

Abstract
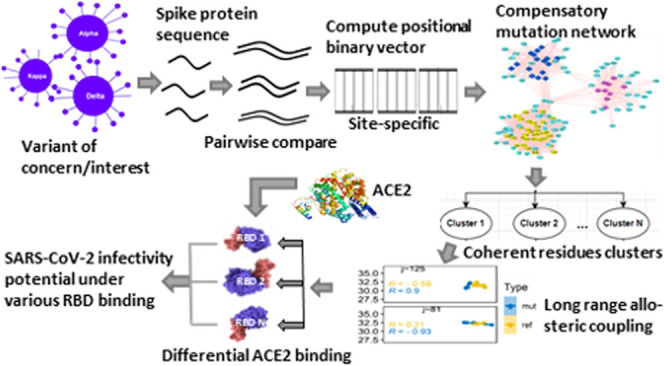
The emergence of a variety of highly transmissible SARS-CoV-2 variants, the causative agent of COVID-19, with multiple spike mutations poses serious challenges in overcoming the ongoing deadly pandemic. It is, therefore, essential to understand how these variants gain enhanced ability to evade immune responses with a higher rate of spreading infection. To address this question, here we have individually assessed the effects of SARS-CoV-2 variant-specific spike (S) protein receptor-binding domain (RBD) mutations E484K, K417N, L452Q, L452R, N501Y, and T478K that characterize and differentiate several emerging variants. Despite the hundreds of apparently neutral mutations observed in the domains other than the RBD, we have shown that each RBD mutation site is differentially engaged in an interdomain allosteric network involving mutation sites from a distant domain, affecting interactions with the human receptor angiotensin-converting enzyme-2 (ACE2). This allosteric network couples the residues of the N-terminal domain (NTD) and the RBD, which are modulated by the RBD-specific mutations and are capable of propagating mutation-induced perturbations between these domains through a combination of structural changes and effector-dependent modulations of dynamics. One key feature of this network is the inclusion of compensatory mutations segregated into three characteristically different clusters, where each cluster residue site is allosterically coupled with specific RBD mutation sites. Notably, each RBD mutation acted like a positive allosteric modulator; nevertheless, K417N was shown to have the largest effects among all of the mutations on the allostery and thereby holds the highest binding affinity with ACE2. This result will be useful for designing the targeted control measure and therapeutic efforts aiming at allosteric modulators.
Introduction
Global efforts are underway toward the development of more effective vaccines and antiviral drugs to efficiently cope with the ongoing COVID-19 pandemic, which has caused 4 170 155 deaths as of July 2021. However, such efforts are being seriously hindered by the rapid emergence of multiple SARS-CoV-2 variants—the causative agent of COVID-19.1−3 There is now growing evidence that mutations that changed the antigenic phenotype of SARS-CoV-2 are capable of evading immune responses and attenuating the neutralizing effects of antibodies.4−6 Recent studies further show that new variants are potentially evolving due to selective pressure exerted by convalescent plasma and mAb treatments.7−9 Under such selective pressure, long-term virus shedders may contribute to sporadic emergence of more intensely mutated variants. There are several variants of concern (VOCs) or variants of interest (VOIs) circulating globally that have been reported by the WHO technical advisory group (Table 1)a,b, demanding immediate attention for setting better control measures. With a noted suite of mutations, these variants are being identified, which have been causing significant community transmission or multiple COVID-19 clusters in multiple countries with increasing relative prevalence alongside increasing numbers of cases over time. Here, we have considered prominent spike receptor-binding domain (RBD) mutations that are commonly observed among the VOI/VOC variants (Table 1), namely, E484K, K417N, L452Q, L452R, N501Y, and T478K. These RBD mutations are noted to characterize and differentiate several emerging variants. VOCs/VOIs with these mutations are distinguished with changes in several virus characteristics such as transmissibility, disease severity, immune escape, diagnostic escape, or therapeutic escape.4,10−13 One such predominant VOC in much of the world that appeared first in India in late 2020 is the delta variant with very high transmissibility. This variant is identified as lineage B.1.617.2, characterized by several spike mutations. Of these, T478K and K417N substitutions in the spike protein make it characteristically different from other VOIs. Although comparative assessments of variant characteristics have been reported in recent studies, it remains unclear how such mutations affect virus characteristics with seemingly uncorrelated sporadic mutations.
Table 1. SARS-CoV-2 Variants of Concern (VOCs) and Variants of Interest (VOIs)a.
| WHO level | Pango linage | spike protein substitutions | name (next strain) | first detected | remarks |
|---|---|---|---|---|---|
| (A) Variant of Concern | |||||
| α | B.1.1.7 | Spike: 69del, 70del, 144del, (E484Ka), (S494Pa), N501Y, A570D, D614G, P681H, T716I, S982A, D1118H (K1191Na) | 20I/501Y.V1 | United Kingdom, Sep 2020 | increase in transmissibility or detrimental change in COVID-19 epidemiology; OR increase in virulence or change in clinical disease presentation; OR decrease in effectiveness of public health and social measures or available diagnostics, vaccines, therapeutics20,33−39 |
| β | B.1.351 | Spike: D80A, D215G, 241del, 242del, 243del, E417K, E484K, N501Y, D614G, A701V | 20H/501.V2 | South Africa, May 2020 | |
| Deltab | B.1.617.2 | Spike: T19R, (G142D), 156del, 157del, R158G, (K417Na) L452R, T478K, D614G, P681R, D950N | 20A/S:478K | India, Oct 2020 | |
| γ | P.1 | Spike: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I | 20J/501Y.V3 | Japan/Brazil, Nov 2020 | |
| (B) Variant of Interest | |||||
| Iota | B.1.526 | L5F, (D80Ga), T95I, (Y144-a), (F157Sa), D253G, (L452Ra), (S477Na), E484K, D614G, A701V, (T859Na), (D950Ha), (Q957Ra) | 20C/S:484K | USA, Nov 2020 | SARS-CoV-2 with genetic changes that are predicted or known to affect virus characteristics such as transmissibility, disease severity, immune escape, diagnostic or therapeutic escape; AND identified to cause significant community transmission or multiple COVID-19 clusters, in multiple countries with increasing relative prevalence alongside increasing numbers of cases over time, or other apparent epidemiological impacts to suggest an emerging risk to global public health36,39−44 |
| Kappa | B.1.617.1 | Spike: (T95I), G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H | 20A/S:154K | India, Dec 2020 | |
| Eta | B.1.525 | A67V, 69del, 70del, 144del, E484K, D614G, Q677H, F888L | 20A/S:484K | UK and Nigeria, Dec 2020 | |
| Lambda | C.37 | G75V, T76I, δ246-252, L452Q, F490S, D614G, and T859N | 21G | Peru, Dec 2020 | |
Indicates presence in some sequences.
Indicates initially designated as VOI; bold marked mutations are spike RBD mutations.
