

INTRODUCTION
Model‐informed drug development (MIDD) tools including physiologically based pharmacokinetic (PBPK) modeling can improve the mechanistic understanding of a drug’s pharmacology and potentially translate into development efficiencies. This article summarizes viewpoints from the November 18, 2019, US Food and Drug Administration (FDA) public workshop titled “Development of Best Practices in Physiologically Based Pharmacokinetic Modeling to Support Clinical Pharmacology Regulatory Decision‐Making,” which discussed best practices in PBPK model development and evaluation, case studies, and research needs. 1
PBPK 360: THE STATE OF THE SCIENCE
The plenary session highlighted current scientific knowledge and gaps from key stakeholders in the PBPK field. Providing an academic perspective, Donald Mager (University of Buffalo) noted that successful PBPK analyses have clearly defined goals and use risk‐informed credibility assessments to guide decision‐making. Scientific challenges in PBPK modeling for drug development include increasing the granularity of organ, tissue, and cellular disposition; refining the models and system parameters for specific populations (e.g., organ impairment); and modeling complex biological therapeutics. Additional considerations in the PBPK modeling of biologics include the effects of receptor binding on a drug’s disposition, potential immunogenicity, and the structural and mechanistic diversity of biologic therapeutics. Some innovative PBPK models for biologics have improved our mechanistic understanding of complex immunotherapies, and further collaborations are critical to advance PBPK modeling for molecules with unique pharmacokinetic (PK) features.
Sharing an industry viewpoint, Stephen Hall (Eli Lilly) highlighted how collaboration through the International Consortium for Innovation and Quality in Pharmaceutical Development helps address questions regarding PBPK model validation. In industry, the application of PBPK occurs throughout drug development in a continuous learn and confirm cycle, including assessing risk during the discovery phase, characterizing the drug’s clinical pharmacology properties, and addressing labeling requirements in late phases. 2 Data from PBPK modeling can be included as part of a regulatory submission for certain areas of application, as illustrated by a PBPK model that helped characterize the drug interaction potential of abemaciclib and informed the label. Industry also uses PBPK modeling to help determine the effects of coadministration with food as well as the effect of hepatic impairment on drug PK, although these areas of application cannot yet replace clinical studies. Hall emphasized that the appropriateness of PBPK for a particular application should be determined on a case‐by‐case basis.
In the past 5 years, PBPK models in regulatory submissions have increased, noted Yaning Wang (FDA). Although the application of PBPK models to assess drug–drug interactions (DDIs) represent the majority of submissions, other areas of PBPK application have expanded. 3 The FDA supports the use of PBPK modeling through a regulatory guidance on submitting PBPK‐related information in regulatory applications and a recent white paper on a proposed framework to assess PBPK model credibility. 4 , 5 Furthermore, the FDA engages with industry on PBPK applications as part of the MIDD paired meeting program. 6 PBPK models submitted as part of a regulatory application contribute to the agency’s understanding of various areas of PBPK application that in turn improve the regulatory evaluation of future submissions and relevant policy development. Challenges in certain areas of PBPK application include a lack of understanding of system parameters, limited confidence in in vitro to in vivo extrapolation (IVIVE), and short review timelines. Early communication with regulatory agencies, accessible knowledge management platforms, and additional technical review staff can promote adoption of PBPK models in drug development.
FDA’S PROPOSED FRAMEWORK FOR CREDIBILITY ASSESSMENT
Colleen Kuemmel (FDA) highlighted the agency’s white paper titled “Consideration of a Credibility Assessment Framework in Model‐Informed Drug Development: Potential Application to Physiologically Based Pharmacokinetic Modeling and Simulation,” 5 which describes the application of an evidentiary framework to assess PBPK model credibility, that could standardize terminology and may offer a uniform approach to model evaluation. The framework was developed by the American Society of Mechanical Engineers and is currently used in medical device development. This framework is flexible and incorporates key model assessment principles, some of which are included in the European Medicines Agency general modeling and simulation framework 7 (Figure 1).
FIGURE 1.
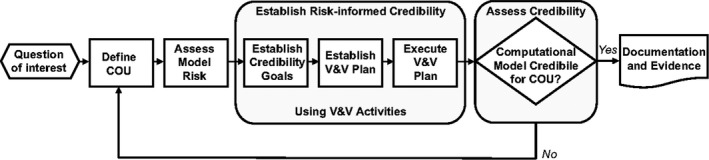
Proposed credibility assessment framework for physiologically based pharmacokinetic modeling and simulation 5 . Credibility assessment is outlined in a five‐step process that can be tailored for a particular physiologically based pharmacokinetic application. Terms defined within this framework include credibility, or the trust in the predictive capability of the model; verification, or the evaluation of the software; and validation, or the evaluation of the model. After identifying the overarching question of interest, it is critical to define the model's context of use (COU). Assessing model risk determines the necessary level of validation and verification (V&V) and allows credibility goals and a V&V plan to be established. If the model’s credibility is not acceptable, more data can be gathered, the influence of the model can be changed, or the COU can be revised. Of note, it requires a team of experts to apply the framework and assess the adequacy of the model
The question‐and‐answer panel discussion was moderated by Ping Zhao (Bill & Melinda Gates Foundation), who engaged with Colleen Kuemmel and panelists Susan Cole (UK Medicines and Healthcare products Regulatory Agency), Tina Morrison (FDA), Million Tegenge (FDA), Yuching Yang (FDA), and Liang Zhao (FDA) to gain insight into the applicability of the framework during drug development and regulatory review. The panelists agreed that the proposed FDA framework is readily applicable during drug development. Furthermore, the majority of the activities and concepts described in this framework are routinely implemented in the regulatory review of PBPK submissions. Providing a common language to discuss regulatory expectations can help facilitate productive discussions regarding modeling strategies early in drug development. To improve the framework, the panelists recommended real‐world use of the framework to develop a shared understanding of the various elements and ensure consistent application.
