

Abstract
OBJECTIVE:
To determine the frequency and characteristics of brainstem or cerebellar involvement in myelin-oligodendrocyte-glycoprotein-antibody-associated-disorder (MOGAD) versus aquaporin-4-IgG-seropositive-neuromyelitis-spectrum-disorder (AQP4-IgG-NMOSD) and multiple sclerosis (MS).
METHODS:
In this observational study, we retrospectively identified 185 Mayo Clinic MOGAD patients with: 1) characteristic MOGAD phenotype, 2) MOG-IgG seropositivity by live-cell-based-assay, and 3) MRI lesion(s) of brainstem, cerebellum, or both. We compared the symptomatic attacks to AQP4-IgG-NMOSD (n=30) and MS (n=30).
RESULTS:
Brainstem or cerebellar involvement occurred in 62/185 (34%) MOGAD patients of which 39/62 (63%) were symptomatic. Ataxia (45%) and diplopia (26%) were common manifestations. The median age in years (range) in MOGAD of 24 (2–65) was younger than MS at 36 (16–65; p=.046) and AQP4-IgG-NMOSD at 45 (6–72; p=.006). Isolated attacks involving the brainstem, cerebellum, or both were less frequent in MOGAD (9/39[23%]) than MS (22/30[73%]; p<0.001) but not significantly different from AQP4-IgG-NMOSD (14/30[47%]; p=0.07). Diffuse middle cerebellar peduncle MRI-lesions favored MOGAD (17/37[46%]) over MS (3/30[10%]; P=.001) and AQP4-IgG-NMOSD (3/30[10%]; p=.001). Diffuse medulla, pons, or midbrain MRI-lesions occasionally occurred in MOGAD and AQP4-IgG-NMOSD but never in MS. CSF oligoclonal bands were rare in MOGAD (5/30[17%]) and AQP4-IgG-NMOSD (2/22[9%]; p=.68) but common in MS (18/22[82%]; p<.001). Disability at nadir or recovery did not differ between the groups.
CONCLUSION:
Involvement of the brainstem, cerebellum, or both is common in MOGAD but usually occurs as a component of a multifocal CNS attack rather than in isolation. We identified clinical, CSF, and MRI attributes that can help discriminate MOGAD from AQP4-IgG-NMOSD and MS.
Search Terms: (1) myelin oligodendrocyte glycoprotein antibody, (2) acute disseminated encephalomyelitis, (3) neuromyelitis optica, (4) MRI
Myelin oligodendrocyte glycoprotein (MOG) antibody associated disorder (MOGAD) is an inflammatory demyelinating disease of the central nervous system (CNS) defined by MOG-IgG seropositivity that is distinct from multiple sclerosis (MS) and aquaporin-4-IgG seropositive neuromyelitis optica spectrum disorder (AQP4-IgG-NMOSD).1 Brainstem and cerebellar involvement are recognized to occur in MOGAD and the clinical syndrome can be severe.2 However, data on brainstem and cerebellar involvement in MOGAD is limited to subsets of case series encompassing all regions of involvement,3–6 with just a few studies with small numbers of patients primarily focused on this region.2 Direct comparisons of MOGAD brainstem and cerebellar involvement to AQP4-IgG-NMOSD and MS are lacking, yet distinguishing MOGAD from those disorders is important as treatment approaches and prognosis differ.7 Characterization of brainstem and cerebellar involvement in MOGAD will allow clinicians to better identify patients that are appropriate for MOG-IgG serologic testing. This is particularly important as ordering in low probability situations may increase the risk of false positives.8 In this study, we sought to determine the frequency of brainstem and cerebellar involvement in MOGAD, the associated symptoms and signs, laboratory, and MRI features and how these differ from AQP4-IgG-NMOSD and MS.
PATIENTS & METHODS
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the institutional review board of Mayo Clinic, Rochester, MN and all patients gave written consent for the passive use of their medical records for research purposes.
Identification of MOGAD patients
We searched for Mayo Clinic patients that were seen at our facility between 1/1/2000–8/31/2019 that had a MOG-IgG seropositive test result from a stored or fresh sample and had a clinical phenotype consistent with MOGAD (N=185). We then retrospectively identified those with inclusion criteria of: 1) MRI lesions in the brainstem, cerebellum, or both attributable to MOGAD and 2) sufficient clinical data available. All patients had a clinical and MRI syndrome compatible with MOGAD as per international diagnostic criteria.8,9
MOG-IgG1 detections
All patients were evaluated for MOG-IgG1 seropositivity in the Mayo Clinic Neuroimmunology Laboratory using a live-cell based assay with full length human MOG in its native conformational form with a cut-off IgG-binding-index of ≥2.5 and dilutions of 1:20, 1:40, 1:100, 1:1000, 1:10,000 to determine the end titer as previously described.9,10 Patients that had retesting 6 months after initial sample were evaluated for persistent positivity.
Comparative groups
A cohort of thirty consecutive patients ordered by medical record number with symptomatic brainstem or cerebellar lesions was identified from a database of all Mayo Clinic patients with AQP4-IgG positivity (fresh or frozen samples thawed for re-analysis) from 1/1/2000–12/31/2019 who fulfilled 2015 international diagnostic criteria.11 Thirty consecutive patients with symptomatic lesions in the brainstem, cerebellum or both were identified from a database of MS patients fulfilling the most recent MS diagnostic criteria12 and consecutively seen by an autoimmune and MS neurologist (E.P.F.) between 1/1/2016–12/31/2019. The patients in both the AQP4-IgG-NMOSD and MS databases were consecutively enrolled beginning with the first symptomatic lesion of the brainstem, cerebellum or both. MOG-IgG was negative in 18 of 18 patients tested with AQP4-IgG-NMOSD and 9 of 9 tested with MS.
Collection of Clinical and Laboratory data
Medical records of patients were reviewed by two neurologists (S.A.B, E.P.F) for demographic, clinical, and laboratory data. Data was collected from each patient’s first brainstem or cerebellar episode and thus patients with multiple brainstem or cerebellar episodes were only evaluated at their first event. A symptomatic episode was defined as a lesion on brain MRI which correlated with symptoms or signs of dysfunction of the brainstem, cerebellum, or both within 30 days. Data collected included: age at attack onset, sex, ethnicity (when available), recent vaccination, and viral prodrome. We evaluated if symptoms were isolated to brainstem or cerebellum, and for symptoms including diplopia from CN III, IV or VI palsy, intranuclear ophthalmoplegia, facial numbness, CNVII palsy including hearing loss and vertigo, dysphagia, dysarthria, intractable nausea or vomiting, ataxia, opsoclonus-myoclonus, and trigeminal neuralgia. Cerebrospinal fluid (CSF) findings were reviewed for elevated white blood cell count (cells/μL; normal, 0–5), markedly elevated white blood cell count (>50 cells/μL) was also documented as it can be an additional useful cut-off, and a positive oligoclonal IgG band result (≥2 unique CSF bands) in MOGAD patients and controls. Unique CSF IgG oligoclonal bands were determined by pairing patient serum and CSF with IgG isoelectric focusing gel assay using Helena SPIFE 3000 platform as previously described.13 Our updated cut-off of ≥2 unique oligoclonal bands for positivity was utilized for all patients. Expanded disability status scale (EDSS) and brainstem and cerebellar functional systems score (FSS) were collected on patients that had complete neurologic exams done at Mayo Clinic during the attack. EDSS and brainstem and cerebellar FSS were also assessed in patients that had complete neurologic exams available at attack recovery. Response to immunotherapy and treatments used were evaluated.
