

Abstract
Evinacumab, an angiopoietin‐like protein 3 (ANGPTL3) inhibitor, has been shown to significantly reduce low‐density lipoprotein cholesterol (LDL‐C) in patients with homozygous familial hypercholesterolemia (HoFH). This work characterized the population pharmacokinetics (PK)/pharmacodynamics (PD) of evinacumab using pooled phase III clinical data. Total evinacumab PK were described by a two‐compartment model with combined linear and saturable (Michaelis–Menten) elimination, and first‐order absorption. At clinically relevant concentrations, plasma drug concentrations were mainly influenced by the linear clearance pathway. Although the maximum target‐mediated rate of elimination (Vmax) parameter for the saturable pathway was found to be positively related to baseline ANGPLTL3, variability in body weight contributed more to the variability in evinacumab exposure than variability in ANGPTL3. An effect of HoFH versus healthy volunteers on Vmax was also identified. Weight‐based dosing regimens resulted in consistent evinacumab exposure across weight ranges. An indirect exposure–response model adequately described the relationship between evinacumab and LDL‐C, where drug concentration is assumed to inhibit LDL‐C production. The final population PK/PD model included two nonclinically significant covariates (race and baseline body weight) on the maximum drug‐induced inhibitory effect (Imax) and one (baseline LDL‐C) on the evinacumab concentration inducing 50% of Imax (IC50). A smaller IC50 was observed in patients with higher baseline LDL‐C, suggesting greater sensitivity to treatment. Population exposure–response analysis permitted estimation of derived PD parameters and individual LDL‐C levels over time for patients with HoFH. The model accurately predicted the proportion of patients with HoFH achieving prespecified LDL‐C goals with evinacumab during the ELIPSE HoFH study, further supporting a dosing strategy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Evinacumab is an angiopoietin‐like protein 3 (ANGPTL3) inhibitor approved in the United States as an adjunct to other low‐density lipoprotein cholesterol (LDL‐C)–lowering therapies for the treatment of adult and pediatric patients aged 12 years and older with HoFH.
WHAT QUESTION DID THIS STUDY ADDRESS?
Can population pharmacokinetic (PK)/pharmacodynamic (PD) analyses using pooled phase III clinical data, including the development of an exposure–response model, be used to characterize the individual variability in treatment response to evinacumab in patients with HoFH?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The indirect exposure–response model adequately characterized the population PK/PD properties of evinacumab in patients with HoFH and permitted the estimation of individual LDL‐C levels over time following evinacumab administration. Moreover, the model accurately predicted the proportion of patients with HoFH achieving prespecified LDL‐C goals with evinacumab during the pivotal phase III ELIPSE HoFH study.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Exposure–response modeling may provide greater understanding of the variability in treatment response and guide evinacumab dose selections for future studies.
INTRODUCTION
Homozygous familial hypercholesterolemia (HoFH) is an autosomal dominant genetic disorder of lipid metabolism characterized by markedly elevated plasma levels of low‐density lipoprotein cholesterol (LDL‐C) from birth, leading to increased risk of premature atherosclerotic cardiovascular disease. Clinical manifestations of HoFH include myocardial infarction and ischemic stroke. 1 , 2 , 3 HoFH arises due to mutations in genes encoding key proteins involved in hepatocellular uptake and catabolism of low‐density lipoprotein (LDL). 4 Loss‐of‐function (LOF) mutations in the LDL receptor (LDLR) gene account for 90% of HoFH cases. 5 Less commonly, functional mutations in the apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and LDL protein receptor adaptor 1 (LDLRAP1) genes may be implicated. 5
Angiopoietin‐like protein 3 (ANGPTL3), a liver‐derived glycoprotein, plays a pivotal role in lipoprotein metabolism via potent inhibition of both lipoprotein lipase and endothelial lipase, resulting in reduced hydrolysis of triglycerides in triglyceride‐rich lipoproteins, and reduced hydrolysis of lipoprotein phospholipids, which ultimately leads to increased circulating plasma concentrations of triglycerides, high‐density lipoprotein cholesterol (HDL‐C), and LDL‐C. 6 , 7 , 8 , 9 , 10 , 11 Preclinical studies in LDLR knock‐out mice suggest that ANGPTL3 reduces LDL‐C independent of the LDLR. 12 Genetic studies have delineated that individuals with heterozygous LOF variants in ANGPTL3 have significantly reduced levels of plasma triglycerides, HDL‐C, and LDL‐C, with no associated adverse effects. 12 In addition, exome sequencing has shown that heterozygous carriers of LOF variants in ANGPTL3 have a 41% lower risk of coronary artery disease. 12 Thus, ANGPTL3 has emerged as a potential target for the treatment of hypercholesterolemia and hypertriglyceridemia, both of which are contributing factors to the development of cardiovascular disease.
