

Abstract
Shotgun proteomics techniques infer the presence and quantity of proteins using peptide proxies produced by cleavage of the proteome with a protease. Most protein quantitation strategies assume that multiple peptides derived from a protein will behave quantitatively similar across treatment groups, but this assumption may be false due to (1) heterogeneous proteoforms and (2) technical artifacts. Here we describe a strategy called peptide correlation analysis (PeCorA) that detects quantitative disagreements between peptides mapped to the same protein. PeCorA fits linear models to assess whether a peptide’s change across treatment groups differs from all other peptides assigned to the same protein. PeCorA revealed that ~15% of proteins in a mouse microglia stress data set contain at least one discordant peptide. Inspection of the discordant peptides shows the utility of PeCorA for the direct and indirect detection of regulated post-translational modifications (PTMs) and also for the discovery of poorly quantified peptides. The exclusion of poorly quantified peptides before protein quantity summarization decreased false-positives in a benchmark data set. Finally, PeCorA suggests that the inactive isoform of prothrombin, a coagulation cascade protease, is more abundant in plasma from COVID-19 patients relative to non-COVID-19 controls. PeCorA is freely available as an R package that works with arbitrary tables of quantified peptides.
Keywords: proteomics, quantification, R, peptides, bioinformatics, computation, proteoform, proteins, linear model, statistics
Graphical Abstract
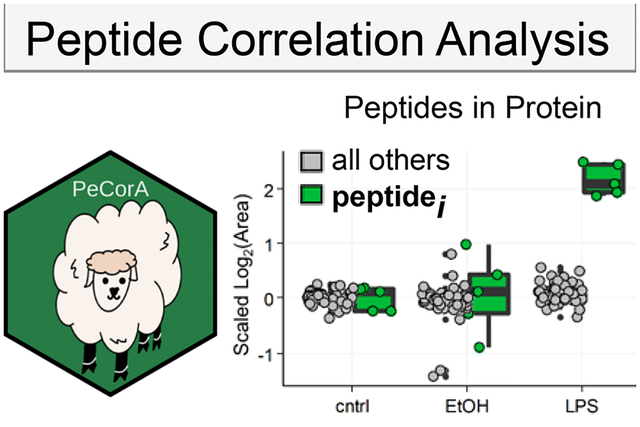
1. INTRODUCTION
Shotgun proteomics relies on the inference of protein identity and quantity from peptide pieces. Although the magnitude of peptide abundance will differ due to different detection sensitivities,1,2 most protein quantitation strategies3,4 assume that multiple peptides derived from a protein will behave quantitatively similar across treatment groups and thus combine multiple peptide quantities into one protein quantity5,6 (Figure 1A). Each protein encoded by a gene exists as a population of unique states, called proteoforms,7 that arise from post-translational modification8 or alternative splicing.9,10 These proteoforms might be influenced differently by biological perturbations. A motivation for top-down proteomics is that proteoform regulation is difficult to capture after the proteolysis required by shotgun proteomics. However, proteoform regulation should be hidden in the differential abundance of peptides mapping to one gene. For example, if a protein quantity is unchanged, but one post-translational modifications (PTM) site is increased with our treatment, then that would cause the apparent disappearance of the peptide sequence that harbors the PTM (Figure 1B). Therefore, strategies to detect discordant peptide quantities among peptides mapped to one gene have the potential to reveal hidden quantitative proteoform information from bottom-up proteomics.
Figure 1.
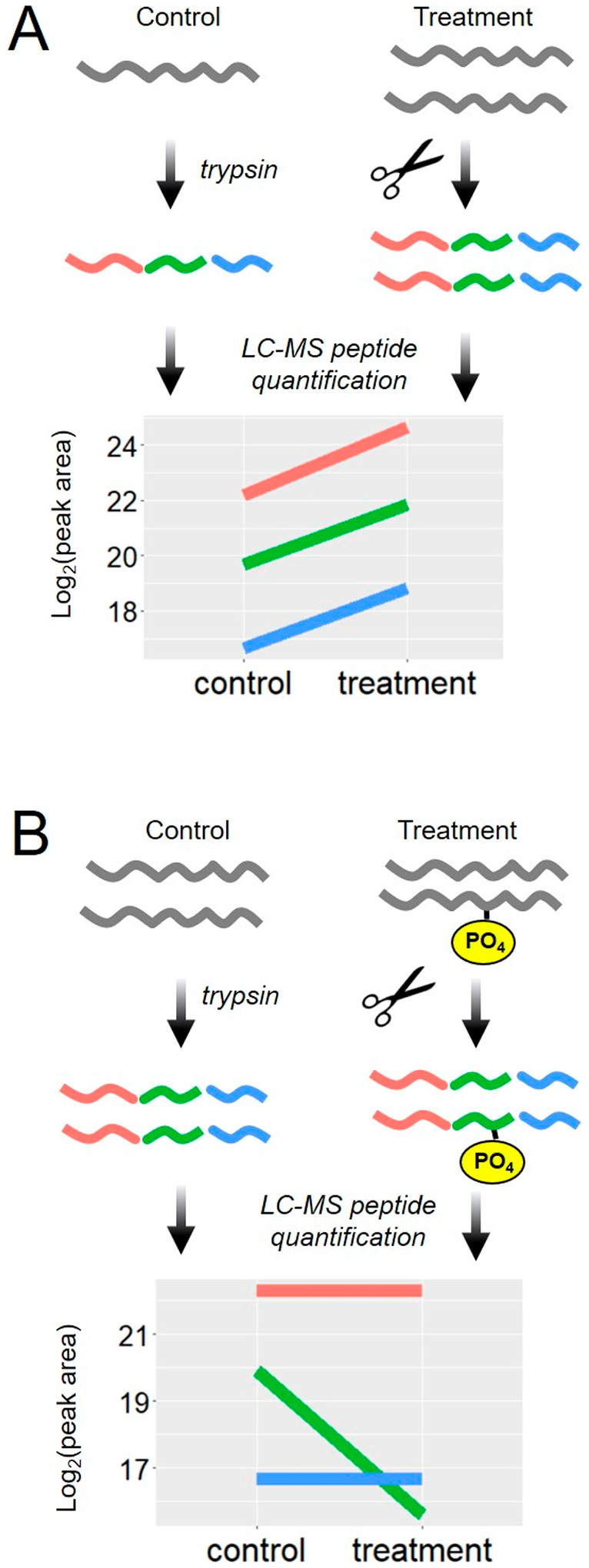
Protein quantification based on multiple measures of peptide parts. In shotgun proteomics, peptides are produced from proteins by proteases; then, the quantities of those peptides are used as a proxy for the protein. (A) Illustration of the case where a protein is increased by a biological treatment. The quantitative values from multiple peptides from one protein change in the same direction across biological treatment groups. (B) Illustration of the case where a protein modification is increased due to a biological treatment, but the protein quantity is unchanged. The quantitative values for multiple peptides from that one protein disagree.
