

Abstract
Cancer immunotherapy has entered a new era with the recent introduction of genetically engineered T cells that express chimeric antigen receptors (CARs) capable of recognizing and destroying tumor cells. Several clinical trials in patients with relapsed or refractory B cell malignancies have demonstrated complete remission rates ranging from 50–90%, with long term data suggestive of a possible curative response. CAR T cell therapy is currently under investigation for earlier use in these disease processes and in various other solid and liquid tumors.
CAR-T cell therapy is associated with a unique post-infusion toxicity profile including cytokine release syndrome and neurotoxicity. These toxicities are usually reversible but can be fatal, requiring close vigilance and prompt treatment often in an Intensive Care Unit setting. CAR T cell therapy is currently restricted to designated centers possessing expertise in acute toxicity management, but wider use is likely if early therapeutic successes are replicated. As perioperative and critical care physicians, anesthesiologists may encounter such patients in the perioperative or ICU setting and should become familiar with this unique and novel therapeutic modality capable of causing extreme cardiovascular and respiratory compromise. This review will describe the immunobiology of CAR-T cells, their relevance to cancer treatment, clinical aspects of their therapeutic use in cancer chemotherapy, toxicities related to CAR T cell use, and their therapeutic management.
Keywords: Immunotherapy, Adoptive/adverse effects, Receptors, Antigen, T-cell/therapeutic use, Cytokines/secretion
What are CAR-T cells?
A T-lymphocyte, sometimes referred to as a T-cell, is a lymphocyte subtype that plays a central role in cell-mediated immunity. As opposed to other lymphocytes, such as B-lymphocytes and natural killer cells, each T-cell expresses a unique antigen receptor on their surface.
CAR-T cells (Chimeric Antigen Receptor T cells) are T-lymphocytes that have been genetically manipulated to express T-cell receptors (TCR) that can recognize tumor-specific antigenes. These receptors are “chimeric” because they contain all the necessary components to activate the lymphocyte, bypassing the need for simultaneous stimulation of multiple additional co-receptors. Once the CAR-T cell is activated, it destroys the tumor cell through secretion of toxic granules and recruitment of other components of the immune system to the tumor site. Clinically, this strategy is remarkably successful at eradicating some types of hematologic malignancies and is currently being studied for other cancer types.