Evinacumab is a recombinant, fully human monoclonal antibody generated using VelocImmunetechnology (Regeneron Pharmaceuticals, Inc.) that specifically inhibits ANGPTL3. 11 , 12 , 13 Evinacumab has been shown to reduce triglycerides, non‐HDL‐C, and LDL‐C in phase I studies comprising healthy volunteers (NCT01749878, NCT03146416), phase I studies comprising patients with hypertriglyceridemia (NCT02107872, NCT01749878), a phase II proof‐of‐concept study comprising patients with HoFH (NCT02265952), and a phase III study comprising patients with HoFH (NCT03399786). 14 , 15 , 16 In February 2021, evinacumab was approved by the US Food and Drug Administration as an adjunct to other LDL‐C‐lowering therapies for the treatment of adult and pediatric patients aged 12 years and older with HoFH. 17
The objectives of these population pharmacokinetics (PK) and PK/pharmcodynamics (PD) analyses using pooled clinical study data were to (1) estimate individual and population PK parameters of evinacumab, (2) estimate variability in PK parameters of evinacumab, (3) characterize clinically relevant covariates as potential sources of variability in PK parameters, (4) quantify the exposure–response relationship of evinacumab on LDL‐C reduction, and (5) characterize clinically relevant covariates as potential sources of variability in PD parameters.
METHODS
Study subjects
The population PK data set used to construct the population PK model were pooled from six evinacumab clinical studies and comprised healthy volunteers (n = 183) and patients with HoFH (n = 95; Table S1). In these clinical studies, evinacumab was administered intravenously (i.v.; 5 to 20 mg/kg, single dose or repeated once weekly [qw], once every 4 weeks [q4w], or once every 12 weeks) or subcutaneously (s.c.; 75 to 450 mg, single or repeated qw or once every 2 weeks) either alone or in combination with other lipid‐lowering therapies (LLTs). The PK analysis set included all subjects who had received any amount of study drug and who had at least one nonmissing total evinacumab measurement following the first dose of study drug or placebo. All studies were conducted in accordance with ethical principles originating from the Declaration of Helsinki and were consistent with the International Conference on Harmonization/Good Clinical Practices and applicable regulatory requirements.
Bioanalytical assays
Serum samples were analyzed for total evinacumab and total ANGPTL3 using validated enzyme‐linked immunosorbent assays. The assay to determine ANGPTL3 included an acid pretreatment of serum samples and used a rat anti‐ANGPTL3 monoclonal antibody as the capture reagent. Captured ANGPTL3 was detected using a sheep polyclonal antibody specific for human ANGPTL3. Both free ANGPTL3 and ANGPTL3 bound to target were measured. The lower limit of quantification (LLOQ) was 0.0195 mg/L in neat human serum.
The assay to determine total evinacumab included an acid pretreatment of the serum samples and used a mouse anti‐evinacumab monoclonal antibody as the capture reagent. Captured evinacumab was detected using a different noncompeting mouse anti‐evinacumab monoclonal antibody. Both free evinacumab and evinacumab bound to one or two molecules of ANGPTL3 were detected. The LLOQ was 0.078 mg/L neat human serum.
Modeling software
Population PK and PK/PD analyses were conducted using the first‐order conditional estimation with interaction method or Monte‐Carlo importance sampling assisted by mode a posteriori method as implemented in the NONMEM software system (version 7.3; ICON Development Solutions) aided by Perl‐speaks‐NONMEM (version 4.6.0; Uppsala University), Xpose 4.5.3, and Xpose 0.4.3 (Uppsala University). A pooled NONMEM‐ready data set was constructed using SAS (version 9.4; SAS Institute). R software (version 3.4.0; R Development Core Team; http://www.r‐project.org/) was used for the preprocessing and postprocessing of NONMEM, output creation of diagnostic plots, graphical visualization, and generation of statistical outputs. Simulations were performed using the R package mrgsolve (0.8.0 or later; Metrum Research Group). The variables used in the final NONMEM data set (Table S2), the NONMEM model code (Supplementary Text), and a sample of the NONMEM data set (Table S3) for the population PK analyses are provided.
Population PK model building
Initial structural model development began with the evaluation of a one‐compartment model with linear elimination. Additional structural models that were evaluated included two‐compartment or three‐compartment models, a target‐mediated drug disposition model, a model using the nonlinear/Michaelis–Menten elimination, and models with parallel linear and nonlinear elimination pathways. The final structural model was determined based on objective function value (OFV), goodness‐of‐fit (GOF) criteria, and a visual inspection of diagnostic plots. Residual error was modeled using a combined additive and proportional error model.
The final structural model was selected for covariate model building. Covariates evaluated as part of the full covariate model included demographic parameters (age, sex, body weight, and race), disease status (HoFH or healthy volunteer), baseline lipid parameters (LDL‐C), concomitant LLTs (PCSK9 inhibitors, statins, and LDL apheresis), baseline albumin, and baseline total ANGPTL3 concentrations. The relationship between empirical Bayesian estimates of PK parameters and patient covariates were screened graphically for relevance. Covariates were then evaluated for significance using a stepwise forward addition and a backward elimination procedure (change in OFV equivalent to p < 0.05 and p < 0.001, respectively). The final population PK parameters were re‐estimated accounting for covariate relationships. Clinical impact was defined as a change in the predicted model parameter from the median covariate value compared with the predicted parameter value at either the 2.5th percentile or the 97.5th percentile of the covariate values of at least 20%. The final population PK model was used to calculate the post hoc estimates of individual PK parameters.