Multiple strategies have focused on the detection of dissonant peptides to remove them and improve protein quantity summarization,11–15 but the concept of gaining proteoform information from discordant peptides is underexplored. The results presented here define a simple strategy called Peptide Correlation Analysis (PeCorA) to detect peptides with discordant quantitation across treatment groups. PeCorA uses linear models to statistically assess the interaction between peptide and treatment groups. In contrast with previous works that discard these peptides, here we focus on the utility of these peptides to reveal quantitative proteoform information. Examples of peptides that reveal proteoform regulation include: direct detection of a regulated methionine oxidation in PKA R1α, indirect detection of a lost phosphorylation in the VAV1 protein, and incorrect peak picking of a peptide from CALR. PeCorA also detected clotting factor and complement system proteoform changes in human plasma proteomics from the SARS-CoV-2 viral response.16 PeCorA can be easily applied to any quantitative proteomics data table and is freely available as a package written in R.
2. EXPERIMENTAL PROCEDURES
2.1. Data
PeCorA was demonstrated with three data sets:
1. Mouse primary microglia:
Raw proteomic data from a published study of mouse primary microglia17 were downloaded from the PRIDE repository18 (identifier PXD014466, https://www.ebi.ac.uk/pride/archive/projects/PXD014466). The data set was composed of five biological replicates each from three sample groups: control, 50 mM ethanol treatment, or 5 ng lipopolysaccharide (LPS) treatment (15 total files). Each sample was analyzed with a 120 min liquid chromatography gradient and online electrospray ionization into a hybrid quadrupole-orbitrap mass spectrometer (Q-Exactive Plus). The data are available from MassIVE (accession MSV000085712; DOI: 10.25345/C57J3B).
2. iPRG DDA benchmark:
Processed proteomic data were downloaded from the 2015 Proteome Informatics Research Group (iPRG) study.19 The data set was composed of three technical replicates acquired in random order each from four complex biological samples containing a constant background of tryptic digests of S. cerevisiae (ATCC strain 204508/S288c) separately spiked with different concentrations of six protein digests (total of 12 analyses). Each sample was analyzed with a 110 min liquid chromatography gradient and online electrospray ionization into a hybrid quadrupole-orbitrap mass spectrometer (Q-Exactive). iPRG DDA benchmark data were downloaded from MassIVE (accession MSV000079843, https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=eccf4bd3e86a4f79af468b0010eb80b0).
3. COVID-19 plasma proteomics:
Processed proteomic data were downloaded from a recent large-scale analysis of COVID-19 severity.16 The data set was composed of over 100 plasma samples from three groups: (1) COVID-19-driven acute respiratory distress syndrome (ARDS) patients, (2) non-COVID-19-driven ARDS patients, and (3) pooled plasma control sample extracted with each batch as quality control. Each sample was analyzed with a 90 min liquid chromatography gradient and online electrospray ionization into a quadrupole–ion trap–Orbitrap hybrid Eclipse. Please see the original publications for more data collection and sample preparation details. In brief, the data in the original publication were analyzed by MaxQuant (version 1.6.10.43) by searching against the UniProt Homo sapiens database containing protein isoforms and computationally predicted proteins (downloaded on 2019–06-18). For our analysis, we did not repeat the database search but used the quantitative values in the peptides.txt file available on MassIVE (accession MSV000085703, DOI: 10.25345/C5F74G).
2.2. Peptide Identification by Database Search of MS/MS Spectra
All raw files from the microglia data set were converted to mzML format using msconvertGUI (part of ProteoWizard)20 and searched against mouse proteins including isoforms downloaded from UniProt (2020–04-08) with MS-Fragger version 2.4.21 Reversed sequences and common contaminants were added the database using the Philosopher toolkit version 2.0.0.22 Searches were performed using the FragPipe user interface with the default closed search settings, except 10 ppm precursor and 20 ppm fragment mass tolerances were used. Search outputs of each separate LC-MS analysis were refined with PeptideProphet23 and combined using iProphet.24
2.3. Peptide Quantification
Filtered peptide identifications from a microglia data set were imported into Skyline for quantification25 by MS1 filtering.26,27 Only protein identifications supported by two peptides were included, and peptides that were not unique to only one protein accession were excluded. A precursor signal within 10 ppm of the theoretical peptide mass for charge states 2–5 was extracted within 5 min of peptide identifications, and peaks were automatically picked by the software.
Quantitative results from the DDA iPRG data set were downloaded from the GitHub repository for the MSstats feature selection paper.11 Those results were processed for quality control into quantitative output for group comparison using the dataProcess function of the MSstats package.28
Peptide-level quantitative data from the COVID-19 data set were downloaded from the massive repository.16 Specifically, the peptides.txt output from MaxQuant (version 1.6.10.43)29 was processed into PeCorA-ready format.
