

ABSTRACT
Phosphatidylinositol 4-kinase IIIα (PI4KIIIα) is the major enzyme responsible for generating phosphatidylinositol (4)-phosphate [PI(4)P] at the plasma membrane. This lipid kinase forms two multicomponent complexes, both including a palmitoylated anchor, EFR3. Whereas both PI4KIIIα complexes support production of PI(4)P, the distinct functions of each complex and mechanisms underlying the interplay between them remain unknown. Here, we present roles for differential palmitoylation patterns within a tri-cysteine motif in EFR3B (Cys5, Cys7 and Cys8) in controlling the distribution of PI4KIIIα between these two complexes at the plasma membrane and corresponding functions in phosphoinositide homeostasis. Spacing of palmitoyl groups within three doubly palmitoylated EFR3B ‘lipoforms’ affects both interactions between EFR3B and TMEM150A, a transmembrane protein governing formation of a PI4KIIIα complex functioning in rapid phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] resynthesis following phospholipase C signaling, and EFR3B partitioning within liquid-ordered and -disordered regions of the plasma membrane. This work identifies a palmitoylation code involved in controlling protein–protein and protein–lipid interactions that affect a plasma membrane-resident lipid biosynthetic pathway.
KEY WORDS: EFR3, Membrane microdomains, Palmitoylation, PI4KIIIα, PI(4)P, Phosphoinositides, TMEM150A
Summary: Different palmitoylation patterns on a lipid kinase adaptor protein control partitioning of the kinase between two spatiotemporally and functionally distinct complexes within the plasma membrane.
INTRODUCTION
Phosphoinositides (PIPs) are low abundance phospholipids synthesized from phosphatidylinositol (PI). Despite their scarcity, PIPs play crucial roles, including signal transduction, proliferation and regulated recruitment of proteins to membranes (Balla, 2013). Their defining structural feature is a myo-inositol headgroup that is phosphorylated by lipid kinases at positions 3, 4 and/or 5, generating a total of seven distinct phosphoinositides (Balla, 2013). The most abundant PIP, phosphatidylinositol 4-phosphate [PI(4)P], is located in multiple organelle membranes and plays diverse cellular roles. At the plasma membrane (PM), it serves as a precursor in the major pathway to produce phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] and phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3]. Beyond this biosynthetic role, PM PI(4)P (Batrouni and Baskin, 2021) regulates transport and homeostasis of phosphatidylserine (Chung et al., 2015a) and other lipids (Timcenko et al., 2019), modulates ion channel activity and establishes PM identity via its anionic charge (Hammond et al., 2012), and serves as a phospholipase C (PLC) substrate (Gil De Rubio et al., 2018; Zhang et al., 2013).
Of the four mammalian phosphatidylinositol 4-kinase (PI4K) isoforms, a single enzyme, PI4KIIIα, encoded by PI4KA, synthesizes the majority of the PM PI(4)P pool, with other non-redundant PI4K isoforms producing biochemically distinct PI(4)P pools on intracellular membranes. Speaking to the vital importance of the PM PI(4)P pool, PI4KIIIα is essential in all model organisms tested, including mice, fruit flies and yeast (Cutler et al., 1997; Nakatsu et al., 2012; Yan et al., 2011). PI4KIIIα function is important to human health, as mutations to PI4KIIIα and some of its non-catalytic accessory proteins cause a variety of disorders, ranging from hypomyelination to severe immunodeficiencies and congenital intestinal obstructions (Baskin et al., 2016; Bigorgne et al., 2014; Gupta et al., 2014; Salter et al., 2021; Traverso et al., 2013; Verdura et al., 2021; Zara et al., 2006). Several viruses also hijack this enzyme to remodel host membranes in support of viral replication (Tai et al., 2009; Vaillancourt et al., 2009). Given these important (patho)physiological functions of PM PI(4)P, the regulation of its levels is critically important.
Because PI4KIIIα lacks membrane-anchoring motifs (Nakatsu et al., 2012), its PM recruitment to access PI requires several additional factors. The six-pass transmembrane protein suppressor of four-kinase 1 (Sfk1) was first identified in yeast as a factor whose overexpression partially suppresses a temperature-sensitive mutant of Stt4, the yeast PI4KIIIα ortholog (Audhya and Emr, 2002). Subsequently, two essential proteins, Efr3 and Ypp1, were characterized to form a complex with Stt4 at the PM (Baird et al., 2008), though the relationship of this complex to Sfk1 remains unclear. The mammalian Efr3 and Ypp1 orthologs, EFR3A and EFR3B, and TTC7A and TTC7B, respectively, similarly recruit PI4KIIIα to the PM (Nakatsu et al., 2012), with the mammalian PI4KIIIα complex containing an additional factor, FAM126A (also known as hyccin) or FAM126B (Baskin et al., 2016; Zara et al., 2006).
Structural, biochemical and imaging studies support a working model for the EFR3–TTC7–FAM126–PI4KIIIα complex, which we term Complex I (Fig. 1A) (Baskin et al., 2016; Chung et al., 2015b). EFR3 constitutively localizes to the PM through electrostatic interactions with a basic patch on its α-helical N-terminal domain and by palmitoylation of an N-terminal Cys-rich motif (Nakatsu et al., 2012). The unstructured EFR3 C-terminal tail binds to TTC7 (Wu et al., 2014), which is part of a globular hexameric complex comprising two copies each of TTC7, FAM126 and PI4KIIIα (Dornan et al., 2018; Lees et al., 2017). As for a potential role of an Sfk1-like protein in mammals, the six-pass transmembrane protein TMEM150A has been identified as a functional Sfk1 ortholog, existing in a second complex with EFR3 and PI4KIIIα, which we term Complex II (Fig. 1A). When overexpressed, Complex II accelerates PI(4,5)P2 resynthesis after acute depletion, presumably via production of PI(4)P, the substrate for phosphatidylinositol 5-kinase (PIP5K) enzymes that synthesize PI(4,5)P2 (Chung et al., 2015b). Interestingly, Complex II forms downstream of Complex I, suggesting a degree of interplay between both complexes wherein PI4KIIIα and EFR3 are handed off from TTC7 and FAM126 to TMEM150A.
Fig. 1.

Dual palmitoylation of EFR3B is required for its PM localization. (A) Diagram illustrating the current understanding of the assembly of PI4KIIIα into two complexes (Complex I and Complex II) at the PM. The sequence of the N-terminal Cys-rich motif of EFR3B is shown, with Cys residue positions highlighted. (B) Representative confocal microscopy images of WT and mutant EFR3B–mCherry expressed in HeLa cells. Traces underneath each image indicate fluorescence intensity along the line indicated in the image, to illustrate the presence or absence of discernible PM fluorescence. Images were considered representative if more than 90% of cells imaged (n=20) showed the same localization. Scale bars: 15 µm. (C) Representative confocal microscopy images of TTC7B–GFP showing its recruitment by WT or CxS mutant EFR3B–mCherry. Images are representative of n=20. Scale bars: 15 µm. (D) APE assay to quantify the extent of palmitoylation of various EFR3B–3×FLAG mutants, with a calnexin positive control (bottom) and omission of hydroxylamine as a negative control. EFR3B or calnexin species with zero, one, two or three PEG groups are indicated by the presence of –, *, **, or ***, respectively. A sample of lysate prior to APE was analyzed to show input (FLAG input). Higher intensity exposures of the same blots shown on the right reveal the palmitoylation state of EFR3B(C5,7,8S). The position of a 100 kDa size marker is indicated. Blots are representative of five experiments. (E,F) Bioorthogonal metabolic labeling pulse-chase experiment with alk-16 [16 h pulse, 0–6 h chase in presence of cycloheximide and lactacystin (Cyclo/Lact) to prevent protein synthesis and degradation] showing that EFR3B–3×FLAG palmitoylation is stable over several hours. (E) In-gel fluorescence (Cy5.5, extent of EFR3B–3×FLAG palmitoylation) and western blot (total EFR3B–3×FLAG). (F) Quantification of normalized data (mean±s.d. of n=3). Statistical significance was determined using a two-tailed unpaired Student's t-test. ns, not significant.
It remains unclear why cells contain two distinct PI4KIIIα-containing complexes within the same membrane to generate PI(4)P. Critically, the mechanisms regulating the balance between both complexes have not been investigated. Given their compositional differences, Complexes I and II may be stabilized in the PM by different types of interactions. Though EFR3 anchors Complex I to the PM, conserved basic residues on TTC7 and PI4KIIIα engage in secondary, stabilizing electrostatic interactions with acidic lipids in the PM inner leaflet (Lees et al., 2017). By contrast, Complex II does not contain TTC7 but includes TMEM150A, which, alongside EFR3, presumably plays important roles in anchoring PI4KIIIα to the PM. The differential mechanisms by which PI4KIIIα is tethered to the PM could form the basis for functionally different complexes.
The palmitoylated protein EFR3 is the only non-catalytic protein shared between both complexes. We reasoned that a detailed understanding of its association with membranes might reveal mechanisms underlying interplay between the two PI4KIIIα complexes. S-acylation is the post-translational modification of Cys residues as fatty acyl thioesters, typically with palmitoyl groups. Palmitoylation of target proteins induces membrane anchoring, and its reversibility makes it a powerful mechanism to regulate protein localization, degradation and function (Abrami et al., 2017; Percherancier et al., 2001; Zhang et al., 2020). Palmitoylation of Cys-containing motifs within proteins can mediate partitioning to liquid-ordered (Lo)-like membrane domains, which are enriched in lipids with saturated tails (Levental et al., 2010). Interestingly, PI(4,5)P2 synthesis may occur in these domains (Myeong et al., 2021), though the mechanisms by which PI(4,5)P2 synthesis is compartmentalized in Lo-like or the complementary liquid-disordered (Ld)-like membrane domains are not fully understood.
The N terminus of EFR3B, one of two mammalian EFR3 proteins implicated in PI4KIIIα function, contains three potentially palmitoylated Cys residues (Cys5, Cys7 and Cys8, referred to hereafter as C5, C7 and C8), and mutation of all three to Ser abolishes the PM localization of EFR3B (Bojjireddy et al., 2015; Nakatsu et al., 2012). However, the contributions of palmitoylation at individual residues to EFR3 localization and PI4KIIIα function have not been examined. Due to the reversibility of palmitoylation and the potential for combinatorial changes to EFR3 palmitoylation to impact its interactions within the two PI4KIIIα complexes, we hypothesized that palmitoylation could represent a regulatory mechanism governing PI4KIIIα recruitment to membrane subregions within distinct complexes.
We have undertaken a detailed examination of the role of EFR3B palmitoylation as a mechanism regulating the assembly of PI4KIIIα into two spatiotemporally and functionally distinct complexes at the PM. A key aspect of our study was understanding how interactions between EFR3B and TMEM150A modulate the biophysical and catalytic properties of the PI4KIIIα complex in the PM. Using biochemistry, chemical biology and biophysical imaging techniques, we found that the majority of EFR3B molecules are heavily palmitoylated and that lipidation of any two of the three Cys residues is sufficient for PM localization. Furthermore, EFR3B palmitoylation was stable over the course of multiple hours, giving rise to several ‘lipoforms’ of EFR3B, representing differentially palmitoylated pools of EFR3B that coexist in the PM. Intriguingly, TMEM150A preferentially interacted with the EFR3B lipoform with adjacent palmitoylation of C7 and C8 [EFR3B(7,8-palm)]. An in situ, imaging-based detergent resistant membrane (iDRM) assay revealed that Complex II preferentially localized to Ld-like PM regions, suggesting a Ld-like specific function for PI(4,5)P2 synthesis. Finally, quantification of PI(4)P and PI(4,5)P2 resynthesis following acute depletion under conditions favoring the formation of Complex I or II solidified a role for Complex II as most efficiently producing PI(4)P for this functional process.