Coronavirus (CoVs) recognize and enter the human host cell by binding its prefusion-form spike protein (S) to the human angiotensin-converting enzyme-2 (ACE2), which is highly expressed on the surface of lungs, heart, kidneys, and intestine cells. To stop this essential entry process and subsequent infection, most of the vaccine development and therapeutic efforts focus on the S-protein.14 The S-protein is a trimer, where each protomer is made of two subunits: an amino(N)-terminal S1 and a carboxyl (C)-terminal S2.15 During proteolytic processing and subsequent membrane fusion, the S1/S2 junctions at Arg685-Ser686 are cleaved by the cellular protease furin. The subunit S1 is mainly involved in prefusion interactions with the human receptor protein ACE2.16 It consists of an N-terminal domain (NTD), the receptor-binding domain (RBD), and two structurally conserved subdomains SD1 and SD2. Several studies have illustrated a variety of conformational arrangements of RBD, switching between the RBD-up position amenable for ACE2 binding and the RBD-down position relatively resistant to receptor binding.17−19 Most VOC/VOI mutations that differentiate one from the others are located in RBD and NTD and affect interactions with ACE2.4 For example, the P.1 lineage detected first in Brazil is characterized by several amino acid (AA) substitutions mostly located either in RBD or in NTD: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y, and T1027I, which are shown to reduce neutralization by some antibodies. However, it is not clear how the NTD mutations affect the interactions with the ACE2 that binds to the RBD. Here, we examine if portions of NTD in part or clustered forms are involved in allosteric communication with VOC/VOI noted in six mutational spots in the RBD.20
Long-range allosteric perturbations are propagated not only by structural changes but also by effector-dependent modulations in dynamics. Studies based on statistical thermodynamics show that cooperative interaction free energies can be generated by ligand binding. Potential changes in macromolecular thermal fluctuations due to ligand binding involve several forms of dynamic communications ranging from highly correlated, low-frequency normal mode vibrations to random local anharmonic motions of molecular domains. This form of dynamic allostery is primarily an outcome of the entropy effect.21−23 The effector-dependent modulations of the structural dynamics play a critical role in propagating long-range allosteric perturbations for which structural changes are not necessary.24 The end-points of this long-range allostery are characterized by comparative analyses between apo- and effector-bound states. Such allosteric propagations of perturbations are performed by a network of AA residues that connect the distal dynamic domain of the protein structure.25 Revelations of such an AA residue network that underpin mutation-induced dynamic allosteric modulations are critical for understanding and predicting differential effects of VOIs. Here, we have used computationally predicted chemical shift data of 1H and 15N, calculated based on SHIFTX2,26 which combines ensemble machine learning methods with sequence alignment-based methods, to probe a variety of VOC/VOI mutation networks differentially connecting NTD and RBD of the SARS-CoV-2 spike protein. In parallel to well-exploring the chemical shift covariance analysis (CHESCA),27 we have designed an integrated methodology combining sequence, structure, and chemical shift data to infer such long-range allosteric connections modulated by SARS-CoV-2 VOC/VOI RBD mutations. The molecular complex detection (MCODE) technique28 was applied in Cytoscape plugins29 to extract highly connected subgraphs of vulnerable mutation sites identified from AA sequences of Spike variants. The chemical shift projection analysis (CHESPA)30 had been implemented on the mutated and nonmutated spike chemical shift data to quantify the effects of the noted RBD mutations. Then, the RBD and ACE2 binding affinity, dissociation constant, H-bond, and salt bridges were calculated using PROtein binDIng enerGY prediction (PRODIGY)31 and PDBePISA webservers.32 It shows that high-frequency correlated mutation sites at NTD are segmented with different combinations of secondary structural components that established a strong allosteric communication with the mutational spot at RBD. In particular, the delta variant consisting of the RBD mutation K417N shows a strong long-range modulated allostery, resulting in higher interactions with ACE2. These results provide important inputs to the awaited development of allosteric modulators inhibiting interactions with ACE2 and thereby that block the viral entry.
Results and Discussion
Compensatory Mutation Network (CMN) across SARS-CoV-2 Spike Variants
Compensatory mutation has beneficial effects on
fitness in the presence of a deleterious mutation, but it, otherwise,
remains neutral or deleterious. It usually occurs nonrandomly over
gene sequences and is more likely than expected by chance to be close
to the site of the actual deleterious mutation. To identify a suite
of such compensatory mutations that are commonly followed by a single
mutation in biological sequences and are responsible for maintaining
conformational and functional stability, we collected SARS-CoV-2 spike
(S) protein sequence variants from the NCBI databasec. We obtained a total of around 91 000 sequence samples, with
each of the sequence variants of length 1273 observed in at least
three different samples (n ≥ 3) for subsequent
alignment analysis (Table S1). Of these,
we found 1784 unique sequences (N = 1784) clustered
among all of the SARS-CoV-2 spike protein sequence samples. Instead
of reference-based comparison, we compared all possible pairwise sequence
variants in each position of the sequence. This creates a binary sequence
variant population of size M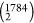 (=1 590 436
sequences), with
0 indicating no substitution and 1 indicating a single AA substitution
(Figure 1).
(=1 590 436
sequences), with
0 indicating no substitution and 1 indicating a single AA substitution
(Figure 1).
Figure 1.

Probabilistic scheme for the identification of compensatory mutations based on amino acid (AA) sequences. The left panel shows the list of mixed AA unique sequences of the S-protein of all SARS-CoV-2 variants, which were then compared pairwise to create a binary population (BP) of the sequence, as shown in the middle panel (N = 1784, L = 1273, M = 1 590 436); “1” stands for the AA substitution, and “0” otherwise. The right panel illustrates how two mutation sites were compared using conditional probability over the S-binary population (BP). The mutation site i is compensatory to the site j or vice versa if both the conditional probability P(si|sj) = 1 and P(sj|si) =1.
This provided a total count of 618 mutation sites (≈50 percent
positions) in the spike protein. With these identified residual sites,
we examined compensatory relationships among these positions by calculating
pairwise joint and conditional probability over the binary population
(( Methods). In pairwise comparison among
the mutation positions, we selected all of the pairs having the highest
equal conditional probability of 1, inferring their strong compensatory
effects. As a result, only 152 sequence sites and 2671 pairwise connections
were identified out of the total 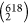 (=190 653) pairwise
mutation sites
(Table S2). It, therefore, provided an
undirected, unweighted network, where each of the 152 mutation sites
represents a network node and each of the 2671 pairwise connections
represents an edge. We, then, formed a component (i.e., a maximally
connected network) by excluding all of the isolated nodes, which eventually
refer to a compensatory mutation network of the SARS-CoV-2 spike protein
(Figure 2A). Analogous
to the usual characteristic of compensatory mutations that tend to
occur more commonly in certain regions of the protein, we have noted
that this entire network lies within the NTD (resi 13–305)
of the spike protein. It is a maximally connected mutation network
including 90% of the mutation sites (136 nodes out of 152 and 2660
edges out of 2671) with a multimodal degree distribution (Figure S1).
(=190 653) pairwise
mutation sites
(Table S2). It, therefore, provided an
undirected, unweighted network, where each of the 152 mutation sites
represents a network node and each of the 2671 pairwise connections
represents an edge. We, then, formed a component (i.e., a maximally
connected network) by excluding all of the isolated nodes, which eventually
refer to a compensatory mutation network of the SARS-CoV-2 spike protein
(Figure 2A). Analogous
to the usual characteristic of compensatory mutations that tend to
occur more commonly in certain regions of the protein, we have noted
that this entire network lies within the NTD (resi 13–305)
of the spike protein. It is a maximally connected mutation network
including 90% of the mutation sites (136 nodes out of 152 and 2660
edges out of 2671) with a multimodal degree distribution (Figure S1).
Figure 2.

SARS-CoV-2 spike protein “AA compensatory mutation network (CMN)”: colored nodes represent the densely connected mutation sites, and the edges denote the compensatory connection. (A) Visual presentation of the CMN-a connected, undirected network entirely located in the N-terminal domain of the S1 unit of a spike protomer, which is visually segregated into three segments that are bridged by three nodes 141, 179, and 180. (B) Three network clusters: C1 (yellow), C2 (pink), and C3 (blue); an outcome of the molecular complex detection (MCODE) technique in cytoscape. (C) Different quantitative characteristics that distinguish the clusters C1, C2, and C3 in the source network (A) and the clustered network.