To understand how CAR T cells work a review of “wild-type” T cell function is helpful. Normal T cells recognize antigens presented to them by antigen presenting cells (APCs). The presentation of antigen to T-cell is the foundation for the cellular adaptive immune system. APCs are a heterogenous group of immunological cells (e.g. macrophages, Langerhans cells, dentritic cells) that can process, and display antigens coupled with a Major Histocompatibility Complex (MHC) molecule. Cancer cells can also express MHC molecules on their surface membranes. Recognition of the antigen by the T-cell is, however, not enough to induce an immune response. For the native T-cell to become “armed and activated”, it also requires co-stimulation by other immune cells to trigger cytokine release, cytotoxic activity, and stimulate proliferation (Figure 1). T-cell receptor activation in the absence of a co-stimulation signal leads to anergy – a state of hibernation and eventual T-cell death. To actively avoid detection by the immune system, tumors employ several techniques including secreting inhibitory cytokines, under-expressing MHC molecules on tumor cell surface, and disabling internal antigen processing mechanisms7–9.
Figure 1.
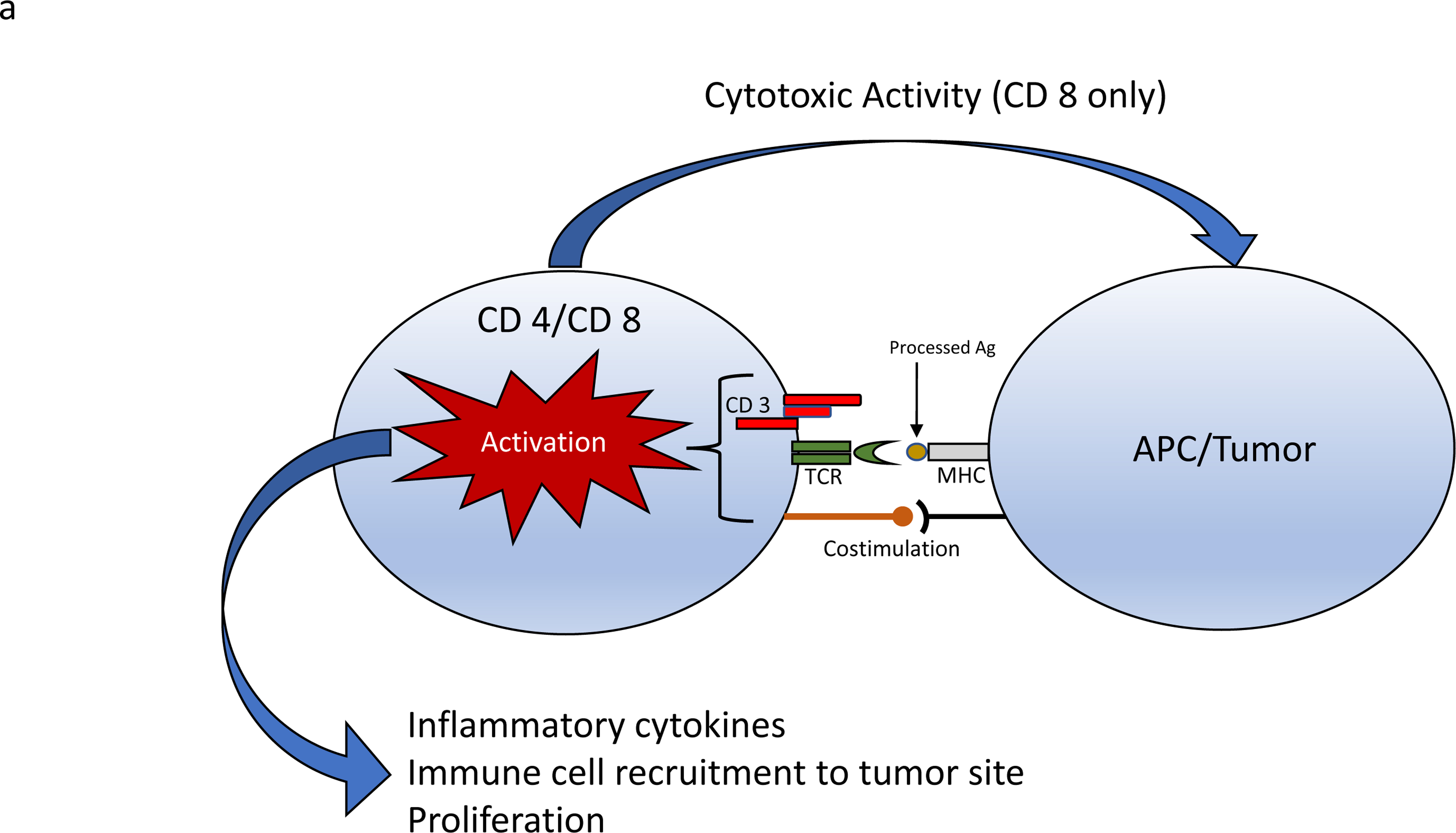
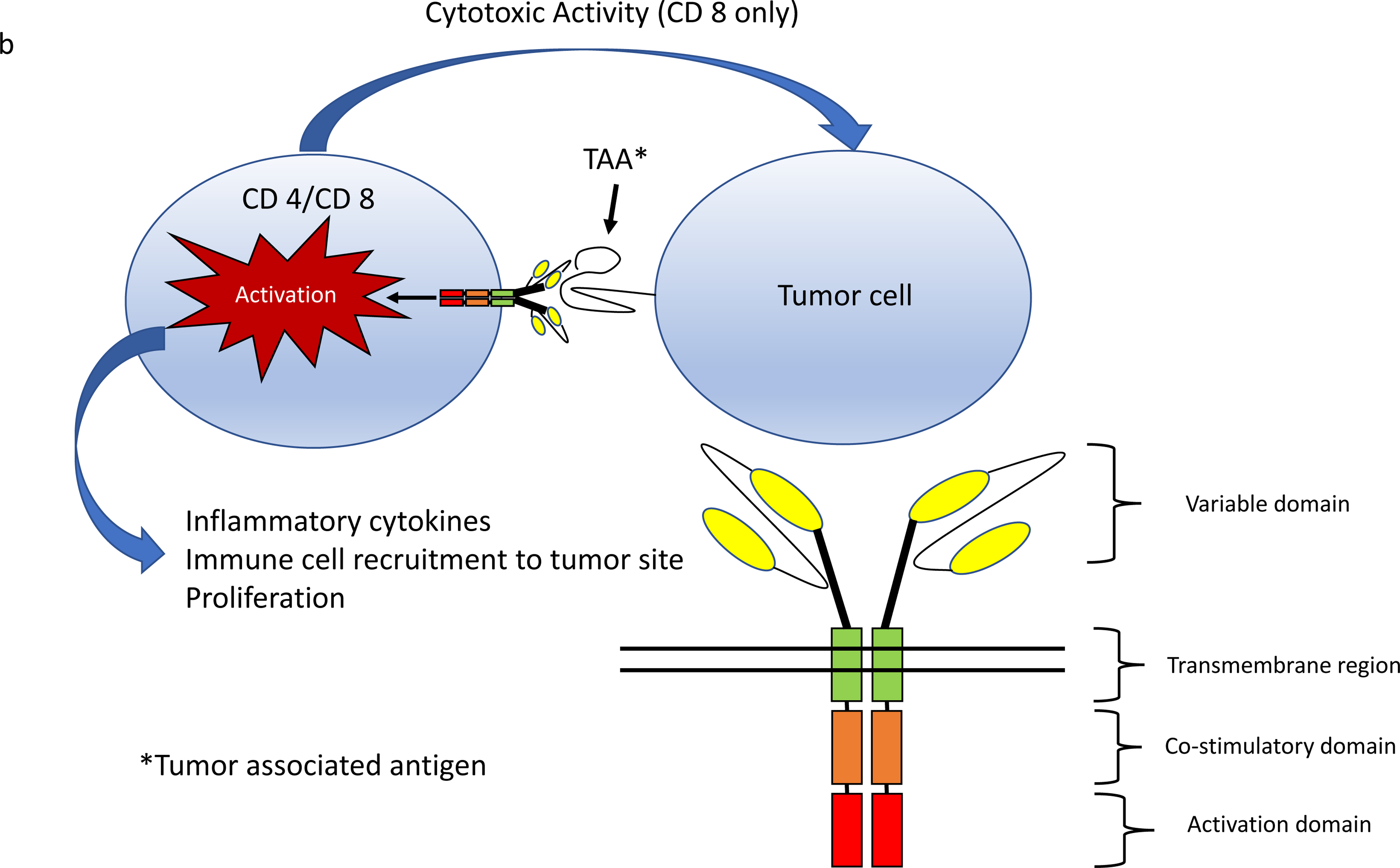
a) T cell receptors recognize antigens presented in MHC molecules by APCs or target cells (tumor). A T cell receptor activation complex is formed in conjunction with CD3. Additional co-stimulation is required for T cell activation leading to proliferation and cytokine release. b) CAR T cells express genetically engineered chimeric receptors capable of recognizing tumor associated antigens independent of MHC and co-stimulation. MHC=major histocompatibility complex, APCs= Antigen presenting cells
To bypass these evasive techniques, cellular engineers have fused various components of the T cell activation complex into a single chain receptor capable of inducing both T cell activation, costimulation and proliferation in response to tumor antigen with high specificity and independent of MHC coupling (Figure 1)10,11. CAR T cells are T CD 4+ and CD 8+ T lymphocytes that have been genetically engineered to express these multifunctional receptors on their surface.
CARs contain at a minimum an antigen recognition moiety capable of recognizing tumor antigen, a hinge and transmembrane segment connecting the extracellular components to the cytoplasmic elements, and an intracellular domain that triggers signaling mechanisms leading to T cell activation (Figure 1). The single chain variable fragment of immunoglobulin is commonly used for antigen recognition due to its high binding specificity, and the ζ chain homodimer of CD 3 containing immunoreceptor tyrosine-based activation motifs acts as a built in T-Cell activation moiety.