CASE STUDIES
Xinyuan Zhang (FDA) presented case studies on the application of PBPK to assess DDIs. For complex cytochrome P450 (CYP) 3A–mediated DDIs, reviewers use a stepwise process to determine the role of each enzyme/transporter in the drug’s absorption, distribution, metabolism, and excretion and if the model is supported by adequate PK and DDI studies as well as sensitivity analyses. A DDI study with a strong inducer tends to provide limited information for a PBPK model on investigational drugs that are sensitive CYP3A substrates and usually leads to an “avoid use” recommendation in the label; therefore, a DDI study with a moderate CYP3A inducer may be more informative for a drug development program. When deciding whether to incorporate metabolite parameters into a PBPK model, the sponsor should evaluate if metabolite exposure changes are clinically relevant and identify safety or efficacy concerns that could result from any proposed dose adjustments to mitigate drug interactions. PBPK models for specific populations such as pediatrics and geriatrics cannot yet replace clinical studies but may help optimize clinical study designs. Due to insufficient IVIVE for transporters, most PBPK analyses where the investigational drug is a transporter substrate or perpetrator are considered exploratory.
From the research perspective, Nina Isoherranen (University of Washington) described the development of a PBPK model of methamphetamine and its metabolite amphetamine to capture the effect of pH‐dependent renal clearance of these compounds on their plasma exposures. 8 During model development, an undocumented spike in amphetamine concentrations was simulated but not corroborated by observed data. Through model optimization, this spike could be eliminated by changing the methamphetamine’s liver Kp value; however, predicted Kp values used in the model diverged significantly from Kp values derived from human positron emission tomography imaging. By examining the structural model, the spike vanished if concentrations in the PBPK model were sampled from a peripheral venous sampling site rather than an arterial site. Therefore, a sampling location discrepancy between the model and observed samples appeared to account for the difference between the observed and predicted data. As such, the sampling site should match between PBPK simulations and experimental studies. Isoherranen noted that it is important to identify sensitive parameters and strategize sensitivity analyses for a particular model.
Providing an industry perspective, Jan Snoeys (Janssen) described how PBPK modeling facilitated the regulatory review of ibrutinib through increased understanding of its DDI potential and the effect of hepatic impairment on the drug’s PK. Key principles in PBPK modeling include clearly defining the model objective, verifying drug‐independent components, confirming with all relevant observed clinical data, and determining the accuracy needed for important decision‐making. For example, with validation from a clinical DDI study, there was high confidence in a PBPK model’s ability to predict DDIs with ibrutinib in fasted subjects with CYP3A inducers or inhibitors. A thorough mechanistic understanding of the known food effect on ibrutinib helped develop a model that could predict the effect of food on ibrutinib DDIs. However, there was low confidence in the PBPK model’s ability to predict the effect of hepatic impairment on the PK of ibrutinib. These PBPK models answered several key scientific questions during the FDA’s regulatory review in 2013 and contributed to labeling regarding DDIs.
KNOWLEDGE GAPS IN PBPK
The last session identified common scientific challenges and potential strategies to advance the field of PBPK. Iain Gardner (Certara‐Simcyp) noted that the applications of PBPK are expanding in drug development, including predicting drug response in specific populations, integrating quantitative systems pharmacology (QSP) models, and informing formulation design. Scientific gaps include the need for thorough validation of models for more COUs and data to better predict transporter DDIs. An open‐science approach including peer review and standardized methods to collect, curate, and analyze data can help advance the field.
Grace Fraczkiewicz (SimulationPlus) highlighted current knowledge gaps in physiologically based disease models, such as the inability to accurately measure in vivo local changes that can affect drug absorption. PBPK modeling cannot yet reliably predict direct food–drug interactions or account for nonoral dosage routes, the impact of excipients, and metabolism and transport in the administering tissue. However, PBPK models have been shown to predict DDIs with acid‐reducing agents, and commercial PBPK platforms have improved model quality, transparency, and reproducibility. Flexible models that combine PBPK and QSP approaches can help integrate the biochemistry of disease pathophysiology to a drug’s PK and pharmacodynamic effects.