Visual analysis and two-dimensional (2D) graphical representations of the SARS-CoV-2 spike compensatory mutation network (Figure 2B) showed clustering of several network nodes (used MCODE28) around the pick of multimodal degree distributions of the whole network. We noted three such clusters represented by subnetworks, namely, C1, C2, and C3, with significantly different network properties (Figure 2C). Moreover, these three subnetworks are bridged within the whole compensatory mutation network by only three nodes: resi 149, 179, and 180; deleting these nodes causes complete disconnection between these three subnetworks. Each of the clusters contains nearly consecutive sites of the primary structure of the spike protein (C1: resi 74–140; C2: resi 159–172; C3: 186–212) (Table S2). When these nodes were mapped on the three-dimensional (3D) structure of the S-protein (Figure 3A), it represented β strands in the NTD of the S-protein (Figure 3B). C1 is characterized by a β sheet of three strands, C2 by a single β strand, and C3 by a β sheet of two strands. Although they are relatively close in the 3D space, they propagate allosteric effects differentially to specific RBD sites under different RBD mutations, resulting in increased or decreased binding affinity with the human receptor ACE2 (described in the following sections). Given the high level of structural plasticity of NTD and RBD domains (Figure 3C), there can be many more combinations of mutations requiring compensatory changes that are compatible with high viral fitness and may contribute to efficient immune escape. For example, a recent study showed that N439K compensated for an RBM mutation K417V that otherwise decreases the receptor-binding affinity and that several mAbs were more sensitive to these mutations in combination versus individually.45
Figure 3.

Mapping of compensatory mutation segments onto the 3D structure of the spike (S) protein protomer with the RBD-up form. (A) Different domains and subdomains belonging to S1 (upper part) and S2 (lower or base part) units of the S-protomer. (B) Mapped structural segments C1, C2, and C3 on the N-terminal domain (NTD) (pink) involving differently composed β strands. (C) Schematic presentation of the functional significance of C1, C2, and C3 that differentially establishes a long-range allosteric communication with the RBD (blue) mutation site, where C1, C2, and C3 residues act as allosteric activators capable of propagating the mutation-induced allosteric signals to a distant allosteric site in RBD through a combination of structural changes and modulations of dynamics.
Mutational Spots in the Receptor-Binding Domain (RBD) Are Driven by Long-Range Dynamic Allostery
To understand the functional significance of the compensatory mutation network, we examined if there are any allosteric communications between the compensatory mutations and the different VOI-specific RBD mutations in the spike protein. Such a long-range allosteric perturbation (>20 Å), arising from mutations or chemical modifications of the ligand effector and propagated to a distant end, can effectively be detected in chemical shift changes of diamagnetic 1H, 13C, and 15N of protein residues. Here, we have used a computer program called SHIFTX2,26 which is capable of accurately predicting 1H, 13C, and 15N chemical shifts from protein coordinate data (Table S3). SHIFTX2 uses a large, high-quality database of training proteins (>190) in advanced machine learning techniques, incorporating many more features (χ2 and χ3 angles, solvent accessibility, H-bond geometry, pH, temperature) and thereby achieving high accuracy by combining sequence-based and structure-based chemical shift prediction techniques. This chemical-shift-based prediction of long-range allostery has further been probed in the Ohm-a computationally efficient network-based method,46,47 which is similar to the well-known NMR chemical shift covariance analysis (CHESCA).27 “Ohm” automatically determines the allosteric network architecture and identifies allosterically coupled residues based on the perturbation propagation algorithm that repeats the stochastic process of perturbation propagating on a network of interacting residues in a given protein. Such residue–residue allosteric coupling is measured by an allosteric coupling intensity (ACI)—a frequency with which each residue is affected by a perturbation. By considering C1, C2, and C3 residue positions in NTD as active sites, we calculated the ACI for all of the VOC-/VOI-specific RBD mutation sites and then compared them with the chemical-shift-based results.
Using 1H and 15N combined chemical shifts, we examined whether the pairwise inter-residue correlation remains linear in different conformational states of RBD bound with/without the ACE2 and further observed if there is any deviation from this linear relationship under different RBD mutations. Such a linear correlation in different states of the same protein indicates inter-residue allosteric coupling. This examination showed that several residues belonging to the C1, C2, or C3 cluster of the compensatory mutation network retained the strong linear correlation with VOC-/VOI-specific RBD mutation sites in all of the given RBD conformational states. Although this linearity is noted in the RBD-mutated and nonmutated states as well, it largely varies among the C1, C2, and C3 clusters with inconsistent differences between the mutated and nonmutated states (Table 2). When compared among the different RBD-mutated states (i.e., E484K, K417N, L452Q, L452R, N501Y, and T478K), it showed strong allosteric signals activated by a suite of residues in C1, C2, and C3 with overall correlation ≥ 0.7 (Figure 4a) that allosterically coupled with the RBD mutation differentially, indicating potential long-range allosteric communication between compensatory mutation sites in NTD and the RBD mutation site (Figure 4b). The number of residues among the compensatory mutation clusters that allosterically coupled with the RBD mutation sites varies significantly, referring to differential effects of RBD mutation sites. For example, spike K417N and the nonmutated states involve 33.33 and 44.44% residues of the C2 cluster, respectively (Table 2), which allosterically coupled with the RBD site 417 with the absolute correlation being greater than 0.7. However, S E484K involves 44.44% residues of the C2 cluster, which is 10% less than its nonmutated form, having allosteric coupling with the RBD mutation site 484. Existence of such coupling has further been observed in a measured allosteric coupling intensity (ACI) in the Ohm webserver. With the inputs of compensatory mutation sites of C1, C2, and C3 as active sites in Ohm separately, we calculated the allosteric coupling intensity (ACI) for the RBD mutation sites (Table S4). It showed that ACIs are more than 0.2 bound with/without the ACE2; however, the bound ACE2 reduces and the RBD mutation with the bound ACE2 enhances the ACI relative to its native RBD-up state. These Ohm outcomes are in agreement with the residue–residue correlation analysis based on the chemical shifts. In particular, S K417N has ACI values of 0.35, 0.31, and 0.28, respectively, when C3, C2, and C1 are considered as the active sites, which is >40% when compared with its native RBD-up conformational form, indicating strong allosteric connections for the K417N S conformation (Figure 5). In parallel to these noted observations, a recent study reported that K417 and N501 residues serve as effector centers of allosteric interactions and anchor distant sites that mediate long-range allosteric communications in the complex.48
Table 2. Percentage of Residues in the Compensatory Mutation Segments C1, C2, and C3 that Showed Allosteric Connection with the RBD Site with/without Mutation, Having the Combined Chemical-Shift-Based Correlation |r| ≥ 0.7.
| RBD mutation | class | mutated concn (%) | reference concn (%) |
|---|---|---|---|
| E484K | C1 | 23.53 | 11.76 |
| K417N | C1 | 8.82 | 17.65 |
| L452Q | C1 | 26.47 | 2.94 |
| L452R | C1 | 17.65 | 2.94 |
| N501Y | C1 | 20.59 | 11.76 |
| T478K | C1 | 29.41 | 20.59 |
| E484K | C2 | 44.44 | 55.56 |
| K417N | C2 | 33.33 | 44.44 |
| L452Q | C2 | 11.11 | 11.11 |
| L452R | C2 | 22.22 | 11.11 |
| N501Y | C2 | 11.11 | 33.33 |
| T478K | C2 | 11.11 | NA |
| E484K | C3 | 37.50 | 25.00 |
| K417N | C3 | 18.75 | 18.75 |
| L452Q | C3 | 18.75 | 12.50 |
| L452R | C3 | 6.25 | 12.50 |
| N501Y | C3 | 18.75 | 25.00 |
| T478K | C3 | 18.75 | 25.00 |
Figure 4.

Residue–residue allosteric coupling between the residues j in C1, C2, C3, and the VOI-/VOC-specific RBD mutation site i. (a) Pearson correlation (r) based on the combined chemical shift of 1H and 15N ppm values in five conformational forms “k” of the S-protein: S-protein with the RBD-down, the RBD-up, bound with ACE2, clockwise and anticlockwise movement of RBD bound with the ACE2 (δik), and under different specific RBD mutations. (b) Best correlation sites (or j-residues) in the compensatory mutation segments C1, C2, and C3 showing allosteric connection with the specific RBD site that is modulated by the VOI-/VOC-specific RBD mutations (mut, mutated; ref, reference).
Figure 5.

VOI-/VOC-specific RBD mutation-induced variation in the allosteric coupling intensity (ACI)—a frequency with which each residue is affected by a perturbation: ACIs were calculated in the “OHM webserver” with the residues in the compensatory mutation segments C1, C2, and C3 as activator sites. It showed that the RBD mutation modulates the ACIs with a significant increase under the mutation K417N.