We propose a comprehensive framework explaining PI4KIIIα association with and function in the PM. First, PI4KIIIα is recruited to the PM by EFR3B (Baskin et al., 2016; Chung et al., 2015b; Nakatsu et al., 2012) within Complex I, which localizes to Lo-like regions of the PM. Subsequently, the preferential interaction of TMEM150A with EFR3B(7,8-palm) associated with PI4KIIIα leads to the formation of Complex II, which localizes to Ld-like PM regions. The PI4KIIIα pool associating with other EFR3B lipoforms largely remains in Complex I. This model provides a basis for understanding how cells maintain two spatiotemporally and functionally distinct PI4KIIIα complexes to facilitate PI(4)P synthesis within different PM subdomains.
RESULTS
Two of the three Cys residues of EFR3B are required for its PM localization
EFR3B contains three Cys residues (C5, C7 and C8) within an N-terminal palmitoylation motif (Fig. 1A). Mutation of all three palmitoylation motif Cys residues in EFR3B (or of all four motif Cys residues in the case of the homolog EFR3A) abolishes PM localization (Nakatsu et al., 2012; Bojjireddy et al., 2015). Yet, the extent of palmitoylation of wild-type (WT) EFR3B, and the palmitoylation requirements for PM localization and function in the PI4KIIIα complex, remained unknown. Therefore, we systematically evaluated the link between EFR3B palmitoylation status and PM localization. We generated all permutations of single and double Cys-to-Ser mutants in EFR3B–mCherry. Confocal microscopy revealed that mutation of any single Cys residue (C5S, C7S and C8S mutants; referred to as CxS or dual-palm mutants, where x indicates the mutated residue position) did not impact EFR3B–mCherry PM anchoring (Fig. 1B). However, mutation of any Cys pair (referred to as CxxS or single-palm mutants) resulted in mislocalization of most of EFR3B–mCherry to the cytosol (Fig. 1B). The triple Cys mutant (C5,7,8S) was absent from the PM (Fig. 1B) (Nakatsu et al., 2012). Therefore, two of the three Cys residues within the N-terminal motif are minimally required for stable PM anchoring of EFR3B.
We next tested the ability of the CxS dual-palm mutants to recruit TTC7B to the PM as a proxy for Complex I formation. Overexpression of WT EFR3B is necessary to recruit transiently transfected TTC7B to the PM (Nakatsu et al., 2012). Here, we examined the localization of TTC7B–GFP in cells co-expressing either WT or CxS EFR3B–mCherry and found that all three CxS mutants recruited TTC7B as efficiently as WT EFR3B (Fig. 1C).
These results indicate that any two of the three N-terminal Cys residues are sufficient for EFR3B localization and function, leading us to ask whether all three Cys residues of WT EFR3B are actually palmitoylated. We used the acyl-PEG exchange (APE) assay (Percher et al., 2016) to assess the degree of palmitoylation in WT and CxS EFR3B. APE allows for tagging of palmitoylated Cys residues with a mass tag (5 kDa PEG), resulting in a molecular weight shift that can be visualized by western blotting. We found that WT EFR3B was roughly split equally between double and triple palmitoylated forms, with very minimal amounts of single- or non-palmitoylated protein observed (Fig. 1D). All three EFR3B CxS mutants were nearly completely doubly palmitoylated. By contrast, a representative CxxS mutant – C7,8S – was minimally palmitoylated. Combined with the imaging results, these data confirm the dual palmitoylation requirement for EFR3B PM localization and that two Cys residues within this motif result in efficient (i.e. near-stoichiometric) palmitoylation. Furthermore, we found that co-expression of EFR3B with TMEM150A had no effect on EFR3B palmitoylation status (Fig. S1A,B). Although the endogenous levels of EFR3B in these cell lines were too low to reliably detect by western blotting, an APE assay revealed similar heavy palmitoylation of endogenous EFR3A (Fig. S1C), suggesting that the EFR3B palmitoylation patterns under overexpression conditions are representative of the endogenous protein. We also confirmed the colocalization of EFR3B and TMEM150A in the plasma membrane (Fig. S1D).
EFR3B palmitoylation is stable
Palmitoylation is a reversible post-translational modification that can dynamically regulate protein–membrane and protein–protein interactions (Levental et al., 2010). We next investigated the dynamics of EFR3B palmitoylation, using metabolic labeling of cells expressing EFR3B–FLAG with a pulse of a bioorthogonal, alkyne-containing palmitate analog (alk-16) (Martin and Cravatt, 2009) and subsequent chase with regular medium in the presence of cycloheximide and lactacystin (protein synthesis and proteasome inhibitors, respectively), followed by lysis, immunoprecipitation, click chemistry tagging and visualization using SDS–PAGE and western blotting. In contrast to other proteins that display very rapid palmitoylation–depalmitoylation dynamics (Goodwin et al., 2005), we found that EFR3B palmitoylation is stable over several hours (Fig. 1E,F). Collectively, these studies of EFR3B palmitoylation indicate that the majority of WT EFR3B is stably doubly or triply palmitoylated – a requirement for its PM localization.
Interactions between EFR3B and TMEM150A modulate their respective membrane dynamics
We next explored how palmitoylation of EFR3B impacts its interactions with TMEM150A, the only other constitutively PM-localized component involved in PI4KIIIα recruitment. EFR3B and TMEM150A have been observed to interact in co-immunoprecipitation (co-IP) assays (Chung et al., 2015b), though no discrete binding sites have been identified. Therefore, the mode of interaction may not rely entirely on protein–protein binding but may also involve protein–lipid interactions. Lipid-dependent, dynamic partitioning into Lo-like nanodomains helps to concentrate selected proteins to facilitate their interactions (Lingwood and Simons, 2010), and palmitoylation is a major mechanism by which PM proteins can partition into Lo-like nanodomains (Levental et al., 2010). We hypothesized that EFR3B and TMEM150A participate in protein–protein and protein–lipid interactions at the PM, and that the EFR3B palmitoylation status might promote these interactions. We thus characterized the PM dynamics of EFR3B and TMEM150A.
We performed fluorescence recovery after photobleaching (FRAP) experiments on the ventral PM of a rat basophilic leukemia cell line (RBL-2H3) expressing fluorescently tagged proteins to quantitively assess their recovery time and mobile fraction. The average fluorescence recovery time (t1/2, a measure of probe diffusion) of WT EFR3B–GFP was 16±5 s (mean±s.d.; Fig. 2A), which was similar to that of other PM inner leaflet probes in RBL-2H3 cells measured under identical conditions (Bag et al., 2021). The mobile fraction was greater than 80% (Fig. 2C), indicating that the diffusing pool represents the majority of EFR3B molecules. Because WT EFR3B exists as a mixture of triply and doubly palmitoylated lipoforms (Fig. 1D), we also examined whether the observed diffusion properties are dictated by one of these species. We performed FRAP experiments on all three fluorescently tagged CxS mutants and found that their recovery dynamics were the same as those of WT EFR3B (Fig. S2A). Overall, these data confirm that the diffusion properties of triply and doubly palmitoylated species are the same.
Fig. 2.

EFR3B interacts with TMEM150A, and the membrane dynamics of WT and CxS mutant EFR3B are similar. (A) FRAP recovery times (t1/2) of EFR3B–GFP with or without TMEM150A–mCherry (TMEM150A–mCh). The increase in the recovery time of EFR3B in the presence of TMEM150A suggests an interaction between the two proteins. (B) FRAP recovery times (t1/2) of TMEM150A–GFP with or without EFR3B–mCherry (EFR3B–mCh). Note that the recovery times of co-expressed TMEM150A and EFR3B move closer to each other (compare A and B). (C) Mobile fraction of EFR3B–GFP or TMEM150A–GFP when expressed alone or together as evaluated by FRAP. (D) Diffusion coefficients of EFR3B–GFP and TMEM150A–GFP, expressed separately, as measured by ImFCS. Data are pooled from three independent experiments, n=15–16 cells. (E) Recovery times (t1/2) of WT EFR3B–GFP and individual EFR3B–GFP or EFR3B–mNG CxS mutants with or without TMEM150A–mCherry. Note that the CxS mutations have no effect on the membrane diffusion of EFR3B and are insensitive to the presence of TMEM150A. (F) Recovery time (t1/2) of TMEM150A–GFP expressed alone or in the presence of WT or CxS mutant EFR3B–mCherry. Note that the CxS mutants have the same effect on TMEM150A–GFP recovery time as WT EFR3B. To improve clarity and facilitate comparisons between effects of single or co-expression, the data in A and B are also plotted in E and F, respectively. Data in A–C,E,F are mean±s.d., n=14–27. **P<0.01; ***P<0.005; ****P<0.001; ns, not significant (two-tailed unpaired t-test in A and B; one-way ANOVA with Tukey–Kramer post-hoc test in C,E and F).
In contrast to EFR3B–GFP, TMEM150A–GFP showed a much slower recovery (t1/2=55±13 s), with a smaller mobile fraction (∼60%) (Fig. 2B,C), consistent with studies indicating that the diffusion of transmembrane proteins is generally slower than that of lipid-anchored proteins (Bag et al., 2020; Kenworthy et al., 2004). As a control, the FRAP recovery properties of TMEM150A–GFP were similar to its homolog TMEM150B–GFP, which is structurally similar but not a PI4KIIIα interactor (Chung et al., 2015b) (Fig. S2B,C). The diffusion of the TMEM150 proteins was, however, slower than that of other multipass transmembrane proteins. For example, the t1/2 of the seven-pass immunoglobulin E receptor, FcεRI, is 18±4 s when measured on an identical instrumental setup (Bag et al., 2021). Such slow diffusion may not be due to clustering, as we found that co-expression of TMEM150A–mCherry with TMEM150A–GFP to increase the overall TMEM150A levels showed similar diffusion properties as the lower amount of TMEM150A–GFP alone (Fig. S2D,E). Assuming that mass action would increase clustering, these experiments suggest that the slow diffusion may not arise from overexpression artifacts, though homo-oligomerization of TMEM150A cannot be ruled out. Furthermore, TMEM150A–GFP may associate with other unknown membrane components that slow down its diffusion. We used imaging fluorescence correlation spectroscopy (ImFCS) on RBL-2H3 cells expressing EFR3B–GFP or TMEM150A–GFP to measure their diffusion coefficients (D) with high precision (Bag and Wohland, 2014; Bag et al., 2021; Krieger et al., 2015). The cumulative distributions constructed from ∼10,000 D values each confirmed the slower diffusion of TMEM150A–GFP relative to EFR3B–GFP, with average D values of 0.15±0.05 μm2/s and 0.28±0.09 μm2/s, respectively (mean±s.d.; Fig. 2D).
Interestingly, the FRAP characteristics of both proteins changed under co-expression conditions. We observed an increase in the t1/2 of EFR3B–GFP in the presence of TMEM150A–mCherry (27±13 s; Fig. 2A), with unaltered mobile fraction (Fig. 2C), suggesting that the mobile population of EFR3B interacts with TMEM150A, leading to slower diffusion, consistent with previously reported co-IP results (Chung et al., 2015b). We observed consistent changes in the diffusion of TMEM150A–GFP co-expressed with EFR3B–mCherry (Fig. 2B). In this case, the t1/2 of TMEM150A–GFP decreased significantly (t1/2=38±14 s), and the mobile fraction was again unaltered (Fig. 2C). This result indicates that associating with EFR3B–mCherry liberates TMEM150A–GFP from its interactions with other membrane components, as speculated above. Notably, the t1/2 values for co-overexpressed EFR3B–GFP and TMEM150A–GFP moved closer to one another compared to those for individually expressed proteins (Fig. 2A,B). The recovery profiles of representative single cells with fitted curves and residuals are shown in Fig. S3.
