

Abstract
Apolipoprotein L1 (APOL1) risk alleles in donor kidneys associate with graft loss, but whether recipient risk allele expression affects transplant outcomes is unclear. To test whether recipient APOL1 risk alleles independently correlate with transplant outcomes, we analyzed genome-wide SNP genotyping data on donors and recipients from 2 kidney transplant cohorts: Genomics of Chronic Allograft Rejection (GOCAR) and Clinical Trials in Organ Transplantation 01/17 (CTOT-01/17). We estimated genetic ancestry (quantified as the proportion of African ancestry, or pAFR) by ADMIXTURE and correlated APOL1 genotypes and pAFR with outcomes. In the GOCAR discovery set, we noted that the number of recipient APOL1 G1/G2 alleles (R-nAPOL1) associated with an increased risk of death-censored allograft loss (DCAL), independent of ancestry (HR = 2.14; P = 0.006), as well as within the subgroup of African American and Hispanic (AA/H) recipients (HR = 2.36; P = 0.003). R-nAPOL1 also associated with an increased risk of any T cell–mediated rejection (TCMR) event. These associations were validated in CTOT-01/17. Ex vivo studies of PMBCs revealed, unexpectedly, high expression levels of APOL1 in activated CD4+/CD8+ T cells and NK cells. We detected enriched immune response gene pathways in risk allele carriers compared with noncarriers on the kidney transplant waitlist and among healthy controls. Our findings demonstrate an immunomodulatory role for recipient APOL1 risk alleles associated with TCMR and DCAL. We believe this finding has broader implications for immune-mediated injury to native kidneys.
Keywords: Nephrology, Transplantation
Keywords: Genetic variation, Organ transplantation, T cells

Introduction
Patients with African ancestry have a significantly higher risk of non-Mendelian focal segmental glomerulosclerosis (FSGS) as well as end-stage renal disease (ESRD) (reviewed in ref. 1). Seminal work identified the risk genotypes of apolipoprotein L1 (APOL1) when present as 2 copies of either or both G1 and G2 alleles (2), which explained the increased risk of FSGS and ESRD observed in African Americans (AAs) (3, 4). Mechanistic data have since focused on the role of APOL1 risk alleles in kidney epithelial cells, including gain-of-function roles in FSGS (5) and preeclampsia (6) and the loss-of-function role in parietal cell biology (7).
In renal transplantation, 2 copies of the APOL1 risk alleles, when present in the donor, have been associated with death-censored allograft loss (DCAL) (8–10). Although donor African ancestry is incorporated into the kidney donor risk index (11), giving weight to donors carrying 2 copies of the APOL1 risk allele versus all others improved the prediction of DCAL (12). Limited mechanistic data suggest the development of FSGS in APOL1 risk genotype–carrying allografts (13, 14). On the other hand, a single retrospective study of kidney transplant recipients reported that recipient carriage of APOL1 risk alleles was not associated with DCAL (15). These data have since led to an emphasis on the role of APOL1 expression in renal cells and outcomes. The role of APOL1 risk alleles in nonrenal tissues, including immune cells, has not, to our knowledge, been specifically examined. A universal mechanism linking APOL1 risk alleles to allograft outcomes has not yet emerged from the literature, and a nationwide prospective study is currently underway (16).
Previous studies by several groups showed associations among self-declared AA race and increased rejection episodes and/or DCAL (17–21). We recently reported that recipient African ancestry expressed as a quantitative variable (defined as the recipient proportion of African ancestry [R-pAFR]) was associated with DCAL in a prospective renal transplant cohort (22). While current concepts implicate altered tacrolimus metabolism (18), specific induction and/or maintenance therapy (19), and socioeconomic factors to account for these observations, these associations do not fully explain the worse outcomes in transplant recipients with African ancestry.
As APOL1 G1/G2 alleles are seen exclusively in AAs and Hispanics (AA/H, with recent African ancestry), here, we studied 2 prospective transplant cohorts (22, 23) to test for associations among the number of recipient APOL1 risk alleles (R-nAPOL1), the R-pAFR, and transplant outcomes. We report the unexpected association of R-nAPOL1 with DCAL in additive models, implying a role for even 1 APOL1 risk allele (either G1 or G2) in recipients, distinct from the previously reported association of 2 risk alleles in the donor with increased DCAL. This association was identified in all recipients as well as in AA/H recipients. We then identified an association of R-nAPOL1 with T cell–mediated rejection (TCMR), independent of recipient AA ancestry, and validated these results externally. Finally, additional analyses implicated an unanticipated role for APOL1 risk alleles in immune activation, specifically in activated CD4+/CD8+ T cells and CD56dim NK cells, pointing to a potential mechanism to account for the observed associations.
Results
Study cohorts.
The Genomics of Chronic Allograft Rejection (GOCAR) (24, 25) and Clinical Trials in Organ Transplantation 01/17 (CTOT-01/17, hereafter referred to as CTOT) (23) were prospective, multicenter observational studies that enrolled crossmatch-negative kidney transplant candidates. We used a subcohort of 385 donor-recipient (D-R) pairs with genome-wide genotype data from GOCAR for discovery (22), and a subcohort of 122 D-R pairs with genome-wide genotype data from CTOT as a validation set (Supplemental Figures 1 and 2; supplemental material available online with this article; https://doi.org/10.1172/JCI146643DS1). Demographic and clinicopathologic characteristics of GOCAR and CTOT cohorts, stratified by R-nAPOL1, are listed in Table 1 and were published elsewhere (22, 23). The clinical characteristics between the 2 cohorts were comparable (Supplemental Table 1), although the CTOT cohort had a higher proportion of AA/H recipients and deceased donors (DDs). The GOCAR cohort had longer follow-up periods according to the United Network for Organ Sharing (UNOS) and the Australian and New Zeland Dialysis and Transplant registry (ANZDATA) databases (mean follow-up of 4.6 years) and thus more DCAL and TCMR events (22), whereas the CTOT program collected information over a period of up to 5 years (mean follow-up of 3.7 years). For each D-R pair from both cohorts, we used genome-wide genotype data excluding the MHC region (22) to estimate pAFR and infer genetic ancestry (Supplemental Table 2). As expected, APOL1 genotyping showed that G1/G2 risk alleles were only detected in genetic AAs or Hispanics (i.e., AA/H) among D-Rs in both cohorts (see Methods and Supplemental Tables 3 and 4). We observed a higher frequency of depletional induction agents in recipients carrying APOL1 risk alleles from both cohorts, without differences in the number of D-R HLA mismatches between recipients carrying APOL1 risk alleles and noncarriers (Table 1).
Table 1. Demographic and clinicopathologic characteristics of donors and recipients in the GOCAR and CTOT cohorts stratified by the number of recipient APOL1 risk alleles.

Recipient APOL1 G1/G2 alleles associate with graft loss.
We investigated the association of R-nAPOL1 with DCAL for all recipients and in the AA/H strata in the GOCAR (discovery) and CTOT (validation) cohorts. Kaplan-Meier survival curve analysis (Figure 1) stratified by the number of APOL1 risk alleles (0, 1, or 2) showed clear differentiation among groups, with the number of risk alleles correlating directly with a higher risk of DCAL in both cohorts (GOCAR cohort: log-rank P < 0.0001; CTOT cohort: log-rank P = 0.0075). Our finding supports an additive effect of the APOL1 risk alleles in recipients on graft survival, i.e., each copy of the risk alleles increases the risk of graft loss, distinct from prior data (3). The additive model was also confirmed to have the best interpretation of the data compared with the dominant and recessive models (Supplemental Table 5). We next adjusted for covariates previously shown to be associated with DCAL (22), including genetic ancestry, induction therapy, and donor type, using a multivariable Cox regression analysis (Table 2). This analysis revealed that in GOCAR, R-nAPOL1 remained associated with DCAL in an additive manner (HR = 2.14 per additional copy of risk alleles, i.e., 0 vs. 1 allele, or 1 vs. 2 alleles; P = 0.006), independent of the recipients’ genetic ancestry. Analysis of the CTOT cohort validated the results (Supplemental Table 6). When we performed a meta-analysis including both cohorts (Figure 2), we found that the HR of each additional risk allele was 2.27 (95% CI: 1.41~3.63; P = 0.0007). When we performed sensitivity analysis within the strata of AA/H recipients, we observed a similar (although marginally significant) pattern of separated Kaplan-Meier survival curves for the 3 R-nAPOL1 groups (Supplemental Figure 3A) as well as a significant association between R-nAPOL1 and DCAL with a similar effect size (Table 2) in the GOCAR cohort. Within the AA/H strata of CTOT, we found that the pattern of differentiated survival curves (Supplemental Figure 3B) and the positive association of R-nAPOL1 with DCAL (Supplemental Table 6) remained, albeit with a diminished significance level due to the limited sample size. To account for the effect on allograft survival for the donor APOL1 risk genotype, where a high-risk genotype in donors is defined as 2 copies of G1/G2 alleles and a low-risk genotype as 0 or 1 copy of G1/G2 allele, we performed additional sensitivity analyses stratified by donor APOL1 risk genotype in both cohorts. Multivariable Cox regression analyses was conducted on the stratum of donors carrying the low-risk genotype, as the sample sizes for the high-risk group in both cohorts were limited. The results from our stratified analyses remained similar to those for the main analysis: the R-nAPOL1 was associated with DCAL, independent of the donor’s APOL1 risk genotype, for all recipients and for AA/H recipients (Supplemental Tables 7 and 8). Together, these data demonstrate that R-nAPOL1 associates with DCAL in an additive manner in both cohorts.