To evaluate the effects of RBD-specific mutations on the allosteric communications and thereby on the interactions with ACE2, we applied chemical shift projection analysis (CHESPA).30 In CHESPA, one of the vectors A is projected on the other vector B to measure the shift along B, where A denotes the residue-wise combined chemical shift differences between the RBD-up S without the bound ACE2 and the mutated ACE2-bound S, and B denotes the differences between RBD-up S and the nonmutated ACE2-bound S. The analysis provides two key residue-specific descriptors of the perturbation caused by the mutations: the fractional shift (X) and the cos θ (Methods). Only residues with absolute cos θ values close to 1 are suitable reporters of the allosteric activity, while the S-protein binds with ACE2. However, if the fractional shift X is positive, it indicates that the RBD mutation effects toward an allosterically more active state, and it is otherwise if X is negative. CHESPA outcomes show that a large number of NTD residues hold positive X values with absolute cos θ greater than 0.9 (Figure 6), indicating the strong allosteric activity of compensatory mutations sites in NTD and thereby coupling with VOI-specific RBD mutation sites. While all of the mutations have significant effects, K417N showed many more residues with very high positive X values. Moreover, many mutation sites in the compensatory mutation segments C1, C2, and C3 are common and highly active (X > 1.0 and cos(θ) > 0.9) across all of the RBD mutation sites: 87, 89, 91, 114, 115, 123, and 128 of C1; 159 of C2; and 187 and 196 of C3, indicating their critical role in maintaining the allosteric communications. In a recent study of dynamic profiling of binding and allosteric propensities of the SARS-CoV-2 spike protein with different classes of antibodies, it was seen that RBD mutation sites K417, E484, and N501 correspond to a group of versatile allosteric centers in which small perturbations can modulate collective motions, alter the global allosteric response, and elicit binding resistance.49
Figure 6.

Chemical shift (CS) projection analysis showing the effects of VOI-/VOC-specific RBD mutations. (a) Fractional shift (X) variation in the compensatory mutation segments C1, C2, and C3 induced by the RBD mutations, E484K, K417N, L452Q, L452R, N501Y, and T478K. There are a select few residues for which X is negative in all of the segments when |cos θ| ≥ 0.9. (b) Projection angle, cos θ, which is an indicator of the direction of chemical shift movement along (+ve values) or against (−ve values) the allosteric activity.
Mutation-Induced Modulation of Spike Protein Interactions with ACE2 through the Dynamic Allosteric Network
Successful entry of SARS-CoV-2 into the human host cell depends on how the S-protein RBD interacts with the human receptor ACE2 and subsequently modulates the proteolytic processing of the S1 unit for membrane fusion. We probe here how the VOC-/VOI-specific RBD mutation affects this interaction with the ACE2. We used PROtein binDIng enerGY prediction (PRODIGY)31—a webserver that predicts the binding affinity from their 3D structures based on intermolecular contacts and properties derived from the noninterface surface. Before giving input into PRODIGY, the individual mutant structure is prepared for all of the VOI-/VOC-specific RBD mutants with the reference pdb 7a94 having one up-RBD bound with ACE2. The rotamer library in UCSF-Chimera50 has been utilized to select the most copious pose for the substitution, and then, this mutated structure has been refined in the 3Drefine program51 that optimizes the hydrogen-bonding network and minimizes the energy using all atom force fields. These energy-minimized structures were used to dock with ACE2 in the Frodock2.0 docking server that includes a complementary knowledge-based protein-docking potential, choosing a dock having minimum energy, and a pose of ACE2 bound with the up-RBD. Finally, this resultant complex was used in PRODIGY.
To calculate structural and chemical properties of ACE2-bound RBD nonmutant- and mutant S-proteins, we used PISA software.32 While comparing the interface interactions, it shows that all of the VOI-specific mutations have significant effects at the interface with an approx. 2 times higher solvent-accessible area (SAA) at the interface for the mutated structure (Table 3), indicating a higher potential of ACE2 and RBD interactions. In particular, delta-specific RBD mutation K417N, T478K, and Lamda-specific L452Q showed a much higher SAA relative to the ACE2-bound active S-protein. When we examined the H-bond network at the interface, we found mutation-specific alterations and unequal distribution of H-bonds. Besides the H-bond, T417N, L452Q, L452R, N501Y, and T478K caused the formation of several salt bridges at the interface. This differential distribution of H-bonding and salt bridges may explain the different binding energy (ΔG) and dissociation coefficient (kd) under dissimilar RBD mutations. When compared with the ACE2-bound nonmutated S-protein, all of the RBD mutations showed reduced ΔG and Kd, indicating higher interface interactions and binding affinity caused by the mutations. Among all of the considered mutations, it was seen that K417N has the highest effect on ΔG and Kd (−17.2 kcal/mol, 2.50 × 10–13 M). Notably, the K417N S-protein holds five H-bonds with the distance cutoff of 3.00 Å and four salt bridges to hold tight interactions with ACE2. It further shows that ACE2-bound K417N S holds 7.7 and 3.3% solvent-accessible area at the interface of ACE2 and spike, respectively, and 182 interface contact residues, which is the largest among all of the mutations. Among all of the interface residues, it is seen that the dominant polar hydrophilic residue SER appeared 24 times on the RBD, while the ACE2 interacting interface dominant hydrophilic residues GLU occurs 39 times, allowing formation of a large number of H-bonds and salt bridges at the interface.
Table 3. Interface Properties of the S-RBD and ACE2 Interactions under VOI-/VOC-Specific Six RBD Mutations with Different RBD Orientations.
| interface interaction properties | wild | E484K | K417N | L452Q | L452R | N501Y | T478K | ||
|---|---|---|---|---|---|---|---|---|---|
| Kd (M) at 25 °C | 2.50 × 10–9 | 1.90 × 10–11 | 2.50 × 10–13 | 1.90 × 10–10 | 4.60 × 10–11 | 1.20 × 10–11 | 4.40 × 10–11 | ||
| ΔG (kcal/mol) | –11.7 | –14.6 | –17.2 | –13.3 | –14.1 | –14.9 | –14.1 | ||
| SAA at the interface (%) | ACE2 | 3.1 | 4.7 | 7.7 | 6.3 | 5.1 | 4.9 | 5.2 | |
| SARS-CoV-2 S | 1.4 | 2.1 | 3.6 | 2.8 | 2.4 | 2.4 | 2.4 | ||
| ACE2-bound stable RBD | no. of intermolecular contacts | 72 | 105 | 182 | 162 | 109 | 124 | 111 | |
| no. of H-bonds (dist. cutoff: 3 Å) | 6 | 2 | 5 | 5 | 2 | 5 | 2 | ||
| no. of salt bridges | 0 | 0 | 4 | 5 | 1 | 4 | 1 | ||
| ACE2-bound RBD moved clockwise | Kd | 3.30 × 10–9 | 6.70 × 10–9 | 2.50 × 10–8 | 1.20 × 10–9 | 1.70 × 10–9 | 2.60 × 10–10 | 2.80 × 10–9 | |
| ΔG | –11.6 | –11.1 | –10.4 | –12.1 | –11.9 | –13.1 | –11.7 | ||
| ACE2-bound RBD moved anticlockwise | Kd | 3.50 × 10–9 | 2.10 × 10–8 | 1.60 × 10–7 | 3.30 × 10–8 | 1.60 × 10–8 | 4.60 × 10–11 | 1.10 × 10–9 | |
| ΔG | –11.5 | –10.5 | –9.3 | –10.2 | –10.6 | –14.1 | –12.2 | ||
Along with the VOC-/VOI-specific mutations, we further examined the effects of different flexible RBD orientations (i.e., clockwise and anticlockwise movements of RBD) that can be caused by several mutations at hinge residues located close to the S1/S2 junction (e.g., D614G).52 Such flexible orientations of RBD were found to have significant diminishing effects, altering the binding affinity to ACE2 (Figure 7A). Interestingly, we observed a significant increase in ΔG and Kd under the flexible RBD orientations in contrast to their lowest values for the K417N S-protein (Figure 7B). Thus, reorientation of RBD with mutations at hinge residues may alter the VOI-/VOC-specific mutation effects. This result, therefore, generally indicates that the increased stability and rigidity of the prefusion confirmation state of the S-protein have a higher affinity to ACE2 under all of the VOC-/VOI-specific RBD mutations.