Early CAR designs failed to sufficiently activate and co-stimulate T-cells in response to antigen12. Co-stimulatory domains were thus built into modern constructs including CD 28, 4–1BB, and OX-40 13. These “second generation” CARs were the first to demonstrate tumor eradication in animal models 14. Modern CAR constructs contain one or more co-stimulatory domains to improve T-cell activation and persistence 15.
Once CAR T-cells are activated, they rapidly proliferate and release inflammatory cytokines, in turn recruiting other immune cells such as macrophages, monocytes and natural killer cells to the tumor site. CD 8 + CAR T-cells also exert cytotoxic effects by releasing granzyme and perforin granules, and directly stimulating apoptosis via fas/fas-L and TNF/TNF-R pathways that lead to tumor cell destruction16.
Identifying suitable tumor targets is a key aspect of CAR construction. An ideal target antigen is abundantly present at the surface of tumor cells and only minimally present or absent in healthy tissue to mitigate non-tumor effects. The CD 19 CAR was constructed with this approach because the CD 19 antigen is highly expressed in a variety of B cell malignancies. In mouse models, targeting CD 19 produced a B cell aplasia considered to be manageable with immunoglobulins if time-limited and potentially advantageous in preventing the formation of anti-CAR antibodies 9.
On target non tumor effects have been observed in humans. In a 2010 case report, administration of CAR T cells designed to target the tumor associated antigen ERBB2 caused immediate respiratory failure from pulmonary edema with death 5 days later. Serum samples suggested a cytokine storm localized to the lung, possibly due to the presence of low levels of ERBB2 on lung epithelial cells 17.
How are CAR T cells created and administered to patients?
CAR T cells are manufactured by harvesting autologous or allogenic peripheral blood lymphocytes via leukocyte apheresis and transfecting them ex-vivo with genes that encode the desired chimeric receptor. This step often represents the first contact between the patient and anesthesia services, as sedation is often requested for leukophereses catheter placement. A variety of different transfection techniques have been used including viral vectors (adenovirus, lentivirus, retrovirus) and non-viral systems such as transposons8. Transfected cells are then expanded in the laboratory utilizing various methods to stimulate cellular division such as artificial antigen presenting cells expressing CD 80 and stimulating cytokines such as IL-1514.