Figure 1. Kaplan-Meier plot of death-censored allograft survival for recipients with different numbers of APOL1 risk alleles.

(A) Kaplan-Meier survival plot for the GOCAR cohort. (B) Kaplan-Meier survival plot for the CTOT cohort.
Table 2. Association of APOL1 risk alleles with DCAL in an additive manner using multivariable Cox regression.
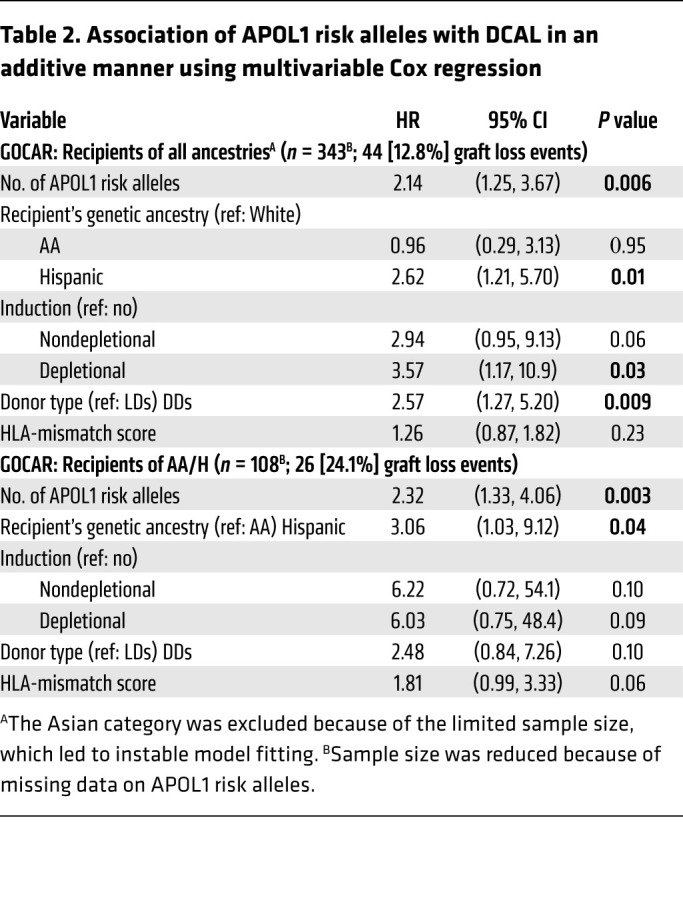
Figure 2. Meta-analysis for the association of APOL1 risk alleles with death-censored allograft survival across the GOCAR and CTOT studies.

HR estimates of the number of APOL1 risk alleles associated with DCAL from the GOCAR and CTOT cohorts for all ancestries were included in a fixed-effect meta-analysis. The HRs and corresponding 95% CIs for individual studies and meta-analysis are presented as forest plots. The size of the squares shown for the individual studies is proportional to the sample size of each study.
Recipient APOL1 G1/G2 alleles associate with clinical and subclinical rejection.
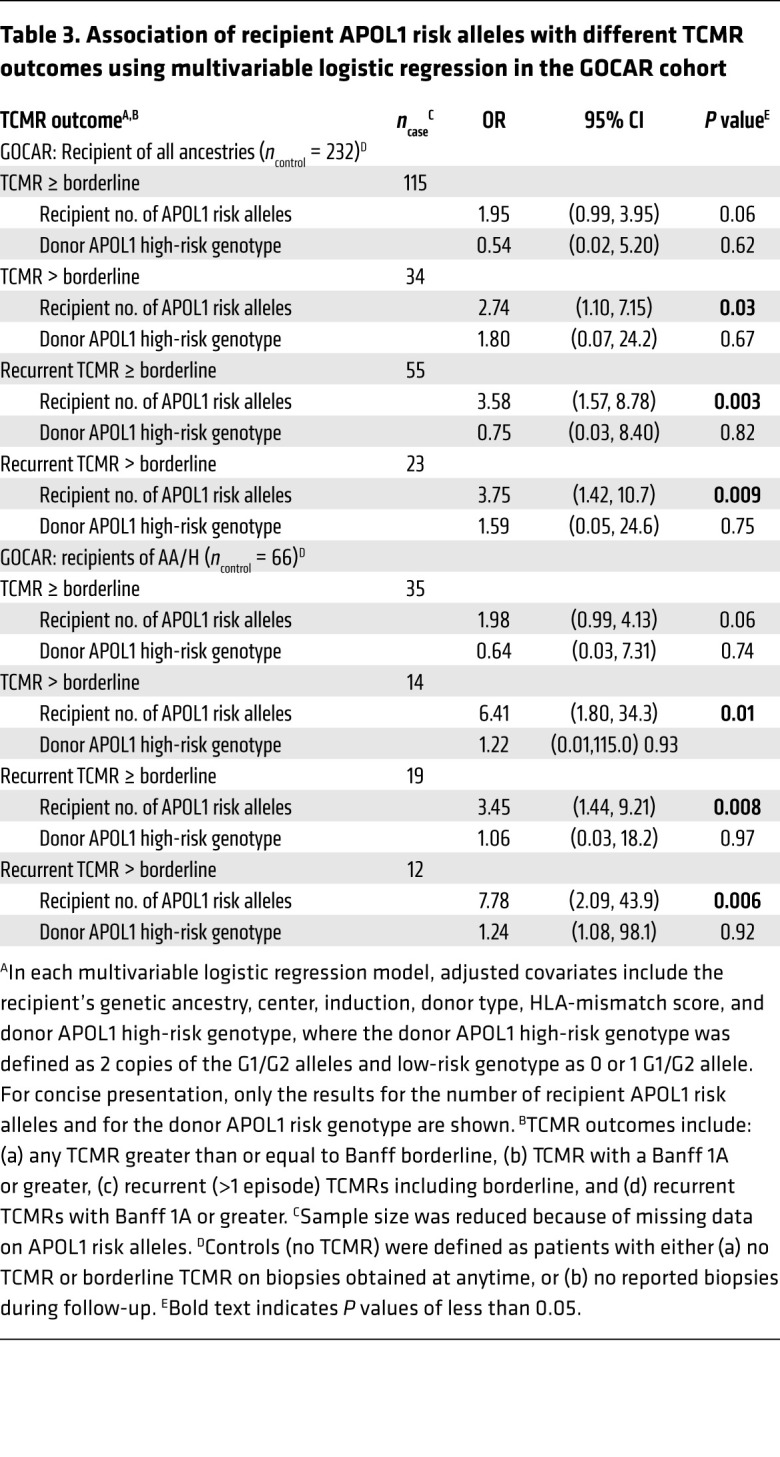
We next tested the strength of the association between R-nAPOL1 and TCMR episodes in the GOCAR and CTOT cohorts. The study designs captured clinical rejection episodes for a period of up to 2 years in both cohorts, as well as subclinical rejections at 3, 12, and 24 months (GOCAR), and at 6 months (CTOT) (23–25). In GOCAR, 126 recipients (32.7%) had at least 1 episode of subclinical or clinical TCMR (with a Banff borderline score or greater) identified among 3 surveillance biopsies (22, 25), whereas in CTOT, 15 recipients (12.3%) had at least 1 TCMR episode including the 6-month surveillance biopsy (23). R-nAPOL1 was significantly associated with various TCMR outcomes in multivariable logistic regression models, independent of donor APOL1 risk genotype, with progressively increasing ORs present with more severe TCMR phenotypes (Table 3). For sensitivity analyses in the subset of AA/H recipients, we found that the association of R-nAPOL1 with different TCMR outcomes remained, with similar increasing ORs with increased severity of TCMR phenotypes. In the CTOT cohort, by logistic regression, we observed that the association of R-nAPOL1 with TCMR was significant for the whole cohort and the AA/H subset in univariate analysis, whereas the direction and magnitude of the association remained with reduced significance in a multivariable analysis (Supplemental Table 9). Taken together, these data indicate a strong association between R-nAPOL1 and TCMR events.