Population PK model evaluation
Model verification was performed by examination of the GOF plots, including observed data (DV) versus predicted data (PRED); DV versus individual predicted data (IPRED); absolute value of weighted residual versus PRED; conditional weighted residuals (CWRES) versus time; and individual DV, PRED, and IPRED versus time. To evaluate predictive performance of the final PK model, visual predictive checks (VPCs) stratified by study, disease type, and administration route were performed. Replicate simulations (n = 1000) were performed using the final model and final population PK parameter estimates. Concentration–time profiles were plotted for the 2.5th, 50th, and 97.5th percentiles of the simulated data and overlaid with the 95% confidence intervals (CIs) of the 2.5th, 50th, and 97.5th percentiles of the observed data.
Population PK/PD model building
The population PK/PD model was developed using individual predicted PK parameters and observed PD. 18 Data were pooled from one phase II and two phase III studies of evinacumab (n = 95; patients with HoFH). Across these three studies, evinacumab dosing ranged from 15 to 20 mg/kg i.v. (single dose or repeated q4w) and 250 to 450 mg s.c. (single dose or repeated qw). Population PK/PD analysis was performed using individual PK parameters to predict the evinacumab profiles needed for the integration of the population PK/PD model. Based on an exploratory analysis and an understanding of the mechanism of action, an indirect PK/PD response model was developed to link decreasing LDL‐C production with serum evinacumab concentrations. The pharmacostatistical PK/PD model was a Type 1 indirect response model 19 and was parameterized with a zero‐order rate constant for production of LDL‐C, the maximum drug‐induced inhibitory effect (Imax), and the evinacumab concentration inducing 50% of Imax (IC50). Interindividual variability was modeled using a log‐normal distribution. Residual error was described using a combined additive and proportional error model.
Covariates were selected based on clinical relevance and biological plausibility, and tested using a stepwise approach. Covariates evaluated included demographic parameters (age, sex, body weight, and race), baseline lipid parameters (LDL‐C, fasting triglycerides), concomitant LLTs (PCSK9 inhibitors, statins, and LDL apheresis), and baseline total ANGPTL3 concentrations. Selected covariates were added sequentially to the model in a forward selection method and tested for significance. Backward elimination was performed to verify that the retained covariates were relevant in the final model. A significance level of α = 0.01 was used in the forward selection, and a significant level of α = 0.001 was used in the backward elimination. The population parameters were then re‐estimated considering the relationship with the covariates.
PK/PD model evaluation
Model verification was performed by examination of the GOF plots including DV versus PRED, DV versus IPRED, CWRES versus PRED, CWRES/individual DV/PRED/IPRED versus time, distribution plots for the empirical Bayes prediction of the interindividual random effect in a PK or PD parameter (η), scatterplots of the η correlations, and individual predicted concentrations, population predicted concentrations, and observed concentrations (overlaid) by subject. To verify that the final model adequately predicts both the central tendency and the variability of the observed data, VPCs were performed. For the VPCs, replicate simulations (n = 1000) were performed using the final model and final model parameters. LDL‐C concentration–time profiles were plotted for the 2.5th, 50th, and 97.5th percentiles of the simulated data and overlaid with the 95% CI of the 2.5th, 50th, and 97.5th percentiles of the observed data. Bootstrap methods were used to assess the robustness of the PK/PD model and the accuracy of the parameter estimates (indicated mainly by 95% CIs).
RESULTS
Population PK analysis
The PK of total evinacumab was described by a two‐compartment model with a combined linear and saturable (Michaelis–Menten) elimination and first‐order absorption (Figure 1). The intercompartment clearance was parameterized with central volume of distribution (second compartment) and intercompartmental rate constants. Interindividual variability was added to systemic clearance from the central compartment and peripheral volume of distribution using a log‐normal distribution. The bioavailability of evinacumab by the s.c. route was estimated to be 71%. An exploratory plot of observed evinacumab concentration versus time (Figure S1) suggested evinacumab has a long half‐life. In addition, exploratory frequency diagrams of categorical covariates and scatterplots of continuous covariates (Figures S2 and S3, respectively) suggested a typical distribution of all covariates, except for baseline LDL and baseline triglycerides.
FIGURE 1.
