b, Cdkn2c, Nf1 and Fbxw7. These data suggested that PHF23 was required for the expressions of tumor suppressor and differentiation-related genes.
To directly test the function of Phf23 in B cell development, we transplanted Phf23+/+ or Phf23−/− hematopoietic stem and progenitor cells together with recipient-type BM cells for irradiation protection into lethally irradiated recipient mice. Given that the Phf23−/− mice were embryonically lethal, Phf23+/+ or Phf23−/− FLCs were used. (Fig. 3G). Four months later, the donor-type BM cells were analyzed. The percentages of both hematopoietic stem and progenitor cells (Lin−Sca1+cKit+, LSK) and common lymphoid progenitor cells (Lin−Sca1lowcKitlow, CLP) were significantly increased in the BM of recipient mice with Phf23−/− FLCs compared to those with Phf23+/+ FLCs (Fig. 3H and Supplementary Fig. S4A). However, the B progenitor cells (B220+CD43+) were significantly less in recipients with Phf23−/− FLCs than those with Phf23+/+ FLCs (Fig. 3I and Supplementary Fig. S4B). In more details, the Phf23−/− pre-pro B cells (B220+CD43+CD24−) were comparable to the wildtype, while the pro-B cells (B220+CD43+CD24+) were significantly reduced in recipients (1.2% vs. 0.2%) with Phf23−/− FLCs compared to those with wildtype FLCs (Fig. 3J–K and Supplementary Fig. S4C). Accordingly, both the percentages and cell numbers of Phf23−/− B220+ cells in BM and spleen were significantly less than those of wildtype (Supplementary Fig. S4D–F). Thus, Phf23 deficiency impaired B cell differentiation, which may contribute to the development of B malignancies.
Identifying a PHF23-SIN3-HDAC (PSH) complex mediated by the N terminus of PHF23
To further understand how PHF23 regulated gene expressions, we performed co-immunoprecipitation mass spectrometry (Co-IP/MS) assay to identify potential PHF23 binding partners. Interestingly, the top hits were SIN3 transcription regulator family member a and b (SIN3A and SIN3B) (Supplementary Table S2). SIN3 is a master scaffold protein in the SIN3-HDAC corepressor complex, which represses transcription and is involved in various biological and cellular processes(35,36). The SIN3-HDAC (histone deacetylase) complex contains multiple tightly associated components, and notably all of them were co-immunoprecipitated with PHF23 in our Co-IP/MS assay, including HDAC1 and HDAC2, the core catalytic proteins of this complex and other 9 factors SAP130, FAM60A, RBBP7, RBBP4, ARID4B, SUDS3, SAP30L, BRMS1L, SAP30 (Fig. 4A and Supplementary Table S2)(37,38). The interactions of PHF23 with SIN3A and HDAC1 were confirmed with Co-IP followed by Western blotting (Fig. 4B). The endogenous PSH complex was further validated by Co-IP with antibody against PHF23 (Fig. 4C). We noticed that ING2, another H3K4me3 reader which has been reported to bind the SIN3-HDAC complex and associated with gene repression(39,40), was not detected by PHF23 Co-IP/MS. These results suggested that PHF23 directly interacted with the SIN3-HDAC complex to form a PHF23-SIN3-HDAC (PSH) complex.
Figure 4 |. PHF23 directly bound and negatively regulated the SIN3-HDAC complex.
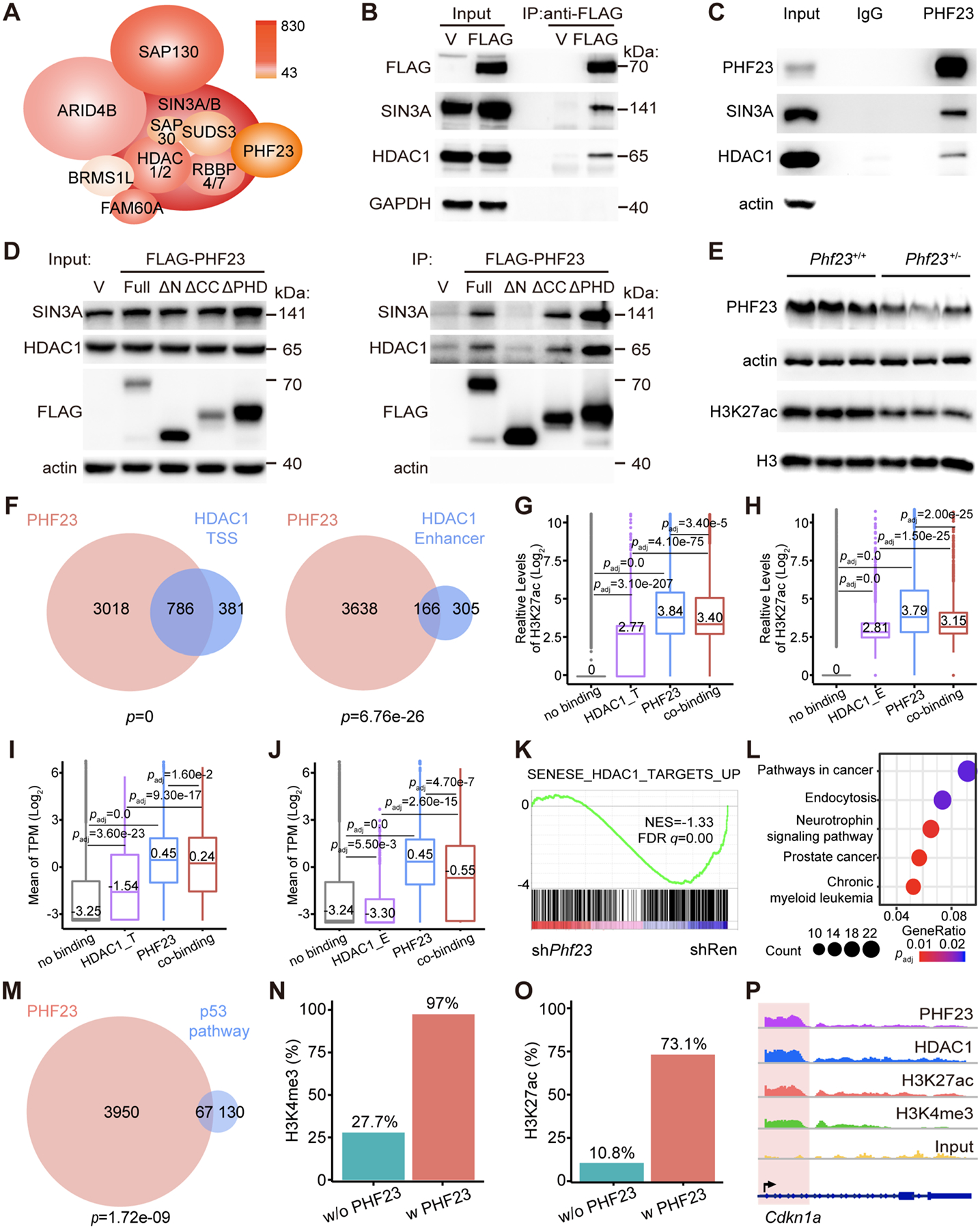
A, Schematic diagram of the PHF23-SIN3-HDAC (PSH) complex. Colors indicated the Mascot scores of each component measured by Co-IP of FLAG-PHF23 and LC/MS. B, Representative Western blotting pictures showing FLAG-PHF23, SIN3A and HDAC1 of input and FLAG-PHF23 Co-IP. C, Representative Western blotting pictures of endogenous PHF23, SIN3A and HDAC1 of input and Co-IP with IgG or PHF23 antibodies. D, Representative Western blotting pictures of FLAG-PHF23, SIN3A and HDAC1 of input and FLAG tagged full length or truncated PHF23 Co-IP. E, Representative Western blotting pictures of PHF23 and H3K27ac in Phf23+/+ and Phf23+/− FLCs. F, Venn diagrams showing overlapping of PHF23 and HDAC1 bound genes, analyzed by ChIP-seq. p, Fisher’s exact test. Left, HDAC1 bound at TSS; Right, HDAC1 bound at enhancer regions. G, Mean log2 H3K27ac levels on genes with no binding (without HDAC1 TSS and PHF23 binding), HDAC1_T (only HDAC1 at TSS), PHF23 only or co-binding (both HDAC1 and PHF23 binding at TSS) in Ba/F3 cells. padj, Wilcoxon signed-rank test. H, Mean log2 H3K27ac levels on genes with no binding (without HDAC1 enhancer and PHF23 binding), HDAC1_E (only HDAC1 at enhancer regions), PHF23 only or co-binding (both HDAC1_E and PHF23 binding). padj, Wilcoxon signed-rank test. I, Mean