Table 3. Association of recipient APOL1 risk alleles with different TCMR outcomes using multivariable logistic regression in the GOCAR cohort.
Recipient AA ancestry associates with creatinine levels up to 24 months after transplantation and not with TCMR.
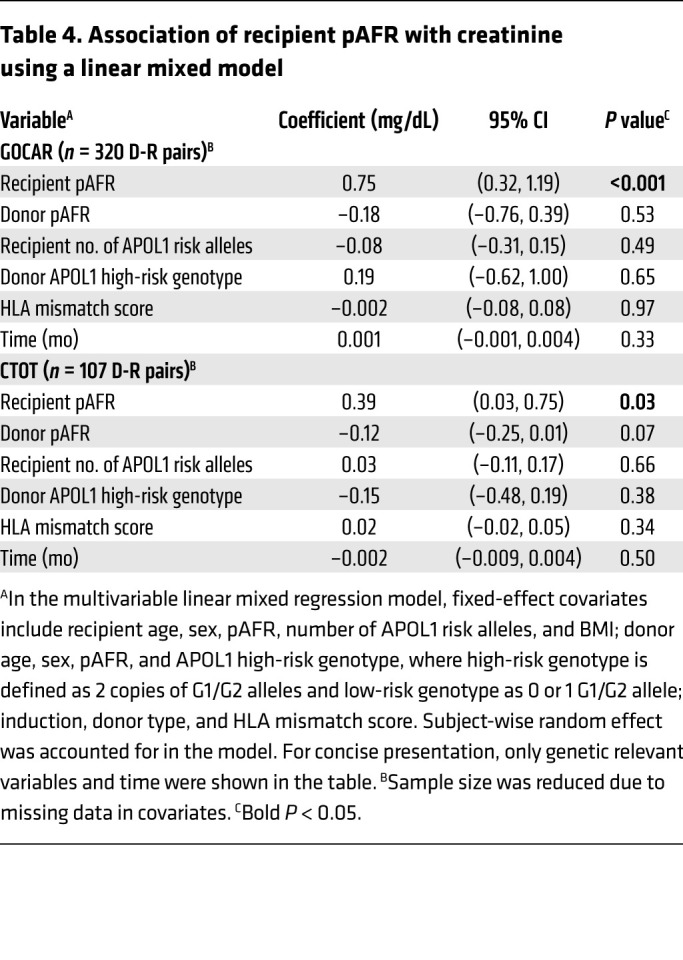
Since we found a correlation between R-pAFR and R-nAPOL1, we tested for associations between R-pAFR and transplant outcomes independent of R-nAPOL1, noting that we previously reported that R-pAFR did not associate with Banff inflammation subscores or TCMR up to 2 years after transplantation (22). This previous work also showed that no other Banff component scores in biopsies obtained within 2 years associated with R-pAFR in GOCAR. Here, including the GOCAR and CTOT cohorts, we used linear mixed models incorporating all available longitudinal creatinine data (to account for intraindividual variability) to determine the association between R-pAFR and creatinine levels (or the estimated glomerular filtration rate [eGFR]) as a measure of kidney function. This analysis revealed that in GOCAR, R-pAFR was significantly associated with serum creatinine levels from 3 to 24 months after transplantation, independent of R-nAPOL1, post-transplant recipient BMI (to account for creatinine generation), donor APOL1 risk genotype, and donor pAFR (to account for AA-to-AA transplants; Table 4 and Supplemental Figure 4). For example, as shown in Table 4, a recipient with 100% of African ancestry had, on average, 0.75 mg/dL higher serum creatinine levels than did a recipient with no African ancestry, or, equivalently, every 10% increment of African ancestry in a recipient would lead to a 0.075 mg/dL increment in creatinine levels. We confirmed this association in the CTOT cohort with creatinine levels between 3 and 12 months after transplantation (Table 4 and Supplemental Figure 4). The eGFR by modified diet in renal disease (MDRD) or Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equations (26) tended to be inversely correlated with R-pAFR in mixed models, but insignificantly (P = 0.06; not shown). These data relayed distinct post-transplantation phenotypic associations of recipient African ancestry and recipient APOL1 risk allele status in our study cohorts.
Table 4. Association of recipient pAFR with creatinine using a linear mixed model.
SNP-based mismatches in APOL1 between D-R pairs do not associate with DCAL.
We further asked whether the association of R-nAPOL1 with DCAL was related to, or independent of, “mismatches” at the APOL1 locus itself, between D-R pairs. This was especially relevant, since 90.3% (GOCAR) and 82.5% (CTOT) of donors had a G0/G0 genotype, while 11.2% (GOCAR) and 21.7% (CTOT) of recipients had either 1 or 2 copies of G1 and/or G2 alleles (Supplemental Table 3), increasing the likelihood of an APOL1 D-R mismatch among recipients with APOL1 risk alleles. We defined an SNP-based mismatch score to quantify the overall mismatch between any given D-R pair at the APOL1 locus, and to reflect the overall effect of the introduction of any new APOL1 variants from the donor kidney into the recipient (see Methods). Multivariable Cox models showed that the APOL1 SNP–based mismatch score had no significant effect on DCAL (Supplemental Table 10), and, conditional on the APOL1 SNP-based mismatch score, the R-nAPOL1 remained associated with DCAL for all the recipients and the AA/H recipients in both cohorts (Supplemental Table 10). This suggests an intrinsic effect of APOL1 risk alleles in recipients on DCAL, independent of APOL1 D-R “mismatches.”
Phenotypic data from immune cell types carrying APOL1 risk alleles in AA/H recipients show immune activation.
Since R-nAPOL1, rather than mismatches at the APOL1 locus between donors and recipients, associated with DCAL and TCMR, we examined the immune cell phenotype and function in AA/H recipients with APOL1 risk alleles using auxiliary data from the public data sets GOCAR and CTOT (see Methods). First, we confirmed APOL1 protein expression in PBMCs using a discarded leukapheresis sample (Figure 3A). Positive and negative controls, respectively, included APOL1 overexpressing human podocytes and a mouse macrophage cell line (see Methods).
Figure 3. Phenotypic data on immune cell types carrying APOL1 risk alleles in AA/H recipients show immune activation.

(A) APOL1 expression at the protein level was confirmed by Western blotting. (B) APOL1 mRNA expression in 15 different types of immune cells in DICE AA/H individuals. (C) Enrichment in immune-related pathways of DEGs identified by comparing DICE AA/H individuals carrying 1 or 2 copies of the G1 or G2 alleles with individuals with the G0/G0 genotype in activated CD4+ T cells and CD56dim NK cells. (D) Enrichment in immune-related pathways of DEGs in CD4+ and CD8+ T cells and CD56dim NK cells; the DEGs were identified by comparing scRNA-Seq data for 2 GOCAR AA recipients with APOL1 G1/G0 and G1/G2 genotypes with the other 2 AA recipients with the G0/G0 genotype. (E) Enrichment of DEGs in immune-related pathways when GOCAR AA/H recipients carrying 1 or 2 copies of G1 or G2 alleles were compared with individuals with the G0/G0 genotype, or when those carrying any 1 risk allele were compared with individuals with the G0/G0 genotype. (F) Panel reactive T cell ELISPOT assay comparing CTOT AA/H recipients with any APOL1 G1 or G2 allele versus G0/G0 genotype. *P < 0.05, by Wilcoxon rank-sum test.
Since PBMCs include a mixture of mononuclear cells, to understand cell-type–specific expression of APOL1, we used the data generated by the Database of Immune Cell Expression, quantitative trait loci (eQTLs), and epigenomics (DICE) project (27), where bulk RNA-Seq data for 15 sorted immune cell types and APOL1 genotype information were available for 91 healthy individuals. We focused on the subset of 22 AA/H individuals,5 of whom carried 1 or 2 copies of G1/G2 alleles. Among the 15 cell types, we discerned that APOL1 mRNA expression was highest in CD56dim NK cells and that ex vivo polyclonally activated CD4+ and CD8+ T cells (but not in unstimulated T cells, Figure 3B). We next performed differential gene expression analysis to identify differentially expressed genes (DEGs) in individuals with any G1/G2 alleles as compared with those with the G0/G0 genotype (see Methods). Our analyses showed a significant enrichment of DEGs in pathways involved in immune activation within activated CD4+ T cells and cytotoxic CD56dim NK cells from participants with any versus no G1/G2 alleles (Figure 3C and Supplemental Table 11). Within activated CD4+ T cells from these healthy controls with APOL1 risk alleles, we observed enrichment of genes involved in allograft rejection and antigen-processing pathways (HLA genes), T cell activation, and differentiation (IL2, IL21, IL21R, IL18R, GATA3), and chemokines (CXCL8, CXCL3, CXCL11). In both CD4+ lymphocytes and cytotoxic NK cells, DEGs included TNF-α–signaling pathway genes and antiviral response genes (Supplemental Table 11).