Figure 7.

Effects of VOI-/VOC-specific RBD mutations on the SARS-CoV-2 S-RBD and ACE2 interactions, with the different RBD orientations. (A) Snapshots of the effects of the RBD mutations, N501Y, T478K, and K417N; interface interactions and their distribution between ACE2 (red) and the RBD (blue) differed among the mutated states with different RBD movements, determining different dissociation constants Kd. Among all six considered RBD mutations, K417N showed the lowest Kd. (B) Zoomed-in snapshots of the H-bond and salt bridge interactions with the K417N mutation. It holds five H-bonds with the distance cutoff of 3.00 Å and four salt bridges and has 182 interface contact (IC) residues, the largest among all six mutations.
In addition to mutations, SARS-CoV-2 S-protein N-glycosylation plays an important role in viral entry to human cell models as the viruses lacking N-glycans enter the host cell less effectively.53 In a recent study, glycosylation profiles and their changes during the course of global transmission were characterized and compared with SARS-CoV. It reported nine predicted N-glycosylation sites and three O-glycosylation sites unique to SARS-CoV-2, but there was no evidently observed variation of the glycan sites so far.54 Further research showed that the glycans at sites N165 and N234 affect the RBD conformational plasticity, as their presence stabilizes the RBD “up” conformation, permitting efficient binding to the human angiotensin-converting enzyme-2 (hACE2) receptor. Deletion of these glycan residues through N165A and N234A mutations significantly reduced binding of the S-protein to ACE2 as a result of a conformational shift of the RBD toward the “down” state, weakening accessibility to ACE2.55
Conclusions
We have described structural and functional significance of SARS-CoV-2 variant-specific spike (S) protein RBD mutations, E484K, K417N, L452Q, L452R, N501Y, and T478K, which characterize and differentiate several variants of interest/concern (VOIs/VOCs) as reported by the World Health Organization (WHO). We have shown here that these mutations are not sporadic but engaged in an interdomain allosteric network differentially affecting interactions with the human receptor ACE2 and thereby are the determining factors of the higher transmissibility and infectivity of SARS-CoV-2. This allosteric network coupled the residues of the N-terminal domain (NTD) and the receptor-binding domain (RBD), which are further modulated by the RBD-specific mutations and are capable of propagating mutation-induced perturbations between these domains through a combination of structural changes and RBD mutation-dependent modulations of dynamics. One key feature of this network is the inclusion of compensatory mutations segregated into three characteristically different clusters, where each cluster residue site is allosterically coupled with specific RBD mutation sites. We mutated specific RBD mutation sites and quantified the changes in the allosteric coupling and then its effects on the interactions between RBD and ACE2. Each mutation was observed to increase interactions with ACE2; however, the extent of effects varies among the RBD mutations through different allosteric connections with the compensatory mutation clusters in the NTD. This result, therefore, provides important clues on how these specific RBD point mutations modulate the spike dynamics and interactions for higher transmissibility.
We have demonstrated highly sensitive hybrid detection methods of compensatory mutation coupling and its allosteric connections with a distant residue in another domain. Notably, this simple sequence-based probabilistic scanning of compensatory mutations deployed for SARS-CoV-2 spike proteins with hundreds of mutations generates segregated patterns in the NTD which are similar to the general features of compensatory mutations. While each cluster separately coupled with the RBD mutation sites through combined chemical shift changes, it shows a linear correlation with a high allosteric coupling intensity (ACI). Mutation effects are quantified by projecting residue–residue chemical shift changes in the RBD-mutated spike on the nonmutated spike bound with ACE2 that are surrogated by two descriptors: fractional shift (X), describing the extent of effects, and the cosine of the angle between the vectors, including mutated and nonmutated residues that describe the activation/inactivation of allosteric states. Among all of the considered mutations that showed positive allosteric modulation, K417N was found to have the largest effects on the allostery and thereby holds the highest binding affinity with ACE2, providing an explanation as to why the delta variant shows higher transmissibility.
Estimate of such mutational pathways by which SARS-CoV-2 will evolve is extremely challenging. Nevertheless, there is a rapidly expanding knowledge base regarding the effect of SARS-CoV-2 spike mutations on antigenicity and attenuation of the neutralizing effects of antibodies. Integration of SARS-CoV-2 sequences with structure-based pharmacological studies facilitates the detection of potential variants of concern at a relatively low frequency. This is useful for targeted control measure and further laboratory characterization and therapeutic efforts, advancing molecular understanding and connections among different mutations and their significance.
Methods
Identifying Vulnerable Mutational Sites across SARS-CoV-2 Spike Variants
Instead of classical reference-based sequence analysis,
we rely on differential sequence comparison among the mutated sequences
to identify vulnerable mutation sites that are prone to frequent alterations
in the available SARS-CoV-2 variants. We emphasized only the sites
that are highly susceptible to mutation, with the hypothesis that
SARS-CoV-2 changes itself structurally for more stronger binding with
the host. We used a binary vector-based comparison for the ease of
computation of such highly vulnerable sites. A pair of sequences is
compared for each amino acid. A match is ignored, scoring 0 in the
position of the resultant vector. However, a mismatch is rewarded
with 1 to indicate the presence of alternation in a particular position.
The same process has been applied to all pairs of N candidate sequences, leading to 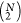 binary vectors. Next,
we calculated the
probability of vulnerability of each site.
binary vectors. Next,
we calculated the
probability of vulnerability of each site.
Assume a set of M = 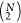 binary
vectors and each vector has L residual sites, S = {s1,s2, ···, sL}, where si =
{b1,b2, ···, bM}. The vulnerability of a
mutation site si can
be calculated as follows.
binary
vectors and each vector has L residual sites, S = {s1,s2, ···, sL}, where si =
{b1,b2, ···, bM}. The vulnerability of a
mutation site si can
be calculated as follows.
| 1 |
A site si is considered potentially prone to mutation if P(si) > τ, a user-defined threshold. Considering all such potentially vulnerable sites, we constructed a network of coexisting vulnerable sites.
Extracting Strongly Cohesive Mutation Sites
We calculated coexisting vulnerable sites that exhibit certain co-occurrence of alternation with respect to all candidate variants. We used the conditional probability score to calculate coexisting vulnerable sites. The coexistence probability of two sites si and sj (si, ···, sj) can be calculated as follows.
| 2 |
All pairs of si, ···, sj are combined to create an adjacency matrix A as follows.
| 3 |
Next, we applied the well-known molecular complex detection (MCODE) technique28 using Cytoscape plugins29 to extract highly connected subgraphs of vulnerable sites with the above network. MCODE is actually designed to identify connected regions in large protein–protein interaction networks that may represent molecular complexes. The method is based on vertex weighting by local neighborhood density and outward traversal from a locally dense seed protein to isolate the dense regions according to given parameters.
Chemical Shift Changes and Allosteric Coupling of Residues between RBD and NTD
It often remains experimentally challenging to define a network of residues that mediate the cross-talk between distal sites. Such clusters of coupled residues are particularly elusive in allosteric processes with a significant dynamically driven component, as in this case the allosteric signal propagation relies on subtle, but critical, conformational and side-chain packing rearrangements that often fall below the resolution of common X-ray or NMR structure determination methods. To cope with this situation, it was noted that chemical shift (CS) data can be explored to examine long-range allosteric communications. Exploring CS data was found to be very effective, as allosterically coupled residues exhibit concerted and correlated chemical shift changes for a given set of perturbations. Such correlations can be observed in a set of allosteric perturbations that induce different degrees of activation with minimal covalent modifications and that are spatially colocalized within a single region of the protein structure.