MRI Review
All available imaging sequences were reviewed of MRIs performed either at Mayo Clinic or other medical centers. Axial T2-weighted, fluid-attenuated inversion recovery (FLAIR), and axial T1-post gadolinium sequences were predominantly utilized for analysis. All MRI images available for 185 MOGAD patients were initially screened in an unblinded fashion for brainstem or cerebellar lesions by a neurologist or neuroradiologist. Medical records were then assessed for accompanying clinical manifestations attributable to dysfunction of the brainstem, cerebellum, or both. Respective MRIs of symptomatic MOGAD patients were then analyzed for deep grey matter, cerebral white matter, spinal cord, and optic nerve lesions. Patients with asymptomatic brainstem lesions or without images available for review were evaluated based upon MRI reports by a neuroradiologist.
A neuroradiologist (P.P.M.) blinded to the antibody seropositivity and clinical information then reviewed the cerebellar and brainstem MRI findings in MOGAD symptomatic attacks and control symptomatic attacks (MS and AQP4-IgG-NMOSD). The following features were evaluated on the initial MRI: number of medullary, pontine, midbrain, and cerebellar peduncle or cerebellar parenchymal T2 hyperintense lesions, presence of diffuse T2 hyperintense lesion (>75% of region involved by visual inspection on axial sequences) in each location, and the presence of enhancement in the cerebellum or brainstem. Two patients with only MRI reports available were included in the text but excluded from statistical comparison of radiographic features to controls which required the available images for comparison. Follow-up MRIs were also evaluated in patients that had an MRI greater than or equal to 6 months after the episode with no interval attacks of any type. Resolution of lesions was assessed on this follow-up MRI.
Statistical analysis
Summary statistics were reported as median (range, minimum-maximum) for continuous variables and as frequencies and percentages for categorical variables. Categorical variables were analyzed and compared by Fisher’s exact test and continuous variables with Mann Whitney U test (IBM SPSS 23 software). A two-sided P-value <.05 was considered statistically significant.
Data Availability Statement
Anonymized data used for this study are available from the corresponding authors on reasonable request.
RESULTS
Frequency of brainstem or cerebellar involvement in MOGAD
Of the 185 total MOGAD patients (Adult, 130; children, 55), 62 (34%) had MRI involvement of the brainstem, cerebellum, or both during their disease course. Among these, 39 of 62 patients (63%) with brainstem or cerebellar lesions had one or more symptomatic brainstem or cerebellar attacks while the remaining 23 patients (37%) had lesions without accompanying symptoms referable to the brainstem or cerebellum (i.e., asymptomatic involvement of this region). Nine of the symptomatic brainstem or cerebellar attacks were isolated and 30 were multifocal attacks occurring in conjunction with: transverse myelitis (TM), 15; acute disseminated encephalomyelitis (ADEM) or attack with supratentorial brain lesions, 13; or optic neuritis (ON), 2. There was a similar proportion of brainstem or cerebellar lesions in children (20 of 55 [36%]) and adults (42 of 130 [32%]) among the MOGAD patients identified (p=0.61) and the proportion with brainstem or cerebellar lesions that were symptomatic in children (15 of 20 [75%]) versus adults (24 of 42 [57%]) did not differ (p=0.26)
Symptomatic brainstem or cerebellar involvement with MOGAD
The demographics and clinical features of the 39 patients with symptomatic brainstem or cerebellar lesions are summarized in Table 1. Viral prodrome was present in 24 of 37 (65%; 15/24 had ADEM), and recent vaccination was noted in 4 of 37 (11%; 4/4 had ADEM) in those with details available. An attack involving the brainstem, cerebellum, or both occurred as a component of the initial manifestation of MOGAD in 21 of the 39 patients (54%) with symptomatic attacks which represented 11% of the total MOGAD cohort. The brainstem or cerebellar involvement occurred as part of a subsequent relapse in 18 of 39 (46%). The most frequently observed signs and symptoms were ataxia followed by diplopia (Table 1). Intractable nausea and vomiting in MOGAD usually occurred with other symptoms, rather than in isolation (Table 1). Other brainstem symptoms or signs such as hearing loss, opsoclonus-myoclonus, trigeminal neuralgia and internuclear ophthalmoplegia (INO) were not encountered although full extraocular movement exam details were available in a minority of MOGAD (Table 1). The patients were followed for a median of 21 months (range, 3–121), and had a median of 3 (range, 0–12) additional symptomatic relapses (cerebellum or brainstem: median of 1 [range, 0–4]; transverse myelitis: median of 1 [range 0–4]; optic neuritis: median of 1 [range, 0–10]; or ADEM or attacks with supratentorial brain lesions: median of 1 [range, 0–4]).
Table 1:
Comparison of clinical characteristics of attacks of MOGAD, AQP-4-IgG-NMOSD, and MS involving the brainstem, cerebellum or both
| MOGAD brainstem or cerebellar attack (n=39)a | AQP4-IgG NMOSD brainstem or cerebellar attack (n=30)a | MOGAD vs AQP4-IgG-NMOSD P-valueb | MS brainstem or cerebellar attack (n=30)a | MOGAD vs MS P-valuec | |
|---|---|---|---|---|---|
| Demographics | |||||
| Median age at attack onset, years (range) | 24 (2 – 65) | 45 (6 – 72) | .006 | 36 (16 – 65) | .046 |
| Children, <18 y (%) | 15/39 (38) | 3/30 (10) | .01 | 2/30 (6.7) | .004 |
| Sex, female (%) | 23/39 (59) | 27/30 (90) | .006 | 22/30 (73) | .31 |
| Ethnicity, Caucasian (%) | 30/37a (81) | 21/29a (72) | .56 | 27/28a (96) | .12 |
| Clinical features | |||||
| Isolated brainstem or cerebellar symptoms/signs during attack (%) | 9/39 (23) | 14/30 (47) | .07 | 22/30 (73) | <.001 |
| Diplopia or CN III, IV, or VI Palsy (%) | 10/38a (26) | 14/30 (47) | .12 | 20/30 (67) | .001 |
| INO (%) | 0/13a (0) | 1/22a (5) | >.99 | 6/21 (29) | .06 |
| Facial numbness (%) | 2/37a (5) | 4/29a (14) | .39 | 9/30 (30) | .009 |
| CN VII Palsy (%) | 7/38a (18) | 4/30 (13) | .74 | 4/30 (13) | .74 |
| Hearing loss (%) | 0/38a (0) | 1/29a (3) | .43 | 2/30 (7) | .19 |
| Vertigo (%) | 7/37a (19) | 7/29a (24) | .76 | 7/30 (23) | .76 |
| Dysphagia (%) | 3/39 (8) | 6/30 (20) | .16 | 6/30 (20) | .16 |
| Dysarthria (%) | 9/39 (23) | 7/29a (24) | >.99 | 0/30 (0) | .004 |
| Intractable Nausea/Vomiting (%) | 10/39 (26) | 18/30 (60) | .006 | 1/30 (3) | .02 |
| Ataxia (%) | 17/38a (45) | 9/28a (32) | .32 | 15/30 (50) | .81 |
| Cerebrospinal fluid findings | |||||
| Elevated CSF white cell count, >5 cells/μl (%) | 23/30dg (77) | 9/19aeh (47) | .06 | 8/20afi (40) | .02 |
| Markedly elevated CSF white cell count, >50 cells/μl (%) | 11/30ad (37) | 1/19ae (5) | .02 | 0/18af (0) | .003 |
| Elevated (≥2) CSF unique oligoclonal bands (%) | 5/30ad (17) | 2/22ae (9) | .68 | 18/22af (82) | <.001 |
Key: AQP4, aquaporin-4; CN, cranial nerve; CSF, cerebrospinal fluid; INO, intranuclear ophthalmoplegia; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis.