of log2 TPM of genes with no binding (without HDAC1 TSS and PHF23 binding), HDAC1_T (only HDAC1 at TSS), PHF23 only or co-binding (both HDAC1 and PHF23 binding at TSS) in immature B lymphoblastic malignancy cells. padj, Wilcoxon signed-rank test. J, Mean of log2 TPM of genes with no binding (without HDAC1 enhancer and PHF23 binding), HDAC1_E (only HDAC1 at enhancer regions), PHF23 only or co-binding (both HDAC1_E and PHF23 binding). K, GSEA showing the negative enrichment of the SENESE_HDAC1_TARGETS_UP gene set in shPhf23 pre-B cells, comparing to shRen cells (NES=−1.33; FDR q=0.00). L, Top 5 KEGG pathways enriched in genes co-bound by both PHF23 and HDAC1. M, Venn diagram showing overlapping of the PHF23 bound genes and the p53 pathway genes. p, Fisher’s exact test. N, Percentages of genes with H3K4me3 binding in p53 pathway genes with PHF23 binding or p53 pathway genes without PHF23 binding. O, Percentages of genes with H3K27ac binding in p53 pathway genes with PHF23 binding or p53 pathway genes without PHF23 binding. P, IGV plots showing PHF23, HDAC1, H3K27ac, and H3K4me3 binding density on Cdkn1a.
PHF23 contains multiple functional domains, including the PHD zinc finger domain at the C terminus that can bind H3K4me3 in a sequence-specific manner and a coiled-coil domain in the middle that might mediate protein-protein interactions, while the N terminus had been less characterized (20) (Supplementary Fig. S5A). We tested which region of PHF23 interacted with the SIN3-HDAC complex with PHF23 truncated mutants. Interestingly, we found that neither coiled-coil nor PHD domain truncations (ΔCC and ΔPHD) affected the binding of PHF23 with SIN3A and HDAC1. However, the N terminus truncation (ΔN) completely disrupted the interactions between PHF23 and SIN3A-HDAC1 (Fig. 4D). Thus, the N terminus of PHF23 was essential for the PSH complex.
The PSH complex coordinates H3K4me3 and H3K27ac for gene activation
Histone deacetylation is one of the gene regulation mechanisms, and here we focused on the deacetylation of H3K27ac, an active histone marker(30,41,42). To test the effect of the PSH complex on the H3K27ac, we measured the total H3K27ac levels of Phf23 deficient cells by Western blotting. As Figure 4E shown, H3K27ac were significantly reduced in Phf23+/− FLCs compared to that in Phf23+/+ FLCs. To further understand the regulation of PHF23 on the functions of the SIN3-HDAC complex, we analyzed the ChIP-seq data with HDAC1 or H3K27ac antibody (Supplementary Fig. S5B–C). Consistent with the interaction between PHF23 and the SIN3-HDAC complex, PHF23 binding sites significantly overlapped with HDAC1 binding peaks at both transcription start sites (TSS) and enhancer regions (Fig. 4F). Importantly, the binding of PHF23 significantly increased the H3K27ac levels, compared to those with only HDAC1 binding at both TSS and enhancer regions (Fig. 4G–H). 79.8% of TSS with both HDAC1 and PHF23 were associated with H3K27ac, while only 31.3% of HDAC1 bound TSS were associated with H3K27ac. Similar correlations were observed in the enhancer regions (Supplementary Fig. S5D). These results suggested that PHF23 repressed the deacetylation activity of HDAC1 on H3K27ac.
Increased H3K27ac levels are associated with the upregulation of target genes(30,42). To analyze the effect of the PSH complex on gene regulation, we analyzed RNA-seq data of shPhf23 tumor cells, compared to those with wildtype Phf23. The expression levels of genes associated with both PHF23 and HDAC1 were significantly higher than those with only HDAC1 in lymphoma/leukemias (Fig. 4I–J). A similar expression pattern was also observed in pre-B cells (Supplementary Fig. S5E–F). H3K27ac binding sites were largely overlapped with those of H3K4me3, and both of them were associated with higher gene expression levels of targets, compared to those without these histone markers (Supplementary Fig. S5G–H). Even among the genes with both H3K4me3 and H3K27ac, those with PHF23 binding had significantly higher expression levels than those without PHF23 binding (Supplementary Fig. S5I–J). Especially, GSEA showed that the SENESE_HDAC1_TARGETS_UP gene set was significantly downregulated in shPhf23 pre-B cells (NES=−1.33, FDR q=0.00), which was consistent with the interaction of PHF23 and the SIN3-HDAC complex (Fig. 4K). Thus, the PSH complex might be a critical regulator of gene expression in both pre-malignant and malignant cells.