Paul Seo (FDA) shared insights on knowledge gaps in the application of PBPK from a regulatory perspective. To ensure regulatory consistency, regulators should use a scientifically sound and logical framework to assess the suitability of PBPK models. In addition, modelers should adequately justify the use of certain data/approaches over others when creating a model for an intended use. Scientific advances are needed to better understand the mechanisms of excipient effects and the impact of the drug‐making processes on a model. The acceptance of PBPK modeling as a drug development tool has increased greatly in the past 2 decades and has the potential to continue to accelerate the pace of drug development.
Knowledge gaps are exacerbated by scientific practices that hinder the reproduction of results from published PBPK models, noted Marc Gastonguay (Metrum Research). A five‐pronged open‐science approach can expand the knowledge base of PBPK modeling and improve reproducibility through independent evaluation of PBPK models from the software; transparent and reproducible models that provide complete specifications; open provenance of derived model parameters; quality software and model development lifecycle management, including peer‐review; and community engagement in model verification and validation.
Tycho Heimbach (Novartis) provided examples of how PBPK modeling can supplement clinical trial data with limited PK data from patient groups. For example, a pediatric PBPK model adequately justified the use of body surface area dosing of nilotinib in children younger than 6 years of age, a population with limited PK data. In addition, PBPK simulations of an experimental drug were able to describe observed PK data from only two patients with severe hepatic impairment. Compilation of individual‐level clinical data across different drugs could improve the predictability of PBPK models for patients with limited PK data.
CONCLUSION
Effective application of PBPK modeling in drug development requires predictability and consistency in how the stakeholder communities view, apply, and evaluate this approach, noted Issam Zineh (FDA). This workshop brought together regulators, industry, academics, and platform developers to discuss the current challenges and knowledge gaps in PBPK modeling and propose solutions to advance the utility of PBPK in drug development (Table 1). These proposed solutions may serve as a starting point for future discussions and collaborations.
TABLE 1.
Proposed solutions to challenges and knowledge gaps in advancing the science of PBPK modeling
| Challenge | Proposed solution |
|---|---|
| Increase effectiveness/efficiency of regulatory review |
|
| Increase data acquisition, analysis, and transparency |
|
| Global harmonization of PBPK modeling in drug development |
|
COU, context of use; FDA, Food and Drug Administration; IND, investigational new drug; PBPK, physiologically based pharmacokinetic; V&V, validation and verification.
DISCLAIMER
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the US Food and Drug Administration, the Bill & Melinda Gates Foundation, the University of Washington, or Eli Lilly and Company. As an Associate Editor of CPT: Pharmacometrics & Systems Pharmacology, Ping Zhao was not involved in the review or decision process for this paper.
CONFLICT OF INTEREST
N.I. reports consultancy agreements with Boehringer‐Ingelheim and Xenon Pharmaceuticals and honoraria from the National Institutes of Health and is an Associate Editor of Clinical and Translational Science and Drug Metabolism and Disposition. All other authors declared no competing interests for this work.
Daphney Jean and Kunal Naik contributed equally to this work.
FUNDING
No funding was received for this work.
REFERENCES
- 1. US Food and Drug Administration . Development of best practices in physiologically based pharmacokinetic modeling to support clinical pharmacology regulatory decision‐making. https://www.fda.gov/drugs/news‐events‐human‐drugs/development‐best‐practices‐physiologically‐based‐pharmacokinetic‐modeling‐support‐clinical. Accessed May 15, 2021. [DOI] [PMC free article] [PubMed]
- 2. Jones HM, Chen Y, Gibson C, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Therapeut. 2015;97:247–262. [DOI] [PubMed] [Google Scholar]
- 3. Grimstein M, Yang Y, Zhang X, et al. Physiologically based pharmacokinetic modeling in regulatory science: An update from the U.S. Food and Drug Administration's Office of Clinical Pharmacology. J Pharmaceut Sci. 2019;108:21–25. [DOI] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration . Physiologically based pharmacokinetic analyses—format and content guidance for industry (September 2018). https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/physiologically‐based‐pharmacokinetic‐analyses‐format‐and‐content‐guidance‐industry. Accessed May 15, 2021.
- 5. Kuemmel C, Yang Y, Zhang X, et al. Consideration of a credibility assessment framework in model‐informed drug development: potential application to physiologically‐based pharmacokinetic modeling and simulation. CPT Pharmacomet Syst Pharmacol. 2020;9(1):21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. US Food and Drug Administration . Model‐informed drug development program. https://www.fda.gov/drugs/development‐resources/model‐informed‐drug‐development‐pilot‐program. Accessed May 15, 2021.
- 7. Manolis E, Rohou S, Hemmings R, Salmonson T, Karlsson M, Milligan PA. The role of modeling and simulation in development and registration of medicinal products: output from the EFPIA/EMA Modeling and Simulation Workshop. CPT Pharmacomet Syst Pharmacol. 2013;2(2):e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang W, Czuba LC, Isoherranen N. Mechanistic PBPK modeling of urine pH effect on renal and systemic disposition of methamphetamine and amphetamine. J Pharmacol Exp Therapeut. 2020;373(3):488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]