The denominator varied due to some patients lacking some aspects of the clinical, laboratory and MRI data
statistical comparison between MOG IgG and AQP4 IgG cases,
statistical comparison between MOG IgG and MS cases,
25/30 (83%) of CSF was acquired at time of brainstem attack in MOG-IgG seropositive patients,
8/22 (36%) of CSF was acquired at the time of brainstem attack in AQP4-IgG seropositive patients,
15/22 (68%) of CSF was acquired at the time of brainstem attack in MS patients,
median CSF white blood cell count for MOG-IgG cases was 32 (range, 1–568),
median CSF white blood cell count for AQP4-IgG cases was 5.5 (range, 1–303),
median CSF white blood cell count for MS cases was 2.5 (range, 1–49),
Laboratory characteristics in symptomatic brainstem or cerebellar MOGAD attacks
Cerebrospinal fluid (CSF) was available in 30 MOGAD patients, the findings are outlined in Table 1.The median MOG-IgG titer of the first sample was 1:100 (range, 1:20–1:1000; normal, <20). There were 24 patients that had retesting 6 months after initial sample, of which 19 (79%) were persistently positive. Of the 19 patients that had persistently positive MOG-IgG, 15 (79%) had at least one more attack of any type, while 2 of 5 patients with subsequent negative MOG-IgG had further attacks. The phenotype of the initial attack was ADEM in 4 of 5 that seroconverted to MOG-IgG negative.
MRI head characteristics in symptomatic brainstem or cerebellar MOGAD attacks
The MRI characteristics of MOGAD patients with symptomatic brainstem and cerebellar lesions with MRI available for re-review are outlined in Table 2. Two patients had MRI reports available (pons, middle cerebellar peduncles T2-hyperintense lesions, 1; medulla T2-hyperintense lesion, 1) but images were not available for the comparison to controls. MRI T2/FLAIR hyperintense lesions in patients with MOGAD were generally large and had poorly-defined borders (Figures 1, 2, & 3). Symptomatic MOGAD lesions of the brainstem, cerebellum or both most commonly involved the pons (26/37, 70%: Figure 1C) and middle cerebellar peduncles (MCP) or cerebellar parenchyma (25/37, 68%; Figures 1B, 2A3) (Table 2). Most T2/FLAIR-hyperintense lesions in MOGAD resolved on follow-up MRI (Figure 3A1–2) (Table 2). Additional MRI lesions in other locations included: cerebral white matter lesions, (26/39, 67%), cerebral deep grey matter lesions (21/ 39, 54%), and spinal cord lesions (19/26, 73%). In addition to the posterior fossa (Table 2), gadolinium enhancement was noted in the following locations: supratentorial region (13/38, 34%), leptomeninges (8/38, 21%), and optic nerve (7/38, 18%).
Table 2:
Comparison of MRI features of attacks of MOGAD, AQP-4-IgG-NMOSD, and MS involving the brainstem, cerebellum or both
| MOGAD brainstem or cerebellar attack (n=37) | AQP4-IgG-NMOSD brainstem or cerebellar attack (n=30) | MOG-IgG vs AQP4-IgG P-value | MS brainstem or cerebellar attack (n=30) | MOG-IgG vs MS P-value | |
|---|---|---|---|---|---|
| MRI head features | |||||
| Medulla lesion present (%) | 18/37 (49) | 25/30 (83) | .005 | 8/30 (27) | .08 |
| Number of lesions in medulla, median (range)f | 0 (0 – 4) | 1 (0 – 4) | .006 | 0 (0 – 8) | .16 |
| Medulla lesion diffuse (%)g | 7/37 (19) | 11/30 (37) | .17 | 0/30 (0) | .01 |
| Pons lesion present (%) | 26/37 (70) | 16/30 (53) | .21 | 21/30 (70) | >.99 |
| Number of lesions in pons, median (range)f | 1 (0 – 7) | 1 (0 – 7) | .09 | 1 (0 – 14) | .69 |
| Pons lesion diffuse (%)g | 8/37 (22) | 3/30 (10) | .32 | 0/30 (0) | .007 |
| Midbrain lesion present (%) | 18/37 (49) | 12/30 (40) | .62 | 7/30 (23) | .04 |
| Number of midbrain lesions, median (range)f | 0 (0 – 4) | 0 (0 – 2) | .21 | 0 (0 – 6) | .047 |
| Unilateral or bilateral diffuse midbrain lesion (%)g | 7/37 (19)a | 4/30 (13)b | .74 | 0/30 (0) | .01 |
| Presence of cerebellar peduncle or parenchymal lesion (%) | 25/37 (68)c | 10/30 (33)d | .007 | 18/30 (60)e | .61 |
| Number of cerebellar parenchymal or peduncle lesions, median (range)f | 2 (0 – 6) | 0 (0 – 3) | .002 | 1 (0 – 8) | .88 |
| Unilateral or bilateral diffuse MCP lesion (%)g | 17/37 (46) | 3/30 (10) | .001 | 3/30 (10) | .001 |
| Enhancement of ≥1 posterior fossa lesion | 22/38 (58) | 17/29 (59) | >.99 | 19/28 (68) | .45 |
| Complete resolution of all prior T2 lesions at ≥6 months (%) | 6/9 (67) | 3/11 (27) | .18 | 3/19 (16) | .01 |
Key: AQP4, aquaporin-4; MCP, middle cerebellar peduncle; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis.