To further investigate the transcriptomes of NK cells and activated T cells, we generated single-cell RNA sequencing (scRNA-Seq) data from PBMCs collected before transplantation from 4 AA GOCAR recipients with a known APOL1 genotype (NCBI, Gene Expression Omnibus [GEO] GSE182916; see Methods). Two recipients carrying APOL1 risk genotypes (G1/G0 and G1/G2) had recurrent TCMR and sustained graft loss during follow-up, while the other 2 were G0/G0 recipients and had surviving allografts without TCMR (Supplemental Table 12). We also used raw sequencing reads aligned to the APOL1 locus to confirm the APOL1 genotypes of all 4 patients and simultaneously demonstrated the expression of APOL1 G1 and G2 alleles at the mRNA level in PBMCs (Supplemental Figure 5). In the scRNA-Seq data, we confirmed the enrichment of differentially expressed genes DEGs in immune-related pathways in CD4+ and CD8+ T cells as well as in CD56dim NK cells (Figure 3D and Supplemental Table 13). In the single-cell transcriptome of CD4+ T cells from the risk allele carriers, similar to healthy controls from DICE, we identified significant enrichment of DEGs associated with allograft rejection, antigen processing, and graft-versus-host disease. In APOL1 risk allele–carrying NK cells and stimulated CD4+ T cells from the DICE data, as well as in NK cells and CD8+ T cells in the scRNA-Seq data, we observed significant upregulation of IFNG transcripts (Supplemental Table 13). Additionally, we assessed PBMC scRNA-Seq data (GEO GSE162470) from 2 patients with ESRD (G2/G0 vs. G0/G0) on the transplant waitlist and identified highly consistent enrichment of immune activation signatures in activated CD4+ and CD8+ T cells and CD56dim NK cells (Supplemental Figure 6), as well as upregulation of IFNG. Hence, these data suggested cell-type–specific immune activation with either G1 or G2 alleles.
To further validate the immune activation signature of APOL1 risk genotypes in our cohorts, we examined pre-transplant peripheral blood transcriptomes from bulk RNA-Seq data on AA/H recipients within GOCAR (28). In GOCAR, APOL1 genotyping and pre-transplant blood transcriptomes were available for 60 recipients (Table 1), who had 1 or 2 copies of G1/G2 alleles (n = 26) or the G0/G0 genotype (n = 34). We performed differential gene expression analysis of whole-blood mRNA samples prior to transplantation (see Methods; ref. 28). Interestingly, recipients with any copy of the G1 or G2 alleles showed DEGs enriched in immune response pathways compared with the G0/G0 recipients (Figure 3E and Supplemental Table 14). We identified DEGs associated with allo- and antiviral-response pathways, similar to the DICE and scRNA-Seq data, as being significantly enriched in peripheral transcriptomes of the GOCAR recipients with 1 or 2 copies of the risk alleles (Supplemental Table 12).
In an effort to assess whether APOL1 risk alleles associated with T cell function, we reanalyzed ELISPOT data from the CTOT cohort, in which AA/H recipients with APOL1 genotype information were tested for frequencies of alloreactive IFN-γ–producing PBMCs cocultured with a panel of 6 HLA-disparate stimulator cell lines (29). These analyses showed stronger responses in AA/H recipients with any APOL1 risk alleles (n = 5) versus G0/G0 recipients (n = 15; Table 1 and Figure 3F).
Discussion
Using 2 large prospective kidney transplant cohorts, we show for the first time to our knowledge that recipient APOL1 risk alleles were associated with long-term death-censored graft survival and clinical and subclinical as well as recurrent TCMR events up to2 years after transplantation. These findings were identified in additive models of APOL1 genotype, showing that even a single risk allele in recipients presented an increased risk for both acute rejection and long-term graft survival. We then used in silico and ex vivo auxiliary data from immune cells to confirm the expression of APOL1 at mRNA and protein levels in PBMCs and demonstrated enrichment of pathways involving generic immune responses as well as IFN-γ ELISPOT responses. We identified higher APOL1 expression in CD56dim NK cells and ex vivo–stimulated CD4+ and CD8+ T cells using the DICE RNA-Seq data from healthy individuals (27). In stimulated CD4+ T cells, we confirmed significant enrichment of immune response pathways among DICE participants with 1 or 2 copies of G1 or G2 alleles versus those with the G0/G0 genotype. Peripheral CD4+ and CD8+ T cells and NK cells in scRNA-Seq data obtained from GOCAR patients before transplantation (and waitlisted ESRD patients) with risk alleles also revealed consistent enrichment of the identified pathways. Together, these data support a role for FSGS-associated G1/G2 APOL1 alleles in immune cells in modulating alloimmune responses.
Although the association of APOL1 G1/G2 risk alleles with the lifetime risk of ESRD and FSGS in AAs and admixed populations with African ancestry has been repeatedly affirmed in clinical data (3, 4), data regarding the mechanism of adverse effects has been focused on the expression of mutant APOL1 protein (5, 6) or mRNA in kidney epithelial cells (30). In renal transplantation, the association of donor APOL1 risk alleles with DCAL has been consistently observed in retrospective data (8–10), possibly via allograft FSGS (14). Similar large-scale examinations of the association of the recipient APOL1 risk allele with graft outcomes have not been reported. A single-center retrospective study of 119 AA renal recipients did not find an association of the recipient APOL1 risk allele with DCAL (15). However, the donor APOL1 genotypes were unknown here. Further, this study reported an unusually high DCAL rate of 25% at 5 years (vs. 5% and 11% for living donor [LD] and DD kidneys in recent Scientific Registry of Transplant Recipients [SRTR] data), which likely contributed to the inability to identify a significant effect of R-nAPOL1 in this data set (17).
A problem with APOL1 association in transplantation studies is the exclusive association of G1/G2 alleles with African ancestry, a potential confounder for transplant outcomes (30). In our data, we addressed this issue in adjusted models using genetic ancestry, which was previously reported by our group (22) and others (31) as being more accurate than self-reported ancestry, or using R-pAFR (a quantitative measurement of ancestry), inferred from genome-wide genotype data. Additionally, we performed sensitivity analyses in the strata of AA/H recipients, further strengthening our findings of an association of APOL1 risk alleles with DCAL. We then identified an association of R-pAFR (and not R-nAPOL1) with serum creatinine levels within 2 years of transplantation. Furthermore, adjustment for longitudinal creatinine levels as a time-varying covariate in the survival models did not significantly attenuate the association of R-nAPOL1 with DCAL, suggesting that creatinine levels were not a mediator for the association of R-nAPOL1 with DCAL (data not shown). Since transplant recipients are dependent on the allograft for creatinine excretion, the association between R-pAFR and post-transplantation creatinine levels may reflect an increased generation of creatinine in recipients with African ancestry. Hence, our data reflective of genetically determined changes in creatinine levels are also timely to the ongoing discussion of the role of ancestry adjustment in eGFR equations (32) and contributory to the use of genetic ancestry for these purposes (33).
We believe our data open new avenues for the investigation of APOL1 in alloimmune responses in renal transplantation: the role of APOL1 gene products in CD4+ and CD8+ T cells and cytolytic NK cells (CD56dim), the role of wild-type versus variant APOL1 protein/mRNA, and gain-of-function versus loss-of-function mechanisms all need to be comprehensively examined. Although renal epithelial cell injury mechanisms from APOL1 risk variants have been the subject of intensive study (5, 6, 30), APOL1 homologs were originally identified as TNF-α–responsive genes in endothelial cells (34, 35). In humans, the APOL1 promoter has binding sequences for STAT2- and IFN-responsive transcription factors, and a role for APOL1 as a cellular immune response gene in antiviral immunity has been postulated to explain its association with HIV-associated nephropathy (36). Collapsing FSGS after viral infection was reported in APOL1 risk allele–carrying allografts (14) and recently in a recipient with the APOL1 risk allele after COVID-19 infection (37). In lupus nephritis, where APOL1 risk genotypes associate with an increased progression of disease, a toxic gain-of-function role for APOL1 variants by disruption of T cell autophagy and IFN signaling has been postulated (38). Consistently, progressive nephritis was found to be worsened with every copy of the risk allele in Brazilian AA/H patients with lupus (39). In this context, our data implicating a role for APOL1 within T cells involved in adaptive immune responses against a donor organ demonstrate that previous data regarding R-nAPOL1 and allograft outcomes need to be reinterpreted, and its role in infiltrating inflammatory mononuclear cells in native kidney glomerulonephritis investigated.