To test the specificity of the diffusion changes of co-expressed EFR3B and TMEM150A, we conducted a series of additional control experiments. First, we measured the diffusion of TMEM150A–GFP co-expressed with Lyn–mRFP, a PM-localized inner leaflet, lipid-anchored kinase. The t1/2 and mobile fraction of TMEM150A–GFP remained unaltered, indicating that Lyn and TMEM150A do not interact non-specifically (Fig. S4A,B). This result suggests that the faster diffusion of TMEM150A in the presence of EFR3B is the result of specific interactions. We performed reciprocal experiments to test whether EFR3B impacts the diffusion of other transmembrane proteins by measuring the diffusion of a YFP-tagged muscarinic M1 receptor (M1R, also known as CHRM1), a seven-pass G protein-coupled receptor (GPCR), with or without co-overexpressed EFR3B–mCherry. The t1/2 of M1R–YFP was ∼23±5 s in both conditions, along with an unaltered mobile fraction (Fig. S4C,D), establishing the specificity of the EFR3B–TMEM150A interaction measured by FRAP.
To assess the importance of EFR3B palmitoylation in the EFR3B–TMEM150A interaction, we monitored the diffusion changes of TMEM150A–GFP in the presence of the dual-palm EFR3B mutants. Co-overexpression with CxS EFR3B–mCherry had a largely similar effect to that of WT EFR3B–mCherry, also decreasing the t1/2 of TMEM150A–GFP, indicating interactions between the doubly palmitoylated species and TMEM150A–GFP (Fig. 2F). By contrast, the diffusion of the CxS mutants, unlike WT EFR3B, did not show reduced diffusion (i.e. t1/2 was unchanged) when co-overexpressed with TMEM150A–mCherry (Fig. 2E).
We note that mNeonGreen (mNG)-tagged EFR3B(C7S) was used throughout because of clustering of the GFP-tagged construct, which was not observed with the mNG- or mCherry-tagged versions. FRAP experiments with the various EFR3B(C7S) constructs indicated that the GFP fusion displayed much slower diffusion kinetics than the other two GFP-tagged CxS constructs, whereas the mNG- and mCherry-tagged EFR3B(C7S) constructs did not (Fig. S4E). Collectively, these results suggest that the WT and dual-palm EFR3B mutants can dissociate TMEM150A from its low-diffusion assembly with other membrane components. The increased recovery time of EFR3B in the presence of TMEM150A may be due to contributions from the triply palmitoylated lipoform, which is absent in all CxS mutant-containing samples. Increased partitioning to Lo-like domains or changes in protein–protein interactions could reduce diffusion of the triply palmitoylated lipoform, which would be reflected in the bulk measurement of EFR3B diffusion by FRAP because it represents a significant fraction of total EFR3B (Fig. 1D).
TMEM150A preferentially interacts with certain EFR3B lipoforms within Ld-like membrane regions
Palmitoylation is a primary mechanism governing PM-localized protein partitioning to Lo-like nanodomains (Sezgin et al., 2017). These nanodomains function to provide optimal spatial compartmentalization for productive interactions between proteins. We recently developed an in situ, imaging-based detergent resistant membrane (iDRM) assay (Fig. 3A) to quantify relative preference of a protein for Lo-like nanodomains (Bag et al., 2021). The underlying principle is that Lo-preferring proteins within the PM are more resistant to mild treatment with the detergent Triton X-100 (TX-100) than proteins in Ld-like nanodomains. Therefore, a higher fraction of Lo-preferring proteins will remain in the PM of intact cells after TX-100 treatment (Fig. 3A). The key metric obtained from iDRM experiments is the characteristic retention (R) value, defined as the ratio of the fluorescence of TX-100-treated (+TX-100) and untreated (−TX-100) cells . A higher R value is consistent with a stronger interaction between the target protein and Lo-like nanodomains, resulting in more protein retention. A lower R value suggests a weaker interaction, as more protein is extracted from the PM by the detergent wash. Note that other factors may lead to detergent resistance, such as interactions with the actin cytoskeleton and other detergent-resistant proteins. The recently reported R values of probes that prefer Lo-like domains or Ld-like domains in RBL cells guide the interpretation of our results (Bag et al., 2021).
Fig. 3.

TMEM150A preferentially interacts with EFR3B(C5S) to form Complex II. (A) Diagram illustrating the workflow of the iDRM assay. All iDRM experiments were performed in RBL-2H3 cells. (B) Representative images (two per condition) of RBL-2H3 cells expressing EFR3B–GFP treated with PBS (−TX-100) or 0.04% TX-100 (+TX-100). Images are for −Tx-100 top, 22×19 µm; for −Tx-100 bottom, 28×19 µm; for +Tx-100 top, 25×21 µm; for +Tx-100 bottom, 28×20 µm. (C) Quantification of iDRM experiments on EFR3B–GFP (WT or CxS mutants) or EFR3B–mNG (for the C7S mutant) in the presence and absence of TMEM150A–mCherry (TMEM150A–mCh), with the R value (fraction of retained fluorescence) plotted. Note that WT EFR3B exhibits a strong increase in detergent resistance in the presence of TMEM150A–mCherry. However, the EFR3B(CxS) palmitoylation mutants do not. (D) Quantification of iDRM experiments on TMEM150A–GFP in the presence of EFR3B–mCherry (EFR3B–mCh; WT or indicated CxS mutants) or the indicated tagged Complex II components. In C and D, each data point represents the mean R value from 30 +TX-100 cells and 30 –TX-100 cells. Mean±s.d. of n=3 is indicated. *P<0.05; **P<0.01; ****P<0.001; ns, not significant (two-tailed unpaired t-test in C; one-way ANOVA with Tukey–Kramer post-hoc test in D).
We performed iDRM experiments using the same (co)-overexpression conditions as our earlier FRAP experiments. Representative images of cells expressing EFR3B–GFP washed with phosphate-buffered saline (PBS) or 0.04% TX-100 (Fig. 3B) show a modest amount of detergent resistance, yielding an R value of 0.37±0.03 (mean±s.d.). The detergent resistance of EFR3B–GFP strongly increased (R=0.71±0.07) when co-expressed with TMEM150A–mCherry. Interestingly, this was not the case for the dual-palm mutants (Fig. 3C). These observations are analogous to the increased t1/2 observed for WT EFR3B, but not for the EFR3B CxS mutants, in the presence of TMEM150A (Fig. 2E) and may be governed by an elevated Lo-like domain partitioning of the triply palmitoylated lipoform under these conditions.
By monitoring TMEM150A–GFP using the iDRM assay, we found that it was more detergent resistant (R=0.54±0.02) than EFR3B–GFP when expressed separately (Fig. 3C,D). Based on evidence that TMEM150A did not form homo-oligomers (Fig. S2D,E), we reasoned that TMEM150A detergent resistance may stem from its Lo-like domain preference and/or heterotypic interactions with other proteins. Interestingly, TMEM150A–GFP became highly susceptible to detergent when co-expressed with EFR3B–mCherry (Fig. 3D; R=0.13±0.04) or EFR3B(C5S)–mCherry (Fig. 3D; R=0.14±0.02). Yet, strikingly, the EFR3B(C7S)–mCherry mutant had no effect on the detergent resistance of TMEM150A–GFP (Fig. 3D; R=0.60±0.09), whereas EFR3B(C8S)–mCherry showed an intermediate effect (Fig. 3D; R=0.37±0.09).
We also tested the detergent resistance of key components of Complexes I and II. The R value of GFP–PI4KIIIα was similar in cells expressing components to assemble either Complex I (R=0.51±0.11) or Complex II (R=0.55±0.07) (Fig. S5). Recent structural studies of Complex I using cryogenic electron microscopy and hydrogen-deuterium exchange mass spectrometry have revealed extensive protein–protein interactions via the formation of a hexamer containing two copies each of TTC7B, FAM126A and PI4KIIIα (and presumably EFR3B) (Dornan et al., 2018; Lees et al., 2017). These strong protein-based interactions could explain the detergent resistance of GFP–PI4KIIIα in Complex I. Interestingly, when TMEM150A, the unique component of Complex II, was co-expressed with PI4KIIIα, EFR3B and TTC7B, it exhibited a very low R value (Fig. 3D; R=0.07±0.05), similar to its R value when co-expressed with WT or C5S mutant EFR3B (Fig. 3D; R=0.13±0.04 and R=0.14±0.02, respectively). This result cannot be explained by different levels of expression, as is indicated by plots showing individual fluorescence intensity values of TX-100-treated and untreated cells (Fig. S5). Overall, these results indicate that Complex II likely has a greater preference for Ld-like domains or has fewer intermolecular interactions than Complex I. Additionally, because the R value of GFP–PI4KIIIα remained unaltered under co-overexpression conditions to form both complexes, these results raise the possibility that both complexes may coexist in the PM.
Our iDRM results suggest that the palmitoylation pattern of EFR3B is key to the formation of Complex II. Specifically, TMEM150A interacts more strongly with EFR3B palmitoylated at adjacent positions (C7 and C8, as occurs in the C5S mutant of EFR3B). By contrast, we detected minimal interaction between TMEM150A and the C7S and C8S mutants of EFR3B, in which the remaining palmitoyl groups are separated by one or two residues (Fig. 3D). Overall, the spacing between palmitoylation sites on EFR3B, as well as the extent of their modification, could modulate the partitioning of EFR3B, leading to preferential interaction of TMEM150A with the C7/C8 doubly palmitoylated lipoform of EFR3B.
Interactions between EFR3B lipoforms and TMEM150A differentially impact the kinetics of PI(4,5)P2 synthesis
Finally, we investigated the functional significance of the preferential interaction between TMEM150A and the EFR3B lipoforms. A key role for PI4KIIIα is to provide PI(4)P for subsequent phosphorylation by PIP5K enzymes to produce PI(4,5)P2, for which demand is particularly high following its transient depletion by agonist-induced PLC signaling. TMEM150A has previously been implicated in PI(4,5)P2 homeostasis (Chung et al., 2015b), and we sought to investigate the role of the EFR3B–TMEM150A interaction in this process.
We used an established system for acutely depleting PI(4,5)P2 and observing its resynthesis in the PM of HeLa cells. This system takes advantage of the activation of the muscarinic M1 receptor (M1R) by its agonist oxotremorine-M (oxo-M), resulting in downstream activation of PLCβ and hydrolysis of a major portion of PM PI(4,5)P2. This signaling event is reversed by addition of the M1R antagonist atropine, allowing resynthesis of PI(4,5)P2 from PI, via the action of PI4KIIIα and PIP5Ks. The reversible nature of this system allows for monitoring of the recovery kinetics of PI(4,5)P2 by co-expression of a PI(4,5)P2-binding fluorescent biosensor, iRFP–PH(PLCδ), which localizes to the PM when it is replete with PI(4,5)P2 and to the cytosol upon PI(4,5)P2 depletion (Fig. 4A).
Fig. 4.

Experimental setup for studies to quantify PI(4,5)P2 recovery kinetics after acute depletion. (A) Diagram representing the M1R-mediated PI(4,5)P2 depletion system. DAG, diacylglycerol; IP3, inositol trisphosphate. (B) Top: representative curve (red line is raw data and black line is smoothed curve) depicting the cytosolic fluorescence of the PI(4,5)P2 biosensor iRFP–PH(PLCδ) over time, with corresponding confocal micrographs of a cell expressing iRFP–PH(PLCδ) at the indicated timepoints. Scale bars: 15 µm. Bottom: the rate of change of fluorescence for the cytosolic fluorescence curve shown above. The region highlighted by the orange box is shown in the expanded panel and depicts the relevant time period, post-addition of atropine, for determination of relevant kinetics parameters, which are defined on the right [initial rate of PI(4,5)P2 resynthesis and maximum rate of PI(4,5)P2 resynthesis]. A.U., arbitrary units.