2.4. Peptide Correlation Analysis (PeCorA)
PeCorA takes as input tabular data that must include the (1) peptide, (2) protein annotation, (3) condition group, and (4) replicate number. Data can be analyzed within R or via a command line using a wrapper R script. The quantitative results table is read into R with the import_processed_data function from the PeCorA package. After data import, the values are initially filtered to include only peptides with a user-defined minimum quantitative value in all samples. Next, multiple charge states of one peptide are summed into one value “all”, the quantitative values of peptides are log-transformed, and the global distribution of all peak areas is scaled to have the same mean center and standard deviation equal to 1 using the scale_by function of the R package standardize (as shown in Figure 2A). After global scaling and centering per replicate, each peptide quantity is centered by subtracting the mean of the control group’s peak area, which aligns all peptides relative to the control group average of 0 (as shown in Figure 2B). These twice-scaled data can be obtained using the PeCorA_preprocessing function. Data are then used for peptide correlation analysis with the PeCorA function. At a high level, the slope of each peptide’s quantity across experimental conditions is compared with the slope of all other peptides measured from that protein. This is achieved by iteratively setting each peptide to its own factor group against the factor group of all other peptides and then fitting a linear model that includes a term for the interaction between the peptide groups. The p value of significance for an interaction is recorded for each peptide group in each iteration. All p values for the peptide interaction terms within one protein are then corrected with the Benjamini–Hochberg procedure.30 Finally, the PeCorA_plotting function generates boxplots of any peptide’s quantity compared with the other peptides in that protein across biological conditions (similar to the boxplots shown in this Technical Note).
Figure 2.
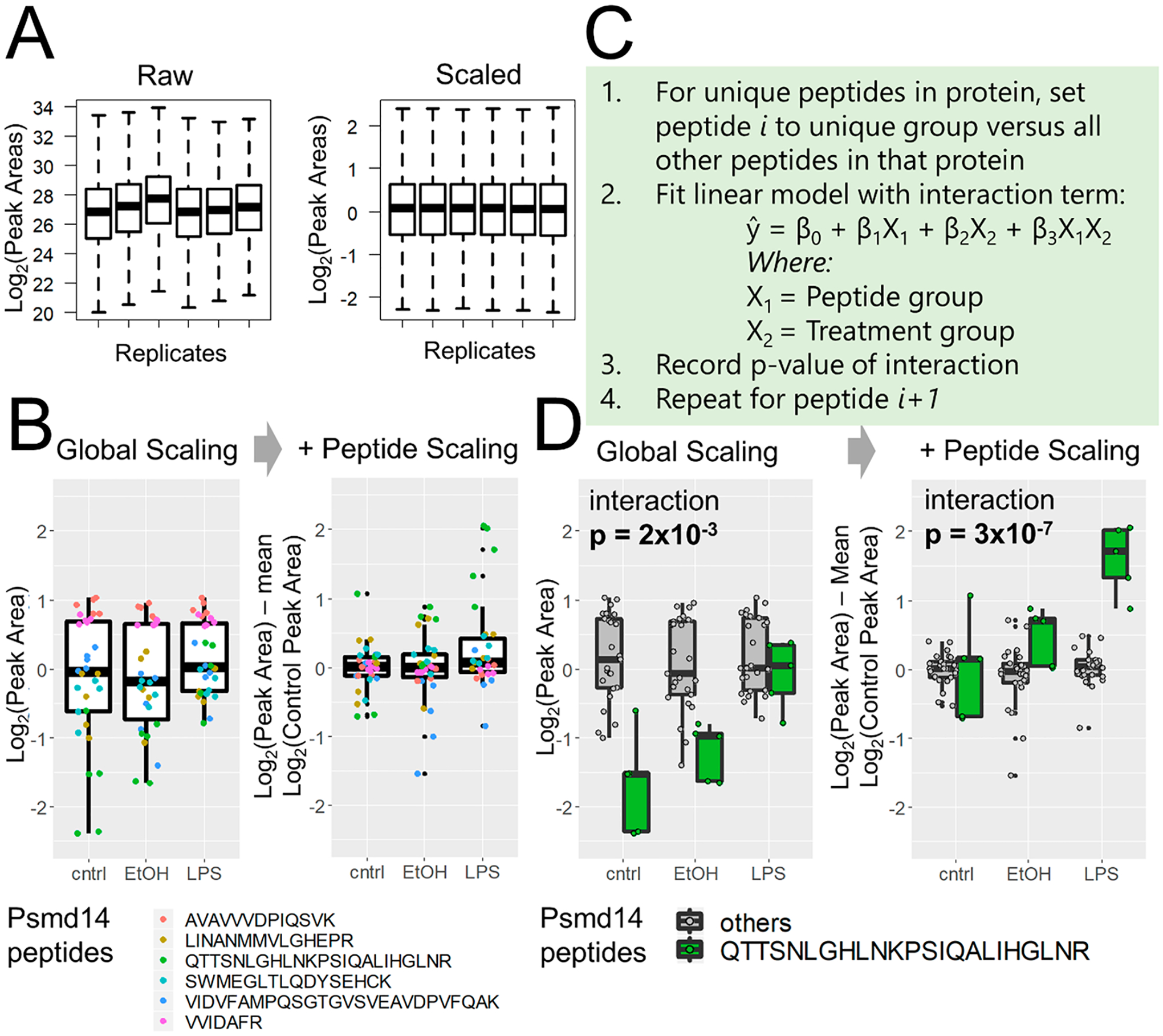
Peptide correlation analysis (PeCorA) to detect differentially modified peptides. (A) First, global data distributions are scaled to be centered around zero. (B) Next, each peptide is scaled to the mean of the control group to produce a unitless relative peptide quantity across groups. This helps correct the problem of differential peptide ionization and detection efficiency to produce more uniform data distributions. (C) Third, the quantitative values of one peptide from a protein are compared with the quantities of all other peptides in that protein using a linear model with a term for the interaction between peptides and biological treatment groups. This is repeated in a loop to compare each peptide to all other peptides. The p value obtained from an ANOVA test of the interaction is used to determine whether the quantity of each peptide is statistically different from that of all other peptides in that protein. (D) Example of one peptide that is statistically different from the quantities of all other peptides. The p value of the interaction between the peptide and the treatment group decreases when peptide scaling aligns the data.
2.5. Code Availability
The peptide correlation analysis code is written in R. The code, documentation, and example data sets are available as an R package on GitHub (https://github.com/jessegmeyerlab/PeCorA).