Although our data provide what we believe to be new insights, we acknowledge several limitations. First, although we adjusted for biologic confounders on the basis of clinical data collected in both cohorts, we cannot eliminate the residual confounding effects of other factors including socioeconomic and behavioral data (e.g., nonadherence), which were not collected. Second, in the CTOT cohort, our validation data on graft loss and acute rejection in the AA/H subcohort did not reach statistical significance because of the limited sample size and paucity of events, although the effect sizes we observed were similar. Notably, the sample sizes for AA/H donors with African ancestry in both cohorts were limited, thus we observed few APOL1 risk alleles in the donors. Larger multiethnic cohorts with adequate sample sizes of APOL1 risk alleles in D-R pairs will allow evaluation of the interaction of APOL1 risk alleles in donor organs and recipients in long-term transplant outcomes (16). Last, the results from DEGs and enrichment analysis were of a nominal significance level, given the limited sample sizes and the burden of multiple-hypothesis testing. Nevertheless, rather than draw firm conclusions here, we aimed to identify directions pointing toward pathways and cell types in which APOL1 risk alleles may affect the transcriptome and transplant outcomes. We believe our findings provide a platform for investigating cell-type–specific immune functions of APOL1 in experimental models such as the human BAC–transgenic mouse strains expressing either G0, G1, or G2 genes at physiological levels, while remaining responsive to endogenous cytokine stimuli.
In summary, using 2 prospective transplant cohorts, we report for the first time to our knowledge the association of recipient APOL1 risk alleles with allograft survival and cellular rejection events. We demonstrate these associations in additive models showing the role of even a single copy of the G1 or G2 allele in the observed outcomes. We show phenotypic data supporting immune effects of APOL1 expression in specific cell types. We also report the association of African ancestry in recipients, quantified as R-pAFR, with serum creatinine after transplantation. We believe our work forms a basis for further mechanistic work to understand the immunologic role of APOL1.
Methods
Discovery cohort.
The GOCAR study is a prospective, multicenter study designed to examine the utility of genomics and genetics to predict the development of chronic allograft injury. Patients included in the study were prospectively enrolled from May 12, 2007, to July 30, 2011. Details of the study were reported elsewhere (22, 24, 25). Clinical data and laboratory samples were collected from the enrolled patients at baseline and 3, 12, and 24 months after renal transplantation.
Validation cohort.
CTOT-01/17 study was a prospective, multicenter, observational study that enrolled crossmatch-negative kidney transplant candidates with 2 years of follow-up (40). Adult and pediatric participants undergoing a primary kidney transplantation and who had a negative flow cytometry crossmatch at the time of transplantation were eligible for enrollment. In the current study, only adult participants aged 18 years or older who had graft survival of more than 1 week were included. Exclusion criteria included plans for multiorgan transplantation and/or clinically significant liver disease. The overall objective of CTOT-01 was to determine the relationships between the immune assay results and a composite primary endpoint (clinically evident or subclinical biopsy-proven cellular acute rejection with a Banff grade ≥1A, an increase in the Banff chronic sum score >2, an increase in interstitial fibrosis >15%, graft loss, or death 6 months after transplantation) and/or a change in renal function (>30% decrease in eGFR) between 6 and 24 months after transplantation. CTOT-17 (extension study of CTOT-01) was designed to collect information on 5-year outcomes in this cohort. Details on this cohort have been published previously (23).
Genotyping, data processing, and quality control
The genotyping and quality control (QC) for the GOCAR cohort have been reported previously (22). After data processing and QC, complete genotype-phenotype data for 385 D-R pairs and 131,035 SNPs remained for statistical analysis. We applied the same procedure used for COGAR to CTOT. Briefly, recipient DNA was obtained from PBMCs, while donor DNA was obtained from either preperfusion allograft biopsies (in DDs) or PBMCs (in LDs). In cases where DNAs from both sources were available, the genotype data was derived from PBMC DNA. The Illumina Infinium Global Screening Array (GSAMD-24v1-0_20011747_A1) was applied on the extracted DNA. The raw genotype data were subjected to a series of QC steps (Supplemental Figure 1). In sample-wise QC, we excluded samples on the basis of the following criteria: (a) the genetically inferred sex was inconsistent with the reported sex; (b) missing genotype rate above 0.03; (c) excessive genome-wide heterozygosity (indicating potential sample contamination); or (d) the individual was of European ancestry but carried APOL1 G1/G2 risk alleles (see APOL1 genotyping section). In SNP-wise QC, we excluded SNPs on the basis of the following criteria: a) missing genotype rate above 0.05; (b) minor allele frequency (MAF) of less than 0.01; or (c) a Hardy-Weinberg equilibrium (HWE) P value of less than 1 × 10–6. The markers with no chromosome information, or with ambiguous alleles (A/T or C/G), or not located on autosomes were excluded as well.
To prepare for downstream analysis (see ADMIXTURE analysis section), the processed genotype data from CTOT samples were merged with the genotype data from the 1000 Genomes Project (KGP) (41) samples at shared SNP loci on autosomal chromosomes. From merged data, common SNPs with a MAF of greater than 0.05 were selected, where the MAF was estimated on the basis of KGP samples. The list of high-density SNP markers was pruned on the basis of pairwise linkage disequilibrium (42), where the pairwise linkage disequilibrium between SNPs was derived from KGP samples. In order to explore the genetic effect beyond HLA, we excluded SNPs located in the MHC region in subsequent genetic analyses. After these steps, there were 122 D-R pairs with complete genotype data and 126,872 SNPs left in the CTOT cohort (Supplemental Figure 1).
ADMIXTURE analysis and genetically inferred ancestry.
We used ADMIXTURE (43) to estimate the proportions of genetic ancestries of donors and recipients and inferred their genetic ancestries for the GOCAR cohort as previously detailed (22). The same analysis pipeline was also applied to the processed genotype data from the CTOT cohort. Briefly, we applied ADMIXTURE on the genome-wide genotype data with 1000 Genomes Project (KGP) Phase I (41) as reference populations to anchor the major ancestral populations. The genetic background of each individual was inferred as a mixture of 4 ancestral components, corresponding to African, White, East Asian, and American Indian ancestry (Supplemental Figure 2). As shown in Supplemental Figure 2A, the estimated pAFR and White (pEUR) ancestry were used to define, in a conventional meaning, the genetic ancestry of the donors and recipients. With simple thresholds, the individuals were categorized as AA if the pAFR was 0.6 or higher, White if the pEUR was 0.9 or higher, Asian if the pAFR plus pEUR was 0.1 or lower (and the proportion of East Asian [pASN] was 0.9 or higher), and Hispanic (i.e., admixed population with a spectral mixture of White, African, and American Indian ancestral components) otherwise (Supplemental Figure 2B).
APOL1 genotyping.
The G1 allele of APOL1 is represented by rs73885319 and rs60910145, two missense SNPs in almost perfect linkage disequilibrium, whereas the G2 allele is represented by a 6 bp microdeletion rs143830837 (or equivalently rs71785313) (2). The allele that does not carry any of these variants is hereafter referred to as G0. In the GOCAR cohort, the 3 allele-representing markers were imputed by the pipeline composed of SHAPIT (44) and IMPUTE2 (45) software packages (see section below). To ensure the quality of imputation, the posterior probability of an imputed genotype was required to be greater than 0.95; otherwise, the imputed genotype was considered as missing data. Among the 385 D-R pairs with genotype information, there were missing data in the APOL1 genotype for 29 recipients and 14 donors (Supplemental Table 3). In the CTOT cohort, fortunately, the 2 representative variants rs73885319 and rs71785313 were genotyped directly by the SNP array platform used, and thus the APOL1 genotype could be defined accordingly. The individuals genetically determined as White but carrying G1/G2 alleles, contradictory to the origin of the risk variants from African ancestry, were excluded (Supplemental Figure 1). In fact, some of the ancestry-of-origin–inconsistent APOL1 genotypes were later confirmed to be genotyping errors by PCR. Among the 122 D-R pairs with genotype information, there were missing data in the APOL1 genotype for 2 recipients and 2 donors due to a failed genotyping effort at these 2 variants (Supplemental Table 3).
APOL1 SNP–based mismatch score.