Allosterically coupled residues exhibit concerted and correlated chemical shift changes for a given set of perturbations. Such correlations can be observed in a set of allosteric perturbations that induce different degrees of activation with minimal covalent modifications and that are spatially colocalized within a single region of the protein structure. According to a two-state activation model with a fast exchange regime, chemical shifts for different residues sufficiently away from the effector binding site56 that senses the same perturbed equilibrium are linearly correlated. In the case of the multistate model, linearity was still maintained through partial reordering of the points that correspond to the same active ligands. Within these premises, the existence of long-range allosteric communication can be probed, noting linear coupling of distant residues by their chemical shifts, as has been successfully implemented in the chemical shift covariance analysis (CHESCA).27 Structural changes and the effector-dependent modulations in dynamics both play critical roles in long-range propagation of allosteric signals. Comparative analyses of the structural and dynamic profiles of apo- and effector-bound states are effectively used to characterize the terminal receptor sites of these allosteric signals. However, defining a network of residues that mediate such cross-talk between distal sites remains technically difficult. In particular, when the allosteric signal propagation primarily relies on conformational and side-chain packing rearrangements, identifying such clusters of coupled residues remains elusive. As chemical shifts are highly sensitive to both structural changes and the effector-dependent modulations, it has been shown to be very effective for determining long-range allostery greater than 20 angstrom. In a chemical-shift-based approach, residues that belong to the same effector-dependent allosteric network exhibit a concerted response to the perturbation set, which may not be true when such a network was solely determined based on the 3D protein structure. Given the five conformational states of the spike protein with active/inactive RBD, RBD bound with ACE2, and clockwise and anticlockwise poses of ACE2-bound RBD, we separately probed such a linear correlation between the residues in segments C1, C2, and C3 of the compensatory mutation network in the NTD and the VOC-/VOI-specific RBD mutation sites away from the ACE2-bound residues.
Residue-wise combined chemical shifts are calculated as the weighted sum of the ppm values of the amide proton 1H and 15N nitrogen
| 4 |
where δik denotes the combined chemical shift of residue i at the kth perturbed state and WN and WH are the weights for the chemical shifts δikN and δik, respectively. If two residues (or sites) i and j belong to the same allosteric network, their perturbation-dependent chemical shift variations exhibit a linear correlation regardless of their magnitude. Therefore, allosterically coupled residues i and j establish a linear equation as follows
| 5 |
The presence of nonlinear terms in eq 5 is neglected based on the observations that a slight RBD reorientation causes correlated perturbation in the local environment of residues i and j. Such correlated perturbations (Pearson correlation (rij)) as calculated in eq 6 are depicted in the maintained correlation |rij| ≥ 0.7, displaying a collective concerted response to perturbed states.
| 6 |
where δik and δjk are two vectors
of equal
length and 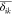 and
and 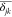 correspond to the means of δik and δjk, respectively.
correspond to the means of δik and δjk, respectively.
To
gauge the direction in which the given RBD mutation affects
the allosteric active states, we have implemented the chemical shift
projection analysis (CHESPA).30 The commonly
used compounded ppm changes, computed as 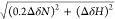 ,57−62 is based only on the magnitude of the chemical shift variations
caused by a mutation and not on the direction in which the given mutation
affects the dynamic equilibrium. The compounded chemical shift difference
between the apo-S and the S-mutant was calculated as the magnitude
of vector A, connecting the apo-S and S-mutant ACE2-bound peaks and
defined in the plane of the 1H and scaled 15N ppm coordinates. The scaling factor of the 15N ppm values
is 0.2.63 Similarly, the compounded chemical-shift
difference between the apo-S and the ACE2-bound S is computed as the
magnitude of vector B, which represents the activation vector joining
the apo/inactive to the allosterically active state. The projection
of vector A onto B is a measure of the shift along the activation
vector caused by a given mutation. To quantify the extent of activation
(or inactivation) achieved by a mutation, the fractional shift (X) is calculated as the ratio of the component of vector
A along vector B and the magnitude of vector B (i.e., |B|).
,57−62 is based only on the magnitude of the chemical shift variations
caused by a mutation and not on the direction in which the given mutation
affects the dynamic equilibrium. The compounded chemical shift difference
between the apo-S and the S-mutant was calculated as the magnitude
of vector A, connecting the apo-S and S-mutant ACE2-bound peaks and
defined in the plane of the 1H and scaled 15N ppm coordinates. The scaling factor of the 15N ppm values
is 0.2.63 Similarly, the compounded chemical-shift
difference between the apo-S and the ACE2-bound S is computed as the
magnitude of vector B, which represents the activation vector joining
the apo/inactive to the allosterically active state. The projection
of vector A onto B is a measure of the shift along the activation
vector caused by a given mutation. To quantify the extent of activation
(or inactivation) achieved by a mutation, the fractional shift (X) is calculated as the ratio of the component of vector
A along vector B and the magnitude of vector B (i.e., |B|).
The fractional shift (X) is scalar and is complemented by the cos θ value. It is based on the relative orientation of vectors A and B. Thus, the projection analysis of the chemical shifts results in two key residue-specific descriptors of the perturbation caused by the mutation, i.e., the fractional shift (X) and the cos θ. The fractional shift, X, is positive if the mutation shifts the equilibrium toward the allosterically active state and negative otherwise. The absolute value of X approaches 0 if the ppm variations caused by the mutation are negligible when the mutation results in ppm changes of comparable magnitude and direction, |X| ∼ 1. The |cos θ| values approach unity (i.e., |cos θ| ∼ 1) with the strong allosteric effect of the mutation. However, |cos θ| < 1 for residues are more significantly affected by the mutation through nearest-neighbor effects or other structural distortions caused by the mutation and not through long-range allostery.
The fractional shift (X) is calculated as the ratio of the component of vector A along vector B and the magnitude of vector B (i.e., |B|)30
| 7 |
where B = [.2ΔδikrN, Δδik′rH], A = [.2ΔδikrN, Δδik′mH], and r and m refer to the reference and mutated conditions, respectively, in the kth (or k′th) state. θ is the angle between vectors A and B.
| 8 |
| 9 |
Interface Analysis Using 3D Protein Structures
RBD and ACE2 binding affinities, dissociation constant, H-bond, and salt bridges were calculated using PROtein binDIng enerGY prediction (PRODIGY)31 and PDBePISA webserversd.32 PRODIGYe predicts the binding affinity and identifies the interfaces from 3D protein structures with the number and type of intermolecular contacts within the 5.5Å distance cutoff. It predicts the binding affinity based on a simple linear regression of interface contacts (ICs) and some properties of noninteracting surfaces (NISs) that were shown to influence the binding affinity
| 10 |
ICs are classified based on the type of contacts within a distance of 5.5 Å, where ICsxxx/yyy is the number of interfacial contacts found at the interface between Interactor1 and Interactor2. The dissociation constant (Kd) is calculated using the formula ΔG = RT ln Kd, where R is the ideal gas constant (in kcal K–1 mol–1), T is the temperature (in K), and ΔG is the predicted free energy. For our calculation, the temperature was set to 25.0 °C.
To extract H-bond and salt bridge interactions at the interface, we used the PDBePISA webserverf. PISA considers whether a H-bond will be present if the distance between the heavy atoms in the donor and acceptor is less than 3.89 Å. In the presence of the H atom, the acceptor–H distance must be ≤ 4 Å and the angle A-H-D lies between 90 and 270. The relevant distance for a salt bridge is 4 Å. ACE2 docking with the RBD-mutated spike was carried out in the FRODOCK webserverg.64 Given the 3D coordinates of two interacting proteins, it uses a fast-rotational docking method. It performed global energy optimization by a six-dimensional (6D) (3D rotations + 3D translations) rigid-body exhaustive search of the orientations of a fixed molecule with respect to a mobile receptor. Considering only the rotational part, each energy term was calculated with a correlation function defined by the interaction of potential parts of the receptor and the ligand. This fast exhaustive rotational search was combined with an implicit translational scan. The translational search was done implicitly by sampling the space uniformly with a fixed step size grid.