The most enriched KEGG pathways in PHF23 and HDAC1 co-binding genes were pathways in cancer and leukemia (Fig. 4L). Because PHF23 was co-deleted with TP53 in human cancers with 17p deletions(3,8) and loss of both synergistically promoted tumorigenesis (Fig. 2H–J), we wondered if the PSH complex would directly regulate the p53 pathway. We found that 67 out of the 197 p53 target genes (p=1.72e-09) were bound by PHF23(43) (Fig. 4M). Consistent with the potential function of the PSH complex to link the two active histone markers H3K4me3 and H3K27ac, the majority of the PHF23-binding p53 target genes were also associated with H3K4me3 and H3K27ac (Fig. 4N–O and Supplementary Fig. S5K–L). As a result, the p53 pathway was significantly negatively enriched in shPhf23 lymphoma/leukemia cells, compared to those with shRen (Fig. 2J). Among these PSH target genes, were multiple well-known p53 targets, including Cdkn1a, Bax, and Mdm2. These genes were co-bound by PHF23 and HDAC1 and also associated with histone markers H3K4me3 and H3K27ac in Ba/F3 cells (Fig. 4P and Supplementary Fig. S5M–N). Taken together, these biochemical and multi-omics data suggested that the PSH complex might link the two active histone markers, H3K4me3 and H3K27ac, for synergistically regulating gene activation, including the p53 pathway genes.
The PSH complex is critical for tumorigenesis and maintenance
Given the importance of the PSH in histone modifications and gene regulation, we wondered whether this complex played significant roles in lymphoma/leukemia genesis and maintenance. SIN3A and its associated HDAC1 and HDAC2 have been shown to be required for normal hematopoiesis(44). We found that knockdown of Sin3a completely blocked the development of Phf23+/−;Myc lymphoma/leukemia (Fig. 5A–B). Similarly, Sin3b deficiency also significantly extended the tumor-free survival of mice transplanted with Ph23+/−;Myc FLCs cells (Fig. 5C–D). The latencies of Ph23+/−;Myc lymphoma/leukemia were correlated with the knockdown efficiencies of shSin3b. These in vivo functional studies suggested that the SIN3-HDAC complex would be essential for lymphomagenesis driven by Phf23 loss.
Figure 5 |. The PSH complex dysregulation was critical for tumorigenesis and maintenance.
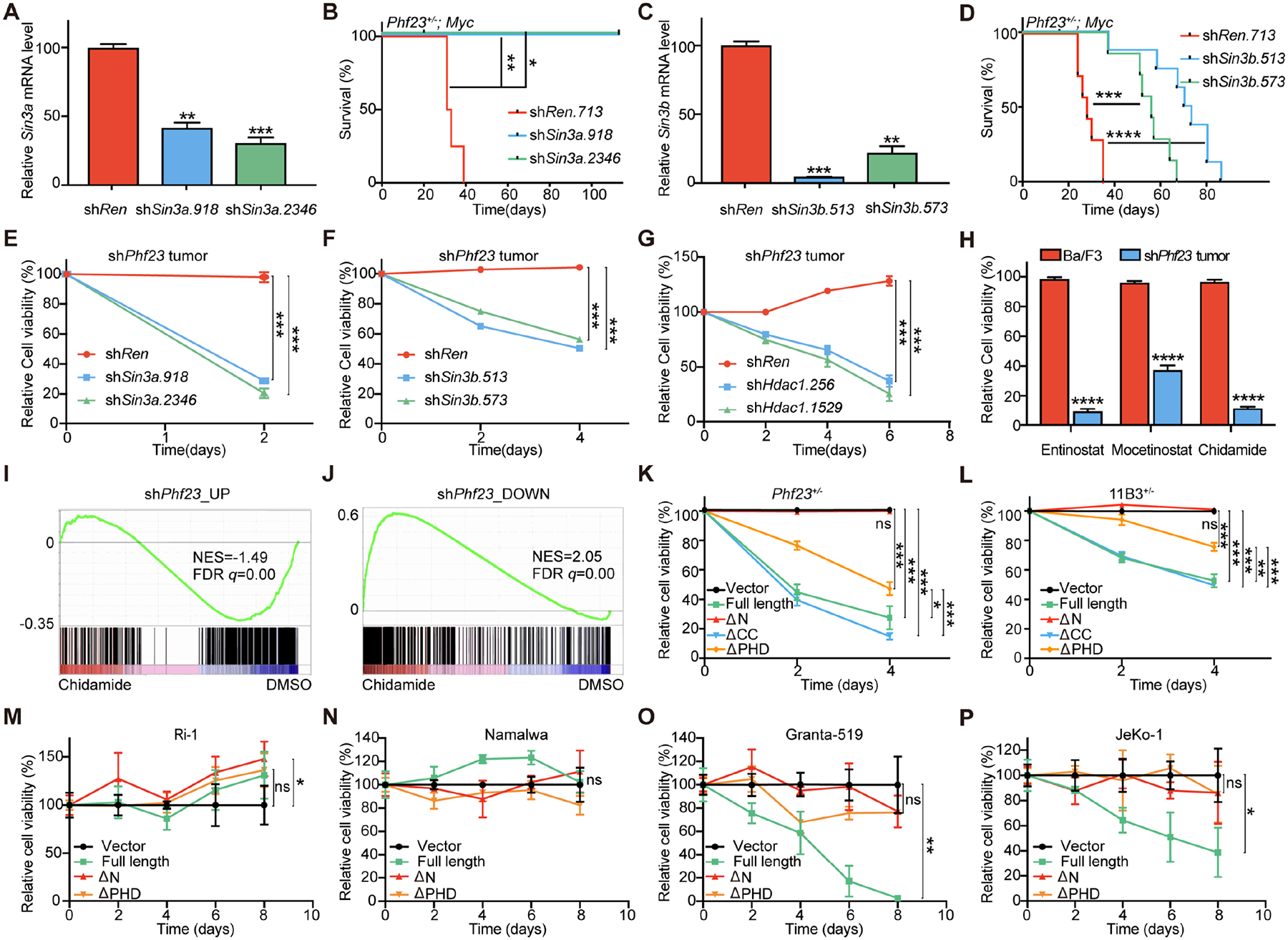
A, Knockdown efficiencies of shSin3a, measured by qPCR. B, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with Phf23+/− FLCs with Myc-linked shSin3a or shRen. n=4. *p<0.05, **p<0.01 (log-rank test). C, Knockdown efficiencies of shSin3b measured by qPCR. D, Kaplan-Meier tumor-free survival curves of recipient mice transplanted with Phf23+/− FLCs infected with Myc-linked shSin3b or shRen. n=8. ***p<0.001, ****p< 0.0001 (log-rank test). E, Relative cell viabilities of shPhf23 lymphoma/leukemia cells with shSin3a. n=3. F, Relative cell viabilities of shPhf23 lymphoma/leukemia cells with shSin3b. n=3. G, Relative cell viabilities of shPhf23 lymphoma/leukemia cells with shHdac1. n=3. H, Relative cell viabilities of Ba/F3 cells and shPhf23 lymphoma/leukemia cells treated with HDAC inhibitors (entinostat, 0.5μM; mocetinostat, 0.1 μM; chidamide, 0.5 μM). n=3. I, GSEA showing the negative enrichment of the shPhf23 upregulated gene set in chidamide treated shPhf23 lymphoma/leukemia cells, comparing to DMSO treated cells (NES=−1.49; FDR q=0.00). J, GSEA showing the positive enrichment of the shPhf23 downregulated gene set in chidamide treated shPhf23 lymphoma/leukemia cells, comparing to DMSO treated cells (NES=2.05; FDR q=0.00). K, Relative cell viabilities of Phf23+/−;Myc lymphoma/leukemia cells transduced with full-length or truncated Phf23. n=3. L, Relative cell viabilities of chromosome 11B3 deleted lymphoma/leukemia cells transduced with full-length or truncated Phf23. n=3. M, Relative cell viabilities of 17p intact Ri-1 human lymphoma cells transduced with full-length or truncated PHF23. n=3. N, Relative cell viabilities of 17p intact Namalwa human lymphoma cells transduced with full-length or truncated PHF23. n=3. O, Relative cell viabilities of 17p deleted Granta-519 human lymphoma cells transduced with full-length or truncated PHF23. n=3. P, Relative cell viabilities of 17p deleted JeKo-1 human lymphoma cells transduced with full-length or truncated PHF23. n=3. A, C, E-H and K-P, *p<0.05, **p<0.01, ***p<0.001, ns, not significant (unpaired two-tailed t-test).