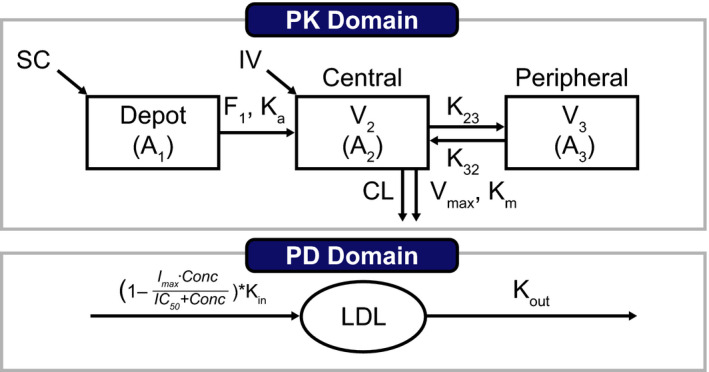
Schematic of the population PK/PD model for both i.v. and s.c. administration of evinacumab. The initial condition is described as baseline LDL‐C = kin/kout. PK equations: ; ; ; PK/PD equation: . A1, amount of evinacumab dosed via the subcutaneous route and subject to bioavailability; A2, amount of evinacumab in the central compartment; A3, amount of evinacumab in the peripheral compartment; C, patient time course of the drug concentration (PK) at time t predicted using empirical Bayes estimates from the population PK model; CL, plasma clearance; F, bioavailability; Conc, concentration of LDL‐C; Imax, maximal inhibitory effect; IC50, drug concentration to reach 50% of Imax; K23 and K32, intercompartmental rate constants; Ka, absorption rate constant; Kin, zero‐order constant of production of response (LDL‐C); Km, Michaelis–Menten constant; Kout, first‐order constant of loss of response (LDL‐C); IV, intravenous; LDL, amount in effect compartment; LDL‐C, low‐density lipoprotein cholesterol; PD, pharmacodynamics; PK, pharmacokinetics; SC, subcutaneous; V2, volume of distribution (central compartment); V3, volume of distribution (peripheral compartment); Vmax, maximum target‐mediated rate of elimination
Baseline covariates for patients included in the analysis are presented in Tables S4 and S5. Linear clearance and volume of distribution was dependent on weight, with an allometric relationship (a power model with exponents of 0.75 [fixed] and 0.875 [estimated], respectively). A fixed theoretical exponent of 0.75 was used for the power relationship between weight and clearance as it was unable to be estimated. In subjects with HoFH, the saturable elimination pathway maximum target‐mediated rate of elimination per unit time (Vmax) was reduced by 25% relative to healthy volunteers. Increases in ANGPTL3 concentrations predicted a higher Vmax. Variability in ANGPTL3, and the effect of HoFH on Vmax, contribute less to variability in exposure (area under the plasma concentration–time curve [AUC], maximum concentration [Cmax]) relative to variability in weight. A weight‐based dosing regimen (e.g., 15 mg/kg) results in consistent evinacumab exposure across a range of body weights for patients with HoFH. Consistent evinacumab exposure was achieved, as the predicted exposure at steady‐state for patients with HoFH with extreme values of baseline ANGPTL3 or body weight did not deviate from a typical patient by more than 30%. In addition, patients with HoFH with body weights of 60 kg or less, 60–80 kg, and more than 80 kg achieved mean (standard deviation) evinacumab trough concentrations at steady state of 171.8 (40.1) mg/L, 245.6 (102.7) mg/L, and 275.8 (91.9) mg/L, respectively.
Between‐subject variability in key parameters was typical for a monoclonal antibody (coefficient of variation [CV%]: 36% for linear clearance and 21% for central volume of distribution [first compartment]). The final population PK parameters estimations and covariate effects are shown in Table 1. The effects of continuous and categorical covariates on population PK parameters are summarized in Tables S6 and S7, respectively. Inspection of GOF plots suggested good agreement between observed and population predicted values for the overall pooled population and when stratified by individual study (Figure S4). VPCs showed that a large majority of the observed concentrations were within the range of the 2.5th to 97.5th predicted percentiles, thus representing a good qualification of the final population PK model (Figure S5).
TABLE 1.
Population PK parameters for the final evinacumab covariate model
| Parameter (units) | Population estimate (% RSE) |
|---|---|
| PK parameter | |
| CL, L/day for 74.1 kg subject | 0.0955 (3.40) |
| V2, L for 74.1 kg subject | 2.56 (1.67) |
| F for s.c. dose | 0.714 (1.55) |
| K23, 1/day | 0.109 (10.4) |
| K32, 1/day | 0.124 (10.4) |
| Vmax, mg/day | 3.16 (2.01) |
| K m, mg/L | 1.02 (9.31) |
| Alag, day | 0.168 (8.20) |
| K a, 1/day | 0.181 (10.6) |
| Covariate | |
| Weight ~ central V | 0.875 (5.37) |
| Weight ~ linear clearance | 0.75 (fixed) |
| ANGPTL3 ~ Vmax | 0.405 (9.98) |
| Disease state ~ Vmax | –0.289 (24.7) |
| OMEGA correlation matrix | |
| σ, η(CL) | 0.355 (10.5) |
| σ, η(2,1) | 0.213 (23.2) |
| σ, η(V) | 0.213 (7.65) |
| σ, η(Ka) | 0.686 (14.8) |
| σ, η(Alag1) | 1.19 (5.55) |
| Residual error | |
| σ additive, mg/L | 0.303 (9.01) |
| σ proportional, CV% | 0.189 (2.02) |
Abbreviations: Alag, absorption lag time; ANGPTL3, angiopoietin‐like protein 3; CL, plasma clearance; CV%, coefficient of variation; F, bioavailability; K23 and K32, intercompartmental rate constants; Ka, absorption rate constant; Km, Michaelis‐Menten constant; PK, pharmacokinetics; RSE, relative standard error; s.c., subcutaneously; V, volume; V2, volume of distribution (central compartment); Vmax, maximum target‐mediated rate of elimination.
The magnitude of the covariate effect on derived PK exposures is illustrated in the tornado plot (Figure 2). The predicted evinacumab exposures (AUC for a dosing interval, Cmax, and minimum concentration) at steady state for patients with extreme baseline values for ANGPTL3 or body weight did not deviate from a typical patient by more than 30%. The typical patient was defined as someone with HoFH with a median weight of 74.1 kg and a median baseline ANGPTL3 value of 0.08 mg/L. In addition, compared with the unexplained between‐subject variation of PK exposures, the magnitude of covariate effect was not pronounced and is not expected to be clinically significant.
FIGURE 2.