Once cells are ready the patient is prepared. Conditioning lymphodepleting chemotherapy typically with cyclophosphamide and/or fludarabine is then given to the patient days three to five days prior to CAR T-cell administration. This pretreatment potentiates the therapeutic effect of CAR T cells by eliminating regulatory immune cell populations which may constrain CAR T-cell expansion. CAR T-cells are then infused into the patient with or without concomitant IL 2 or supplemental immunotherapy depending on the protocol.
Outcomes to date from CAR T cell therapy
By far the most successful and widely studied CAR T-cell products in clinical trials target CD 19 in acute leukemias and non-Hodgkin’s lymphomas18–35. Response rates range from 50–90% in large trials focusing on patients with relapsed or refractory disease even after hematopoietic stem cell transplantation36,37. Long term follow-up suggests that CAR T therapy may be curative for some patients 25,32,35,36. Both Tisagenlecleucel and Axicabtagene ciloleucel, the products commercially available today, are anti-CD 19 CAR T-cells indicated for the treatment of relapsed or refractory B-ALL in pediatrics and young adults and B cell lymphoma in adults, respectively 38–40. Other hematologic malignancies treated with CAR T-cells in humans include chronic lymphocytic leukemia41 and multiple myeloma42 with less robust results. Trials are currently underway to assess the safety and efficacy of CARs in the treatment of glioblastoma, mesothelioma, breast and ovarian carcinoma 16,36.
At the time of manuscript preparation several hundred active clinical trials involving CAR T-cell therapy are registered in clinicaltrials.gov. An abundance of clinical data is thus likely to emerge in the coming months to years. Several trials address obstacles identified in the treatment of solid tumors by including concurrent administration of synergistic agents such as immune checkpoint inhibitors, incorporating CARs that simultaneously target multiple tumor associated antigens, and improving recruitment of other components of the immune system to attack antigen-negative cancer cells. As such, the indications for and use of CAR T-cell therapy may expand to the community hospital setting.
Toxicities from CAR T Therapy
CAR T cell therapy is associated with dangerous and potentially life-threatening complications affecting all major organ systems (Table 1). This toxicity profile remains a major barrier towards broader utilization of CAR T-cell strategies and has led to the early termination of large clinical trials of some CAR constructs. CAR T-cell toxicities include an exaggerated immune response from T cell activation, on-target off-tumor effects such as B cell aplasia seen with CD 19 CARs, and pulmonary edema with ERBB2 CARs.
Table 1.
Toxicities from CAR T cell therapy
| Constitutional | Fevers |
| Malaise | |
| Arthralgias | |
| Myalgias | |
| CNS | Headache |
| Encephalopathy | |
| Aphasia (expressive and receptive) | |
| Tremors | |
| Myoclonus | |
| Paresis | |
| Seizures | |
| Cerebral edema | |
| Cerebral Herniation | |
| Cardiovascular | Tachycardia |
| Arrhythmias | |
| Heart block | |
| Cardiomyopathy | |
| Troponemia | |
| Hypotension | |
| Refractory vasoplegia/shock | |
| Pulmonary | Hypoxia |
| Tachypnea | |
| Bronchospasm | |
| Pulmonary edema | |
| Pleural effusions | |
| ARDS | |
| Renal/Electrolytes | Hyponatremia |
| Hypo/Hyperkalemia | |
| Hypophosphatemia | |
| Hypomagnesemia | |
| AKI/ATN | |
| Oliguria | |
| Renal Failure requiring CRRT | |
| GI | Nausea/vomiting |
| Enterocolitis | |
| Cholangitis | |
| Hyperbilirubinemia | |
| Transaminitis | |
| GI hemorrhage | |
| Hematologic | Neutropenia |
| Anemia | |
| Thrombocytopenia | |
| Hyperferritinemia | |
| Hypofibrinogenemia | |
| DIC | |
| HLH/MAS | |
| Other | Anaphylaxis |
| Tumor Lysis Syndrome | |
| Infection |
Cytokine release syndrome (CRS), seen in 70–94% of patients, is the most common serious side effect of CAR T-cell therapy33–35,43. The median onset of CRS is two days following CAR T-cell administration, but symptoms can be delayed by up to 10–14 days43,44. This clinical syndrome can rapidly progress from fever, tachycardia, tachypnea and hypoxia to hypotension, coagulopathy, hypoalbuminemia, hypoproteinemia, respiratory failure, refractory shock and multiorgan failure37,43,44. The exact pathophysiological mechanism is not known, though aberrant activation of the vascular endothelial system likely plays a significant role. Activated CAR T-cells, monocytes, myeloid and peripheral tissue cells cause release of pro-inflammatory cytokines such as IL-6, TNF α and INF γ which lead to endothelial activation via modulation of the angiopoietin (Ang)-TIE2 axis and release of nitric oxide43,45,46. This activation in turn leads to loss of vascular integrity, capillary leak, consumptive coagulopathy and vascular smooth muscle dysfunction. Serum biomarkers of endothelial activation such as von Willebrand Factor (VWF), Ang-2, as well as Ang-2:Ang-1 ratio are elevated in patients with severe CRS, which supports this hypothesis43,45.