Further, we went to test the potential roles of the PSH complex in tumor maintenance. We reduced the expression of Sin3a by shRNA in shPhf23 lymphoma/leukemia cells and as well as Ba/F3 cells, which worked as a control to rule out generally cytotoxicity of Sin3a deficiency. The results showed that Sin3a knockdown specifically impaired the lymphoma/leukemia cell growth with Phf23 loss, but the mild effect on Ba/F3 cells (Fig. 5E and Supplementary Fig. S6A). Sin3b suppression had a similar effect on shPhf23 lymphoma/leukemia cells as Sin3a deficiency, but little effect on untransformed Ba/F3 cells (Fig. 5F and Supplementary Fig. S6B). Further, Hdac1 shRNAs significantly reduced the survival of Phf23 deficient lymphoma/leukemia than shRen (Fig. 5G). HDAC inhibitors, entinostat, mocetinostat and chidamide, specifically induced the death of shPhf23 lymphoma/leukemia cells but not the control Ba/F3 cells at the given concentrations, suggesting a potential therapeutic window of these small molecules for Phf23 deficient cancers (Fig. 5H).
To explore the molecular effects of HDAC inhibitors on Phf23 deficient tumor cells, we performed RNA-seq analyses of shPhf23 lymphoma/leukemia cells treated with or without entinostat or chidamide. Interestingly, the upregulated genes in HDAC inhibitors-treated cells significantly outnumbered the down-regulated genes, just opposite to those in shPhf23 cells (35 and 176 genes upregulated and downregulated, respectively, by shPhf23 vs. 384 and 33 genes upregulated and downregulated, respectively, by HDAC inhibitors. Log2 FoldChange>1 or <−1, padj<0.05) (Supplementary Fig. S6C–D and Supplementary Table S3–4). GSEA showed that the upregulated genes in shPhf23 cells were significantly negatively enriched in lymphoma/leukemia cells treated with either chidamide or entinostat (chidamide: NES=−1.49, FDR q=0.00; entinostat: NES=−1.34, FDR q=0.04), while the downregulated genes in shPhf23 cells were significantly positively enriched (chidamide: NES=2.05, FDR q=0.00; entinostat: NES=1.89, FDR q=0.00) (Fig. 5I–J and Supplementary Fig. S6E). Thus, treatment with HDAC inhibitors reversed the effects of Phf23 loss on gene regulation.
Because the N terminus of PHF23 was the key domain mediating the formation of the PSH complex, we introduced full-length or other PHF23 mutants into Phf23 deficient tumor cells. Here, we used Phf23+/− lymphoma/leukemia cells instead of shPhf23 cells to avoid the interference between shRNAs and exogenous cDNAs. Full-length PHF23 specifically repressed Phf23+/− lymphoma/leukemia while did not exhibit significant effect on Ba/F3 cells, which further confirmed Phf23 as a bona fide TSG (Fig. 5K and Supplementary S6F). In contrast, ΔN PHF23 completely lost its tumor suppression function, which strongly suggested that dysfunction of the PSH complex, mediated by the N terminus of PHF23, was required for tumor maintenance. ΔPHD PHF23 had a significantly reduced repression effect on Phf23+/− lymphoma/leukemia, indicating that the H3K4me3 binding activity was required for the tumor suppression function of PHF23, while ΔCC had the minimal effect (Fig. 5K). Taken together, these results indicated that deficiency of the PSH complex was critical for the maintenance of tumors with Phf23 loss.
Given the large number of genes located on chromosome 17p and the seemingly dominant effect of TP53, we wondered if the PSH complex played significant roles in del(17p) cancers. We transduced full-length, ΔN, ΔCC and ΔPHD PHF23 into mouse immature B lymphoblastic malignancy cells with deletion of chromosome 11B3, which highly resembled human cancers with 17p deletions(8). PHF23 restoration significantly reduced the survival of chromosome 11B3 deleted tumor cells, emphasizing that PHF23 significantly contributed to the pathology of chromosome 17p deleted cancers (Fig. 5L). Further, ΔPHD PHF23 had impaired tumor suppression function on chromosome 11B3 deleted lymphoma/leukemia, suggesting the importance of the PSH complex for the function of chromosome 17p. To further validate the functions of the PSH complex in human cancers, we transduced wildtype or mutant PHF23 into human lymphoma cells with or without 17p deletions. We found that wildtype PHF23 significantly inhibited the growth of 17p deleted Granta-519 and JeKo-1 cells, while it had no effect on 17p intact Ri-1 and Namalwa cells (Fig. 5M–P). Further, consistent with our findings in murine lymphoma/leukemia cells, ΔN PHF23 completely lost its tumor suppression activity. These data strongly suggested that PHF23 deficiency significantly contributed to the pathology of del(17p) cancers, which gave rise to an unexpected susceptibility for treatments targeting the dysregulated PSH complex.