We evaluated two kinetic parameters of the PI(4,5)P2 resynthesis process: initial rate and maximum rate (Fig. 4B). Rates were measured as the slope of the tangent to the recovery curve at a specified time point. The initial rate corresponds to the rate of loss of the cytosolic fluorescence signal of iRFP–PH(PLCδ) at t=0 of the recovery (i.e. at the time of addition of atropine). The maximum rate corresponds to the highest rate of loss of iRFP–PH(PLCδ) from the cytosol (i.e. the point of the recovery curve for which the slope of the tangent is greatest). These parameters afforded a comprehensive view of the recovery of the PI(4,5)P2 biosensor to the PM. Because phosphorylation of PI to form PI(4)P is the rate-limiting step in the resynthesis of PI(4,5)P2 (Falkenburger et al., 2010), monitoring PI(4,5)P2 resynthesis serves as a useful proxy for the PI4K activity in this system.
We began by examining the initial rates of PI(4,5)P2 resynthesis in control cells or cells overexpressing TMEM150A and/or EFR3B (either WT or a dual-palm mutant). We found that overexpression of TMEM150A alone did not result in a statistically significant increase in the initial rate of PI(4,5)P2 synthesis, though it showed a trend in that direction, in general agreement with published data (Chung et al., 2015b) (Fig. 5A). Compared to TMEM150A, however, overexpression of EFR3B led to a much larger increase in the initial rate of PI(4,5)P2 resynthesis. Although none of the dual-palm mutants had the same effect as WT EFR3B, overexpression of the C7S and C8S mutants resulted in a small but significant increase in the initial rate, consistent with a role in the formation of Complex I. Furthermore, we found that co-expression of TMEM150A with WT, C7S or C8S EFR3B had no effect on the initial rate of PI(4,5)P2 synthesis (Fig. 5B); however, co-expression of the EFR3B(C5S) mutant with TMEM150A had a stronger effect on the initial rate than expression of the EFR3B(C5S) mutant or TMEM150A alone. Based on these comparisons, we conclude that PI4K activity from Complex I impacts the initial rate more strongly than that from Complex II, which may form on a slower timescale following atropine treatment. However, the co-overexpression of the EFR3B(C5S) mutant, which preferentially interacts with TMEM150A (Fig. 3), may generate Complex II on a faster timescale, resulting in a statistically significant increase in the initial rate of PI(4,5)P2 resynthesis (Fig. 5B).
Fig. 5.

EFR3B(C5S)–TMEM150A interactions facilitate a more rapid recovery of PI(4)P and PI(4,5)P2 homeostasis than those of other palmitoylated forms of EFR3B. (A–D) HeLa cells were transfected with iRFP–PH(PLCδ), M1R–3×FLAG, and the indicated combination of TMEM150A–GFP and EFR3B–mCherry (EFR3B–mCh) variant (WT or CxS mutant). A time-lapse movie was acquired, recording iRFP fluorescence over time. Shown are kinetics parameters during the post-atropine phase (see Fig. 4): the initial rate of PI(4,5)P2 recovery (A,B) and maximum rate of PI(4,5)P2 recovery (C,D). Note that, to improve clarity and facilitate comparisons between effects of either EFR3B mutants alone or co-expressed with TMEM150A, the EFR3B–mCh only data in A are also plotted in B, and all data in C are also plotted in D. (E) Maximum rate of PI(4)P recovery in the presence of the indicated EFR3B and TMEM150A proteins, monitored by the recovery of the PI(4)P biosensor iRFP–P4M to the PM after M1R-mediated depletion. Mean±s.d. are indicated, n=9–21. *P<0.05; **P<0.025; ***P<0.01; ****P<0.001; ns, not significant (two-tailed unpaired t-test in B,D,E for comparisons between two groups; one-way ANOVA with Tukey–Kramer post-hoc test in A,C,E for comparisons between multiple groups).
We next investigated additional kinetic parameters of the recovery of the PM pool of the PI(4,5)P2 biosensor to glean insight into potential effects of both complexes. First, we found that the maximum rate of PI(4,5)P2 recovery was strongly increased in cells expressing WT EFR3B and was more modestly increased in cells expressing the dual-palm mutants (Fig. 5D, filled shapes). Second, overexpression of TMEM150A modestly increased the maximum rate of PI(4,5)P2 recovery compared to that in control cells (Fig. 5C, open versus filled circles). Compared to this modest increase, systematic co-overexpression of TMEM150A with either WT EFR3B or each of the dual-palm mutants revealed that only EFR3B(C5S) co-overexpression led to a further increase in maximum rate of PI(4,5)P2 recovery (Fig. 5C,D).
To further support these conclusions, we directly monitored the production of PI(4)P, the PI4KIIIα product and biosynthetic precursor of PI(4,5)P2, using the iRFP–P4M biosensor (Brombacher et al., 2009; Hammond et al., 2014; Schoebel et al., 2010). Co-expression of EFR3B(C5S) and TMEM150A resulted in a significant increase in the maximum rate of PI(4)P recovery (Fig. 5E), relative to their expression alone. We also observed significant increases in the maximum rate of recovery when EFR3B(C7S) or EF3B(C8S) were co-expressed with TMEM150A. However, those increases were most likely due to the expression of TMEM150A, because the maximum rates under these conditions were not statistically different from the maximum rate when TMEM150A was expressed alone (Fig. 5E). Overall, these observations support the idea that the EFR3B(C5S)- and TMEM150A-containing Complex II is most efficient at promoting PI(4)P production for PI(4,5)P2 resynthesis after acute depletion and strengthens a proposed role for TMEM150A in PI(4,5)P2 homeostasis.
DISCUSSION
PI4KIIIα, the enzyme responsible for the majority of PI(4)P synthesis at the PM, is thought to exist in two distinct complexes, containing either EFR3–TTC7–FAM126 (Complex I) or EFR3–TMEM150A (Complex II) (Fig. 1A) (Chung et al., 2015b). The goals of this study were to elucidate mechanisms underlying the distribution of PI4KIIIα between these two complexes and reveal functional differences between them in support of PI(4)P synthesis at the PM. Because the sole shared component between these two PI4KIIIα complexes is EFR3, whose membrane anchoring requires palmitoylation of a Cys-rich motif, we posited that differential localization within Lo-like and Ld-like regions of the PM may be a defining difference between Complexes I and II that is critical to their unique functional properties.
PI(4,5)P2 is enriched in Lo-like regions of the PM, and some evidence points to its local synthesis in these membrane domains (Calloway et al., 2011; Johnson et al., 2008; Myeong et al., 2021; Vasudevan et al., 2009; Wang and Richards, 2012). However, these studies have also detected substantial pools of PI(4,5)P2 in Ld-like membrane domains. The mechanisms that lead to compartmentalization of PI(4,5)P2 in Lo or Ld domains are not well understood. Clearly, multiple lines of evidence point to a primary role of PI4KIIIα in the synthesis of PI(4,5)P2, via production of PI(4)P, in the PM (Bojjireddy et al., 2014; Nakatsu et al., 2012). The link between TMEM150A and PI(4,5)P2 resynthesis following acute depletion may shed some light on the compartmentalization of PI(4,5)P2 in the PM. Specifically, the capacity of EFR3 for clustered palmitoylation at its N terminus and the well-established connection between palmitoylation and Lo partitioning of proteins (Levental et al., 2010) prompted us to investigate the membrane partitioning properties of EFR3 and their impact on its interaction with TMEM150A. We addressed the above questions using a multidisciplinary approach involving chemical biology, biochemistry and fluorescence-imaging techniques. Our results provide evidence for a palmitoylation code on EFR3B, the membrane anchor of both PI4KIIIα complexes, which drives the selective formation of the TMEM150A-containing Complex II (Fig. 6).
Fig. 6.

Model for the regulation of PI4KIIIα complexes. (A) Schematic of the EFR3B N terminus and its multiple lipoforms (PTM, post-translational modification). (B) Model for the regulation of the formation of the two distinct PI4KIIIα complexes. PI is shown in yellow; PI(4)P is shown in orange. Palm, palmitoylation.
Using acyl-PEG exchange, we found that EFR3B exists as dual and triple palmitoylated forms and that mutation of any one Cys in the palmitoylation motif resulted in near-quantitative palmitoylation of the remaining ones. These results show that all three Cys residues can be palmitoylated and that palmitoylation of any two is sufficient for PM localization of EFR3B. Using pulse-chase metabolic labeling with a bioorthogonal palmitate analog, we further showed that the palmitoylation of EFR3B is highly stable, allowing for robust, long-term PM anchoring. Collectively, these results suggest that the dual palmitoylated pool of WT EFR3B observed by acyl-PEG exchange is likely a heterogenous mix of three lipoforms (C5/C7, C5/C8 and C7/C8). We note that the pulse-chase experiments do not preclude the enzymatic or non-enzymatic migration of palmitoyl groups within this motif in these lipoforms.
The palmitoylation status of EFR3B affected its biophysical organization within the PM. Palmitoylation of peripheral membrane proteins is a primary regulator of their preference for Lo-like nanodomains (Levental et al., 2010). Indeed, our quantitative iDRM results reveal that WT EFR3B is moderately detergent resistant, supporting an Lo preference. However, its Lo preference was not as strong as other well-known lipidated protein probes that prefer Lo domains, such as Lyn kinase (Bag et al., 2021). The detergent resistance of WT EFR3B (consisting of multiple lipoforms) was very similar to those of the dual palmitoylated CxS mutants, indicating that the triply palmitoylated lipoform had a similar Lo preference to its doubly palmitoylated counterparts. This finding is also reflected in the similar t1/2 values of WT and all three CxS EFR3B mutants (Fig. 2E). The moderate Lo preference of EFR3B also indicates that a substantial minority fraction of EFR3B is present in Ld-like domains at steady state. Having pools of EFR3, the membrane anchor for all known PI4KIIIα complexes, in both Lo and Ld domains could be functionally relevant, because PI(4,5)P2 synthesis, which likely occurs in both domains (Calloway et al., 2011; Johnson et al., 2008; Myeong et al., 2021), requires recruitment of PI4KIIIα.
Our iDRM results demonstrated that TMEM150A is resistant to detergent, indicating its Lo preference. Transmembrane proteins had been previously thought to be excluded from the Lo-like domains due to unfavorable energetics (Van Duyl et al., 2002). However, some recent examples of Lo-preferring transmembrane proteins have been reported, in which the biochemical basis of the Lo preference depends on protein–lipid interactions (Holowka et al., 2018; Lin and London, 2013; Lorent et al., 2017; Marinko et al., 2020). Molecular dynamics simulations using realistic lipid bilayer models have revealed that membrane proteins can organize the lipid environment around themselves to maximize favorable interactions, further suggesting a preference for certain lipids (Corradi et al., 2018). TMEM150A may be a new addition to this category of membrane protein, although the features governing its Lo preference remain unknown and may involve association with other proteins. A recent study has demonstrated that Sfk1, the yeast homolog of TMEM150A, acts to retain ergosterol, the yeast cholesterol analog, in the PM (Kishimoto et al., 2021). This property may translate to TMEM150A in mammalian cells, where cholesterol is a key regulator of Lo domain formation (Kusumi et al., 2020). Notably, the detergent resistance of TMEM150A is highly sensitive to the presence of WT and C5S EFR3B, suggesting that the Lo domain partitioning of TMEM150A, or its protein–protein interactions, are modulated by interactions with EFR3B. The EFR3B C7S mutant showed no effect on TMEM150A partitioning, and the EFR3B C8S mutant showed an intermediate effect, leading us to hypothesize that the adjacent palmitoylation sites on the EFR3B C5S mutant result in favorable interactions with TMEM150A, thus driving the formation of Complex II in Ld-like membrane regions.
Interestingly, we observed an increase in the detergent resistance of WT EFR3B in the presence of TMEM150A. Separately, we confirmed that the relative abundance of the doubly and triply palmitoylated lipoforms of WT EFR3B are unchanged by the presence of TMEM150A. Because the EFR3B CxS mutants did not exhibit any differences, relative to WT EFR3B, in the detergent-resistant fraction (as assessed using the iDRM assay) or diffusion kinetics and mobile fraction (as assessed using FRAP), we hypothesize that the triply palmitoylated EFR3B lipoform partitions more strongly into Lo-like domains in the presence of TMEM150A. However, the underlying mechanism that confers stronger Lo preference to this lipoform under these conditions is currently unknown.