Details of the functions included in this package can be found in the package vignette. The raw mass spectrometry data from the microglia data set is available from the PRIDE repository of the original publication (PXD014466) and also on massive.ucsd.edu (data set MSV000085712). The Skyline file used to generate peptide areas from microglia data is available on Panorama at https://panoramaweb.org/HRvw2O.url.
The GitHub repository also contains the Skyline report template (skyr), the skyline report from the microglia data used here, and the PeCorA outputs from the analysis of the microglia, iPRG, and COVID data used here. Additional scripts including those to reproduce the exact comparisons in Supplementary Figure 1 are available from https://github.com/demar01/PeCorA-addin.
3. RESULTS
3.1. PeCorA Method Overview
PeCorA starts by scaling and centering the per-sample peptide intensity distributions (Figure 2A); then, each peptide is scaled to the center of the control condition (Figure 2B). Peptide-level scaling removes the spread of peptide quantities that results from different ionization efficiencies, and results in a tighter distribution of peptide quantities for each protein. After the data are scaled, the PeCorA algorithm iteratively compares each peptide to all other peptides in the same protein (pseudocode in Figure 2C). At a high level, the slope of each peptide quantity is compared with the slope of all other peptide quantities across biological conditions using a linear model that includes an interaction term between the treatment and peptide groups. This enables the calculation of a p value testing whether each peptide agrees quantitatively with the group of all other peptides in that protein. All p values for all peptides are recorded; then, multiple hypothesis testing is applied on a per-protein basis. Comparing the interaction p value with and without peptide scaling demonstrates the improved statistical power (Figure 2D). Peptide quantities from the same protein (Psmd14) are used in Figure 2B,D to illustrate these concepts. Thus PeCorA statistically determines whether a peptide’s quantity across experimental treatments disagrees with other peptides assigned to the same protein.
The frequency of quantitative peptide disagreements in a data set is a useful first analysis to understand the potential utility of PeCorA. We started with a reanalysis of data from mouse primary microglia.17 After excluding peptides that map to multiple proteins and requiring two peptides per protein, a total of 27 685 peptides from 2918 proteins were identified. The table was filtered to keep only peptides with peak areas over 100 and only peptides with quantities in all replicates, which left 26 444 peptides from 2858 proteins. Of those, 416 proteins were found to harbor at least one peptide that disagreed with the other peptides in that protein, which corresponds to ~15% of quantified proteins. From the perspective of quantified peptides, the frequency of disagreement is much lower; 489 of the 26 444 peptides, or 1.9%, were found to disagree quantitatively with the other peptides mapped to the same protein. Overall, these results suggest that quantitative disagreements are rare at the peptide level but are relatively common from the protein perspective.
3.2. PeCorA Discovers Multiple Types of Peptide Disagreements
3.2.1. Direct Evidence of Regulated PTMs.
As an example of direct evidence of a regulated PTM discovered by PeCorA, the peptide with the most significant adjusted p value in the microglia data set was from PKA R1α and contained methionine 331 oxidation (Table S1, Figure 3A). This peptide was detected with and without the oxidation, but only the oxidized peptide was increased due to LPS stimulation. PKA R1α is thought to be regulated by oxidative stress,31–33 and the finding that LPS can induce this site-specific oxidation may prove to be yet another mechanism by which cellular metabolism is tuned. This site M331 is on the surface of a second nucleotide binding domain (CNB-B) near the cAMP binding site34 and could regulate cAMP binding or protein–protein interactions (Figure 3B). This example of the direct observation of this regulated PTM site uncovered by PeCorA would be missed using standard protein quantity summaries based on grouped peptide quantities.
Figure 3.
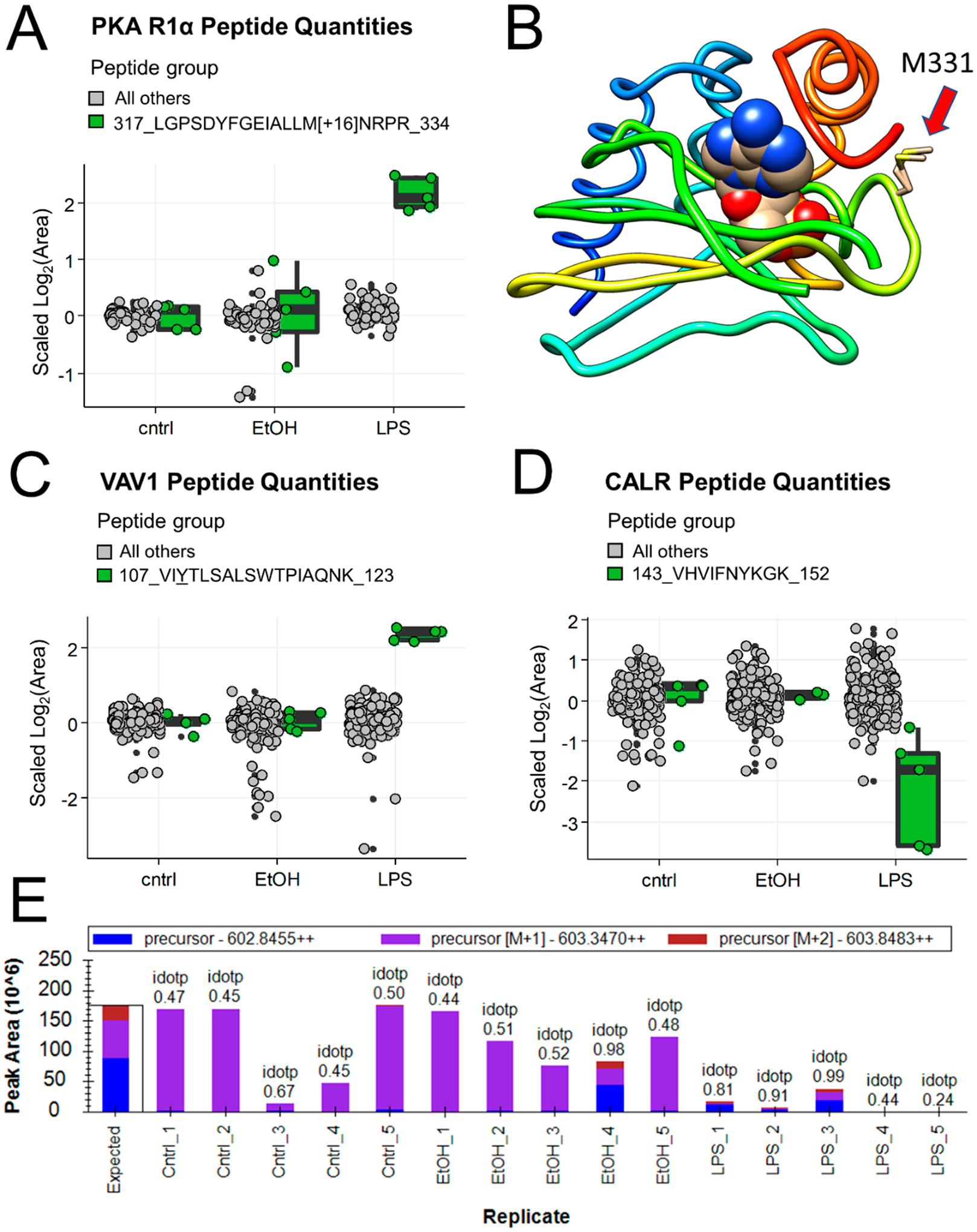
Examples of interesting peptides revealed by PeCorA. (A) PKA R1α peptide quantities comparing the sequence containing oxidized methionine in green on the right with all other peptides in the same protein in gray on the left. (B) Crystal structure (PDB: 5KJZ) of the second nucleotide binding domain of PKA R1α bound to cGMP showing the location of the oxidized methionine 331 with a red arrow. (C) VAV1 peptide quantities comparing the sequence with an inferred change in phosphorylation in green on the right versus all other peptides in gray on the left. (D) CALR peptide quantities showing the peptide with problematic quantitation in green on the right with all other peptides in gray on the left. (E) Peak area summary plot from Skyline for the peptide from CALR in panel D showing the poor isotopic dot product (idotp) reflecting incorrect peak picking across most samples.