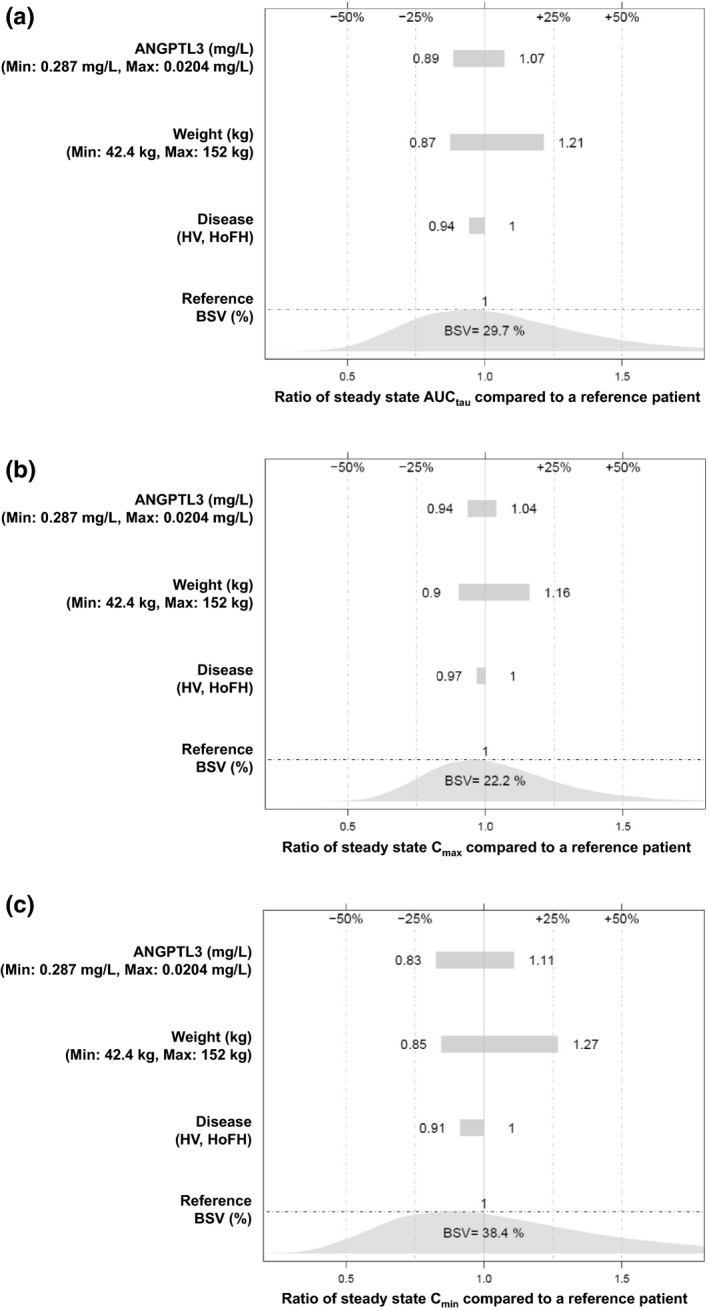
Tornado plots showing the effect of statistically significant covariates on post hoc steady‐state evinacumab exposures: (a) AUCtau, (b) Cmax, and (c) Cmin. The reference patient is a patient with HoFH with a median weight of 74.1 kg and a median ANGPTL3 value of 0.08 mg/L. ANGPTL3, angiopoietin‐like protein 3; AUCtau, area under the plasma concentration–time curve for a dosing interval; BSV, between‐subject variability; Cmax, maximum concentration; Cmin, minimum concentration; HoFH, homozygous familial hypercholesterolemia; HV, healthy volunteers; Min, minimum; Max, maximum
Population PK/PD analysis
The PD data set comprised 1203 LDL‐C concentration timepoints from patients with HoFH. Nine LDL‐C data points with CWRES more than 5 were removed as outliers. Thus, a total of 1194 LDL‐C data points from 95 patients with HoFH were included in the final model estimation. The amount of missing continuous covariate and categorical covariate data was limited (less than 5%).
The baseline covariates evaluated in the PK/PD model are summarized in Tables S8 and S9. The final population PK/PD model included two covariates on Imax and one on IC50. In patients with higher baseline LDL‐C, a smaller IC50 was observed, suggesting greater sensitivity to treatment. An increase in potential Imax was observed in patients who were White and/or had a lower baseline weight. The impact of these covariates is reflected in the estimated 18% residual variability observed for the final PK/PD model. Compared with the base model, the inclusion of the covariates reduced parameter variability (CV%) of Imax and IC50 by 0.3% and 14.4%, respectively.
The final population PD parameters for the final covariate model are presented in Table 2 together with the bootstrap means, medians, and 95% CIs. The bootstrap means and medians were concordant with the population predicted values. Inspection of GOF plots of the final PK/PD model suggested good agreement between the observed and population predicted LDL‐C values for the overall pooled population (Figure S6) and when stratified by individual study (Figure S7). Additional GOF plots of observed, population predicted, and individual predicted LDL‐C concentrations by individual over time for the base model are presented in Figure S8.
TABLE 2.