The iDRM measurements on PI4KIIIα in the fully assembled Complex I revealed strong detergent resistance, suggesting an Lo-like membrane environment surrounding this complex. This observation is consistent with the inherent Lo preference of EFR3B, the sole membrane anchoring component of this complex. Complex II has two constitutively PM-localized components, EFR3B and TMEM150A (Chung et al., 2015b), resulting in TMEM150A susceptibility to detergent (and Ld preference). The R values from iDRM experiments of PI4KIIIα under conditions favoring formation of Complex I or Complex II are strikingly similar (Fig. S5). These observations suggest that PI4KIIIα is not restricted to a single complex when all four components are co-transfected and that the two complexes likely co-exist in the PM, each with different preferences for Lo-like (Complex I) and Ld-like membrane domains (Complex II) at steady state (Fig. 6).
In addition to revealing the biophysical organization of PI4KIIIα at the PM, our data also support a functional role for an EFR3B palmitoylation code in regulating the activity of PI4KIIIα in the context of PI(4,5)P2 resynthesis after acute depletion. Among the three EFR3B dual-palm mutants, the C5S mutant interacts most favorably with TMEM150A, resulting in more efficient resynthesis of PI(4,5)P2 after acute depletion. Our data are consistent with a known role for TMEM150A in PI(4,5)P2 homeostasis (Chung et al., 2015b). We show that TMEM150A-expressing cells, upon co-expression with EFR3B(C5S), exhibit increases in initial and maximum rates of PI(4,5)P2 resynthesis not observed upon co-expression with EFR3B(C7S) or EFR3B(C8S). Therefore, the specific interaction of TMEM150A with EFR3B(C5S) in Ld-like domains has the strongest effect on PI(4,5)P2 production after acute hydrolysis. Importantly, the use of PI(4,5)P2 recovery experiments here is supported by previous findings indicating that phosphorylation of PI to PI(4)P, rather than the subsequent conversion of PI(4)P to PI(4,5)P2, is the rate-limiting step in PI(4,5)P2 resynthesis following agonist-mediated M1R stimulation (Falkenburger et al., 2010; Myeong et al., 2020), and by direct measurements of PI(4)P recovery.
These results support a model wherein Complex II is in part responsible for the synthesis of PI(4,5)P2 in Ld-like membrane regions, whereas Complex I preferentially partitions to Lo-like membrane regions, where the PI(4)P it produces locally may be used to replenish PI(4,5)P2. We propose that an equilibrium exists between both complexes that is in part dictated by the relative abundance of the different lipoforms of EFR3B, with the C7/C8 lipoform preferentially forming Complex II, and the other two lipoforms with palmitoyl groups spaced farther apart preferentially forming Complex I. Considering the well-known preference of palmitoylation for Lo-like membrane domains (Levental et al., 2010), it is likely that Complex I shows a preference for Lo-like membrane domains. Relative to Complex I, Complex II shows an increased affinity for Ld-like nanodomains. PI(4,5)P2 pools in Lo and Ld domains, synthesized by different PI(4)P 5-kinase isoforms, play different functional roles in calcium signaling (Calloway et al., 2011; Vasudevan et al., 2009). Specifically, pools of PI(4,5)P2 in Ld domains reduce store-operated calcium entry (SOCE). In light of these findings, our results raise the possibility that Complex II may contribute to PI(4,5)P2 synthesis in Ld domains to regulate calcium signaling in response to SOCE, which is activated by GPCRs that signal via Gq, such as M1R.
The idea of Complex I favoring Lo-like domains has support from several lines of indirect evidence from the literature. First, yeast Stt4 exists at the PM in so-called PIK patches with the EFR3 and TTC7 orthologs, Efr3 and Ypp1, which contain ∼30 copies of each protein and which recover minimally from photobleaching on the minute timescale (Baird et al., 2008). Second, the plant orthologs of EFR3, TTC7 and FAM126 have been recently identified, and their roles in recruiting the plant PI4KIIIα ortholog, PI4Kα1 (encoded by PI4KA1), to the PM are conserved. Interestingly, the EFR3 ortholog EFOP (EFR3 OF PLANTS) is PM-localized via palmitoylation of multiple Cys residues, and the complex appears to associate with specific membrane nanodomains (Noack et al., 2021).
Many questions remain about the function of TMEM150A and its interactions with EFR3. The binding determinants between TMEM150A and EFR3B, including how the former can distinguish different EFR3B lipoforms, are unknown, although the C-terminal cytosolic tail of TMEM150A has been implicated in this interaction and in PI(4,5)P2 homeostasis (Chung et al., 2015b). Additionally, TMEM150A and TMEM150B structural predictions using two different algorithms (AlphaFold and trRosetta; Jumper et al., 2021; Yang et al., 2020) suggest that the C-terminal tail of TMEM150A may have a partial helical character and interact with both the membrane interface and one of its transmembrane helices. This feature is predicted to be absent from TMEM150B, which does not interact with PI4KIIIα, and could explain the importance of the tail of TMEM150A in the recovery of PI(4,5)P2. This structural feature could also be the source of the selectivity of TMEM150A for specific lipoforms of EFR3B. Thus, further investigation is warranted into the molecular and functional roles of the tail of TMEM150A in the context of Complex II.
Furthermore, the palmitoylation state of EFR3A, which contains four adjacent Cys residues within its N-terminal palmitoylation motif, has not been studied in detail (Bojjireddy et al., 2015). We show that endogenous EFR3A is heavily palmitoylated, existing as a mixture of doubly, triply and quadruply palmitoylated lipoforms. The existence of a similar palmitoylation code on EFR3A would be an interesting area for future study. Moreover, further experiments are needed to establish the relative abundance of each EFR3B lipoform at endogenous expression levels. In yeast, Efr3 is not palmitoylated (Nakatsu et al., 2012) and relies exclusively on its polybasic patch to interact with the PM, highlighting major mechanistic differences in the assembly of the PI4KIIIα complex in this organism. Though Sfk1, the yeast homolog of TMEM150A, is clearly involved in kinase recruitment and function (Audhya and Emr, 2002), the underlying mechanism, and its relationship to the Efr3–Ypp1–Stt4 complex, remain unknown. The roles of Sfk1 in retention of ergosterol in the PM (Kishimoto et al., 2021) and maintenance of PM integrity and impermeability (Mioka et al., 2018) may or may not be related to its role in Stt4 function. Reciprocally, it is unknown whether TMEM150A plays similar roles in PM cholesterol homeostasis, transbilayer movement of lipids, or membrane integrity in mammalian cells.
We show that formation of Complex II results in faster PI(4)P and PI(4,5)P2 recovery kinetics after acute depletion. Many cellular processes rely on PI(4,5)P2 for proper function, such as PI(4)P–phosphatidylserine counter-transport at ER–PM contact sites, mediated by the human ORP5 and ORP8 proteins (also known as OSBPL5 and OSBPL8, respectively; Chung et al., 2015a). These transport proteins bind to PI(4)P and PI(4,5)P2, both of which are essential for proper PM anchoring. Acute depletion of PI(4,5)P2 has been shown to eliminate ORP5 and ORP8 localization at ER–PM contact sites (Sohn et al., 2018). Considering the function of TMEM150A in PI(4,5)P2 homeostasis, it is possible that TMEM150A plays a role in the re-establishment of lipid transport by ORP5 and ORP8 at ER–PM contact sites after acute depletion. More broadly, the general function of Complex II may be two-fold: re-establishing PI(4,5)P2 homeostasis at the PM for future signaling events and, by doing so, minimizing disturbances to other PI(4,5)P2-dependent processes.
In conclusion, we propose that a palmitoylation code within EFR3B, the membrane anchor for PI4KIIIα in the PM, dictates the distribution of PI4KIIIα between two distinct multicomponent complexes responsible for production of PI(4)P. Our model suggests unique roles for distinct lipoforms of EFR3B, which have very subtle differences in their palmitoylation patterns, in influencing interactions with TMEM150A and localization of PI4KIIIα to Lo-like and Ld-like domains. We show that these subtle changes in EFR3B palmitoylation impact the function of PI4KIIIα in producing PI(4)P required for restoration of PI(4,5)P2 levels at the PM following its acute depletion by Gq–PLC signaling. These findings reinforce the value of evaluating the detailed mechanisms of post-translational lipidation in the spatiotemporal regulation of protein–protein and protein–lipid interactions within the PM, and we anticipate that similar functionally important palmitoylation codes on multiply palmitoylated proteins will emerge in diverse contexts.
MATERIALS AND METHODS
General methods
Sources of chemical reagents, primers, antibodies and plasmids are listed in Table S1.
Cell culture
All cell lines were cultured at 37°C in 5% CO2. HeLa cells were grown in DMEM (Corning) supplemented with 10% FBS and 1% penicillin/streptomycin (P/S). RBL-2H3 cells were maintained in MEM (Corning) supplemented with 20% FBS and 10 mg/l gentamicin sulfate. Cells were obtained from ATCC and regularly tested for contamination.
Transfection
Chemical transfection of HeLa cells
Cells were seeded on #1.5 glass-bottom, 35 mm imaging dishes (MatTek and Matsunami) at a density of 150,000 cells per dish and left to grow overnight. Transfections were carried out the next day. In a polystyrene tube, 150 µl of Transfectagro (Corning) was added per transfected dish and 1.5 µl of Lipofectamine 2000 (Thermo Fisher) per dish was mixed in. In a separate transfection tube, the same volume of Transfectagro was added and mixed with 1.5 µg of each transfected plasmid. Tubes were left to incubate for 5 min at room temperature and were then mixed together. After mixing by gentle pipetting, tubes were left at room temperature for 20 min. The medium from the imaging dishes was aspirated and replaced with 1.7 ml of Transfectagro supplemented with 10% FBS. 300 µl of the appropriate transfection mix was added to each dish. Dishes were placed in an incubator at 37°C. After 7 h, Transfectagro was aspirated and replaced with growth medium, and the cells were left in the incubator for imaging the next day.
The transfection procedure for 60 mm dishes was the same, except that dishes were seeded with 800,000 cells and 375 µl of Transfectagro was used, for a total of 750 µl per dish. 4.5 µg of each plasmid and 4 µl of Lipofectamine 2000 were used for each dish.
Chemical transfection of RBL-2H3 cells
For transfection, 20,000 cells were suspended in 2 ml of growth medium and placed, homogeneously spread, in a 35 mm glass-bottom imaging dish (MatTek). After overnight growth, the cells were transfected using FuGENE HD transfection kit (Promega). For one imaging dish, plasmid DNA (2 μg of each plasmid) and FuGENE (3 μL FuGENE/μg DNA) were mixed thoroughly in 100 μL Opti-MEM (Thermo Fisher) and incubated at room temperature for 15 min. Next, cells were washed once and covered with 1 ml Opti-MEM. The DNA–FuGENE complex was spread evenly over the cells and incubated for 1 h. Then, 1 ml of pre-warmed phorbol 12,13-dibutyrate (PDB, 0.1 μg/ml; diluted in Opti-MEM from a 10,000× stock prepared in DMSO) was then added to each imaging dish, and cells were incubated for 3 h at 37°C. Finally, 2 ml of growth medium was added to each imaging dish after discarding Opti-MEM. The transfected cells were cultured for 18–22 h before FRAP measurements.
Electroporation of RBL-2H3 cells
RBL-2H3 cells in a confluent 75 cm2 flask were washed and trypsinized for 8 min at 37°C with 3 ml of 0.05% Trypsin-EDTA (Thermo Fisher). The detached cells were resuspended in 7 ml of growth medium and centrifuged to remove the medium. The cell pellet (1.5×106 cells) was resuspended in 1.5 ml of cold electroporation buffer (137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1 mg/ml glucose and 20 mM HEPES; pH 7.4). Next, 5 μg of plasmid DNA corresponding to each protein of interest was thoroughly mixed with 500 μl of the resuspended cells in an electroporation cuvette (Bio-Rad). This cuvette was subject to an electroporation pulse (280 V, 950 µF) using a Gene Pulser X (Bio-Rad) electroporation module. The electroporated cells were then added to 6 ml of growth medium, mixed thoroughly, and deposited in imaging dishes (2 ml/dish). The cells were allowed to attach on the dish for 3 h at 37°C, following which the medium was replaced with fresh growth medium. The cells were cultured for 24 h to recover before proceeding to the next sample preparation steps for iDRM.