Elevation of other pro-inflammatory cytokines including IL-1, IL-2, IL-6, IL-10, TNF, IFN, MCP-1, and gp 130 have also been observed in severe CRS34,43. Peak serum IL-6 concentrations correlate positively with CRS severity and anti-IL 6 therapies effectively reverse CRS symptomatology 32,37,44. IL-6 exerts its effect either via “cis-signaling” through membrane bound IL-6 receptors or “trans-signaling” through soluble IL-6 receptors (sIL6r) that interact with membrane bound gp 130 receptors leading to activation of the JAK/STAT signaling pathway. Circulating IL-6/sIL6r complexes are cleared when bound to peripheral gp 130 through tissue uptake37,47. CAR T-cells do not directly produce IL-6 and neither elevated IL-6 levels nor CRS are required for an effective anti-tumor response47.
Treatment of CRS is mostly supportive. The IL-6 receptor antagonist Tocilizumab is the only approved agent for the treatment of CRS. Evidence to date suggests that treatment of CRS with Tocilizumab does not affect CAR T-cell efficacy48. Corticosteroids can also be effective at mitigating the exaggerated immune response but their effect on CAR T-cell function is less certain37,44.
Patients with mild CRS initially feature fever and tachycardia at presentation. Severe CRS can develop rapidly, however, often requiring treatment in an intensive care unit (ICU) for institution of vasoactive and ionotropic agents and aggressive respiratory support 44. It is not yet possible to predict which patients will develop mild or severe CRS, however several host and treatment risk factors have been identified. Patient factors include high disease burden in the bone marrow, tumor type (ALL being most prominent), presence of thrombocytopenia, and elevated markers of endothelial activation prior to treatment with conditioning chemotherapy. Treatment factors include higher CAR T cell dose, manufacturing techniques and choice of conditioning chemotherapy 35,43,47.
In some case of severe CRS, laboratory data suggestive of hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) may also be seen. HLH presents as an acute febrile illness with elevated serum ferritin levels, cytopenias in multiple lineages and coagulopathy37,47. The presence of HLH in a patient that has received CAR T-cell therapy suggests a worse prognosis. It is unclear if HLH represents a separate toxic entity or exists within the clinical syndrome of exaggerated immune activation leading to a massive systemic inflammation. Marrow examination in several patients exhibiting these laboratory abnormalities was not consistent with excessive hemophagocytosis, a non-specific finding in HLH/MAS 47. Peripheral blood examination also does not demonstrate presence of red blood cell fragmentation in the microcirculation43. Thus, while these additional features may be of prognostic significance, they do not affect our approach or management of these patients.
Neurotoxicity is a second, distinct adverse effect of CAR T cell therapy. This complication typically manifests as an encephalopathy with or without expressive and/or receptive aphasia44–47. The median time of presentation is about 5 days post infusion, and median duration is around 10 days33. Neuroimaging including head CT and MRI usually show non-specific findings such as diffuse vasogenic edema46 and EEG testing typically features patterns of diffuse slowing. CSF analysis of patients with neurotoxicity show elevated protein, increased white blood cell count (including CAR T-cells and myeloid cells), and elevated levels of inflammatory cytokines higher in some patients than serum concentrations suggestive of blood-brain barrier disruption with concomitant local cytokine production 45,46. Patients can progress to obtundation, seizure activity, status epilepticus, increased ICP and brain herniation44.
The mechanisms of CAR T-cell induced neurotoxicity are unknown. Patient risk factors associated with severe neurotoxicity include high disease burden and high peak CAR T cell expansion. Neurotoxicity is almost always preceded by CRS46. Much like in CRS, markers of endothelial activation such as vWF, Ang 2 and Ang 2:Ang 1 ratio are often elevated in patients who experience severe neurotoxicity 45. Autopsy tissue from patients who died from severe neurotoxicity showed multifocal microhemorrhages, platelet microthrombi and disrupted endothelium with reactive microglia in a perivascular distribution46. CAR T-cells infiltrate the brain parenchyma but the presence of CAR T-cells in the brain has not correlated with severity of neurotoxicity46. Tocilizumab and steroids have been used to ameliorate symptoms, although they are less effective at reversing symptoms of neurotoxicity than symptoms of CRS33,45. In fact, use of tocilizumab increases serum IL-6 concentration by interfering with IL-6 clearance through uptake in peripheral tissues37. Tocilizumab does not readily penetrate the central nervous system (CNS) when administered systemically 49. Thus, its use in patients experiencing neurotoxicity may be detrimental46,50.