Population pharmacodynamic parameters and bootstrap confidence intervals for the final covariate model
| Parameter | Point estimate (CV%) | Bootstrap median (2.5th, 97.5th percentiles) |
|---|---|---|
| Typical value | ||
| Kin, 1/day | 38.99 (14.2) | 39.17 (31.44, 50.16) |
| Imax, % | 0.74 (1.5) | 0.74 (0.7, 0.8) |
| IC50, mg/L | 57.4 (20.3) | 56.68 (33.22, 89.17) |
| Covariate | ||
| IC50 ~ baseline LDL (power) | –1.17 (22.1) | –1.14 (–1.99, –0.51) |
| Imax ~ weight (power) | –0.27 (12.3) | –0.3 (–0.68, –0.04) |
| Imax ~ race | 0.83 (3.9) | 0.83 (0.72, 0.9) |
| Interindividual variability, η | ||
| σ, η(IC50) | 3.11 (20.3) | 3.12 (1.78, 5.44) |
| σ, η(Kin) | 0.47 (38.6) | 0.41 (0.15, 0.78) |
| Residual error | ||
| σ proportional | 0.18 (4.1) | 0.18 (0.13, 0.22) |
| σ additive, mg/dl | 17.97 (4.7) | 17.63 (5.53, 23.69) |
For presentation, the covariate of race on Imax is the exponent of −0.19 from the estimates. The expressions including the effects of the covariates are: IC50 = 57.3976 * (baseline LDL/211)**(–1.16801); Imax = 0.743459 * (weight/71)**exp(RAC1*–0.191248), where race was coded as White (RAC1 = 0) and others (RAC1 = 1). *denotes multiplication; **denotes mean exponent (raised to a power).
Abbreviations: CV%, coefficient of variance (100% × standard deviation/parameter estimates); exp, exponent; IC50, drug concentration to reach 50% of Imax; Imax, maximal inhibitory effect; Kin, zero‐order constant of production of response; LDL, low‐density lipoprotein; RAC, race.
VPC evaluations showed that most observed LDL‐C concentrations were generally within the 95% prediction intervals, thus indicating concordance between model‐based simulated data and the observed data (Figure S9). Considering the heterogeneity of LLT used in these patients, VPC evaluations indicated that the model adequately predicted the median, 2.5th, and 97.5th percentiles of the concentration–time profile of LDL‐C after dosing with evinacumab 15 mg/kg q4w.
Post hoc PD parameter estimations for 95 patients with HoFH were performed using the final population PK/PD model to predict the LDL‐C response at Week 24 following administration of evinacumab 15 mg/kg q4w. The percentage of patients predicted to achieve LDL‐C reductions of 30% or more and 50% or more from baseline at Week 24 was 86.3% and 56.8%, respectively (Table 3). As the predicted values were comparable to the 83.7% and 55.8% of patients in the pivotal trial achieving LDL‐C reductions of 30% or more and 50% or more, respectively, it provided further confirmation of model performance. The predicted percentages of patients reaching LDL‐C reduction were close to the observed values reported in pivotal trial, confirming the model performance. 15 The magnitude of the covariate effect on derived PD end points is illustrated in Figure 3. Baseline LDL‐C concentration showed the highest magnitude of effect among the statistically significant covariates. Trial simulations showed that the incremental benefit of increasing the evinacumab i.v. q4w dose from 15 mg/kg to 20 mg/kg was modest, and a dose of 5 mg/kg is expected to achieve lower efficacy (Table 4).
TABLE 3.
Post hoc prediction of patients with HoFH achieving LDL‐C goals at Week 24 following administration of evinacumab i.v. q4w
| Evinacumab i.v. q4w | ∆LDL‐C ≥30%, % | ∆LDL‐C ≥50%, % | ∆LDL‐C ≥70%, % | LDL‐C <100 mg/dl, % |
|---|---|---|---|---|
| 15 mg/kg | 86.3 | 56.8 | 8.42 | 52.6 |
Abbreviations: ∆LDL‐C, percentage low‐density lipoprotein cholesterol change from baseline; HoFH, homozygous familial hypercholesterolemia; i.v., intravenously; LDL‐C, low‐density lipoprotein cholesterol; q4w, every 4 weeks.
FIGURE 3.
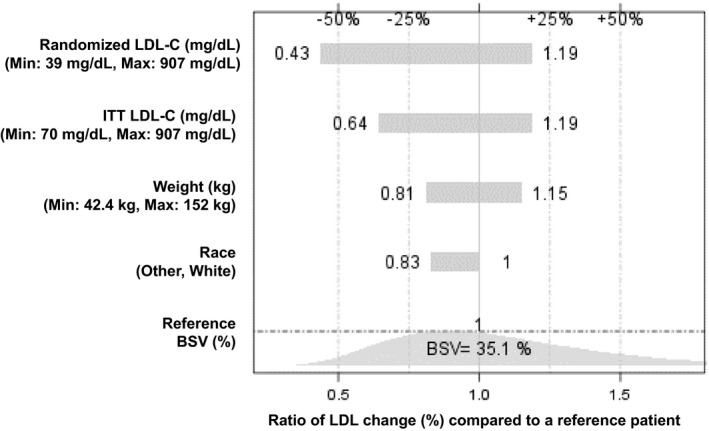
Tornado plot of significant covariates on percentage of LDL‐C reduction from baseline at Week 24 compared with a reference patient. The reference patient is a White patient with HoFH with a baseline LDL‐C of 211 mg/dl and a weight of 71 kg. “Randomized” corresponds to all patients who were randomly assigned in the pivotal phase III ELIPSE HoFH study. BSV, between‐subject variability; HoFH, homozygous familial hypercholesterolemia; ITT, intention‐to‐treat population (baseline LDL more than 70 mg/dl); LDL, low‐density lipoprotein; LDL‐C, low‐density lipoprotein cholesterol; Min, minimum; Max, maximum
TABLE 4.