Cloning
WT EFR3B–3×FLAG, EFR3B(C5,7,8S)–GFP (mouse) (Nakatsu et al., 2012) and TMEM150A–GFP (human) (Chung et al., 2015b) are identical to constructs used in the referenced studies (Chung et al., 2015b) and were cloned in the p3×FLAG-CMV-14 and pEGFP-N1 vectors, respectively. EFR3B CxS and CxxS mutants were generated by using Quikchange PCR with the primers listed in Table S1 and were validated by sequencing. C7S, C8S and C7,8S mutants were generated using WT EFR3B as a template. C5S, C5,7S and C5,8S mutants were generated using EFR3B(C5,7,8S) as a template. M1R–3×FLAG was subcloned from M1R–YFP (obtained from the Pietro De Camilli laboratory, Yale University, USA), using NotI and EcoRI. Subcloning was performed using appropriate restriction enzyme pairs compatible with donor and target plasmids according to standard procedures.
Acyl-PEG exchange palmitoylation assay
Acyl-PEG exchange experiments were performed essentially as previously reported (Percher et al., 2016). Briefly, cells were lysed in lysis buffer [50 mM triethanolamine (TEA), 4% SDS, 150 NaCl, pH 7.3 with 1× cOmplete Protease Inhibitor cocktail (Roche)]. Cell lysates were sonicated to ensure complete lysis and to fragment nucleic acids. Protein concentration was determined by BCA assay, and protein concentration was normalized to 2 mg/ml. For the assay, 92.5 µl of protein was used. 5 µl of 200 mM TCEP (neutralized to pH 7 by addition of 800 mM NaOH) was added to all samples, which were then left on the nutator at room temperature for 30 min. 2.5 µl of 1 M N-ethylmaleimide (NEM) in ethanol was added to all samples. Samples were then incubated for 2 h at room temperature on the nutator. A protein precipitation was then performed by adding pre-chilled methanol, water and chloroform at a 400 µl:150 µl:300 µl ratio. Samples were inverted and spun at 17,000 g for 5 min at 4°C. The aqueous layer (top) was gently removed and replaced with 1 ml of methanol. Samples were spun again in the same conditions for 3 min, and the supernatant removed and replaced with 800 µl of methanol. Samples were spun again for 3 min. The methanol was decanted, and samples dried on a benchtop concentrator (CentriVap) at 37°C until no methanol remained. Samples were resuspended in 100 µl of lysis buffer in a 37°C water bath for 10 min, or until the protein pellet was no longer visible. Samples were then gently sonicated in a sonicating water bath for 10 s to ensure total dissolution of the pellet. This protein extraction was performed a total of three times. After the third time, samples were resuspended in 60 µl of lysis buffer with 5 mM EDTA and without protease inhibitor. After pellet dissolution, 90 µl of 1 M hydroxylamine (pH 7) dissolved in 50 mM TEA 0.2% TX-100 were added to each sample. For a no hydroxylamine control, buffer with 50 mM TEA and 0.2% TX-100 was used. Samples were placed on the nutator for 1 h at room temperature, and then a protein precipitation was performed, after which pellets were solubilized in 30 µl of lysis buffer and treated with 90 µl of a solution of 1.33 mM mPEG-maleimide in 50 mM TEA and 0.2% TX-100. Samples were left on the nutator at room temperature for 2 h. A final protein precipitation was performed, after which samples were resuspended in 50 µl of lysis buffer and resolubilized. SDS–PAGE loading buffer [6× Laemmli buffer; 0.1 g/ml SDS, 0.5 mg/ml Bromophenol Blue, 50 mM Tris-HCl pH 6.8, 8% (v/v) β-mercaptoethanol, 40% (v/v) glycerol] was added, and samples incubated at 95°C for 5 min and stored at −20°C until analysis using SDS–PAGE and western blotting, as described below.
Bioorthogonal metabolic labeling pulse-chase assay for palmitoylation stability
HeLa cells were seeded on 60 mm dishes (800,000 cells per dish). The next day, cells were transfected according to the above protocol with EFR3B–3×FLAG. After 24 h, cells were rinsed with PBS, and the growth medium changed to DMEM supplemented with 10% dialyzed FBS containing 50 µM 17-ODYA (alk-16). The next day, the 17-ODYA was rinsed out of the dishes, and the growth medium was replaced with regular growth medium (i.e. DMEM with standard FBS and P/S) in the presence of lactacystin (1 µM) and cycloheximide (50 µg/ml) for 1, 2, 4 or 8 h. After each time point, cells were harvested, rinsed three times in PBS by pelleting at 4°C in a benchtop centrifuge (900 g, 2 min) between washes. Cells were either flash frozen in liquid nitrogen for temporary storage or lysed in 200 µl of lysis buffer (50 mM TEA, pH 7.5, 150 NaCl, 1% NP-40, 0.25% sodium deoxycholate and cOmplete EDTA-free protease inhibitor cocktail). Samples were then sonicated while taking care to keep them on ice. Protein concentration was normalized to 2 mg/ml by performing a BCA assay. An anti-FLAG immunoprecipitation (IP) to recover labelled EFR3B was performed using 500 µg of sample and 20 µl of anti-FLAG bead slurry (EZview Red ANTI-FLAG M2 Affinity gel from Millipore Sigma). Samples were left to rotate for 2 h at 4°C. The beads were then recovered and rinsed three times with wash buffer (25 mM TEA, 150 mM NaCl, 0.2% NP-40, pH 7.4) with pelleting in between each step (900 g, 2 min). After the last rinse, beads were resuspended in 16 µl of the wash buffer. Cy5.5-azide was then tagged onto the 17-ODYA by the Cu-catalyzed azide–alkyne cycloaddition (click chemistry). The reaction requires final concentrations of 100 µM Cy5.5-azide, 1 mM CuSO4, 600 µM tris-hydroxypropyltriazolylmethylamine (THPTA) and 50 mM sodium ascorbate. 20× stocks were made in DMSO for Cy5.5-azide and in water for CuSO4, THPTA and sodium ascorbate. The CuSO4 and sodium ascorbate solutions were made fresh every time. To carry out the reaction, the CuSO4 and THPTA solutions were first mixed in a 1:1 ratio. Then, 1 µl of the Cy5.5-azide solution was added to the samples, followed by 2 µl of the pre-mixed CuSO4/THPTA solution, and finally 1 µl of the sodium ascorbate solution. The reactions were left at room temperature in the dark for 1 h. After the reaction was completed, beads were rinsed three times with the wash buffer to remove excess fluorophore. Samples were then denatured using 6× SDS–PAGE Laemmli loading buffer diluted down to 2×. Samples were boiled at 100°C for 5 min and stored at −20°C until analysis using SDS–PAGE and western blotting, as described below.
SDS–PAGE and western blotting
SDS–PAGE was performed using the Mini-Protean system (Bio-Rad), running at 160 V, using Precision Plus molecular weight standards (Bio-Rad). For western blotting, transfers to nitrocellulose were carried out for 2 h at 70 V (constant voltage) at 4°C. Membranes were stained using Ponceau S and scanned on an imager (ChemiDoc MP, Bio-Rad). Blocking was performed for 1 h with rocking at room temperature using 5% milk in TBS with 0.1% Tween-20 (TBS-T). Primary antibody staining was performed overnight at 4°C with rocking, with primary antibody diluted in blocking buffer. Primary antibodies were used at 1:1000 for FLAG and calnexin, and 1:5000 for GAPDH (for western blot in the supplementary information). Following three 5 min rinses in TBS-T, secondary antibody incubation was performed with the appropriate HRP-conjugated secondary antibody (1:5000 dilution in blocking buffer) for 1 h at room temperature with rocking. The membranes were then rinsed three times for 7 min in TBS-T, then for 7 min in TBS. Membranes were then exposed to Clarity ECL solution (Bio-Rad) for 5 min and then imaged (Chemidoc MP, Bio-Rad). Western blots were quantified using ImageLab software (Bio-Rad).
iDRM experiments
Sample preparation
All iDRM preparations used RBL-2H3 cells that were electroporated with a given set of plasmids and allowed to recover overnight under physiological conditions. Cells in imaging dishes (MatTek) were washed with 1 ml of buffered salt solution (BSS: 135 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.6 mM glucose and 20 mM HEPES, pH 7.4) followed by addition of another 1 ml of fresh BSS. The dishes were placed on ice for 10 min. From the dishes designated as ‘with TX-100’ (+TX-100 sample), the BSS was removed slowly, and 1 ml of BSS with 0.04% TX-100 was added dropwise before incubation for 10 min in ice. A control dish (–TX-100) was prepared in which 1 ml of BSS without TX-100 was added dropwise before incubation for 10 min in ice. After this incubation step, the solutions were gently removed from both dishes to minimize any mechanical perturbation of the cells. 1 ml of fixing solution containing 4% paraformaldehyde and 0.1% glutaraldehyde in PBS was added dropwise to both dishes at room temperature for 20 min. The fixation was quenched by addition of 0.5 ml of 10 mg/ml bovine serum albumin (BSA) in PBS for 10 min, and samples were stored at 4°C until imaging.
Imaging
iDRM was imaged with a home-built total internal reflection fluorescence microscope (TIRFM) comprising a home-built microscope (DMIRB, Leica Microsystems) with an oil immersion objective (PlanApo, 100×, NA 1.47; Leica Microsystems) and an electron-multiplying charge-coupled device camera (black illuminated Andor iXON 897DU, pixel size 16 µm; Andor Technology). To measure the fluorescence of proteins tagged with green fluorescent protein (GFP), a 488 nm laser (Coherent) was used. To measure the fluorescence of proteins tagged with mCherry, a 561 nm laser (Coherent) was used. Each image was taken with a data acquisition of 100 frames with 0.01 s per frame. Approximately 30 cells per dish were imaged. All images were saved in TIFF format in 16-bit grayscale with a data range of 0 to 65535. Image acquisition was performed with Andor Solis software. 30 cells each of the –TX-100 and +TX-100 samples were imaged for one biological replicate for a given set of probes. For each condition, at least three biological replicates were performed.
Data analysis
Quantification and analysis were done using FIJI (Schindelin et al., 2012). Cellular fluorescence was measured by manually drawing a region of interest (ROI) around the cell and using the measure feature of FIJI. To account for background fluorescence, a ROI was drawn outside of the cell, and the difference between the cell fluorescence and the background fluorescence was calculated to return a background-corrected average fluorescence. This process was performed for each cell in the samples with or without TX-100 for each construct. All images are the average of the 100 frames and are 200×200 pixels, with a pixel size of 160 nm. The background-corrected fluorescence of a probe in individual cells from –TX-100 and +TX-100 samples (∼30 cells each) for given biological replicates yields a distribution of probe fluorescence values for each category. We used the Mann–Whitney U-test to determine the statistical significance between –TX-100 and +TX-100 samples. Furthermore, we defined a parameter, called the fluorescence retention (R) value, to quantitatively determine the extent of detergent resistance for a given probe using the following equation:
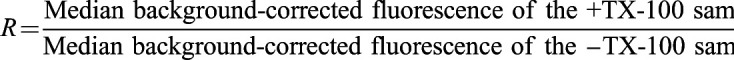 |
An R value of 1 means the probe is detergent-insoluble (i.e. the probe is surrounded by a Lo-like environment), whereas a value of zero indicates complete solubility of the probe and an Ld-like environment. Based on our previous studies, we interpret R values >0.6 as corresponding to Lo-preferring lipid probes and <0.3 as corresponding to Ld-preferring lipid probes (Bag et al., 2021).