We evaluated the SNP-wise mismatches at the APOL1 locus for both cohorts following the procedures similar to those described in recent reports (46). First, genome-wide genotype imputation was performed for both cohorts. For GOCAR, the imputation was done by the pipeline composed of SHAPIT (44) and IMPUTE2 (45) software packages using the 1000 Genomes Project Phase I data (47) as a reference panel; while for CTOT, the imputation was done by the Michigan Imputation Server (https://imputationserver.sph.umich.edu) (48) using the Haplotype Reference Consortium (HRC) reference panel (Release 1.1; ref. 49). Second, at each SNP locus, a mismatch score of 1 for a D-R pair was assigned when the donor introduced any allele(s) that did not appear in the recipient, and a score of 0 otherwise. Third, the SNP-wise mismatch scores of SNPs within the range of the APOL1 locus (chromosome 22: 36649117–36663577) were summed as a measure of the total mismatch at the APOL1 locus, and then the raw values of the APOL1 mismatch score for each D-R pair was normalized by the IQR across D-R pairs within each cohort.
PBMC RNA-Seq data analysis for a subgroup of GOCAR patients.
The details of PBMC isolation for a subgroup of GOCAR patients for RNA-Seq experiments and the data analysis pipeline have been reported previously by our group (28). Briefly, total RNA was extracted from whole blood drawn from the transplant recipients before transplantation, and mRNA sequencing was performed on an Illumina HiSeq 4000 sequencer. Gene expression data were obtained from the NCBI’s GEO database (GEO GSE112927). In this study, we focused on the subgroup of 60 AA/H patients with genotype information available as well. Differential gene expression analysis was carried using an R package limma (50), comparing recipients carrying 1 or 2 copies of APOL1 risk alleles (n = 26) versus zero copies (n = 34), and comparing those with 1 copy (n = 14) versus zero copies (n = 34). DEGs were initially identified at a P value of less than 0.05. Biological functional pathways enriched for DEGs were determined by Fisher’s exact test at a P value of less than 0.05 using the “biological process” category in Gene Ontology (GO) resource (51) and pathways curated in several pathway databases (Kyoto Encyclopedia of Genes and Genomes [KEGG], ref. 52; Ingenuity Pathway Analysis [IPA; QIAGEN, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis]; BioCarta, ref. 53; Panther, ref. 54; Pathway Interaction Database [PID], ref. 55; REACTOME, ref. 56; and WikiPathways, ref. 57).
Panel of reactive T cell assay for a subgroup of CTOT patients.
Details and standardization of the IFN-γ ELISPOT assay have been published by our group before (40, 58). IFN-γ production by recipient PBMCs against isolated ex vivo–stimulated B cells from the respective donor, randomly chosen third party, and a standardized 6-donor panel were evaluated before transplantation. The results were respectively reported as donor-specific, third-party, and panel of reactive T cell (PRT) assay. ELISPOT data for a subgroup of CTOT AA/H recipients with 1 or 2 copies of the APOL1 G1/G2 alleles (n = 5) were compared data on individuals with the G0/G0 genotype (n = 15). The experimental procedures are described briefly as follows. Blood samples from recipients were collected in heparinized green-top tubes, and PBMCs were isolated by Ficoll separation at each site within 6 hours of collection and frozen using a standard operating procedure. Blood samples were obtained from LDs and processed in a similar manner. PBMCs or splenic cells obtained from DDs were sent to the Mount Sinai core laboratory, where they were processed and frozen. Recipient PBMCs (300,000 per well) were stimulated against the respective stimulator cells (100,000 per well) in triplicate. The resulting spots were counted with an ImmunoSpot computer-assisted ELISPOT image analyzer (Cellular Technology Ltd.). Results are shown as the mean number of IFN-γ spots per 300,000 recipient peripheral blood lymphocytes based on duplicate or triplicate measurements in a given assay.
DICE RNA-Seq data analysis.
To explore the expression of APOL1 risk alleles and associated gene signatures in various immune cell types, we used the RNA-Seq data generated by the DICE project (https://dice-database.org/). Access to the DICE data sets located in the NCBI’s database of Genotypes and Phenotypes (dbGaP) was requested (request 97206-2) and approved (dbGaP study accession number phs001703), and these data sets were analyzed in this study. A description of the data set has been detailed in the literature (27). Briefly, whole transcriptomic data were generated by bulk RNA-Seq from immune cell types isolated from leukapheresis samples provided by 91 healthy subjects. The cell types surveyed included 3 innate immune cell types (CD14hiCD16– classical monocytes, CD14–CD16+ nonclassical monocytes, and CD56dimCD16+ NK cells); 4 adaptive immune cell types (naive B cells, naive CD4+ T cells, naive CD8+ T cells, and naive Tregs); 6 CD4+ memory or more differentiated T cell types (Th1, Th1/Th17, Th17, Th2, follicular helper T cell [Tfh], and memory Tregs); and 2 activated cell types (naive CD4+ and CD8+ T cells that were stimulated ex vivo) (27). In this study, the analysis was mainly focused on a subgroup of 22 AA/H individuals. The gene expression data were measured as transcripts per million reads (TPM). Genes with mean a TPM of less than 1 across all samples were excluded from further analysis. Raw TPM expression profiles were log2-transformed by log2(TPM + 1), where a value of 1 was added to account for 0 values in TPM. DEGs between the group of individuals with 1 or 2 copies of APOL risk alleles (n = 5) and the group of individuals without any risk alleles (n = 17) were identified using the functions “contrasts.fit” and “eBayes” implemented in an R package limma (version 3.38.3; ref. 59). Genes with a P value of less than 0.05 were considered nominally significant. Pathway enrichment analysis for DEGs was performed using clusterProfiler (60), based on the KEGG pathway database (52). A P value of 0.05 in enrichment analysis was considered nominally significant. An adjusted P value using the Benjamini-Hochberg method (61) and a q value quantifying the FDR using an R package q value (62) were also provided for multiple hypothesis testing control.
Generation and analysis of scRNA-Seq data from 4 GOCAR recipients.
Within our GOCAR cohort, we generated scRNA-Seq data from PBMCs collected before transplantation from 4 AA recipients with a known APOL1 genotype. Among these 4 recipients (all allografts from DDs), 2 with APOL1 risk alleles (G1/G0 and G1/G2) later developed recurrent TCMR and graft loss, while the other 2 with the G0/G0 genotype had no TCMR or graft loss during the study follow-up (Supplemental Table 12). PBMCs were isolated from ethylenediaminetetraacetic acidanticoagulated blood of the recipients using a Ficoll-Hypaque density solution according to standard density-gradient centrifugation methods. The viability of all PBMC samples assessed exceeded 80%. scRNA-Seq libraries were prepared according to the Chromium Single Cell 3′ Reagents Kit V3 User’s Guide (10x Genomics).
The generated scRNA-Seq data were deposited in the NCBI’s GEO database (GEO GSE182916). Raw sequencing reads were aligned using CellRanger (version 5.0.0) (10x Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). Cell QC, intergradation, and clustering were performed using Seurat (version 3.1.5; ref. 63). Genes expressed in fewer than 3 cells were filtered. Cells expressing fewer than 200 genes or with more than 20% of the reads coming from mitochondrial genes were considered poor quality and removed. Cells expressing more than 5000 genes were considered doublets and removed. Cells from the 4 recipients were integrated using the “IntegrateData” function with the first 30 dimensions. Unsupervised clustering of cells was done with the “FindClusters” function using the first 15 principal components (PCs) with a resolution of 0.8. Cell types were identified using classic immune markers as described in other PBMC studies (Supplemental Table 15 and ref. 64). To confirm the APOL1 genotype, short reads generated from the 4 single-cell samples were aligned to the human reference genome (GRCh37) by STAR (2.7.5b; ref. 65). The genotype of APOL1 was identified using the “mpileup” command from bcftools (version 1.9) based on the reads mapped to the APOL1 locus (66). G1 and G2 alleles were identified on the basis of the genotyped variants described previously (22). DEGs between recipients with and without APOL1 risk alleles in each cell type were identified using the “FindMarkers” function from the Seurat package with the default testing method, Wilcoxon rank-sum test. Genes with Bonferroni’s adjusted P values of 0.01 or less were considered significant. Pathway enrichment analysis for DEGs was performed the same way as described above for the DICE data analysis.
scRNA-Seq data analysis for 2 patients with ESRD.
We used scRNA-Seq data from PBMC samples collected from 2 patients with ESRD (GEO GSE162470). These data were downloaded and subjected to analysis similar to that described above for the GOCAR recipients.
APOL1 overexpression podocytes.
Human Apol1 was amplified by PCR using cDNA synthesized with human podocyte mRNA as a template. The FLAG peptide sequence was incorporated into the antisense primer containing a terminal Xbal site. The sense primer contained a terminal EcoRV site. The primer sequence was as follows: forward, GATATCATGGAGGGAGCTGCTTTGCTGAGAG; reverse, TCTAGATCACTTGTCGTCATCGTCTTTGTAGTCCAGTTCTTGGTCCGCCTGC.