Acknowledgments
The authors thank all colleagues and project students of the School of Physical Sciences, Sikkim University, for their support while completing computational work in the numerical lab. The authors are also grateful to Professor Lawrence M Nogee, Johns Hopkins University School of Medicine, for his encouragement while working on this manuscript. The project was funded by the National Natural Science Foundation of China under Grant no. 12022113 and the Henry Fok foundation for young teachers (171002).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05155.
Multimodal degree distribution of the amino acid compensatory mutation network (CMN) (Figure S1) (PDF)
Collected protein sequence data from the NCBI virus database; there are a total of 1784 clustered (based on exact sequence similarity) protein sequence samples, with each cluster having a sample frequency ≥ 3 and corresponding accession numbers (Table S1); a total of 618 mutation sites (or residues), with each with amino acid residue and associated clusters count (out of 1784) from Table S1 (Table S2); predicted 1H and 15N chemical shifts from protein coordinate data for five pdb structures (6VXX, 6VSB, 7A94, 7A95, 7A96) in each sheet for six RBD mutations (Table S3); predicted ACI data considering reference pdb structures (7a94) and six RBD mutations (Table S4) (XLSX)
Author Present Address
○ Biomedical Research Center, National Institute of Aging, National Institutes of Health, Bethesda, Maryland 20814, United States
Author Contributions
J.K.D., S.R., and A.C. conceived the study. J.K.D., B.T., and A.C. conducted computational experiments and analytical analysis. J.K.D., K.M.K., S.R., G.-Q.S., and A.C. carried out statistical analysis. J.K.D., S.R., G.-Q.S., A.S., and A.C. prepared the initial draft of the manuscript. All authors have reviewed and edited the final version.
The authors declare no competing financial interest.
Footnotes
Supplementary Material
References
- Gómez C. E.; Perdiguero B.; Esteban M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243 10.3390/vaccines9030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Guo H.; Zhou P.; Shi Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Das J.; Tradigo G.; Veltri P.; H Guzzi P.; Roy S. Data science in unveiling COVID-19 pathogenesis and diagnosis: evolutionary origin to drug repurposing. Briefings Bioinf. 2021, 22, 855–872. 10.1093/bib/bbaa420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. T.; Carabelli A. M.; Jackson B.; Gupta R. K.; Thomson E. C.; Harrison E. M.; Ludden C.; Reeve R.; Rambaut A.; Peacock S. J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A. J.; Starr T. N.; Gilchuk P.; Zost S. J.; Binshtein E.; Loes A. N.; Hilton S. K.; Huddleston J.; Eguia R.; Crawford K. H.; et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 2021, 29, 44–57. 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M.; de Marco A.; Lempp F. A.; Tortorici M. A.; Pinto D.; Walls A. C.; Beltramello M.; Chen A.; Liu Z.; Zatta F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. 10.1016/j.cell.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell H. F.; Agius R. M.; Kendrick D.; Stewart M. Vaccines, convalescent plasma, and monoclonal antibodies for covid-19. Br. Med. J. 2020, 370, m2722 10.1136/bmj.m2722. [DOI] [PubMed] [Google Scholar]
- Tang X.; Ying R.; Yao X.; Li G.; Wu C.; Tang Y.; Li Z.; Kuang B.; Wu F.; Chi C.; et al. Evolutionary analysis and lineage designation of SARS-CoV-2 genomes. Sci. Bull. 2021, 66, 2297–2311. 10.1016/j.scib.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll J. G.; van Helmond N.; Senefeld J. W.; Wiggins C. C.; Klassen S. A.; Baker S. E.; Larson K. F.; Murphy B. M.; Andersen K. J.; Ford S. K.; et al. Convalescent plasma for infectious diseases: Historical framework and use in COVID-19. Clin. Microbiol. Newsl. 2021, 43, 23–32. 10.1016/j.clinmicnews.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D.; Dejnirattisai W.; Supasa P.; Liu C.; Mentzer A. J.; Ginn H. M.; Zhao Y.; Duyvesteyn H. M.; Tuekprakhon A.; Nutalai R.; et al. Evidence of escape of SARS-CoV-2 variant B. 1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348–2361. 10.1016/j.cell.2021.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm E.; Kronig I.; Neher R. A.; Eckerle I.; Vetter P.; Kaiser L.; et al. Novel SARS-CoV-2 variants: the pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. 10.1016/j.cmi.2021.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J. K.; Roy S.; Guzzi P. H. Analyzing Host-Viral Interactome of SARS-CoV-2 for Identifying Vulnerable Host Proteins during COVID-19 Pathogenesis. Infect., Genet. Evol. 2021, 93, 104921 10.1016/j.meegid.2021.104921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J. K.; Chakrobarty S.; Roy S. A Scheme for Inferring Viral-Host Associations based on Codon Usage Patterns Identifies the Most Affected Signaling Pathways during COVID-19. J. Biomed. Inf. 2021, 118, 103801 10.1016/j.jbi.2021.103801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori G.; Luberto L.; Maffei M.; Aurisicchio L.; Roscilli G.; Palombo F.; Marra E. SARS-CoV-2 SPIKE PROTEIN: an optimal immunological target for vaccines. J. Transl. Med. 2020, 18, 222 10.1186/s12967-020-02392-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satarker S.; Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. 10.1016/j.arcmed.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Tsybovsky Y.; Gorman J.; Rapp M.; Cerutti G.; Chuang G.-Y.; Katsamba P. S.; Sampson J. M.; Schön A.; Bimela J.; et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe 2020, 28, 867–879. 10.1016/j.chom.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztain T.; Ahn S.-H.; Bogetti A. T.; Casalino L.; Goldsmith J. A.; Seitz E.; Mc- Cool R. S.; Kearns F. L.; Acosta-Reyes F.; Maji S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 963–968. 10.1038/s41557-021-00758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R.; Edwards R. J.; Mansouri K.; Janowska K.; Stalls V.; Gobeil S. M.; Kopp M.; Li D.; Parks R.; Hsu A. L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. 10.1038/s41594-020-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateeb J.; Li Y.; Zhang H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Critical Care 2021, 25, 244 10.1186/s13054-021-03662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.; Dryden D. Allostery without conformational change. Eur. Biophys. J. 1984, 11, 103–109. 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- Bozovic O.; Zanobini C.; Gulzar A.; Jankovic B.; Buhrke D.; Post M.; Wolf S.; Stock G.; Hamm P. Real-time observation of ligand-induced allosteric transitions in a PDZ domain. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 26031–26039. 10.1073/pnas.2012999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid K. M.; Yu X.; Leitner D. M. Change in vibrational entropy with change in protein volume estimated with mode Grüneisen parameters. J. Chem. Phys. 2021, 154, 055102 10.1063/5.0039175. [DOI] [PubMed] [Google Scholar]
- Bu Z.; Callaway D. J. Proteins move! Protein dynamics and long-range allostery in cell signaling. Adv. Protein Chem. Struct. Biol. 2011, 83, 163–221. 10.1016/B978-0-12-381262-9.00005-7. [DOI] [PubMed] [Google Scholar]
- Gerek Z. N.; Ozkan S. B. Change in allosteric network affects binding affinities of PDZ domains: analysis through perturbation response scanning. PLoS Comput. Biol. 2011, 7, e1002154 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.; Liu Y.; Ginzinger S. W.; Wishart D. S. SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton S.; Selvaratnam R.; Ahmed R.; Melacini G.. Protein NMR; Springer, 2018; pp 391–405. [DOI] [PubMed] [Google Scholar]