3.2.2. Indirect Evidence of Regulated PTMs.
As an example of indirect evidence of an altered PTM, a peptide from the proto-oncogene Vav1 was found to increase due to LPS treatment (Figure 3C). Vav1 is known to signal downstream of tyrosine kinases, and this peptide revealed by PeCorA contained two known phosphorylation sites, pY110 and pS113. Prior literature provides strong evidence that this change in the unmodified peptide quantity may reflect a change in the abundance of the phosphorylated peptide form. Vav1 pY110 was decreased with IL-33 stimulation by 33%,35 and IL-33 has been suggested as the signal mediating microglial response to LPS.36 A decrease in the phosphorylation of this peptide would appear as an increase in the unmodified peptide observed here. Further work is needed to verify if the peptide containing this phosphotyrosine is indeed altered in microglia in response to LPS stimulation. This example shows how PeCorA enables inference of PTM changes from unmodified peptides.
3.2.3. Incorrect Peak Picking.
A third example shows the utility of PeCorA for quality control of peptide quantification. Because of the large number of peptides in any proteomics experiment, automated peak picking is required for quantification, which undoubtedly leads to errors. In fact, active research is ongoing to develop methods that detect and exclude poorly quantified signals.11,15 One peptide assigned to the protein Calreticulin (CALR) appeared to decrease due to LPS treatment (Figure 3D), but manual inspection of this peptide’s areas in Skyline revealed that the wrong peak was chosen for most replicates (Figure 3E).
3.2.4. PeCorA as a Peptide Filter before Protein Quantitation.
Although the main goal of PeCorA is to discover differentially regulated proteoforms, based on the example in Figure 3E, we wondered if PeCorA could improve protein-level quantitation if used as a prefilter for peptides before statistical testing. To test this, we used a benchmark data set from the iPRG composed of data from four samples spiked with six different proteins at varied concentrations.19 PeCorA prefiltration of the peptides input to MSstats reduced false-positives (FPs) compared with no feature filtration (FDR < 0.05, as described in Tsai et al.,11 Supplemental Figure 1A). The quantitative qualities produced by both feature selection methods were similar (Supplementary Figure 1B–E). An example of how PeCorA can eliminate FPs is highlighted in Supplementary Figure 1F. The inclusion of all-features resulted in a false-positive detection of differential protein abundance (adjusted p value ≈ 0.0015). The exclusion of discordant peptides detected by PeCorA corrected this artifact in accordance with the feature selection method by Tsai et al.11 Overall, these results demonstrate the potential for PeCorA to improve protein-level quantitation by acting to prefilter poorly quantified peptides before statistical summary.
3.3. PeCorA Reveals Coagulation Proteoform Changes Resulting from COVID-19 Disease
To show that the detection of differential proteoforms is generalizable to additional data set types, we performed PeCorA on a recently published data set containing the plasma proteome signatures of patients with COVID-19-driven acute respiratory distress syndrome (ARDS) or non-COVID-19 ARDS controls.16 A prevalent characteristic of COVID-19 severity is elevated prothrombin time,37 which measures how quickly a prothrombin undergoes cleavage by prothrombinase to change to active thrombin that can stop the bleeding. Like many proteases, thrombin is translated as an inactive precursor, and it contains an N-terminal pro sequence, a signal peptide, and two fragment peptides F1 and F2 that must be removed to produce active thrombin38 (Figure 4A). PeCorA identifies a peptide from the N-terminal of prothrombin mapping to the F1 region that indicates a significantly elevated level of the inactive prothrombin form in COVID-19 ARDS patients relative to controls (Figure 4B). In contrast, PeCorA shows that all other peptides from other regions of prothrombin were unchanged between groups (Figure 4B and Supplementary Figure 2). This result suggests that the differential regulation of thrombin isoforms in COVID-19 patients may partially explain their elevated risk of thrombosis.39 However, the interpretation of this result is difficult without additional experiments because thrombin can act as both a pro- and anticoagulant.40
Figure 4.