4 of the diffuse midbrain lesions in MOGAD were unilateral and 3 were bilateral,
3 of the diffuse midbrain lesions in AQP4-IgG seropositive NMOSD were unilateral and 1 was bilateral,
There were 12 unilateral MCP lesions seen in MOGAD versus 13 bilateral,
There were 4 unilateral MCP lesions in AQP4 seropositive NMOSD versus 4 bilateral,
There were 11 unilateral MCP lesions in MS versus 4 bilateral,
Some lesions traversed two locations and were counted in both locations,
Diffuse lesions were defined as involving greater than or equal to 75% of the parenchyma as viewed on axial sequence by visual inspection.
Figure 1. Representative examples of MRI findings in MOGAD brainstem and cerebellar lesions.
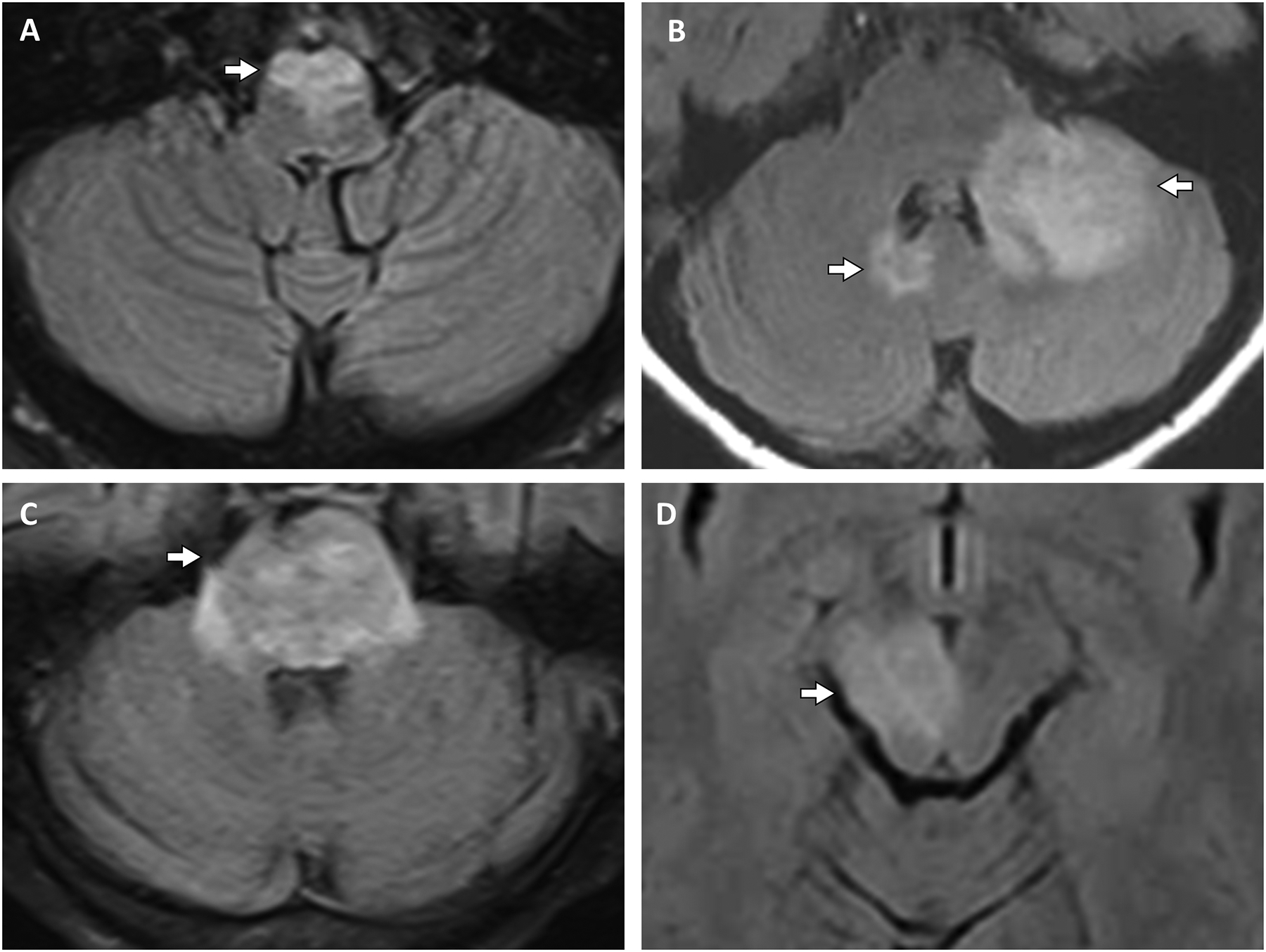
A: An adult patient with an axial T2 FLAIR hyperintense lesion (A, arrow) involving the ventral medulla. B: An adult patient with axial T2 FLAIR hyperintense lesions (B, arrows) involving the left middle cerebellar peduncle, dentate nucleus and cerebellar hemisphere, and the right dentate nucleus with mild mass effect on the left side of the fourth ventricle. C: A pediatric patient with a diffuse pontine T2 FLAIR hyperintense lesion (C, arrow). D: An adult patient with an asymptomatic T2 FLAIR hyperintense lesion involving the right cerebral peduncle (D, arrow).
Key: FLAIR, fluid-attenuated inversion recovery; MOGAD, myelin oligodendrocyte glycoprotein antibody associated disorder.
Figure 2. Comparison of representative brainstem lesions in MOGAD, AQP4-IgG-NMOSD, and MS.