The most severe manifestations of neurotoxicity include refractory seizure activity and life-threatening cerebral edema44–46. Multiple fatalities from brain herniation lead to the early termination of the 2016 anti-CD19 CAR ROCKET trial37.
The grading of CRS and neurotoxicity severity is inconsistent in existing literature. Various scales have been proposed, with the most widely adopted that of Lee et al 51. Recently, the National Cancer Institute convened a meeting of key clinicians and scientists with extensive clinical experience to develop a universal grading system that is clinically relevant and easily applied using bedside evaluation and chart review (Table 2)52.
Tabel 2.
Consensus grading of CRS and Neurotoxicity
| Toxicity | Features | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|---|
| CRS | |||||
| Fever | Present | Present | Present | Present | |
| Hypoxia | requiring low FiO2 | requiring high FiO2 | requiring positive pressure ventilation | ||
| Hypotension | Fluid responsive | requiring one vasopressor | requiring multiple vasopressors | ||
| Neurotoxicity | |||||
| SCAN SCORE* | SCAN 7–9 | SCAN 3–6 | SCAN 0–2 | Unable to perform | |
| Level of Consciousness | normal | Mild lethargy | Severely lethargic | Obtunded/comatose | |
| Weakness | Deep focal motor weakness | ||||
| Seizures | Any seizure (focal or generalized) resolving with intervention | Life-threatening prolonged seizures with no return to baseline | |||
| Cerebral Edema | Focal edema or hemorrhage on neuroimaging | Decorticate/decerebrate posturing Cushing’s triad Diffuse cerebral edema on neuroimaging |
|||
Orientation to year, month, city, hospital: 4 points; Name 3 objects: 3 points; Following complex commands: 1 point; Ability to write a standard sentence: 1 point; Attention: Count backwards from 10: 1 point52
Other toxicities associated with CAR T-cell therapy include prolonged cytopenias, anaphylaxis, tumor lysis syndrome, infections, and toxicities related to on target-off tumor effect seen in various CAR T-cell constructs37,47. Infectious complications are common and can be fatal as most patients are neutropenic44.
Considerations in acute care and perioperative management of CAR T-cell patients
Our adult oncologic ICU has been treating patients with CAR T-cell associated toxicities for the past seven years and our approach has evolved with experience. Febrile episodes can present as early as the day of infusion and usually coincide with a period of rapid CAR T-cell expansion. All patients are cultured (blood, urine, sputum) and covered with broad spectrum antimicrobials until an infectious etiology can be ruled out and neutropenia resolves. Antipyretics such as acetaminophen and non-steroidal anti-inflammatory agents are used to treat CRS mediated fevers. External cooling is useful in refractory cases though it can be quite uncomfortable for patients. Fever is usually accompanied or preceded by tachycardia which may not be tolerated in patients with cardiovascular disease and can lead to arrhythmias, ventricular dysfunction and serum troponin elevation. Prolonged fevers can lead to dehydration and electrolyte depletion. All patients are started on maintenance crystalloids and any electrolyte abnormalities are promptly corrected. As CRS progresses, capillary leak from the pulmonary bed leads to pulmonary edema which manifests clinically as an increase in oxygen requirement, worsening congestion, and/or new pleural effusions. Once patients develop pulmonary congestion, most patients progress to hypotension from vasodilation and loss of preload from capillary leak into interstitial spaces. Initial management including fluid resuscitation is similar to that for sepsis although caution is warranted as fluid administration can quickly lead to respiratory compromise. We limit fluid boluses to less than 30 ml/kg and discontinue them with any sign of worsening oxygenation opting at that point to initiate vasopressor agents.
We consider Tocilizumab appropriate for patients who develop persistent fevers for longer than 72 hours that respond poorly to antipyretics or those who develop hypotension and worsening oxygenation with repeated fluid boluses. If repeated doses of tocilizumab are required, patients are supplemented with moderate dose corticosteroids (dexamethasone 10 mg every 6–8 hours) to help modulate the immune response.