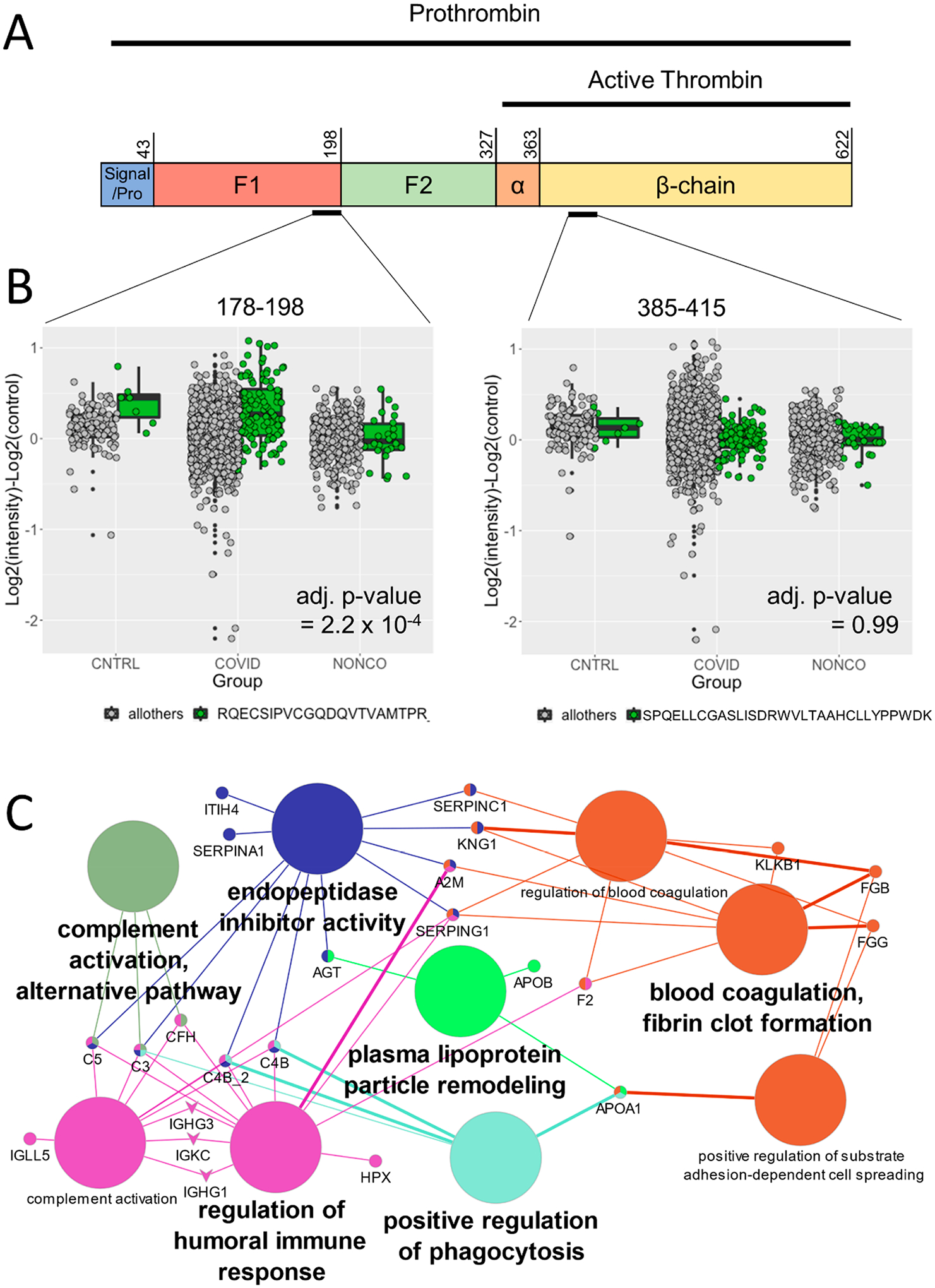
PeCorA detects altered plasma proteoforms associated with COVID-19 infection. (A) Schematic representation of the prothrombin primary sequence, which is the precursor of active thrombin. Prothrombin contains N-terminal pro and signal sequences (residues 1–43) followed by two fragment peptides that must be removed to activate thrombin, F1 (residues 44–198) and F2 (residues 199–327). The active protease domain comprises residues 328–622. Once in its active form, thrombin promotes coagulation. (B) Left: Prothrombin peptide covering residues 177–198 of the inactive F1 region identified as significantly elevated by PeCorA in COVID-19 ARDS patients relative to non-COVID-19 ARDS patients. Right: Unchanged thrombin peptide from the active β-chain 385–415. (C) Enriched GO Biological Process terms from the 26 proteins with altered proteoforms based on the PeCorA adjusted p value < 0.01. The big circles are the GO Biological Process terms, the small circles are the proteins, and the edges show how the proteins are members of the various GO terms. GO terms were filtered to show only the minimal subset needed to make all of the connections between proteins.
To understand the biology of differential proteoforms associated with COVID-19 ARDS, gene ontology (GO) term enrichment analysis was performed using the ClueGO plugin41 within Cytoscape.42 Twenty-six proteins with at least one discordant peptide were input (PeCorA adj. p value < 0.01, Table S1). This analysis revealed that the pathways with differential proteoforms were related to pathways that are dependent on proteolytic processing cascades, including multiple proteins from the complement immune pathway (C3, C5, CFH, C4B, C4B2) and blood coagulation (FGB, FGG, F2, KLKB1; Figure 4C). Notably, multiple peptides from fibrinogen, which is cleaved to form the protein component of clots, were differentially regulated according to PeCorA. This analysis also revealed that APOB and APOA1 isoforms may be differentially regulated as part of the plasma lipoprotein particle remodeling process. In summary, PeCorA reveals multiple potentially dysregulated protease-driven pathways where the subtle differences in proteoforms were previously missed due to peptide quantity aggregation.
4. CONCLUSIONS
PeCorA is a new strategy to detect biologically interesting proteoform changes based on discordant peptide quantification across treatment groups. PeCorA revealed that discordant peptides are present in ~15% of proteins but represent only ~2% of the total peptides. Therefore, one conclusion is that the common practice of obtaining protein quantity summaries from aggregating peptide quantities is accurate in most cases. However, in agreement with other studies,11–15 we provide evidence that the exclusion of poorly quantified peptides can improve protein quantity summarization. In contrast with previous works, we show how discordant peptides revealed by PeCorA extract interesting biology from existing data, including: (1) direct and indirect evidence of regulated PTMs and (2) differential regulation of clotting and complement cascade proteoforms likely resulting from post-translational proteolytic processing.