Taken together, we identified PHF23 as a 17p TSG, loss of which was critical for both tumorigenesis and tumor maintenance and significantly contributed to the pathology of 17p deleted cancers. PHF23 colocated with active histone marker H3K4me3, directly bound and repressed the SIN3-HDAC complex through its N terminus. The PSH complex provided a synergistic link between H3K4me3 and H3K27ac for gene activation, especially TSGs and differentiation genes (Fig. 6A–B).
Figure 6 |. Schematic diagram showing the working model for the PSH complex in normal and tumor cells.
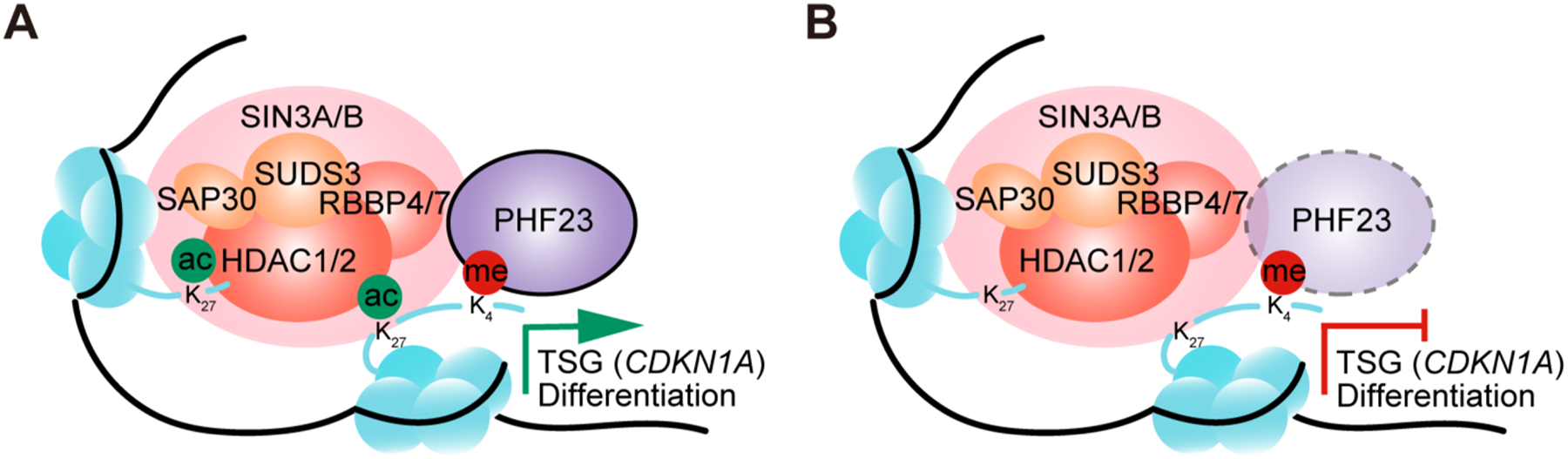
A, Schematic diagram showing the working models of the PSH complex in B cell differentiation and TSG regulation. B, Schematic diagram showing the working models of the PSH complex in tumor cells with PHF23 deficiency.
Discussion
Given the prevalence of CNVs in human cancers and their association with poor prognosis, it is important to understand the underlying mechanisms of CNVs in tumorigenesis and treatment. In this study, we identify PHF23 on chromosome 17p13.1 as a haploinsufficient TSG with multiple in vivo functional genetic strategies in this study. Remarkably, PHF23 deficiency always happens together with chromosome 17p loss while there are very few independent mutations in human cancers, we wonder that PHF23 loss would contribute to tumorigenesis in the context of loss of other co-occurring 17p genes, especially TP53. we demonstrate that PHF23 can cooperate with TP53, which is generally co-deleted with PHF23 in human cancers, through co-repressing the expressions of their common downstream target genes. We propose that, while haploinsufficiencies of either PHF23 or TP53 would have mild effects on the p53 pathway, the cooperation of both on the same pathway may have synergistic effects to give rise to significant repression of this pathway and thus promote tumorigenesis. This model would help to explain the significance of 17p deletions and potentially other CNVs with multiple TSGs, as a whole, during tumorigenesis. Altogether, our studies on PHF23 provide an example for identifying other potential TSGs and exploring their mechanisms involved in CNVs in human cancers.
PHF23 has been proposed to be an H3K4me3 reader(20), our study demonstrates that PHF23 can form a new PSH complex together with the SIN3-HDAC complex, an eraser of H3K27ac. PHF23 directly binds with the SIN3-HDAC complex through its N terminus and restrains its deacetylation capacity on H3K27ac. Hence, the PSH complex provides a direct link between the two active histone markers for their synergistic regulation on gene activation. The PSH complex is distinguishable from the ING2-SIN3-HDAC complex(39,40). ING2, also a PHD finger protein and H3K4me3 reader, directly binds and enhances the deacetylation activity of the SIN3-HDAC complex, which leads to gene repression(39). It would be interesting to dissect the potential competitions between the PSH complex and the ING2-SIN3-HDAC complex for H3K4me3 sites for gene regulation and their paradoxical potential cooperations for biological processes including tumorigenesis.
It is intriguing that, in contrast to the haploinsufficient loss of PHF23 in human cancers with 17p deletions, PHF23 is also involved in AML through chromosome translocation to form an oncofusion protein NUP98-PHF23(8,18,19). NUP98-PHF23 binds to specific H3K4me3 sites and drives an AML program with upregulated HOX genes. Paradoxically, PHF23 loss in del(17p) cancers seems to have dysregulation of the SIN3-HDAC complex and results in decreased expressions of B-cell differentiation genes and other TSGs. Therefore, distinct mechanisms underly different PHF23 genetic alterations in AML and lymphoma. We believe that the caveat might be the N terminus of PHF23, which is critical for the formation of the PSH complex and the tumor suppression function of PHF23. The N terminus of PHF23 is deleted in AML-associated NUP98-PHF23(18,19), which can also at least partially contribute to the pathology of human AML. Interestingly, the translocation of NUP98-PHF23 results in not only an oncogenic fusion protein (NUP98-PHF23), but also loss of one copy of NUP98 and PHF23. It is very interesting to test whether the disruption of one allele of PHF23 in NUP98-PHF23 AML would contribute to the disease. Our studies, together with others, illustrate the complexity of the molecular mechanisms underlying genetic abnormalities in human cancers.