FRAP assays
Transfected cells were rinsed once with 1 ml of BSS to remove leftover growth medium. FRAP experiments were carried out in 1 ml of BSS buffer on a Zeiss LSM 800 confocal microscope equipped with a 1.4 NA, 40×, oil immersion objective and GaAsP detectors. Five frames were acquired before bleaching, after which cells were photobleached on their ventral surface using 488 nm laser illumination at 100% power for 50 iterations, resulting in strong visible bleaching in the target region of interest (bleached ROI). A separate, unbleached ROI of the same dimensions was used to control for photobleaching of the GFP tag due to image acquisition and a third ROI was used to account for background fluorescence of the system (positioned outside of the cell). Time-lapse imaging was performed, with images acquired every 1.3 s for 3 min. All ROIs were circles of 3.5 μm diameter. FRAP data from the bleached ROI was normalized after accounting for photobleaching during the recovery process and background using the fluorescence traces from the control ROI (inside cell but away from bleached ROI) and background ROI (located outside cell) using the FRAPanalyzer software (Halavatyi et al., 2008). The normalized FRAP data were such that the fluorescence before photobleaching is 1 [i.e. Fnormalized=1 at recovery time (t) <0], while fluorescence at the time of bleaching is zero (i.e. Fnormalized=0 at t=0). During and after completion of the recovery the Fnormalized is greater than zero (i.e. Fnormalized>0 at t>0). The normalized FRAP data (Fnormalized) as a function of recovery time (t) is then fitted using the following exponential function:
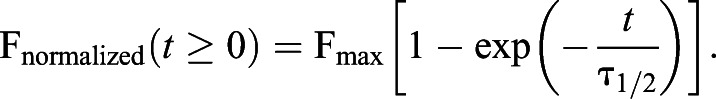 |
The saturation value of the fitted curve at extended time (i.e. when recovery is completed), Fmax, is the mobile fraction, whereas the timescale of diffusion is given by the recovery time (τ1/2) of the bleached spot.
Imaging fluorescence correlation spectroscopy
The instrumental setup, data acquisition and processing have been described previously (Bag et al., 2020). Briefly, we collected time-lapse images (80,000 images at 3.5 ms frame rate) of ventral plasma membrane of fluorescently labeled RBL-2H3 cells using a home-built TIRFM setup equipped with a high NA (1.49) oil-immersion objective and EMCCD camera (Andor iXon 897). Fluorescence fluctuations embedded in the image series were further processed using a FIJI (Schindelin et al., 2012) macro (http://www.dbs.nus.edu.sg/lab/BFL/imfcs_image_j_plugin.html) to determine the D values.
Imaging of PI(4)P and PI(4,5)P2 resynthesis kinetics after acute depletion
Acquisition
HeLa cells were transfected with M1R–YFP or M1R–3×FLAG, iRFP–P4M(SidM) [Addgene #51470 for PI(4)P], iRFP–PH(PLCδ) [for PI(4,5)P2] and other constructs, as necessary. On the day of the experiment, imaging buffer (BSS+1 mg/ml BSA) was warmed to 37°C and used to rinse the cells once before the experiment. Confocal microscopy imaging was carried out on a Zeiss LSM 800 confocal microscope. Time-lapse acquisitions were started prior to addition of oxo-M, with frames acquired every 1.2 s. After 30 s, oxo-M diluted in water was added to the cells at a final concentration of 10 μM. 60 s after the addition of oxo-M, atropine was added to the dish to a final concentration of 50 μM. During the addition of oxo-M and atropine, care was taken to add the drug drop by drop throughout the imaging dish in an effort to reduce error due to diffusion and to reduce the effect on the focal plane during image acquisition. The time-lapse acquisition was continued for a total of 15 min for PI(4)P depletion and 10 min for PI(4,5)P2 depletion. Recovery curves were fitted to an exponential function using the Igor Pro software (Version 8; WaveMetrics). From the fitted curves, a value for t1/2 for recovery was obtained.
Data analysis
The time-dependent decrease of cytosolic fluorescence of iRFP–PH(PLCδ) was monitored after atropine addition. First, the fluorescence versus time curve was smoothed to eliminate noise (rolling time average smoothing). The reliability of the smoothing process was confirmed by overlaying the smoothed curve on the raw curve. The PI(4,5)P2 synthesis rate at each point after agonist or antagonist addition was calculated from the derivative of smoothed fluorescence intensity with respect to time. Because we monitor cytosolic fluorescence, which decreases over time as PI(4,5)P2 is synthesized at the PM, the sign of the derivative values is negative. The derivative value at the first timepoint after atropine addition is termed as the initial rate. The minimum of the derivative plot (i.e. the most negative value) corresponds to the maximum rate. The time difference between atropine addition and reaching the maximum rate is defined as time to reach maximum rate. Because the rate becomes zero at very long times after atropine addition, as no new net PI(4,5)P2 is being synthesized, this phenomenon is represented in the derivative plot as a plateau with average value of zero. All analysis was done in the Igor Pro software.
Statistical methods
All experiments were performed in at least three biological replicates on different days. Exact numbers of replicate experiments and sample sizes are provided in each figure legend. Statistical significance was determined by the appropriate test based on number of compared samples: pairwise samples were compared using two-tailed Student's t-test with unequal variance and comparisons between more than two samples were made using one-way ANOVA followed by Tukey–Kramer post-hoc test, as indicated in each figure legend.
Supplementary Material
Acknowledgements
We thank Chris Diaz, Allan Lee and Alice Wagenknecht-Wiesner for technical assistance; the Fromme and Emr labs for equipment; and David Holowka, Maurine Linder, Scott Emr, and the Baskin and Baird-Holowka labs for helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.G.B., N.B., B.A.B., J.M.B.; Investigation: A.G.B., N.B., H.T.P.; Resources: B.A.B., J.M.B.; Writing - original draft: A.G.B., N.B., J.M.B.; Writing - review & editing: A.G.B., N.B., B.A.B., J.M.B.; Supervision: N.B., B.A.B., J.M.B.; Project administration: B.A.B., J.M.B.; Funding acquisition: B.A.B., J.M.B.
Funding
We acknowledge support from the National Institutes of Health (R00GM110121 and R01GM131101 to J.M.B.; R01GM117552 to B.A.B.) and the Alfred P. Sloan Foundation (Sloan Research Fellowship to J.M.B.). Deposited in PMC for release after 12 months.
References
- Abrami, L., Dallavilla, T., Sandoz, P. A., Demir, M., Kunz, B., Savoglidis, G., Hatzimanikatis, V. and van Der Goot, F. G. (2017). Identification and dynamics of the human ZDHHC16-ZDHHC6 palmitoylation cascade. Elife 6, 1-24. 10.7554/eLife.27826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya, A. and Emr, S. D. (2002). Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev. Cell 2, 593-605. 10.1016/S1534-5807(02)00168-5 [DOI] [PubMed] [Google Scholar]
- Bag, N. and Wohland, T. (2014). Imaging fluorescence fluctuation spectroscopy: new tools for quantitative bioimaging. Annu. Rev. Phys. Chem. 65, 225-248. 10.1146/annurev-physchem-040513-103641 [DOI] [PubMed] [Google Scholar]
- Bag, N., Holowka, D. A. and Baird, B. A. (2020). Imaging FCS delineates subtle heterogeneity in plasma membranes of resting mast cells. Mol. Biol. Cell 31, 709-723. 10.1091/mbc.E19-10-0559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bag, N., Wagenknecht-wiesner, A., Lee, A., Shi, S. M. and Holowka, D. A. (2021). Lipid-based and protein-based interactions synergize transmembrane signaling stimulated by antigen clustering of IgE receptors. Proc. Natl. Acad. Sci. USA 118, 1-12. 10.1073/pnas.2026583118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, D., Stefan, C., Audhya, A., Weys, S. and Emr, S. D. (2008). Assembly of the PtdIns 4-kinase Stt4 complex at the plasma membrane requires Ypp1 and Efr3. J. Cell Biol. 183, 1061-1074. 10.1083/jcb.200804003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla, T. (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019-1137. 10.1152/physrev.00028.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin, J. M., Wu, X., Christiano, R., Oh, M. S., Schauder, C. M., Gazzerro, E., Messa, M., Baldassari, S., Assereto, S., Biancheri, R.et al. (2016). The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol. 18, 132-138. 10.1038/ncb3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batrouni, A. G. and Baskin, J. M. (2021). The chemistry and biology of phosphatidylinositol 4-phosphate at the plasma membrane. Bioorg. Med. Chem. 40, 116190. 10.1016/j.bmc.2021.116190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigorgne, A. E., Farin, H. F., Lemoine, R., Mahlaoui, N., Lambert, N., Gil, M., Schulz, A., Philippet, P., Schlesser, P., Abrahamsen, T. G.et al. (2014). TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clin. Invest. 124, 328-337. 10.1172/JCI71471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojjireddy, N., Botyanszki, J., Hammond, G., Creech, D., Peterson, R., Kemp, D. C., Snead, M., Brown, R., Morrison, A., Wilson, S.et al. (2014). Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J. Biol. Chem. 289, 6120-6132. 10.1074/jbc.M113.531426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojjireddy, N., Guzman-Hernandez, M. L., Reinhard, N. R., Jovic, M. and Balla, T. (2015). EFR3s are palmitoylated plasma membrane proteins that control responsiveness to G-protein-coupled receptors. J. Cell Sci. 128, 118-128. 10.1242/jcs.157495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brombacher, E., Urwyler, S., Ragaz, C., Weber, S. S., Kami, K., Overduin, M. and Hilbi, H. (2009). Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284, 4846-4856. 10.1074/jbc.M807505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloway, N., Owens, T., Corwith, K., Rodgers, W., Holowka, D. and Baird, B. (2011). Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P2 between distinct membrane pools. J. Cell Sci. 124, 2602-2610. 10.1242/jcs.084178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, J., Torta, F., Masai, K., Lucast, L., Czapla, H., Tanner, L. B., Narayanaswamy, P., Wenk, M. R., Nakatsu, F. and De Camilli, P. (2015a). PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER - Plasma membrane contacts. Science 349, 428-432. 10.1126/science.aab1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, J., Nakatsu, F., Baskin, J. M. and De Camilli, P. (2015b). Plasticity of PI4KIIIα interactions at the plasma membrane. EMBO Rep. 16, 312-320. 10.15252/embr.201439151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi, V., Mendez-Villuendas, E., Ingólfsson, H. I., Gu, R. X., Siuda, I., Melo, M. N., Moussatova, A., Degagné, L. J., Sejdiu, B. I., Singh, G.et al. (2018). Lipid-protein interactions are unique fingerprints for membrane proteins. ACS Cent. Sci. 4, 709-717. 10.1021/acscentsci.8b00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler, N. S., Heitman, J. and Cardenas, M. E. (1997). STT4 is an essential phosphatidylinositol 4-kinase that is a target of wortmannin in Saccharomyces cerevisiae. J. Biol. Chem. 272, 27671-27677. 10.1074/jbc.272.44.27671 [DOI] [PubMed] [Google Scholar]
- Dornan, G. L., Dalwadi, U., Hamelin, D. J., Hoffmann, R. M., Yip, C. K. and Burke, J. E. (2018). Probing the architecture, dynamics, and inhibition of the PI4KIIIα/TTC7/FAM126 complex. J. Mol. Biol. 430, 3129-3142. 10.1016/j.jmb.2018.07.020 [DOI] [PubMed] [Google Scholar]