Patients who develop respiratory failure despite these precautions may require endotracheal intubation and mechanical ventilation. However, hemodynamic management in the peri-intubation period can be challenging as CRS-induced shock is often refractory to vasoconstrictor and fluid therapy. As a result, because CRS is a transient phenomenon that self-terminates, we prefer when possible to support patients with alternative modalities of high oxygen delivery such as high flow nasal cannula, non-rebreather masks or non-invasive positive pressure ventilation. If endotracheal intubation and mechanical ventilation become necessary, induction agents with favorable hemodynamic profiles are preferred. In our experience, epinephrine can be an effective agent for respiratory and hemodynamic support in severe CRS as it treats prominent features of bronchospasm, vasodilatory shock and ventricular dysfunction. Severe hemodynamic collapse warrants escalation of immune suppressive agents including pulse dose methylprednisolone (1 gram up to every 8 hours has been used) and off-label use of the free IL-6 binder Siltuximab (11 mg/kg one time dose), the IL-1 receptor antagonist Anakinra50, intravenous immunoglobulin, lymphocytic agents such as cyclophosphamide, cytokine hemofiltration, or suicide gene activation if present in the CAR T-cell construct. Clinicians should be aware that pulse dose steroids may precipitate bradycardia and hypothermia. Patients may progress to shock refractory to multiple vasopressors, multiorgan failure, coagulopathy and death despite maximum support.
The onset of neurotoxicity symptoms typically coincides with peak or resolving CRS symptomatology. Early signs of neurologic dysfunction are non-specific for CAR T-cell mediated toxicity and include lethargy, confusion and/or verbal deficits. We avoid all neuroleptic or sedative agents in CAR T-cell patients when possible as they may obfuscate neurologic exams. Diagnostic imaging is obtained to rule out bleeding or stroke. A standardized objective neurotoxicity scoring system is used for repeated assessments of mental capacity (Table 2). As symptoms progress, specific features characteristic of CAR T-cell mediated neurotoxicity begin to manifest. These features range from mild word finding difficulty to marked expressive and receptive aphasia in awake and alert patients. These are not life threatening but require moderately aggressive interventions with corticosteroids as above. Antiepileptic agents (Levetiracetam 750 mg every 12 hours) are initiated prophylactically. Lumbar puncture is performed when possible to measure opening pressure, protein level and cell counts, and to rule out infectious etiologies. The appropriate management of concurrent CRS and neurotoxicity is controversial, with some groups advocating continued use of Tocilizumab to ameliorate CRS symptomatology. However, emerging evidence from animal models and clinical trials suggests this approach may lead to worsening neurotoxicity presumably by increasing circulating IL-6 levels while not sufficiently penetrating the CNS50. Therefore, we recommend that any manifestation of neurologic symptoms should be treated with CNS penetrating steroids such as dexamethasone and Tocilizumab should preferentially be avoided.
Severe neurotoxicity progresses with the development of seizures without return to neurologic baseline, obtundation, signs of increased ICP, evidence of blood brain barrier disruption, and posturing. Treatment of these features requires accelerating doses of immune suppressive agents as in severe CRS above (with the exclusion of additional Tocilizumab) until clinical response is noted to avoid the catastrophic progression of cerebral edema. Strategies for lowering intracranial pressure include head of bed elevation, hyperventilation, and osmolar therapy. Patients with severe neurotoxicity are continuously monitored with hourly neurologic exams and neurotoxicity scoring, serial funduscopic exams, and continuous EEG. We opt not to use invasive monitoring of intracranial pressure, rather rely on serial neurologic assessments, opening pressure during lumbar punctures, and imaging findings to guide our management. Patients may require endotracheal intubation and mechanical ventilation for airway protection due to loss of mental status. Propofol is the preferred sedative agent in these patients when required as it allows rapid discontinuation for neurologic assessments and helps suppress seizure activity in conjunction with anti-epileptic agents. Despite maximum support including treatment with pulse dose corticosteroids as above, death from cerebral herniation can occur.
When CAR T-cell patients require anesthetics, it is crucial that the anesthesia team understand the timing, indication and type of CAR T cell product administered. Any elective or invasive procedure should be avoided in the setting of coagulopathy or refractory cytopenias due to excessive risk of bleeding and infection. Corticosteroids should never be administered without consultation with the oncology team as they may affect T-cell function as outlined above. The presence and extent of CRS and neurotoxicity should be noted when developing an anesthetic plan, and anesthesiologists should be familiar with treatment modalities that have been instituted including anti-IL-6 therapies, corticosteroids, and anticonvulsants. Vasopressors and ionotropic agents should be readily available, as well as rapid escalation of respiratory support including mechanical ventilation. Increased intracranial pressure precautions should be assumed when severe neurotoxicity is present, particularly in patients that are obtunded or display epileptiform activity on EEG.
As discussed above, CAR T-cell products are constructed differently depending on manufacturer and indication. Both commercially available and experimental products used today are second through fourth generation CARs. From our experience, each product exhibits a unique toxicity profile – some relatively benign, some with more prominent CRS, and others with profound neurotoxicity. Factors such as pre-treatment disease burden and tumor type are considered when developing management strategies so that symptomatology can be ameliorated while not compromising the benefit of this potentially curative therapy.