Importantly, we show that, though only one of the several hundreds genes involved in del(17p), PHF23 haploinsufficiency is required for the maintenance of human cancers with 17p deletions, which suggests unexpected susceptibility for these refractory diseases. Future works on testing whether HDAC inhibitors and other potential drugs related to the PSH complex would be beneficial for patients in clinical practice.
Materials and Methods
Mice.
All mice experiments were approved by the Institutional Animal Care and Use Committees of Sichuan University. Eμ-Myc mice were from Jackson Laboratories(22). Pre-B cells were enriched from 6–8-week-old C57BL/6 (Charles River, Cat# 219) bone marrow by autoMACS purification with Biotin anti-mouse/human CD45R/B220 (BioLegend Cat# 103204, RRID:AB_312989) and anti-Biotin microbeads (Miltenyi Biotec Cat# 130-090-485, RRID:AB_244365). Purified pre-B cells were cultured in RPMI1640 medium supplemented with 20% FBS and 10ng/ml IL-7 (R&D Cat# 407-ML-200). For in vivo tumorigenesis, pre-B cells or E13.5 FLCs were transduced with retroviruses carrying shRNA or CRISPR/Cas9, respectively, and injected (i.v.) into sub-lethally irradiated (4.5Gy) C57BL/6 recipient mice (6–8-week old, male). All recipient mice were randomly divided into each group before transplantation and monitored twice per week by palpation. The tumor monitoring process was done as blinded experiments. The immunophenotypes of resulting lymphomas were analyzed by flow cytometry using antibodies purchased from Biolegend. Statistical analysis of all survival data was accomplished with the log-rank test from Prism 8.
Phf23 knockout mouse was generated in the Genetically Engineered Mouse Facility of State Key Laboratory of Biotherapy with CRISPR/Cas-mediated gene editing technology as described before(45). Briefly, Phf23 sgRNA (targeting sequence: ATCATGGGCCGCCCCGCAA) and Cas9 mRNA were delivered into C57BL/6 zygotes by microinjection, followed by transplanted into the uterus of surrogate CD-1(ICR) female mice. A 14bp insertion leading to a premature stop codon was occurred at the sgRNA targeted sites. The resulting Phf23 knockout mouse has been backcrossed to wildtype C57BL/6 mouse for more than 8 generations to get rid of potential off-target mutations.
Bone marrow transplantation.
The bone marrow cells from femurs and tibia of CD45.1 donor mice and E14.5 FLCs harvested from CD45.2 Phf23 knockout mice were mixed and transplanted into lethally irradiated CD45.1 C57BL/6 recipient mice. Four months after transplantation, recipient mice were sacrificed for analyses.
Retroviral constructs.
MirE-based shRNAs were cloned into retroviral constructs including MSCV-shRNA-SV40-Myc-IRES-GFP, MSCV-shRNA-IRES-mCherry and MSCV-shRNA-PGK-Puro-IRES-GFP as previously reported(8,13,25). The sequences of shRNAs targeting Renilla or Trp53 are from published reports(8,13,25) and the sequences of shRNAs targeting Phf23, Sin3a, Sin3b and Hdac1 are listed in Supplementary Table S5. sgPhf23: ATCATGGGCCGCCCCGCAA. Retrovirus packaging and infection were done as previously reported(8,13,25).
Cell line culture.
Ba/F3 (#HB-283), HeLa (#CCL-2), 293T (#CRL-1573), JeKo-1 (#CRL-3006), and Namalwa (#CRL-1432) were from ATCC, and Granta-519 (#ACC342) and Ri-1 (#ACC585) were from DSMZ. The 17p status of JeKo-1, Granta-519, Ri-1 and Namalwa have been confirmed by qPCR. All human lymphoma cell lines were cultured at 37 °C with 5% CO2 in RPMI-1640 medium supplemented with 10% v/v fetal bovine serum (FBS) and penicillin (100 units/ml)/streptomycin (0.1 mg/ml). Ba/F3 cells were maintained in RPMI-1640 medium supplemented with 10% v/v fetal bovine serum (FBS) and 2ng/ml IL3 (R&D Cat# 403-ML-050). All cell lines were routinely tested for mycoplasma by PCR. Experiments were performed within four weeks after fresh viable cells were thawed.
Co-immunoprecipitation and immunoblotting.
3xFLAG-tagged full-length, ΔN (2–119 aa deleted), Δcoiled-coil (225–283 aa deleted) and ΔPHD domain (331–393 aa deleted) Phf23 cDNA were cloned into retroviral construct MSCV-IRES-mCherry, and transduced into Ba/F3 cells. mCherry+ cells were sorted out by FACS (Aria III, BD Biosciences). Then, the whole cell lysates were extracted with cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitor cocktail (Roche) and incubated with anti-FLAG M2 mAb-conjugated agarose beads (Sigma-Aldrich Cat# A2220, RRID:AB_10063035). For endogenous PHF23 co-immunoprecipitation, 107 Hela cells were lysed and incubated with IgG or PHF23 antibody. The horseradish peroxidase (HRP)-conjugated FLAG antibody was purchased from Sigma (Sigma-Aldrich Cat# A8592, RRID:AB_439702), the anti-SIN3A antibody was from Thermo Fisher Scientific (Cat# PA1–870, RRID:AB_2187625), the anti-HDAC1 antibody was from Cell Signaling Technology (Cat# 34589, RRID:AB_2756821), the anti-PHF23 antibody, recognizing its C-terminus, was from Bethyl (Cat# A302–320A, RRID:AB_1850230) for Co-IP and western blotting, the anti-IgG antibody was from Abcam (Cat# ab171870, RRID:AB_2687657), the anti-GAPDH antibody was from BioXcell (BX-008) and horseradish peroxidase (HRP)-conjugated actin antibody from Thermo Fisher Scientific (Cat# MA5–32540, RRID:AB_2809817).
Liquid chromatography-mass spectrometry (LC-MS).
The preparation of samples for mass spectrometric analysis was conducted according to procedures reported previously(46). In brief, the immunoprecipitated protein products were prepared as described above and eluted with 0.1% trifluoroacetic acid (TFA). After condensed and dried in the speed vacuum, the lyophilized protein mixtures were digested into peptides using sequence grade trypsin resolved in triethylammonium bicarbonate (TEAB). The tryptic peptides were further reduced by 10mM Dithiothreitol (DTT) at 56°C for 1h, alkylated with 55mM iodoacetamide (IAA) in the dark at room temperature for additional 45min, again dried in the speed vacuum. The lyophilized peptides were desalted with C18 ZipTip (Millipore). Firstly, the ZipTips were equilibrated successively with 100% acetonitrile (ACN), 50% ACN/ddH2O and 0.1% fluoroacetic acid (FA). The peptides resolved in approximately 20ul 0.1% FA were then loaded onto ZipTips and washed extensively with 0.1% FA and eventually eluted with 50% ACN/ddH2O. The eluates were analyzed by a Q-Exactive Plus instrument available at the State Key Laboratory of Biotherapy.