- Falkenburger, B. H., Jensen, J. B. and Hille, B. (2010). Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 81-97. 10.1085/jgp.200910344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil De Rubio, R., Ransom, R. F., Malik, S., Yule, D. I., Anantharam, A., Smrcka, A. V., De Rubio, R. G., Ransom, R. F., Malik, S., Yule, D. I.et al. (2018). Phosphatidylinositol 4-phosphate is a major source of GPCR-stimulated phosphoinositide production. Science 1210, eean1210. 10.1126/scisignal.aan1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, J. S., Drake, K. R., Rogers, C., Wright, L., Lippincott-Schwartz, J., Philips, M. R. and Kenworthy, A. K. (2005). Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 170, 261-272. 10.1083/jcb.200502063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A. R., Pirruccello, M., Cheng, F., Kang, H. J., Fernandez, T. V., Baskin, J. M., Choi, M., Liu, L., Ercan-Sencicek, A. G., Murdoch, J. D.et al. (2014). Rare deleterious mutations of the gene EFR3A in autism spectrum disorders. Mol. Autism 5, 31. 10.1186/2040-2392-5-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halavatyi, A., Medves, S., Hoffman, C., Apanasovich, V., Yatskou, M. and Friederich, E. (2008). Mathematical model and software FRAPAnalyser for analysis of actin- cytoskeleton dynamics with FRAP experiments. Proc. FEBS/ECF Work. Mech. Dyn. Cytoskelet. 2008, 50. [Google Scholar]
- Hammond, G. R. V., Fischer, M. J., Anderson, K. E., Holdich, J., Koteci, A., Balla, T. and Irvine, R. F. (2012). PI(4)P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727-730. 10.1126/science.1222483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, G. R. V., Machner, M. P. and Balla, T. (2014). A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 205, 113-126. 10.1083/jcb.201312072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka, D., Thanapuasuwan, K. and Baird, B. (2018). Short chain ceramides disrupt immunoreceptor signaling by inhibiting segregation of Lo from Ld Plasma membrane components. Biol. Open 7, 1-9. 10.1242/bio.034702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, C. M., Chichili, G. R. and Rodgers, W. (2008). Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J. Biol. Chem. 283, 29920-29928. 10.1074/jbc.M805921200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A.et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy, A. K., Nichols, B. J., Remmert, C. L., Hendrix, G. M., Kumar, M., Zimmerberg, J. and Lippincott-Schwartz, J. (2004). Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 165, 735-746. 10.1083/jcb.200312170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto, T., Mioka, T., Itoh, E., Williams, E. D., Anderson, J. R. and Tanaka, K. (2021). Phospholipid flippases and Sfk1 are essential for the retention of ergosterol in the plasma membrane. Mol. Biol. Cell 32, 1374-1392. 10.1091/mbc.E20-11-0699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger, J. W., Singh, A. P., Bag, N., Garbe, C. S., Saunders, T. E., Langowski, J. and Wohland, T. (2015). Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. Nat. Protoc. 10, 1948-1974. 10.1038/nprot.2015.100 [DOI] [PubMed] [Google Scholar]
- Kusumi, A., Fujiwara, T. K., Tsunoyama, T. A., Kasai, R. S., Liu, A.-A., Hirosawa, K. M., Kinoshita, M., Matsumori, N., Komura, N., Ando, H.et al. (2020). Defining raft domains in the plasma membrane. Traffic 21, 106-137. 10.1111/tra.12718 [DOI] [PubMed] [Google Scholar]
- Lees, J. A., Zhang, Y., Oh, M. S., Schauder, C. M., Yu, X., Baskin, J. M., Dobbs, K., Notarangelo, L. D., De Camilli, P., Walz, T.et al. (2017). Architecture of the human PI4KIIIα lipid kinase complex. Proc. Natl. Acad. Sci. USA 114, 13720-13725. 10.1073/pnas.1718471115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental, I., Lingwood, D., Grzybek, M., Coskun, Ü. and Simons, K. (2010). Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. USA 107, 22050-22054. 10.1073/pnas.1016184107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Q. and London, E. (2013). Transmembrane protein (perfringolysin O) association with ordered membrane domains (rafts) depends upon the raft-associating properties of protein-bound Sterol. Biophys. J. 105, 2733-2742. 10.1016/j.bpj.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood, D. and Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46-50. 10.1126/science.1174621 [DOI] [PubMed] [Google Scholar]
- Lorent, J. H., Diaz-Rohrer, B., Lin, X., Spring, K., Gorfe, A. A., Levental, K. R. and Levental, I. (2017). Structural determinants and functional consequences of protein affinity for membrane rafts. Nat. Commun. 8, 1-9. 10.1038/s41467-017-01328-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinko, J. T., Marinko, J. T., Kenworthy, A. K., Kenworthy, A. K., Sanders, C. R., Sanders, C. R. and Sanders, C. R. (2020). Peripheral myelin protein 22 preferentially partitions into ordered phase membrane domains. Proc. Natl. Acad. Sci. USA 117, 14168-14177. 10.1073/pnas.2000508117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, B. R. and Cravatt, B. F. (2009). Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135-138. 10.1038/nmeth.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mioka, T., Fujimura-Kamada, K., Mizugaki, N., Kishimoto, T., Sano, T., Nunome, H., Williams, D. E., Andersen, R. J. and Tanaka, K. (2018). Phospholipid flippases and Sfk1p, a novel regulator of phospholipid asymmetry, contribute to low permeability of the plasma membrane. Mol. Biol. Cell 29, 1203-1218. 10.1091/mbc.E17-04-0217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeong, J., de la Cruz, L., Jung, S. R., Yeon, J. H., Suh, B. C., Koh, D. S. and Hille, B. (2020). Phosphatidylinositol 4,5-bisphosphate is regenerated by speeding of the PI 4-kinase pathway during long PLC activation. J. Gen. Physiol. 152, e202012627. 10.1085/jgp.202012627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeong, J., Park, C. G., Suh, B. C. and Hille, B. (2021). Compartmentalization of phosphatidylinositol 4,5-bisphosphate metabolism into plasma membrane liquid-ordered/raft domains. Proc. Natl. Acad. Sci. USA 118, 1-10. 10.1073/pnas.2025343118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu, F., Baskin, J. M., Chung, J., Tanner, L. B., Shui, G., Lee, S. Y., Pirruccello, M., Hao, M., Ingolia, N. T., Wenk, M. R.et al. (2012). Ptdins4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003-1016. 10.1083/jcb.201206095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack, L. C., Bayle, V., Armengot, L., Rozier, F., Mamode-Cassim, A., Stevens, F. D., Caillaud, M. C., Munnik, T., Mongrand, S.et al. (2021). A nanodomain-anchored scaffolding complex is required for the function and localization of phosphatidylinositol 4-kinase alpha in plants. Plant Cell, koab135. 10.1093/plcell/koab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percher, A., Ramakrishnan, S., Thinon, E., Yuan, X., Yount, J. S. and Hang, H. C. (2016). Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proc. Natl. Acad. Sci. USA 3, 201602244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percherancier, Y., Planchenault, T., Valenzuela-Fernandez, A., Virelizier, J. L., Arenzana-Seisdedos, F. and Bachelerie, F. (2001). Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 REceptor. J. Biol. Chem. 276, 31936-31944. 10.1074/jbc.M104013200 [DOI] [PubMed] [Google Scholar]
- Salter, C. G., Cai, Y., Lo, B., Helman, G., Taylor, H., Mccartney, A., Leslie, J. S., Accogoli, A., Zara, F., Fasham, J.et al. (2021). Biallelic PI4KA variants cause neurological, intestinal and immunological disease. Brain awab313, 3-46. 10.1093/brain/awab313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel, S., Blankenfeldt, W., Goody, R. S. and Itzen, A. (2010). High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep. 11, 598-604. 10.1038/embor.2010.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin, E., Levental, I., Mayor, S. and Eggeling, C. (2017). The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 18, 361-374. 10.1038/nrm.2017.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, M., Korzeniowski, M., Zewe, J. P., Wills, R. C., Hammond, G. R. V., Humpolickova, J., Vrzal, L., Chalupska, D., Veverka, V., Fairn, G. D.et al. (2018). PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER–PM contact sites. J. Cell Biol. 217, 1797-1813. 10.1083/jcb.201710095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, A. W., Benita, Y., Peng, L. F., Kim, S. S., Sakamoto, N., Xavier, R. J. and Chung, R. T. (2009). A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5, 298-307. 10.1016/j.chom.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timcenko, M., Lyons, J. A., Januliene, D., Ulstrup, J. J., Dieudonné, T., Montigny, C., Ash, M. R., Karlsen, J. L., Boesen, T., Kühlbrandt, W.et al. (2019). Structure and autoregulation of a P4-ATPase lipid flippase. Nature 571, 366-370. 10.1038/s41586-019-1344-7 [DOI] [PubMed] [Google Scholar]
- Traverso, M., Assereto, S., Gazzerro, E., Savasta, S., Abdalla, E. M., Rossi, A., Baldassari, S., Fruscione, F., Ruffinazzi, G., Fassad, M. R.et al. (2013). Novel FAM126A mutations in hypomyelination and congenital cataract disease. Biochem. Biophys. Res. Commun. 439, 369-372. 10.1016/j.bbrc.2013.08.077 [DOI] [PubMed] [Google Scholar]
- Vaillancourt, F. H., Pilote, L., Cartier, M., Lippens, J., Liuzzi, M., Bethell, R. C., Cordingley, M. G. and Kukolj, G. (2009). Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387, 5-10. 10.1016/j.virol.2009.02.039 [DOI] [PubMed] [Google Scholar]
- Van Duyl, B. Y., Rijkers, D. T. S., Kruijff, B. De , and Killian, J. A. (2002). Influence of hydrophobic mismatch and palmitoylation on the association of transmembrane α-helical peptides with detergent-resistant membranes. FEBS Lett. 523, 79-84. 10.1016/S0014-5793(02)02939-3 [DOI] [PubMed] [Google Scholar]
- Vasudevan, L., Jeromin, A., Volpicelli-Daley, L., De Camilli, P., Holowka, D. and Baird, B. (2009). The β- and γ-isoforms of type I PIP5K regulate distinct stages of Ca2+ signaling in mast cells. J. Cell Sci. 122, 2567-2574. 10.1242/jcs.048124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdura, E., Rodríguez-Palmero, A., Vélez-Santamaria, V., Planas-Serra, L., de la Calle, I., Raspall-Chaure, M., Roubertie, A., Benkirane, M., Saettini, F., Pavinato, L.et al. (2021). Biallelic PI4KA variants cause a novel neurodevelopmental syndrome with hypomyelinating leukodystrophy. Brain awab124, 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. and Richards, D. A. (2012). Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol. Open 1, 857-862. 10.1242/bio.20122071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Chi, R. J., Baskin, J. M., Lucast, L., Burd, C. G., DeCamilli, P. and Reinisch, K. M. (2014). Structural insights into assembly and regulation of the plasma membrane phosphatidylinositol 4-Kinase complex. Dev. Cell 28, 19-29. 10.1016/j.devcel.2013.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y., Denef, N., Tang, C. and Schüpbach, T. (2011). Drosophila PI4KIIIalpha is required in follicle cells for oocyte polarization and Hippo signaling. Development 138, 1697-1703. 10.1242/dev.059279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., Anishchenko, I., Park, H., Peng, Z., Ovchinnikov, S. and Baker, D. (2020). Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 117, 1496-1503. 10.1073/pnas.1914677117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zara, F., Biancheri, R., Bruno, C., Bordo, L., Assereto, S., Gazzerro, E., Sotgia, F., Wang, X. B., Gianotti, S., Stringara, S.et al. (2006). Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet. 38, 1111-1113. 10.1038/ng1870 [DOI] [PubMed] [Google Scholar]
- Zhang, L., Malik, S., Pang, J., Wang, H., Park, K. M., Yule, D. I., Blaxall, B. C. and Smrcka, A. V. (2013). Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216-227. 10.1016/j.cell.2013.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M., Zhou, L., Xu, Y., Yang, M., Xu, Y., Komaniecki, G. P., Kosciuk, T., Chen, X., Lu, X., Zou, X.et al. (2020). A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 586, 434-439. 10.1038/s41586-020-2799-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.