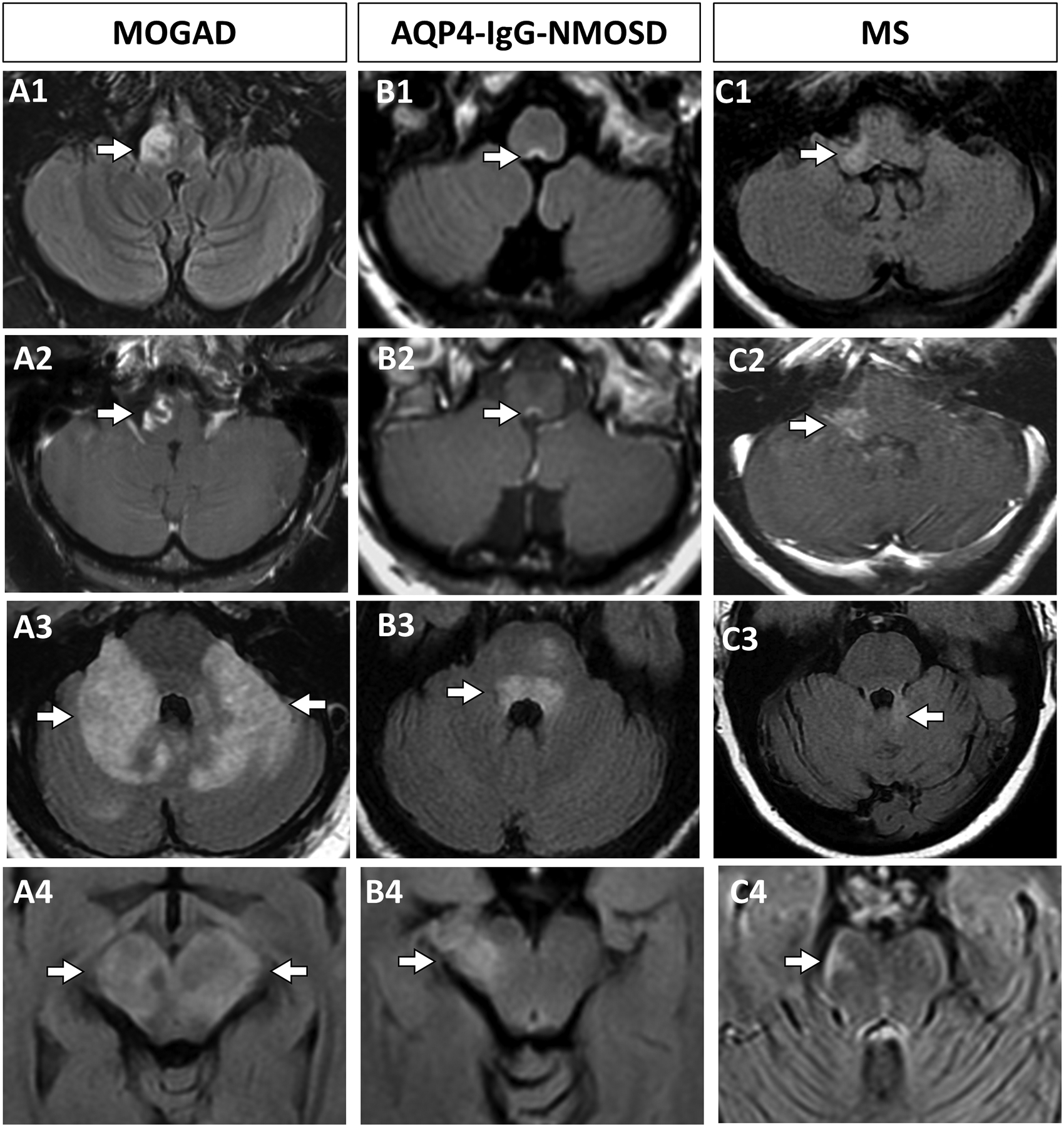
A: An adult patient with MOGAD has a large lesion of the medulla on axial T2 FLAIR (A1, arrow) with associated enhancement on post-gadolinium T1-weighted images (A2, arrow). An adult patient with MOGAD has T2-hyperintense lesions involving bilateral middle cerebellar peduncles diffusely and extending into the cerebellar parenchyma and pons on axial T2 FLAIR images (A3, arrows). A pediatric patient with MOGAD has a midbrain hyperintense lesion diffusely involving both cerebral peduncles on axial T2 FLAIR images (A4, arrows). B: An AQP4-IgG-NMOSD patient has a hyperintense lesion involving the area postrema region of the dorsal medulla on axial T2 FLAIR images (B1, arrow) with associated gadolinium enhancement on T1 images (B2, arrow). An AQP4-IgG-NMOSD pediatric patient has a hyperintense pontine lesion involving the dorsal pons adjacent to the fourth ventricle on T2 FLAIR images (B3, arrow). An AQP4-IgG-NMOSD adult has a hyperintense midbrain lesion involving the right cerebral peduncle on axial T2 FLAIR images (B4, arrow). C: An adult patients with MS associated axial T2 FLAIR hyperintense brainstem hyperintense lesion involving the right posterior-lateral medulla (C1, arrow) with associated enhancement on axial T1-weighted sequences post-gadolinium (C2, arrow). An adult MS patient with a hyperintense lesion involving the medial cerebellum and superior cerebellar peduncle on axial T2-FLAIR images (C3, arrow). An adult MS patient with a hyperintense lesion in the peripheral midbrain on axial T2-FLAIR images (C4, arrow).
Key: AQP4-IgG-NMOSD, aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; FLAIR, fluid-attenuated inversion recovery, MOGAD, myelin oligodendrocyte glycoprotein antibody associated disorder; MS, multiple sclerosis.
Figure 3. Evolution of brainstem lesions in MOGAD, AQP4-IgG-NMOSD, and MS.
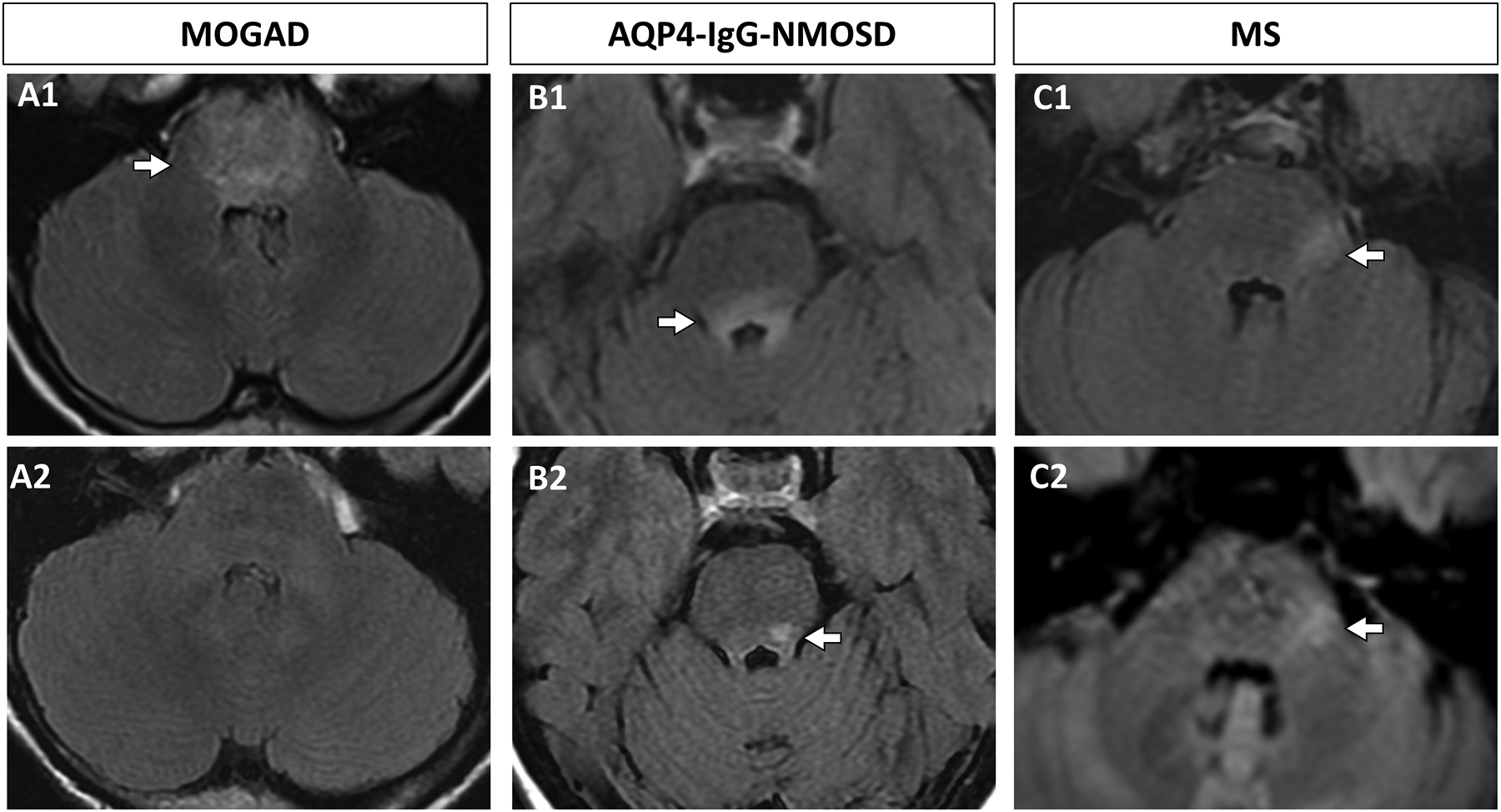
A: A MOGAD pediatric patient had a diffuse T2 FLAIR hyperintense pontine lesion on axial images (A1, arrow) that completely resolved at 6 month follow up (A2). B: An AQP4-IgG-NMOSD seropositive adult patient had a T2 FLAIR hyperintense dorsal pontine lesion on axial images (B1, arrow) that partially resolved at 9 months follow-up (B2, arrow). C: An adult patient with MS had a hyperintense lateral pontine lesion on axial T2 FLAIR images (C1, arrow) that persisted at 11 months follow up (C2, arrow).