Quantitative PCR.
All primers used for qPCR analysis are listed in Supplementary Table S6. To detect the knockdown efficiencies of shRNAs targeting Phf23/Sin3a/Sin3b/Hdac1, Ba/F3 cells were infected with MSCV-mirE-PGK-Puro-IRES-GFP retroviruses, followed by selected with 1ug/ml puromycin (Gibco). For detection of Phf23 expression level in eventual malignant cells, tumor cells were isolated from lymph nodes of diseased mice once upon sacrifice. shTrp53;Myc lymphoma/leukemia cells were used as control. For qPCR, RNA was isolated with Trizol, cDNA was synthesized with M-MLV (Invitrogen), SYBR Green PCR Master Mix (Applied Biosystems) was used and qPCR was performed on a Quantstudio3 (Applied Biosystems).
In vitro drug response assays.
Lymphoma/leukemia cell lines generated from Phf23 knockdown or 11B3 deletion tumor-bearing mice were cultured in BCM medium (45% DMEM, 45% IMDM, 10% FBS, 2mM glutamine, 50μM β-mercaptoethanol, penicillin (100 units/ml)/streptomycin (0.1 mg/ml)) and were plated in 96-well plates with a cell density of 10000 cells per well. The plated cells were treated with the indicated concentrations of HDAC inhibitors (Selleck) or DMSO. The number of living cells was counted by BD Accuri C6.
Rescue assay.
Tumor cells were infected with the indicated retroviruses and the percentages of mCherry+ populations were measured every other day with LSRFortessa flow cytometry. And the cell numbers were counted by BD Accuri C6 every other day. The relative cell viability of mCherry+ or GFP+ cell was calculated by dividing by the percentages of those with empty vectors.
RNA-seq analysis.
Pre-B cells isolated from mouse bone marrow were firstly infected with eMLM-shRen or eMLM-shPhf23 respectively, 48h after infection, the GFP positive cells were sorted out by FACS AriaIII for RNA-sequencing. Phf23 knocked down or control shRen lymphoma/leukemia cells were harvested from enlarged lymph nodes of recipient mice for RNA-seq. shPhf23 lymphoma/leukemia cells treated with HDAC inhibitors, chidamide (0.5 μM), entinostat (0.5 μM) or DMSO as a control for 12 hours were harvested for RNA-seq. RNA-seq libraries were prepared by NEBNext® Ultra™ RNA Library Prep Kit for Illumina®, Lymphoma/leukemia cells’ RNA-seq and HDAC inhibitors treated cells’ RNA-seq were sequenced by Illumina NovaSeq 6000 sequencing machine with 150-bp paired-end reads, pre-B RNA-seq were sequenced by BGISEQ500 sequencing machine with 50-bp single-end reads. The RNA-seq reads were aligned to the reference genome (GRCm38) by STAR_2.6.0a. Transcript abundance was normalized and measured by Transcripts Per Kilobase Million (TPM). Differential gene expression was analyzed by DESeq2 (RRID:SCR_000154). Genes with an absolute fold change greater than 1 and padj≤0.05 were counted as differentially expressed genes. The heatmaps of differentially expressed genes were done by pheatmap (RRID:SCR_016418) and normalized by z-score. The TPM data was used for GSEA. To identify functional categories of differentially expressed genes, Gene Oncology (GO) enrichment analysis was performed using the R package clusterProfiler (RRID:SCR_016884).
Chromatin immunoprecipitation (ChIP)-seq.
ChIP assays were performed according to a modified Micrococcal nuclease (MNase)-based ChIP-seq protocol reported previously(47). Briefly, 10 million Ba/F3 cells stably expressing flag tagged Phf23 were harvested, resuspended in PBS, and cross-linked in 4% formaldehyde solution. Nuclei were then washed, followed by digested using MNase (NEB Cat# M0247S, 2000 gel units/ul) and then the samples were sonicated in the Diagenode Bioruptor (30s on, 30s off for 15 cycles). Incubation with the indicated antibodies was performed overnight at 4°C. FLAG M2 mAb-conjugated agarose beads and HDAC1 antibody were mentioned above and H3K4me3 (Cat# ab8580, RRID:AB_306649) and H3K27ac (Cat# ab4729, RRID:AB_2118291) antibodies were from Abcam. DNA was eluted with 100 ul ChIP elution buffer, followed by purification with Qiagen MinElute PCR Purification Kit and sequenced by Novogene. Adaptor sequences were trimmed for all paired-end reads and mapped against the mm10 reference genome with STAR_2.6.0a. Bam files were transformed to bigwig files by Deeptools (RRID:SCR_016366)(48), and normalized by BPM. Integrative Genome Viewer (IGV) visualization was used for peaks visualization. The heatmap and Average Profile of chip peaks were performed by Deeptools. Peaks were called by macs2 2.1.1 with qvalue<0.01. The DiffBind (RRID:SCR_012918) was used to annotation the peaks, only the genes binding in both biological repeats will be counted. Venn diagrams were generated with the R package of VennDiagram (RRID:SCR_002414). The overlap genes were as an input list to calculate enrichment score in all gene sets of KEGG (RRID:SCR_012773) pathways via clusterProfiler.
Statistical analyses.
Statistical test methods, sample sizes, and p values were indicated in the corresponding figure legends. Statistical analyses were performed using GraphPad Prism (RRID:SCR_002798). The log-rank test was used to compare survival differences among groups for Kaplan-Meier tumor-free survival curve. Data were tested for statistical significance by unpaired two-tailed t-test, except the data in Fig. 3H–K and Supplementary Fig. S4E–F were tested by unpaired one-tailed t-test. The relative protein gray scale was analyzed by ImageJ (RRID:SCR_003070). p values for the Venn diagram overlap analysis were based on hypergeometric tests. Box plots were generated using ggpubr with the horizontal center lines denoting the median, box edges denoting the interquartile range (IQR). padj were calculated by Wilcoxon signed-rank test and adjusted by holm.
Data availability.
The RNA-seq and ChIP-seq datasets can be accessed under GEO accession number GSE145190. The process files and code can be found in GitHub (https://github.com/CCLYLabBioinformatics/epigenetic-mechanism-of-PHF23)
Supplementary Material
Significance.