PCR-amplified products were cloned into the pGEM-T vector (Promega, A3600). The APOL1 sequence was confirmed by DNA sequencing using the T7 primer. cDNAs of APOL1 were released from T vectors with EcorV and XbaI restriction enzymes and inserted into EcorV- and XbaI-digested PCDNA4B vectors. Lentiviral transduction and stably infected human podocyte lines were created. APOL1-FLAG expression was confirmed. A mouse macrophage cell line was used as a negative control for APOL1 expression, since mouse cells do not express APOL1.
Western blotting.
PBMCs from a leukapheresis sample or podocyte cell lines were lysed with a buffer containing 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 5% glycerol, a protease inhibitor mixture, and tyrosine and serine/threonine phosphorylation and phosphatase inhibitors. Lysates were subjected to immunoblot analysis using anti-APOL1 antibody (Abcam, catalog ab231523) and rabbit polyclonal anti-GAPDH antibody (Cell Signaling Technology, catalog 5174).
Statistics.
Descriptive statistics (mean, SD, median, and range) were used to summarize the baseline characteristics of the GOCAR and CTOT cohorts. When comparing the baseline characteristics between groups of recipients carrying different numbers of APOL1 risk alleles, Fisher’s exact test was used to calculate the P value for categorical variables, ANOVA for continuous variables, and the Kruskal-Wallis test for ordinal variables. A Kaplan-Meier plot was used to visualize and compare the death-censored graft survival curves between groups of recipients carrying different numbers of APOL1 risk alleles, and a log-rank test was used to calculate the P value. The association of time-to-event outcome (DCAL) with risk factors was evaluated with Cox regression. The association of dichotomous outcomes (different TCMR outcomes) with risk factors was evaluated with logistic regression. The association of the longitudinal creatinine levels with risk factors was evaluated with linear mixed models, implemented in the R package lme4 (67). The fixed-effect meta-analysis of the GOCAR and CTOT results was conducted using the R package metafor (68). In each regression analysis, the samples with missing data in relevant covariates were omitted. A 2-sided P value of less than 0.05 was considered statistically significant unless otherwise specified. These statistical procedures were implemented in R (69).
Study approval.
For the GOCAR study, written informed consent was obtained from all participants from the individual clinical sites at the time of enrollment in the original study. IRB approval was obtained from all participating institutions (see list below for GOCAR and CTOT). For the CTOT study, written informed was obtained from all participants from the individual clinical sites at the time of enrollment in the original study. IRB approval was obtained from all participating institutions. Consent included the use of deidentified genetic data for research purposes and retrospective data reporting.
Participating institutions for GOCAR: Icahn school of Medicine at Mount Sinai, New York, New York, USA; University of Sydney, Westmead Hospital, Sydney, New South Wales, Australia; University of Wisconsin-Madison, Madison, Wisconsin, USA; Northwestern University, Northwestern Memorial Hospital, Chicago, Illinois, USA; University of Michigan, Ann Arbor, Michigan, USA; Massachusetts General Hospital–Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
Participating institutions for CTOT: Icahn school of Medicine at Mount Sinai; University Hospitals of Cleveland, Cleveland Clinic Foundation, Cleveland, Ohio, USA; Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, USA; Yale University School of Medicine, New Haven, Connecticut, USA; University of Manitoba, Children’s Hospital of Winnipeg, Winnipeg, Manitoba, Canada; Emory University Hospital, Emory Children’s Center, Atlanta, Georgia, USA.
Author contributions
ZZ, JCH, PJO, PSH, BM and MCM designed the study and conducted research. ZS, ZZ, ZY, KH, HC, and WZ performed bioinformatics analyses. QL, RL, and CW performed experiments. KB, MP, and JF prepared and processed samples. MCM, PSH, PC, CJH, SI, and BM provided reagents. KC, SGC, ZZ, ZS, and MCM performed clinical data analyses. FS, IWG, and RBC performed histopathology studies. ZZ, ZS, and PSH, and MCM wrote the manuscript. All authors contributed to the editing of the manuscript.
Supplementary Material
Acknowledgments
We thank all of the patients, the donors, and their families, the participating clinical sites, and the investigators in the GOCAR and CTOT studies. MCM and ZZ acknowledge the Translational Collaborative Research Initiative Grant “Non-HLA donor-recipient differences and allograft survival,” provided by the Department of Medicine, and the computational resources and staff expertise provided by the Scientific Computing department at the Icahn School of Medicine at Mount Sinai. MCM acknowledges funding from the American Heart Association (AHA) (15SDG25870018), the NIH (R01DK122164), and pilot funding from the CTOT-19 study (PSH, principal investigator; NIH grant U01AI063594) to study non-HLA D-R genetic differences. The data reported here are substudies of the GOCAR study (BM, principal investigator), supported by NIH grant U01AI070107-03, and the CTOT-01 study (PSH, principal investigator), supported by NIH grant U01AI63594-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors sincerely thank the CTOT/GOCAR site investigators and staff for their efforts in collecting samples from the study participants. KH is partially supported by grants from the National Institute of Environmental Health Sciences (NIEHS), NIH (1R01ES029212-01); the National Institute on Aging (NIA), NIH (AG058635); and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH (U24DK062429 and DK106593). Funding for the study “Impact of genetic polymorphisms on human immune cell gene expression” (DICE data) was provided by the NIAID (R24-AI108564). Data from the study were provided by Pandurangan Vijayanand on behalf of his collaborators at the La Jolla Institute for Allergy and Immunology.
Version 1. 09/09/2021
In-Press Preview
Version 2. 11/15/2021
Electronic publication
Footnotes
Conflict of interest: BM reports personal fees from ITBMed Biopharmaceuticals and holds stock in RenalytixAI. WZ reports personal fees from RenalytixAI. In addition, BM and WZ are inventors on the following patents: “Method for identifying kidney allograft recipients at risk for chronic injury” (US Provisional Patent Application no. 27527-0134P01, serial number 61/951,651); “Methods for diagnosing risk of renal allograft fibrosis and rejection (miRNA)” (US Provisional Patent Application no. 20190345556); “Method for diagnosing subclinical acute rejection by RNA-Seq analysis of a predictive gene set” (US Provisional Patent Application no. WO2015200887A2); and “Pretransplant prediction of post-transplant acute rejection” (US Provisional Patent Application no. WO2017100259). RBC reports personal fees from Shire/Takeda, CSL Behring, Alexion, and eGenesis; in addition, RBC reports royalties from Takeda Pharmaceuticals for Entyvio (for inflammatory bowel disease). KH receives financial compensation from Sema4 (an Icahn School of Medicine at Mount Sinai spin-off company). Sema4 is currently majority owned by the Icahn School of Medicine at Mount Sinai.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(22):e146643.https://doi.org/10.1172/JCI146643.
See the related Commentary at APOL1 risk variants in kidney transplantation: a modulation of immune cell function.
Contributor Information
Zhongyang Zhang, Email: zhongyang.zhang@mssm.edu.
Zeguo Sun, Email: zeguo.sun@mssm.edu.
Jia Fu, Email: jia.fu@mssm.edu.
Qisheng Lin, Email: linqisheng074@hotmail.com.
Khadija Banu, Email: khadija.banu@yale.edu.
Kinsuk Chauhan, Email: kinsuk.chauhan@mssm.edu.
Marina Planoutene, Email: marina.planoutene@mssm.edu.
Chengguo Wei, Email: chengguo.wei@mssm.edu.
Fadi Salem, Email: fadi.salem@mountsinai.org.
Zhengzi Yi, Email: zhengzi.yi@mssm.edu.
Ruijie Liu, Email: ruijie.liu@mssm.edu.
Paolo Cravedi, Email: paolo.cravedi@mssm.edu.
Haoxiang Cheng, Email: haoxiang.cheng@mssm.edu.
Ke Hao, Email: kehao11@gmail.com.
Philip J. O’Connell, Email: philip.oconnell@sydney.edu.au.
Shuta Ishibe, Email: shuta.ishibe@yale.edu.
Weijia Zhang, Email: weijia.zhang@mssm.edu.
Steven G. Coca, Email: steven.coca@mssm.edu.
Ian W. Gibson, Email: IGibson@dsmanitoba.ca.
Robert B. Colvin, Email: colvin@helix.mgh.harvard.edu.
Peter S. Heeger, Email: peter.heeger@mssm.edu.
Madhav C. Menon, Email: madhav.menon@mssm.edu.