Key: AQP4-IgG-NMOSD, aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; FLAIR, fluid-attenuated inversion recovery, MOGAD, myelin oligodendrocyte glycoprotein antibody associated disorder; MS, multiple sclerosis.
Response to immunotherapy and long-term outcomes
The majority of brainstem or cerebellar attacks in MOGAD responded to acute immunotherapy (38/39, 98%). Treatments included one or more of: IV steroids with or without an oral taper (37/39, 95%), plasma exchange (11/39, 28%), and IVIG (11/39, 28%). One or more maintenance steroid-sparing immunotherapies were utilized in 26 of 39 (67%) including: rituximab (10/39, 26%), IVIG (8/39, 21%), azathioprine (8/39, 21%), mycophenolate (6/39, 15%), or other (8/39: one or more of: methotrexate, cyclophosphamide, ocrelizumab, glatiramer acetate, fingolimod, dimethyl fumarate, interferon β−1a; some of these treatments were in patients initially treated as MS). No patients were maintained on corticosteroid monotherapy. The attack severity and long-term outcomes are summarized in Supplementary table 1.
Comparison of symptomatic infratentorial lesions associated with MOGAD to AQP-4–IgG NMOSD and Multiple Sclerosis
MOGAD patients were less likely to have isolated brainstem attacks than MS, but did not differ from AQP4-IgG-NMOSD (Table 1). MOGAD patients were also younger (Table 1). MOGAD had a similar CSF profile to AQP4-IgG-NMOSD, but higher median white blood cell count and lower frequency of oligoclonal IgG bands than MS (Table 1). Diffuse lesions of medulla, pons, or midbrain were similar in MOGAD and AQP4-IgG-NMOSD but more frequent than MS where short peripheral lesions predominated (Table 2) (Figure 2). Diffuse middle cerebellar peduncle lesions (Figure 1B, 2A3) predominated in MOGAD versus AQP4-IgG-NMOSD and MS (Figure 2) while gadolinium enhancement did not differ (Table 2). MOGAD lesions resolved significantly more frequently than MS lesions (Table 2) (Figure 3) and in a higher proportion of MOGAD than AQP4-IgG-NMOSD without reaching significance (Table 2) (Figure 3). The EDSS, brainstem and cerebellar FSS at attack nadir and in recovery across diseases did not differ with most recovering completely (Supplementary table 1).
Asymptomatic brainstem or cerebellar involvement
The median age in patients with exclusively asymptomatic brainstem or cerebellar lesions was 24 years (range, 8–60) and 5 of 23 (22%) were children and 14 of 23 were female (61%). The median antibody titer in these patients was 1:40 (range, 1:20–1:1000) and 8 of 11 (73%) who were tested at 6 months remained persistently positive. All 23 patients who had asymptomatic brainstem or cerebellar lesions were discovered in the context of clinical attacks involving other structures (100%: TM: 52%, ON: 35%, ADEM/supratentorial attack: 13%). T2 FLAIR hyperintensities involving the medulla were found in 11 of 23 (48%), pons in 12 of 23 (52%), midbrain in 5 of 23 (22%) (Figure 1D), cerebellar peduncle in 10 of 23 (43%), and cerebellar parenchyma in 3 of 23 (23%). These lesions exhibited gadolinium enhancement in 8 of 22 (36%). There were also T2/FLAIR hyperintense lesions seen in the deep grey matter in 6 of 23 (26%), white matter in 16 of 23 (69%), and spinal cord in 12 of 19 (63%). Enhancement of the optic nerve occurred in 7 of 22 (32%), of a supratentorial lesion in 4 of 22 (18%), and of the leptomeninges in 1 of 22 (5%). When asymptomatic brainstem or cerebellar involvement was compared to those with symptomatic attacks in this location we found no significant difference in age, proportion of children, antibody titer, persistent seropositivity or MRI findings.
DISCUSSION
Brainstem or cerebellar involvement occurs in approximately one third of MOGAD patients, and is a component of the heralding attack in 11%. Isolated brainstem and cerebellar attacks in MOGAD are rare and the episodes usually occur in conjunction with other manifestations (TM, ON, ADEM/other supratentorial attack) which differs from MS in which brainstem and cerebellar symptoms often occur in isolation. We identified a number of clinical, CSF, and MRI characteristics that can discriminate MOGAD brainstem or cerebellar attacks from those with AQP4-IgG-NMOSD and MS and help select those at highest risk in whom MOG-IgG testing should be strongly considered. This is important as indiscriminate ordering of rare serologic biomarkers such as MOG-IgG in low probability situations, despite its high specificity, may result in a high false positive rate and has led to published guidelines on when to test MOG-IgG.8 These guidelines had limited data on brainstem or cerebellar features and the extensive detail on these aspects and direct comparison to MS and AQP4-IgG-NMOSD in this study helps both select those for MOG-IgG testing and ensure those with a positive MOG-IgG identified have a compatible syndrome.