PeCorA is available as an R package (www.github.com/jessegmeyerlab/PeCorA) to facilitate its interoperability with other quantitative proteomic tools. The results presented here show that PeCorA is applicable to proteomic data from different species (including human, mouse, and yeast) processed with different tools (including Skyline, Progenesis, and MaxQuant). In summary, PeCorA should find widespread application to proteomic data sets for the detection of differentially regulated proteoforms.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following people for sparking the idea of this paper via discussions on Twitter: Karthik Kamath (@karthikskamath), Mike MacCoss (@mjmaccoss), Kareem Carr (@kareem_carr), Chelsea Parlett-Pelleriti (@ChelseaParlett), James Umbanhowar (@jumbanho), and Phil Wilmarth (@pwilmarth). Ronald Beavis (@NorSivaeb) helped find the microglia data set. We also thank Amanda Momenzadeh for help interpreting the COVID-19 plasma results. We thank Josh Coon and Deane Mosher for support and helpful discussions. This research was supported by the following NIH grants: T15LM007359, 5T32HG002760, and 5P41GM108538.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00602.
Figure S1. PeCorA as a peptide filter before protein quantitation using a benchmark data set. Figure S2. PeCorA comparison plots for all peptides in prothrombin showing that peptides from none of the other regions are differentially regulated (PDF)
Table S1. PeCorA output tables for the three data sets analyzed here (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00602
The authors declare no competing financial interest.
The code, documentation, and example data sets are available as an R package on GitHub (https://github.com/jessegmeyerlab/PeCorA ).
Contributor Information
Maria Dermit, Centre for Cancer Cell and Molecular Biology, Barts Cancer Institute, Queen Mary University of London, London EC1M 6BQ, United Kingdom.
Trenton M. Peters-Clarke, Department of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, United States
Evgenia Shishkova, National Center for Quantitative Biology of Complex Systems, Madison, Wisconsin 53706, United States; Department of Biomolecular Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, United States.
Jesse G. Meyer, Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, United States; Department of Biomolecular Chemistry, University of Wisconsin-Madison, Madison, Wisconsin.
REFERENCES
- (1).Tang L; Kebarle P Dependence of Ion Intensity in Electrospray Mass Spectrometry on the Concentration of the Analytes in the Electrosprayed Solution. Anal. Chem 1993, 65 (24), 3654–3668. [Google Scholar]
- (2).Cech NB; Enke CG Relating Electrospray Ionization Response to Nonpolar Character of Small Peptides. Anal. Chem 2000, 72 (13), 2717–2723. [DOI] [PubMed] [Google Scholar]
- (3).Arul AB; Robinson RAS Sample Multiplexing Strategies in Quantitative Proteomics. Anal. Chem 2019, 91 (1), 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Meyer JG; Schilling B Clinical Applications of Quantitative Proteomics Using Targeted and Untargeted Data-Independent Acquisition Techniques. Expert Rev. Proteomics 2017, 14 (5), 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Krey JF; Wilmarth PA; Shin J-B; Klimek J; Sherman NE; Jeffery ED; Choi D; David LL; Barr-Gillespie PG Accurate Label-Free Protein Quantitation with High- and Low-Resolution Mass Spectrometers. Journal of Proteome Research 2014, 13 (2), 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Silva JC; Gorenstein MV; Li G-Z; Vissers JPC; Geromanos SJ Absolute Quantification of Proteins by LCMS E: A Virtue of Parallel Ms Acquisition. Mol. Cell. Proteomics 2006, 5 (1), 144–156. [DOI] [PubMed] [Google Scholar]
- (7).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How Many Human Proteoforms Are There? Nat. Chem. Biol 2018, 14 (3), 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mair W; Muntel J; Tepper K; Tang S; Biernat J; Seeley WW; Kosik KS; Mandelkow E; Steen H; Steen JA FLEXITau: Quantifying Post-Translational Modifications of Tau Protein in Vitro and in Human Disease. Anal. Chem 2016, 88 (7), 3704–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lau E; Han Y; Williams DR; Thomas CT; Shrestha R; Wu JC; Lam MPY Splice-Junction-Based Mapping of Alternative Isoforms in the Human Proteome. Cell Rep 2019, 29 (11), 3751–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Blencowe BJ The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem. Sci 2017, 42 (6), 407–408. [DOI] [PubMed] [Google Scholar]
- (11).Tsai T-H; Choi M; Banfai B; Liu Y; MacLean B; Dunkley T; Vitek O Selection of Features with Consistent Profiles Improves Relative Protein Quantification in Mass Spectrometry Experiments. Mol. Cell. Proteomics 2020, 19, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang B; Pirmoradian M; Zubarev R; Käll L Covariation of Peptide Abundances Accurately Reflects Protein Concentration Differences. Mol. Cell. Proteomics 2017, 16 (5), 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Forshed J; Johansson HJ; Pernemalm M; Branca RMM; Sandberg A; Lehtiö J Enhanced Information Output From Shotgun Proteomics Data by Protein Quantification and Peptide Quality Control (PQPQ). Mol. Cell. Proteomics 2011, 10 (10), M111.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schwarz E; Levin Y; Wang L; Leweke FM; Bahn S Peptide Correlation: A Means to Identify High Quality Quantitative Information in Large-Scale Proteomic Studies. J. Sep. Sci 2007, 30 (14), 2190–2197. [DOI] [PubMed] [Google Scholar]