Simulated percentage of patients with HoFH achieving LDL‐C goals at Week 24 following administration of evinacumab i.v. q4w
| Evinacumab i.v. q4w | LDL‐C % reduction from baseline | LDL‐C <100 mg/dl | ||||||
|---|---|---|---|---|---|---|---|---|
| 30% | 50% | 70% | ||||||
| Median, % | Range, % | Median, % | Range, % | Median, % | Range, % | Median, % | Range, % | |
| 5 mg/kg | 53.7 | 36.8–68.4 | 29.5 | 14.7–42.1 | 4.2 | 0.0–11.6 | 20.0 | 8.4–33.7 |
| 15 mg/kg | 84.2 | 70.5–94.7 | 63.2 | 46.3–76.8 | 14.7 | 6.3–27.4 | 36.8 | 23.2–51.6 |
| 20 mg/kg | 88.4 | 74.7–96.8 | 69.5 | 52.6–81.1 | 17.9 | 6.3–28.4 | 40.0 | 25.3–55.8 |
Simulations were performed for 1000 trials with 95 patients/trial using the distribution of observed covariates.
Abbreviations: HoFH, homozygous familial hypercholesterolemia; i.v., intravenously; LDL‐C, low‐density lipoprotein cholesterol; q4w, every 4 weeks.
DISCUSSION
These analyses characterized the PK of evinacumab using data pooled from phase I, II, and III clinical studies. A two‐compartment model with parallel linear and Michaelis–Menten clearance adequately described the time course of s.c. and i.v. evinacumab. Like most monoclonal antibodies, at higher‐than‐saturation concentrations drug elimination was best described by a linear, first‐order elimination pathway. 20 The Michaelis–Menten constant for the saturable clearance of evinacumab was estimated to be 1.02 mg/L, which was well below the observed median concentration of 88 mg/L at steady state of the therapeutic dose (15 mg/kg). In the range of evinacumab plasma concentrations seen with the dosing regimens assessed in the evinacumab clinical program, the saturable route will eliminate approximately 3.12 mg/day.
The exponent for the power relationship between weight and clearance could not be estimated and so was fixed to the theoretical value of 0.75. Sensitivity analysis for this assumption (exponent = 0.75) was performed, which suggested that other PK parameter estimates were insensitive to this assumption, consistent with the inability to estimate this number. The PK parameter estimates for clearance, volume, Michaelis–Menten constant, and Vmax varied less than 20% from the estimated values at an exponent of 0.75. Thus, the assumption of a value using an exponent of 0.75 does not significantly influence the estimates of other PK parameters. The central volume of distribution was estimated to be 2.56 L, which is consistent with that reported for other monoclonal antibodies. 21 , 22 , 23 Steady‐state total volume estimate for evinacumab is approximately 4.8 L, which is also consistent with other monoclonal antibodies. 24 Patients with HoFH were found to have a clinically significantly lower Vmax than healthy volunteers (25% reduction). Although the effect of HoFH on Vmax met the criteria for clinical significance (more than 20%), the effect of HoFH on exposure (AUC, Cmax) is expected to be less as the saturable elimination is a minor component of total elimination. As baseline ANGPTL3 was comparable between the two populations, no biological explanation or hypotheses are available for this observation. In subjects with HoFH, the saturable elimination pathway Vmax was reduced by 25% versus healthy volunteers, and was deemed clinically significant. Increases in ANGPTL3 concentrations were predictive of a higher Vmax. The relationship between Vmax and ANGPTL3 is biologically plausible, as evinacumab binds to ANGPTL3 and therefore may be cleared along with ANGPTL3.
A population PK/PD analysis was conducted using a data set composed of 95 patients enrolled in three patient studies. The population PK/PD model developed adequately described the relationship of evinacumab concentrations with LDL‐C concentration in patients with HoFH. The PD effect of evinacumab on LDL‐C was linked by an indirect response model with an inhibitor effect on the production of response (LDL‐C). Baseline LDL‐C, weight, and race were statistically significant covariates. The baseline LDL‐C concentration showed the highest magnitude of effect among the statistically significant covariates, with a lower IC50 and higher predicted LDL‐C reduction associated with higher baseline LDL‐C. A lower baseline body weight, as well as being of White race, is associated with a higher maximum inhibitory effect. Compared with the base model, the inclusion of the covariates reduced parameter variability (CV%) of Imax and IC50 by 0.3% and 14.4%, respectively. The results from the PK/PD analysis suggested that no dose adjustment is needed for baseline LDL‐C, weight, and race, as the magnitude of impacts are small. Simulations based on the final population PK/PD model showed that a lower 5 mg/kg i.v. q4w dose of evinacumab is expected to be less efficacious than the proposed dose regimen of 15 mg/kg i.v. q4w, whereas the incremental benefit from 15 mg/kg to 20 mg/kg is expected to be modest as the ranges are overlapping. As the indirect exposure–response model adequately characterized patients with HoFH and permitted the estimation of individual LDL‐C levels following evinacumab administration, the model may help understand the variability in treatment response and guide evinacumab dose selection in future studies.