Brainstem and cerebellum involvement was seen in 34% (62 of 185) of MOGAD patients, similar to a prior study with a smaller cohort in which 30% (15 of 50 patients) had such lesions.2 Prior small series have reported variable frequencies of involvement of brainstem (10–67%),3–6 cerebellar peduncles (33–39%),3,4 and cerebellar hemispheres/vermis (14–53%)3–5 in MOGAD. The brainstem or cerebellar manifestations occurred in conjunction with attacks involving other regions in 77% in this study and 76% in a prior study,2 highlighting that isolated brainstem or cerebellar attacks are uncommon in MOGAD. The frequency of asymptomatic lesions in the brainstem or cerebellum was approximately a third in our cohort and a prior study.2 Asymptomatic brainstem or cerebellar MOGAD lesions always occurred in conjunction with symptomatic attacks in other CNS regions (ON, ADEM/supratentorial attack, TM) which differs from MS in which development of new lesions between attacks is common.
The most frequent manifestations of MOGAD brainstem or cerebellar attacks were ataxia or diplopia accompanied by CN III, IV or VI palsy followed by CN VII palsy and vertigo. Intractable nausea and vomiting occurred in 25% usually in the setting of ADEM with multifocal lesions and potentially the viral prodrome, vertigo and leptomeningitis contributing in some.14,15 This differs from the discrete area postrema syndrome of AQP4-IgG-NMOSD.16 Previous studies have found hearing loss, INO,2 trigeminal neuralgia,17 and paroxysmal dysarthria18 in MOGAD but were not encountered in our study although limited by lack of extraocular movement exam details in a large proportion of MOGAD patients. Others have reported combined clinico-radiologic trigeminal, vestibulocochlear, and oculomotor nerve involvement but this was rare in our study.19,20 MOGAD patients had severe attacks as evidenced by high scores on EDSS and FSS, but complete symptoms resolution was typical in follow-up consistent with its favorable long term outcome.21 MOGAD patients often had CSF pleocytosis and few had positive oligoclonal bands consistent with the lierature.1,6,22 Similar to prior reports, large, poorly demarcated unilateral or bilateral middle cerebellar peduncle MRI T2-lesions were common in MOGAD while single diffuse medulla, pons or midbrain lesions occurred in approximately 20% sometimes extending to the internal capsules and mimicking neuro-Behçet’s.4,23,24 MOGAD brainstem or cerebellar lesions often resolved completely in follow up, similar to prior studies23,25 and we speculate this may reflect enhanced remyelination potential in MOGAD.
Both overlapping and distinguishing features were observed in comparing MOGAD with AQP4-IgG-NMOSD. Patients with MOGAD were younger and had equal sex distribution while AQP4-IgG-NMOSD patients were more often older and women. We found that 34% of MOGAD patients had brainstem or cerebellar involvement and previous studies found 31% of AQ4-IgG-NMOSD had involvement of these regions.26 Area postrema syndrome attacks predominated in AQP4-IgG-NMOSD over MOGAD.27 The severity of brainstem or cerebellar attacks at nadir in MOGAD and AQP4-IgG-NMOSD were similar and both had very good recovery. MOGAD and AQP4-IgG-NMOSD had similar CSF parameters. MOGAD lesions were less frequent in the medulla but more frequent in the middle cerebellar peduncles. The enrichment of the target antigen AQP4 in the area postrema may partly explain this higher frequency of medulla lesions in AQP4-IgG-NMOSD while the middle cerebellar peduncle predominance in MOGAD may reflect an abundance of MOG in these highly myelinated white matter tracts.28,29 Lesion resolution was more frequent in MOGAD (67%) than AQP4-IgG-NMOSD (27%) consistent with the latter being more destructive but this difference was not significant likely due to insufficient power from small numbers.
Despite some overlap, a number of characteristics discriminated brainstem and cerebellar attacks in MOGAD from MS. While we noted brainstem or cerebellar involvement in 34% of MOGAD, it has been reported in up to 65% of MS.30 Brainstem and cerebellar attacks were often a component of a multisystem event in MOGAD, whereas they usually occurred in isolation in MS. Facial numbness and diplopia including INO were more common in MS. Complete recovery was common with both MOGAD and MS. MOGAD CSF parameters differed from MS with higher white blood cell counts and absence of oligoclonal bands in most, similar to recent CSF reports in MOGAD.31,32 Diffuse medulla, pons, or midbrain lesions occurred in approximately 20% of MOGAD patients but never in MS, and diffuse middle cerebellar peduncle lesions were more common in MOGAD consistent with prior studies.4 MOGAD lesions resolved completely on follow up MRI at 6 months more often than MS lesions providing a useful radiologic discriminator when serial imaging is available.
This study is limited by its retrospective nature and potential referral bias but to our knowledge represents the largest study focusing only on brainstem and cerebellar involvement in MOGAD and the first to compare these attacks to MS and AQP4-IgG-NMOSD. Not all AQP4-IgG-NMOSD or MS controls were tested for MOG-IgG. However, AQP4-IgG and MOG-IgG seropositivity coexist in just 0.06%.33 Furthermore, MS patients fulfilled updated McDonald criteria12 and had typical features of MS including INO in 29%, elevated oligoclonal bands in 82%, and lesion persistence over time in 84%. Our data predominantly included patients of Caucasian ethnicity. There were also more children in the MOGAD cohort than AQP4-IgG-NMOSD and MS, consistent with its typical demographic and while merging children and adult MOGAD could bias our data, in subgroup analyses we found brainstem or cerebellar involvement in pediatric and adult MOGAD was broadly similar. Brainstem or cerebellar symptoms and signs in MOGAD may be under-reported or under-detected on exam in children or those with ADEM. We selected patients based on MRI involvement and cannot exclude that brainstem or cerebellar attacks without accompanying MRI lesions may occur, as noted occasionally in MOG-IgG myelitis.34
CONCLUSION
Brainstem and cerebellar involvement are common in MOGAD and usually occurs as a component of a multifocal presentation, although up to a third may be asymptomatic. Clinical, laboratory and MRI attributes identified in this study can help differentiate MOGAD from AQP4-IgG-NMOSD and MS.
Supplementary Material
Financial Disclosures:
Samantha A. Banks: Reports no disclosures
Padraig P. Morris: Reports no disclosures
John C. Chen: Reports no disclosures
Sean J. Pittock: Reports grants, personal fees and non-financial support from Alexion Pharmaceuticals, Inc.; grants from Grifols, Autoimmune Encephalitis Alliance; grants, personal fees, non-financial support and other from MedImmune, Inc.; Dr. Pittock has a patent # 9,891,219 (Application#12-573942) “Methods for Treating Neuromyelitis Optica (NMO) by Administration of Eculizumab to an individual that is Aquaporin-4 (AQP4)-IgG Autoantibody positive”. Dr Pittock also has patents pending for the following IgGs as biomarkers of autoimmune neurological disorders (septin-5, Kelch-like protein 11, GFAP, PDE10A and MAP1B
Elia Sechi: Reports no disclosures
Amy Kunchok: Reports no disclosures
Jan-Mendelt Tillema: Reports no disclosures
James P. Fryer: Reports no disclosures
Brian G. Weinshenker: Receives royalties from RSR Ltd, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr. Volkmann und Kollegen GbR for a patent of NMO-IgG as a diagnostic test for neuromyelitis optica spectrum disorders, served on adjudication committee for clinical trials in neuromyelitis optica spectrum disorders being conducted by MedImmune/VielaBio and Alexion, and consulted for Chugai/Roche/Genentech and Mitsubishi-Tanabe regarding clinical trials for neuromyelitis optica spectrum disorders.