- (15).Teo G; Kim S; Tsou C-C; Collins B; Gingras A-C; Nesvizhskii AI; Choi H MapDIA: Preprocessing and Statistical Analysis of Quantitative Proteomics Data from Data Independent Acquisition Mass Spectrometry. J. Proteomics 2015, 129, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Overmyer KA; Shishkova E; Miller IJ; Balnis J; Bernstein MN; Peters-Clarke TM; Meyer JG; Quan Q; Muehlbauer LK; Trujillo EA; He Y; Chopra A; Chieng HC; Tiwari A; Judson MA; Paulson B; Brademan DR; Zhu Y; Serrano LR; Linke V; Drake LA; Adam AP; Schwartz BS; Singer HA; Swanson S; Mosher DF; Stewart R; Coon JJ; Jaitovich A Large-Scale Multi-Omic Analysis of COVID-19 Severity. Cell Systems 2020, DOI: 10.1016/j.cels.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Guergues J; Wohlfahrt J; Zhang P; Liu B; Stevens SM Deep Proteome Profiling Reveals Novel Pathways Associated with Pro-Inflammatory and Alcohol-Induced Microglial Activation Phenotypes. J. Proteomics 2020, 220, 103753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Pérez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yılmaz Ş; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaíno JA The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Choi M; Eren-Dogu ZF; Colangelo C; Cottrell J; Hoopmann MR; Kapp EA; Kim S; Lam H; Neubert TA; Palmblad M; Phinney BS; Weintraub ST; MacLean B; Vitek O ABRF Proteome Informatics Research Group (IPRG) 2015 Study: Detection of Differentially Abundant Proteins in Label-Free Quantitative LC-MS/MS Experiments. Journal of Proteome Research 2017, 16 (2), 945–957. [DOI] [PubMed] [Google Scholar]
- (20).Kessner D; Chambers M; Burke R; Agus D; Mallick P ProteoWizard: Open Source Software for Rapid Proteomics Tools Development. Bioinformatics 2008, 24 (21), 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).da Veiga Leprevost F; Haynes SE; Avtonomov DM; Chang H-Y; Shanmugam AK; Mellacheruvu D; Kong AT; Nesvizhskii AI Philosopher: A Versatile Toolkit for Shotgun Proteomics Data Analysis. Nat. Methods 2020, 17 (9), 869–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem 2002, 74 (20), 5383–5392. [DOI] [PubMed] [Google Scholar]
- (24).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI IProphet: Multi-Level Integrative Analysis of Shotgun Proteomic Data Improves Peptide and Protein Identification Rates and Error Estimates. Mol. Cell. Proteomics 2011, 10 (12), M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schilling B; Rardin MJ; MacLean BX; Zawadzka AM; Frewen BE; Cusack MP; Sorensen DJ; Bereman MS; Jing E; Wu CC; Verdin E; Kahn CR; MacCoss MJ; Gibson BW Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline: Application to Protein Acetylation and Phosphorylation. Mol. Cell. Proteomics 2012, 11 (5), 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meyer JG Fast Proteome Identification and Quantification from Data-Dependent Acquisition-Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods and Protocols 2019, 2 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Choi M; Chang C-Y; Clough T; Broudy D; Killeen T; MacLean B; Vitek O MSstats: An R Package for Statistical Analysis of Quantitative Mass Spectrometry-Based Proteomic Experiments. Bioinformatics 2014, 30 (17), 2524–2526. [DOI] [PubMed] [Google Scholar]
- (29).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Benjamini Y; Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995, 57 (1), 289–300. [Google Scholar]
- (31).Haushalter KJ; Schilling JM; Song Y; Sastri M; Perkins GA; Strack S; Taylor SS; Patel HH Cardiac Ischemia-Reperfusion Injury Induces ROS-Dependent Loss of PKA Regulatory Subunit RIα. American Journal of Physiology-Heart and Circulatory Physiology 2019, 317 (6), H1231–H1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brennan JP; Bardswell SC; Burgoyne JR; Fuller W; Schröder E; Wait R; Begum S; Kentish JC; Eaton P Oxidant-Induced Activation of Type I Protein Kinase A Is Mediated by RI Subunit Interprotein Disulfide Bond Formation. J. Biol. Chem 2006, 281 (31), 21827–21836. [DOI] [PubMed] [Google Scholar]
- (33).Srinivasan S; Spear J; Chandran K; Joseph J; Kalyanaraman B; Avadhani NG Oxidative Stress Induced Mitochondrial Protein Kinase A Mediates Cytochrome C Oxidase Dysfunction. PLoS One 2013, 8 (10), No. e77129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lorenz R; Moon E-W; Kim JJ; Schmidt SH; Sankaran B; Pavlidis IV; Kim C; Herberg FW Mutations of PKA Cyclic Nucleotide-Binding Domains Reveal Novel Aspects of Cyclic Nucleotide Selectivity. Biochem. J 2017, 474 (14), 2389–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pinto SM; Nirujogi RS; Rojas PL; Patil AH; Manda SS; Subbannayya Y; Roa JC; Chatterjee A; Prasad TSK; Pandey A Quantitative Phosphoproteomic Analysis of IL-33-Mediated Signaling. Proteomics 2015, 15 (2–3), 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cao K; Liao X; Lu J; Yao S; Wu F; Zhu X; Shi D; Wen S; Liu L; Zhou H IL-33/ST2 Plays a Critical Role in Endothelial Cell Activation and Microglia-Mediated Neuroinflammation Modulation. J. Neuroinflammation 2018, 15 (1), 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).The Lancet Haematology. COVID-19 Coagulopathy: An Evolving Story. The Lancet Haematology 2020, 7 (6), e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Esmon CT; Owen WG; Jackson CM The Conversion of Prothrombin to Thrombin. II. Differentiation between Thrombin- and Factor Xa-Catalyzed Proteolyses. J. Biol. Chem 1974, 249 (2), 606–611. [PubMed] [Google Scholar]
- (39).Ackermann M; Verleden SE; Kuehnel M; Haverich A; Welte T; Laenger F; Vanstapel A; Werlein C; Stark H; Tzankov A; Li WW; Li VW; Mentzer SJ; Jonigk D Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med 2020, 383 (2), 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Narayanan S Multifunctional Roles of Thrombin. Ann. Clin. Lab. Sci 1999, 29 (4), 275–280. [PubMed] [Google Scholar]
- (41).Bindea G; Mlecnik B; Hackl H; Charoentong P; Tosolini M; Kirilovsky A; Fridman W-H; Pagès F; Trajanoski Z; Galon J ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25 (8), 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003, 13 (11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.