We identify PHF23, encoding an H3K4me3 reader, as a new TSG on chromosome 17p, which is frequently deleted in human cancers. Mechanistically, PHF23 forms an unprecedented histone-modifying complex, the PHF23-SIN3-HDAC (PSH) complex, which regulates gene activation through a synergic link between H3K4me3 and H3K27ac.
Acknowledgments
We thank Dr. Yuquan Wei for generous support. We thank Dr. Junhong Han and Dr. Dan Xie for their invaluable advices and help on ChIP-seq. We thank Dr. Yinglan Zhao and Ruizhan Tong for their technical support. We thank all the lab members in the Chen and Liu laboratory for their insightful advice and kindly support. This work was supported by the National Key R&D Program of China (2017YFA0505600), the National Natural Science Foundation of China (81670182, 81770157, 81522003, 81570150) and the Sichuan Science and Technology Program (2018JZ0077, 2017TJPT0005).
Footnotes
Declaration of interests: The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature 2020;578(7793):82–93 doi 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature 2020;578(7793):122–8 doi 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578(7793):112–21 doi 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zabarovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 2002;21(45):6915–35 doi 10.1038/sj.onc.1205835. [DOI] [PubMed] [Google Scholar]
- 6.Kallioniemi A, Kallioniemi OP, Piper J, Tanner M, Stokke T, Chen L, et al. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci U S A 1994;91(6):2156–60 doi 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez MA, Ford RJ, Goodacre A, Selvanayagam P, Cabanillas F, Deisseroth AB. Chromosome 17- and p53 changes in lymphoma. Br J Haematol 1991;79(4):575–82 doi 10.1111/j.1365-2141.1991.tb08084.x. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016;531(7595):471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171(2):481–94 e15 doi 10.1016/j.cell.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science (New York, NY) 2012;337(6090):104–9 doi 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013;155(4):948–62 doi 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, et al. A tumour suppressor network relying on the polyamine–hypusine axis. Nature 2012;487(7406):244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68(4):820–3 doi 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 2001;27(2):172–80 doi 10.1038/84808. [DOI] [PubMed] [Google Scholar]
- 16.Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res 1998;58(11):2339–42. [PubMed] [Google Scholar]
- 17.Reader JC, Leng Q, Rassool FV, Ning Y. Regulation of differentiation by a PHD domain in the NUP98-PHF23 fusion protein. Leukemia research 2010;34(8):1094–7. [DOI] [PubMed] [Google Scholar]
- 18.Gough SM, Lee F, Yang F, Walker RL, Zhu YJ, Pineda M, et al. NUP98–PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer discovery 2014;4(5):564–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reader J, Meekins J, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t (11; 17)(p15; p13) in acute myeloid leukemia. Leukemia 2007;21(4):842–4. [DOI] [PubMed] [Google Scholar]
- 20.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009;459(7248):847–51 doi 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa M, Tsuzuki S, Honma K, Taguchi O, Seto M. Synergistic effect of Bcl2, Myc and Ccnd1 transforms mouse primary B cells into malignant cells. Haematologica 2011;96(9):1318–26 doi 10.3324/haematol.2011.041053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams J, Harris A, Pinkert C, Corcoran L, Alexander W, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985;318(6046):533–8. [DOI] [PubMed] [Google Scholar]
- 23.Kato MV, Shimizu T, Ishizaki K, Kaneko A, Yandell DW, Toguchida J, et al. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer letters 1996;106(1):75–82. [DOI] [PubMed] [Google Scholar]
- 24.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hosteller R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989;342(6250):705–8. [DOI] [PubMed] [Google Scholar]
- 25.Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer cell 2014;25(5):652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNerney ME, Brown CD, Wang X, Bartom ET, Karmakar S, Bandlamudi C, et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood 2013;121(6):975–83 doi 10.1182/blood-2012-04-426965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 2005;106(7):2452–61. [DOI] [PubMed] [Google Scholar]
- 28.Ho H, Skaist AM, Pallavajjala A, Yonescu R, Batista D, Wheelan SJ, et al. NUP98-PHF23 fusion is recurrent in acute myeloid leukemia and shares gene expression signature of leukemic stem cells. Leukemia research 2016;45:1–7. [DOI] [PubMed] [Google Scholar]
- 29.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol 2007;19(1):55–60 doi 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 30.Jenuwein T, Allis CD. Translating the histone code. Science (New York, NY) 2001;293(5532):1074–80. [DOI] [PubMed] [Google Scholar]
- 31.Aikaterini N, Chrisavgi T, Lavigne MD, Vassiliki L, Jeroen D, Triantafillos P, et al. The dual role of LSD1 and HDAC3 in STAT5-dependent transcription is determined by protein interactions, binding affinities, motifs and genomic positions. Nucleic Acids Research 2016(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal R, Janz M, Galson DL, Gries M, Li S, Jöhrens K, et al. C/EBPβ regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood, The Journal of the American Society of Hematology 2009;114(18):3890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coerver KA, Gray SM, Barnes JE, Armstrong DL, McCabe ER. Developmental expression of hexokinase 1 and 3 in rats. Histochem Cell Biol 1998;109(1):75–86 doi 10.1007/s004180050204. [DOI] [PubMed] [Google Scholar]
- 34.Gough SM, Lee F, Yang F, Walker RL, Zhu YJ, Pineda M, et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer discovery 2014;4(5):564–77 doi 10.1158/2159-8290.Cd-13-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta 2009;1789(6–8):443–50 doi 10.1016/j.bbagrm.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev 2005;19(13):1581–95 doi 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fleischer TC, Yun UJ, Ayer DE. Identification and characterization of three new components of the mSin3A corepressor complex. Molecular and cellular biology 2003;23(10):3456–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Dufau ML. Silencing of transcription of the human luteinizing hormone receptor gene by histone deacetylase-mSin3A complex. Journal of Biological Chemistry 2002;277(36):33431–8. [DOI] [PubMed] [Google Scholar]
- 39.Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 2006;442(7098):100–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006;442(7098):96–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol Cell Biol 2002;22(3):835–48 doi 10.1128/mcb.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518(7539):317–30 doi 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heideman MR, Lancini C, Proost N, Yanover E, Jacobs H, Dannenberg J-H. Sin3-aassociated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. haematologica 2014;99(8):1292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013;153(4):910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science (New York, NY) 2007;318(5849):447–50 doi 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 47.Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013;153(5):1149–63 doi 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 2016;44(W1):W160–W5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ChIP-seq datasets can be accessed under GEO accession number GSE145190. The process files and code can be found in GitHub (https://github.com/CCLYLabBioinformatics/epigenetic-mechanism-of-PHF23)