Karl N. Krecke: Reports no disclosures
Alfonso Lopez Chiriboga: Reports no disclosures
Adam Nguyen: Reports no disclosures
Tammy Greenwood: Reports no disclosures
Claudia F. Lucchinetti: Reports no disclosures
Nicholas L. Zalewski: Reports no disclosures
Steven A. Messina: Reports no disclosures
Eoin P. Flanagan: Dr Flanagan is a site principal investigator in a randomized placebo-controlled clinical trial of Inebilizumab (A CD19 inhibitor) in neuromyelitis optica spectrum disorders funded by MedImmune/Viela Bio.
Study Funding:
This study was made possible using funding from the NIH (R01NS113828).
References
- 1.Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nature Reviews Neurology. 2019;15(2):89–102. [DOI] [PubMed] [Google Scholar]
- 2.Jarius S, Kleiter I, Ruprecht K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement-frequency, presentation and outcome. Journal of neuroinflammation. 2016;13(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waters P, Fadda G, Woodhall M, et al. Serial Anti–Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes. JAMA neurology. 2020;77(1):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurynczyk M, Geraldes R, Probert F, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. 2017;140(3):617–627. [DOI] [PubMed] [Google Scholar]
- 5.Baumann M, Grams A, Djurdjevic T, et al. MRI of the first event in pediatric acquired demyelinating syndromes with antibodies to myelin oligodendrocyte glycoprotein. Journal of neurology. 2018;265(4):845–855. [DOI] [PubMed] [Google Scholar]
- 6.de Mol C, Wong Y, van Pelt E, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Multiple Sclerosis Journal. 2019:1352458519845112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flanagan EP. Neuromyelitis optica spectrum disorder and other non–multiple sclerosis central nervous system inflammatory diseases. CONTINUUM: Lifelong Learning in Neurology. 2019;25(3):815–844. [DOI] [PubMed] [Google Scholar]
- 8.Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. Journal of neuroinflammation. 2018;15(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG–associated disorders. JAMA neurology. 2018;75(11):1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waters PJ, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. 2019;92(11):e1250–e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wingerchuk DM, Lennon V, Pittock S, Lucchinetti C, Weinshenker B. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485–1489. [DOI] [PubMed] [Google Scholar]
- 12.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. The Lancet Neurology. 2018;17(2):162–173. [DOI] [PubMed] [Google Scholar]
- 13.Gurtner KM, Shosha E, Bryant SC, et al. CSF free light chain identification of demyelinating disease: comparison with oligoclonal banding and other CSF indexes. Clinical Chemistry and Laboratory Medicine (CCLM). 2018;56(7):1071–1080. [DOI] [PubMed] [Google Scholar]
- 14.Budhram A, Kunchok A, Flanagan E. Adding FUEL to the FLAMES: FLAIR-variable Unilateral Enhancement of the Leptomeninges (FUEL) in MOG-IgG-Associated Disease (862). In: AAN Enterprises; 2020. [Google Scholar]
- 15.Kunchok A, Krecke KN, Flanagan EP, et al. Does area postrema syndrome occur in myelin oligodendrocyte glycoprotein-IgG–associated disorders (MOGAD)? Neurology. 2020;94(2):85–88. [DOI] [PubMed] [Google Scholar]
- 16.Shosha E, Dubey D, Palace J, et al. Area postrema syndrome: Frequency, criteria, and severity in AQP4-IgG–positive NMOSD. Neurology. 2018;91(17):e1642–e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020. [DOI] [PubMed] [Google Scholar]
- 18.Kollmann P, Van Pesch V. MOG antibody-related isolated rhombencephalitis revealed by paroxysmal dysarthria. Journal of the neurological sciences. 2019;405. [DOI] [PubMed] [Google Scholar]
- 19.Cobo-Calvo A, Ayrignac X, Kerschen P, et al. Cranial nerve involvement in patients with MOG antibody–associated disease. Neurology-Neuroimmunology Neuroinflammation. 2019;6(2):e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimori J, Nakashima I. Linear pontine trigeminal root lesion in a patient with anti-MOG antibody-associated encephalitis. Clinical and Experimental Neuroimmunology. [Google Scholar]
- 21.Lopez-Chiriboga S, Sechi E, Buciuc M, et al. Long-term Outcomes in Patients With Myelin Oligodendrocyte Glycoprotein Immunoglobulin G–Associated Disorder. 2020. [DOI] [PMC free article] [PubMed]
- 22.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128–3138. [DOI] [PubMed] [Google Scholar]
- 23.Lechner C, Baumann M, Hennes E-M, et al. Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease. J Neurol Neurosurg Psychiatry. 2016;87(8):897–905. [DOI] [PubMed] [Google Scholar]
- 24.Fujimori J, Takahashi T, Matsumoto Y, et al. Two Japanese cases of anti-MOG antibody-associated encephalitis that mimicked neuro-Behçet’s disease. Journal of neuroimmunology. 2019;334:577002. [DOI] [PubMed] [Google Scholar]
- 25.Juryoczyk M, Tackley G, Kong Y, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. Journal of Neurology, Neurosurgery & Psychiatry. 2017;88(2):132–136. [DOI] [PubMed] [Google Scholar]
- 26.Kremer L, Mealy M, Jacob A, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Multiple Sclerosis Journal. 2014;20(7):843–847. [DOI] [PubMed] [Google Scholar]
- 27.Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82(6):474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Archives of neurology. 2006;63(3):390–396. [DOI] [PubMed] [Google Scholar]
- 29.Solly S, Thomas JL, Monge M, et al. Myelin/oligodendrocyte glycoprotein (MOG) expression is associated with myelin deposition. Glia. 1996;18(1):39–48. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima I, Fujihara K, Okita N, Takase S, Itoyama Y. Clinical and MRI study of brain stem and cerebellar involvement in Japanese patients with multiple sclerosis. Journal of Neurology, Neurosurgery & Psychiatry. 1999;67(2):153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jarius S, Lechner C, Wendel EM, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 2: Results from 108 lumbar punctures in 80 pediatric patients. Journal of neuroinflammation. 2020;17(1):1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarius S, Pellkofer H, Siebert N, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation. 2020;17(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA neurology. 2020;77(2):257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sechi E, Krecke KN, Pittock SJ, et al. Frequency and characteristics of MRI-negative myelitis associated with MOG autoantibodies. Multiple Sclerosis Journal. 2020:1352458520907900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data used for this study are available from the corresponding authors on reasonable request.