CONFLICT OF INTEREST
X.P. was an employee of and stockholder in Regeneron Pharmaceuticals, Inc. F.Y. was an employee of and stockholder in Regeneron Pharmaceuticals, Inc. Y.Z., J.D.D., and N.A.‐H. are employees of and stockholders in Regeneron Pharmaceuticals, Inc. M.S. is a compensated consultant to Regeneron Pharmaceuticals, Inc.
AUTHOR CONTRIBUTIONS
X.P., M.S., F.Y., Y.Z., J.D.D., and N.A.‐H. wrote the manuscript and performed the research. X.P., J.D.D., and N.A.‐H. designed the research. X.P., M.S., J.D.D., and N.A‐H. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients, their families, and all investigators involved in the studies used in this analysis. Fareya Zubair of Regeneron Pharmaceuticals, Inc. assisted the authors with the preparation of the manuscript. Medical writing support was provided by Atif Riaz, PhD, of Prime, Knutsford, UK, supported by Regeneron Pharmaceuticals, Inc., according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M15‐0288). The sponsor was involved in the study design and collection, analysis, and interpretation of data as well as data checking of information provided in the manuscript. The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this publication.
Pu X, Sale M, Yang F, Zhang Y, Davis JD, Al‐Huniti N. Population pharmacokinetics and exposure–response modeling for evinacumab in homozygous familial hypercholesterolemia. CPT Pharmacometrics Syst Pharmacol. 2021;10:1412–1421. doi: 10.1002/psp4.12711
Funding information
This analysis was funded by Regeneron Pharmaceuticals, Inc.
REFERENCES
- 1. Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146‐2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Widhalm K, Benke IM, Fritz M, et al. Homozygous familial hypercholesterolemia: summarized case reports. Atherosclerosis. 2017;257:86‐89. [DOI] [PubMed] [Google Scholar]
- 3. Al‐Shaikh AM, Abdullah MH, Barclay A, Cullen‐Dean G, McCrindle BW. Impact of the characteristics of patients and their clinical management on outcomes in children with homozygous familial hypercholesterolemia. Cardiol Young. 2002;12:105‐112. [DOI] [PubMed] [Google Scholar]
- 4. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478‐3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. France M, Rees A, Datta D, et al. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis. 2016;255:128‐139. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 reduces hepatic VLDL‐triglyceride secretion. J Lipid Res. 2015;56:1296‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220‐2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimamura M, Matsuda M, Yasumo H, et al. Angiopoietin‐like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366‐372. [DOI] [PubMed] [Google Scholar]
- 9. Kersten S. Angiopoietin‐like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13:731‐739. [DOI] [PubMed] [Google Scholar]
- 10. Wu L, Soundarapandian MM, Castoreno AB, Millar JS, Rader DJ. LDL‐cholesterol reduction by ANGPTL3 inhibition in mice is dependent on endothelial lipase. Circ Res. 2020;127:1112‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adam RC, Mintah IJ, Alexa‐Braun CA, et al. Angiopoietin‐like protein 3 governs LDL‐cholesterol levels through endothelial lipase‐dependent VLDL clearance. J Lipid Res. 2020;61:1271‐1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaudet D, Gipe DA, Pordy R, et al. ANGPTL3 inhibition in homozygous familial hypercholesterolemia. N Engl J Med. 2017;377:296‐297. [DOI] [PubMed] [Google Scholar]
- 14. Ahmad Z, Banerjee P, Hamon S, et al. Inhibition of angiopoietin‐like protein 3 with a monoclonal antibody reduces triglycerides in hypertriglyceridemia. Circulation. 2019;140:470‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711‐720. [DOI] [PubMed] [Google Scholar]
- 16. Harada‐Shiba M, Ali S, Gipe DA, et al. A randomized study investigating the safety, tolerability, and pharmacokinetics of evinacumab, an ANGPTL3 inhibitor, in healthy Japanese and Caucasian subjects. Atherosclerosis. 2020;314:33‐40. [DOI] [PubMed] [Google Scholar]
- 17. Regeneron Pharmaceuticals Inc . EVKEEZA™ (evinacumab‐dgnb) injection, for intravenous use. Published 2021. Last accessed August 16, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
- 18. Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best‐case performance. J Pharmacokinet Pharmacodyn. 2003;30:387‐404. [DOI] [PubMed] [Google Scholar]
- 19. Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6:576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovalenko P, DiCioccio AT, Davis JD, et al. Exploratory population PK analysis of dupilumab, a fully human monoclonal antibody against IL‐4Ralpha, in atopic dermatitis patients and normal volunteers. CPT Pharmacometrics Syst Pharmacol. 2016;5:617‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu C, Su Y, Paccaly A, Kanamaluru V. Population pharmacokinetics of sarilumab in patients with rheumatoid arthritis. Clin Pharmacokinet. 2019;58:1455‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinez JM, Brunet A, Hurbin F, DiCioccio AT, Rauch C, Fabre D. Population pharmacokinetic analysis of alirocumab in healthy volunteers or hypercholesterolemic subjects using a Michaelis‐Menten approximation of a target‐mediated drug disposition model‐support for a biologics license application submission: Part I. Clin Pharmacokinet. 2019;58:101‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ovacik M, Lin K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin Transl Sci. 2018;11:540‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material