Little evidence informs the appropriate use of targeted therapies and corticosteroids in treating CAR T-cell related toxicities. Well-designed prospective clinical trials are needed to answer important questions as they relate to treatment efficacy and interference with CAR T-cell anti-tumor effect. Clarifying the mechanisms leading to the development of CRS and neurotoxicity will help in the development of more specific therapies. For example, studies have suggested that long lasting response positively correlates with a high ratio of CAR T-cell expansion to tumor burden but it is unclear if long term CAR T cell persistence is requisite for durable remission33–35. The effect of our interventions on peak expansion and persistence of CAR T-cells is also not known. Though treatment protocols for managing toxicities have been advocated by various manufacturers and institutions including ours, a simple algorithmic approach for all CAR T-cell related toxicities does not sufficiently encapsulate the factors that must be accounted for when managing these patients. Ultimately, until more data is available, the best management decisions are made at the bedside by a dedicated multidisciplinary team of clinicians well-versed in the features of a given CAR T-cell product and its unique side effect profile.
Conclusion
Two CAR T-cell products are commercially available and highly effective for the treatment of some liquid tumors, and others will follow as indications expand. As a result, the number of patients afflicted by CAR T-cell toxicities will rapidly increase in the coming years. Anesthesiologists and intensivists who may care for patients receiving CAR T therapy should familiarize themselves with these novel agents as they will play a major role in their institution and expansion into hospitals across the general medical community.
Footnotes
Conflicts of Interest: None
References:
- 1.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 1988;319:1676–80. [DOI] [PubMed] [Google Scholar]
- 2.Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J Exp Med 1955;102:157–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurnick JT, Kradin RL, Blumberg R, Schneeberger EE, Boyle LA. Functional characterization of T lymphocytes propagated from human lung carcinomas. Clin Immunol Immunopathol 1986;38:367–80. [DOI] [PubMed] [Google Scholar]
- 4.Weiden PL, Flournoy N, Thomas ED, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med 1979;300:1068–73. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg S Lymphokine-activated killer cells: a new approach to immunotherapy of cancer. J Natl Cancer Inst 1985;75:595–603. [PubMed] [Google Scholar]
- 6.Rosenberg SA, Mule JJ. Immunotherapy of cancer with lymphokine-activated killer cells and recombinant interleukin-2. Surgery 1985;98:437–44. [PubMed] [Google Scholar]
- 7.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer 2003;3:35–45. [DOI] [PubMed] [Google Scholar]
- 9.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature 2017;545:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 1993;90:720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989;86:10024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med 1995;181:1653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009;21:215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 2003;9:279–86. [DOI] [PubMed] [Google Scholar]
- 15.Park JH, Brentjens RJ. Are all chimeric antigen receptors created equal? J Clin Oncol 2015;33:651–3. [DOI] [PubMed] [Google Scholar]
- 16.Yu S, Li A, Liu Q, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol 2017;10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116:4099–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011;365:725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013;368:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 2013;122:2965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013;122:4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014;6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385:517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016;8:355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brudno JN, Somerville RP, Shi V, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol 2016;34:1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126:2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kebriaei P, Singh H, Huls MH, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest 2016;126:3363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 2017;377:2545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377:2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JH, Riviere I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 2018;15:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Food and Drug Administration. FDA approves tisagenlecleucel for adults with relapsed or refractory large B-cell lymphoma. United States Food and Drug Administration. Available from https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm606540.htm.) (Accessed 25 October 2018) [Google Scholar]
- 39.Food and Drug Administration. KIMRYAH package insert. Available from https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM573941.pdf.) (Accessed 25 October 2018)
- 40.Food and Drug Administration. Yescarta package insert. Available from https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM581226.pdf.) (Accessed 25 October 2018)
- 41.Turtle CJ, Hay KA, Hanafi LA, et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol 2017;35:3010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 2017;130:2594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017;130:2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gutierrez C, McEvoy C, Mead E, et al. Management of the Critically Ill Adult Chimeric Antigen Receptor-T Cell Therapy Patient: A Critical Care Perspective. Crit Care Med 2018;46:1402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santomasso BD, Park JH, Salloum D, et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov 2018;8:958–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gust J, Hay KA, Hanafi LA, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov 2017;7:1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Titov A, Petukhov A, Staliarova A, et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis 2018;9:897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh N, Hofmann TJ, Gershenson Z, et al. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 2017;19:867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nellan A, McCully CML, Cruz Garcia R, et al. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 2018;132:662–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018;24:739–48. [DOI] [PubMed] [Google Scholar]
- 51.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee DA, Santomasso B, Locke FL, et al. Consensus Grading for Chimeric Antigen Receptor T cell Induced Cytokine Release Syndrome and Neurological Toxicity. American Society for Blood and Marrow Transplantation (In press). [Google Scholar]