References
- 1.Shah S, et al. APOL1 high-risk genotypes and renal transplantation. Clin Transplant. 2019;33(6):e13582. doi: 10.1111/ctr.13582. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, et al. APOL1 G2 risk allele-clarifying nomenclature. Kidney Int. 2017;92(2):518–519. doi: 10.1016/j.kint.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsa A, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369(23):2183–2196. doi: 10.1056/NEJMoa1310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckerman P, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med. 2017;23(4):429–438. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruggeman LA, et al. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol. 2016;27(12):3600–3610. doi: 10.1681/ASN.2015111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar V, et al. Role of apolipoprotein L1 in human parietal epithelial cell transition. Am J Pathol. 2018;188(11):2508–2528. doi: 10.1016/j.ajpath.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reeves-Daniel AM, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11(5):1025–1030. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedman BI, et al. APOL1 genotype and kidney transplantation outcomes from deceased African American donors. Transplantation. 2016;100(1):194–202. doi: 10.1097/TP.0000000000000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman BI, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant. 2015;15(6):1615–1622. doi: 10.1111/ajt.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao PS, et al. A comprehensive risk quantification score for deceased donor kidneys: the kidney donor risk index. Transplantation. 2009;88(2):231–236. doi: 10.1097/TP.0b013e3181ac620b. [DOI] [PubMed] [Google Scholar]
- 12.Julian BA, et al. Effect of replacing race with apolipoprotein l1 genotype in calculation of kidney donor risk index. Am J Transplant. 2017;17(6):1540–1548. doi: 10.1111/ajt.14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zwang NA, et al. APOL1-Associated end-stage renal disease in a living kidney transplant donor. Am J Transplant. 2016;16(12):3568–3572. doi: 10.1111/ajt.14035. [DOI] [PubMed] [Google Scholar]
- 14.Santoriello D, et al. Donor APOL1 high-risk genotypes are associated with increased risk and inferior prognosis of de novo collapsing glomerulopathy in renal allografts. Kidney Int. 2018;94(6):1189–1198. doi: 10.1016/j.kint.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee BT, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant. 2012;12(7):1924–1928. doi: 10.1111/j.1600-6143.2012.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freedman BI, et al. APOL1 long-term kidney transplantation outcomes network (APOLLO): design and rationale. Kidney Int Rep. 2020;5(3):278–288. doi: 10.1016/j.ekir.2019.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart A, et al. OPTN/SRTR 2015 annual data report: kidney. Am J Transplant. 2017;17(suppl 1):21–116. doi: 10.1111/ajt.14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taber DJ, et al. African-American race modifies the influence of tacrolimus concentrations on acute rejection and toxicity in kidney transplant recipients. Pharmacotherapy. 2015;35(6):569–577. doi: 10.1002/phar.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padiyar A, et al. Influence of African-American ethnicity on acute rejection after early steroid withdrawal in primary kidney transplant recipients. Transplant Proc. 2010;42(5):1643–1647. doi: 10.1016/j.transproceed.2010.02.081. [DOI] [PubMed] [Google Scholar]
- 20.Crowson CN, et al. Lymphocyte-depleting induction therapy lowers the risk of acute rejection in African American pediatric kidney transplant recipients. Pediatr Transplant. 2017;21(1):e12823. doi: 10.1111/petr.12823. [DOI] [PubMed] [Google Scholar]
- 21.Liu A, et al. Racial disparity in kidney transplant survival relates to late rejection and is independent of steroid withdrawal. Clin Transplant. 2018;32(9):e13381. doi: 10.1111/ctr.13381. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, et al. Genome-wide non-HLA donor-recipient genetic differences influence renal allograft survival via early allograft fibrosis. Kidney Int. 2020;98(3):758–768. doi: 10.1016/j.kint.2020.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faddoul G, et al. Analysis of biomarkers within the initial 2 years posttransplant and 5-year kidney transplant outcomes: results from clinical trials in organ transplantation-17. Transplantation. 2018;102(4):673–680. doi: 10.1097/TP.0000000000002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connell PJ, et al. Biopsy transcriptome expression profiling to identify kidney transplants at risk of chronic injury: a multicentre, prospective study. Lancet. 2016;388(10048):983–993. doi: 10.1016/S0140-6736(16)30826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W, et al. A peripheral blood gene expression signature to diagnose subclinical acute rejection. J Am Soc Nephrol. 2019;30(8):1481–1494. doi: 10.1681/ASN.2018111098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levey AS, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmiedel BJ, et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175(6):1701–1715. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, et al. Pretransplant transcriptomic signature in peripheral blood predicts early acute rejection. JCI Insight. 2019;4(11):e127543. doi: 10.1172/jci.insight.127543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poggio ED, et al. Panel of reactive T cells as a measurement of primed cellular alloimmunity in kidney transplant candidates. J Am Soc Nephrol. 2006;17(2):564–572. doi: 10.1681/ASN.2005030293. [DOI] [PubMed] [Google Scholar]
- 30.Okamoto K, et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol. 2018;1:188. doi: 10.1038/s42003-018-0188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Udler MS, et al. Effect of genetic African Ancestry on eGFR and kidney disease. J Am Soc Nephrol. 2015;26(7):1682–1692. doi: 10.1681/ASN.2014050474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vyas DA, et al. Hidden in plain sight - reconsidering the use of race correction in clinical algorithms. N Engl J Med. 2020;383(9):874–882. doi: 10.1056/NEJMms2004740. [DOI] [PubMed] [Google Scholar]
- 33.Haas Pizarro M, et al. Glomerular filtration rate estimated by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation in type 1 diabetes based on genomic ancestry. Diabetol Metab Syndr. 2020;12(1):71. doi: 10.1186/s13098-020-00578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monajemi H, et al. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002;79(4):539–546. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 35.Page NM, et al. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics. 2001;74(1):71–78. doi: 10.1006/geno.2001.6534. [DOI] [PubMed] [Google Scholar]
- 36.O’Toole JF, et al. The cell biology of APOL1. Semin Nephrol. 2017;37(6):538–545. doi: 10.1016/j.semnephrol.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shetty AA, et al. COVID-19–associated glomerular disease. J Am Soc Nephrol. 2021;32(1):33–40. doi: 10.1681/ASN.2020060804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blazer AD, Clancy RM. ApoL1 and the immune response of patients with systemic lupus erythematosus. Curr Rheumatol Rep. 2017;19(3):13. doi: 10.1007/s11926-017-0637-9. [DOI] [PubMed] [Google Scholar]
- 39.Vajgel G, et al. Effect of a single apolipoprotein L1 gene nephropathy variant on the risk of advanced lupus nephritis in Brazilians. J Rheumatol. 2020;47(8):1209–1217. doi: 10.3899/jrheum.190684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hricik DE, et al. Interferon gamma ELISPOT testing as a risk-stratifying biomarker for kidney transplant injury: results from the CTOT-01 Multicenter Study. Am J Transplant. 2015;15(12):3166–3173. doi: 10.1111/ajt.13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genomes Project Consortium. et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexander DH, et al. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delaneau O, et al. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9(2):179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 45.Schork NJ, et al. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reindl-Schwaighofer R, et al. Contribution of non-HLA incompatibility between donor and recipient to kidney allograft survival: genome-wide analysis in a prospective cohort. Lancet. 2019;393(10174):910–917. doi: 10.1016/S0140-6736(18)32473-5. [DOI] [PubMed] [Google Scholar]
- 47.Delaneau O, Marchini J. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. 2014;5:3934. doi: 10.1038/ncomms4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Consortium HR. A reference panel of 64,976 haplotypes for genotype imputation. Nature Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reich D, et al. Reconstructing Native American population history. Nature. 2012;488(7411):370–374. doi: 10.1038/nature11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang DW, et al. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protoc. 2008;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 52.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimura D. BioCarta. Biotech Softw Internet Rep. 2001;2(3):117–120. doi: 10.1089/152791601750294344. [DOI] [Google Scholar]
- 54.Thomas PD, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaefer CF, et al. PID: the pathway interaction database. Nucleic Acids Res. 2009;37(suppl_1):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu G, Haw R. Functional interaction network construction and analysis for disease discovery. Methods Mol Biol. 2017;1558:235–253. doi: 10.1007/978-1-4939-6783-4_11. [DOI] [PubMed] [Google Scholar]
- 57.Kelder T, et al. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2011;40(d1):D1301–D1307. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashoor I, et al. Standardization and cross validation of alloreactive IFNγ ELISPOT assays within the clinical trials in organ transplantation consortium. Am J Transplant. 2013;13(7):1871–1879. doi: 10.1111/ajt.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 62.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng GXY, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bates D, et al. Fitting linear mixed-effects models Usinglme4. J Stat Softw. 2015;67(1):1–48. [Google Scholar]
- 68.Viechtbauer W. Conducting meta-analyses inRwith themetaforPackage. J Stat Softw. 2010;36(3):1–48. [Google Scholar]
- 69. R Foundation for Statistical Computing. Version 4.1.1. The R Foundation; 2021. https://www.r-project.org/ Accessed September 15, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.