- Bader G. D.; Hogue C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003, 4, 1–27. 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R.; VanSchouwen B.; Fogolari F.; Mazhab-Jafari M. T.; Das R.; Melacini G. The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys. J. 2012, 102, 630–639. 10.1016/j.bpj.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L. C.; Rodrigues J. P.; Kastritis P. L.; Bonvin A. M.; Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Grabowski F.; Preibisch G.; Giziński S.; Kochańczyk M.; Lipniacki T. SARS-CoV-2 variant of concern 202012/01 has about twofold replicative advantage and acquires concerning mutations. Viruses 2021, 13, 392 10.3390/v13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Nair M. S.; Liu L.; Iketani S.; Luo Y.; Guo Y.; Wang M.; Yu J.; Zhang B.; Kwong P. D.; et al. Antibody resistance of SARS-CoV-2 variants B. 1.351 and B. 1.1. 7. Nature 2021, 593, 130–135. 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- Caniels T. G.; Bontjer I.; van der Straten K.; Poniman M.; Burger J. A.; Appelman B.; Lavell A. H.; Oomen M.; Godeke G.-J.; Valle C.; et al. Emerging SARS-CoV-2 variants of concern evade humoral immune responses from infection and vaccination. medRxiv 2021, 257441 10.1101/2021.05.26.21257441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P. S.; Viswapriya V.; Rajakumari M. L.; Murugan S. Probiotics mediated control and immunomodulation of SARS-CoV-2 associated pathological conditions. Ann. Phytomed. 2021, 10, 130–135. [Google Scholar]
- Davies N. G.; Abbott S.; Barnard R. C.; Jarvis C. I.; Kucharski A. J.; Munday J. D.; Pearson C. A.; Russell T. W.; Tully D. C.; Washburne A. D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science 2021, 372, 3055 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal J. L.; Andrews N.; Gower C.; Gallagher E.; Simmons R.; Thelwall S.; Stowe J.; Tessier E.; Groves N.; et al. Effectiveness of Covid-19 vaccines against the B. 1.617. 2 (Delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Singh J.; Hasnain S. E.; Sundar D. Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity. Int. J. Mol. Sci. 2021, 22, 9131 10.3390/ijms22179131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangra S.; Ye C.; Rathnasinghe R.; Stadlbauer D.; Alshammary H.; Amoako A. A.; Awawda M. H.; Beach K. F.; Bermúdez-González M. C.; Chernet R. L.; et al. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, e283–e284. 10.1016/S2666-5247(21)00068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese M.; Jiang B.; Iwai N.; Mar M.; Sahoo M. K.; Yamamoto F.; Mfuh K. O.; Miller J.; Wang H.; Zehnder J.; et al. Identification of a SARS-CoV-2 variant with L452R and E484Q neutralization resistance mutations. J. Clin. Microbiol. 2021, 59, JCM-00741 10.1128/JCM.00741-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr R.; Dimartino D.; Marier C.; Zappile P.; Wang G.; Lighter J.; Elbel B.; Troxel A. B.; Heguy A.; et al. Dominance of Alpha and Iota variants in SARS-CoV-2 vaccine breakthrough infections in New York City. J. Clin. Invest. 2021, 131, e152702 10.1172/JCI152702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Rojas C.; Jimenez-Vasquez V.; Hurtado V.; Mestanza O.; Molina I. S.; Barcena L.; Ruiz S. M.; Acedo S.; Lizarraga W.; Bailon H.; et al. Genomic analysis reveals a rapid spread and predominance of Lambda (C. 37) SARS-COV-2 lineage in Peru despite circulation of variants of concern. J. Med. Virol. 2021, 93, 6845–6849. 10.1002/jmv.27261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty C.; Bhattacharya M.; Sharma A. R. Present variants of concern and variants of interest of severe acute respiratory syndrome coronavirus 2: Their significant mutations in S-glycoprotein, infectivity, re-infectivity, immune escape and vaccines activity. Rev. Med. Virol. 2021, e2270 10.1002/rmv.2270. [DOI] [Google Scholar]
- Thomson E. C.; Rosen L. E.; Shepherd J. G.; Spreafico R.; da Silva Filipe A.; Wojcechowskyj J. A.; Davis C.; Piccoli L.; Pascall D. J.; Dillen J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187. 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Jain A.; McDonald L. R.; Gambogi C.; Lee A. L.; Dokholyan N. V. Mapping allosteric communications within individual proteins. Nat. Commun. 2020, 11, 3862 10.1038/s41467-020-17618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan N. V. Controlling allosteric networks in proteins. Chem. Rev. 2016, 116, 6463–6487. 10.1021/acs.chemrev.5b00544. [DOI] [PubMed] [Google Scholar]
- Verkhivker G. M.; Agajanian S.; Oztas D. Y.; Gupta G. Comparative perturbation-based modeling of the SARS-CoV-2 spike protein binding with host receptor and neutralizing antibodies: Structurally adaptable allosteric communication hotspots define spike sites targeted by global circulating mutations. Biochemistry 2021, 60, 1459–1484. 10.1021/acs.biochem.1c00139. [DOI] [PubMed] [Google Scholar]
- Verkhivker G.; Agajanian S.; Oztas D.; Gupta G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. 10.1021/acs.jctc.1c00372. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Bhattacharya D.; Nowotny J.; Cao R.; Cheng J. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. 10.1093/nar/gkw336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S. M.-C.; Janowska K.; McDowell S.; Mansouri K.; Parks R.; Manne K.; Stalls V.; Kopp M. F.; Henderson R.; Edwards R. J.; et al. D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. 2021, 34, 108630 10.1016/j.celrep.2020.108630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.; Hughes T. A.; Kelkar A.; Yu X.; Cheng K.; Park S.; Huang W.-C.; Lovell J. F.; Neelamegham S. Inhibition of SARS-CoV-2 viral entry upon blocking N-and O-glycan elaboration. eLife 2020, 9, e61552 10.7554/eLife.61552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Wang M.; Yu D.; Zhang X. Variations in SARS-CoV-2 spike protein cell epitopes and glycosylation profiles during global transmission course of COVID-19. Front. Immunol. 2020, 11, 2222 10.3389/fimmu.2020.565278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020, 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola L.; Hadi-Alijanvand H.; Song X.; Hu G.; Giuliani A. The discovery of a putative allosteric site in the SARS-CoV-2 spike protein using an integrated structural/dynamic approach. J. Proteome Res. 2020, 19, 4576–4586. 10.1021/acs.jproteome.0c00273. [DOI] [PubMed] [Google Scholar]
- Das R.; Melacini G. A model for agonism and antagonism in an ancient and ubiquitous cAMP-binding domain. J. Biol. Chem. 2007, 282, 581–593. 10.1074/jbc.M607706200. [DOI] [PubMed] [Google Scholar]
- Das R.; Esposito V.; Abu-Abed M.; Anand G. S.; Taylor S. S.; Melacini G. cAMP activation of PKA defines an ancient signaling mechanism. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 93–98. 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhab-Jafari M. T.; Das R.; Fotheringham S. A.; SilDas S.; Chowdhury S.; Melacini G. Understanding cAMP-dependent allostery by NMR spectroscopy: comparative analysis of the EPAC1 cAMP-binding domain in its apo and cAMP-bound states. J. Am. Chem. Soc. 2007, 129, 14482–14492. 10.1021/ja0753703. [DOI] [PubMed] [Google Scholar]
- Das R.; Mazhab-Jafari M. T.; Chowdhury S.; SilDas S.; Selvaratnam R.; Melacini G. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J. Biol. Chem. 2008, 283, 19691–19703. 10.1074/jbc.M802164200. [DOI] [PubMed] [Google Scholar]
- Das R.; Chowdhury S.; Mazhab-Jafari M. T.; SilDas S.; Selvaratnam R.; Melacini G. Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J. Biol. Chem. 2009, 284, 23682–23696. 10.1074/jbc.M109.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholl E. T.; Das R.; SilDas S.; Taylor S. S.; Melacini G. Communication between tandem cAMP binding domains in the regulatory subunit of protein kinase A-Iα as revealed by domain-silencing mutations. J. Biol. Chem. 2010, 285, 15523–15537. 10.1074/jbc.M110.105783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R.; Chowdhury S.; VanSchouwen B.; Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6133–6138. 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Aportela E.; López-Blanco J. R.; Chacón P. FRODOCK 2.0: fast protein-protein docking server. Bioinformatics 2016, 32, 2386–2388. 10.1093/bioinformatics/btw141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.