

Abstract
Alpelisib selectively inhibits the p110α catalytic subunit of PI3Kα and is approved for treatment of breast cancers harboring canonical PIK3CA mutations. In head and neck squamous cell carcinoma (HNSCC), 63% of PIK3CA mutations occur at canonical hotspots. The oncogenic role of the remaining 37% of PIK3CA noncanonical mutations is incompletely understood. We report a patient with HNSCC with a noncanonical PIK3CA mutation (Q75E) who exhibited a durable (12 months) response to alpelisib in a phase II clinical trial. Characterization of all 32 noncanonical PIK3CA mutations found in HNSCC using several functional and phenotypic assays revealed that the majority (69%) were activating, including Q75E. The oncogenic impact of these mutations was validated in 4 cellular models, demonstrating that their activity was lineage independent. Further, alpelisib exhibited antitumor effects in a xenograft derived from a patient with HNSCC containing an activating noncanonical PIK3CA mutation. Structural analyses revealed plausible mechanisms for the functional phenotypes of the majority of the noncanonical PIK3CA mutations. Collectively, these findings highlight the importance of characterizing the function of noncanonical PIK3CA mutations and suggest that patients with HNSCC whose tumors harbor activating noncanonical PIK3CA mutations may benefit from treatment with PI3Kα inhibitors.
Keywords: Oncology
Keywords: Head and neck cancer

Introduction
Head and neck squamous cell carcinomas (HNSCCs) are generally characterized by a high mutational load and frequent copy number alterations (1). PIK3CA is the most commonly mutated oncogene in HNSCC, with mutations detected in 13.7% of cases in The Cancer Genome Atlas (TCGA, Firehose Legacy). PIK3CA encodes p110α, the catalytic subunit of class 1A PI3K. When aberrantly activated, PI3K stimulates multiple downstream pathways, leading to unregulated cellular proliferation, survival, and migration, thereby contributing to cancer progression (2).
Of the 97 tumors with PIK3CA mutations reported in TCGA for HNSCC tumors, 63% occur at 1 of 3 “hotspot” locations in the p110α subunit, E542, E545, and H1047, collectively referred to as canonical mutations. E542 and E545 are located in the helical domain and H1047 resides in the kinase domain (3, 4). Activation of p110α by these canonical mutations induces signaling through the PI3K/AKT/mTOR pathway (5). The remaining 37% of HNSCC-associated PIK3CA mutations are distributed throughout the p110α subunit and are collectively known as noncanonical mutations (4). Although canonical mutations are known to activate PI3K pathway signaling, the biological and biochemical impact of the 32 noncanonical mutations detected in HNSCC are incompletely understood. Of note, 10 of the 32 noncanonical mutations are unique to HNSCC and have not been identified in other cancers.
Several studies have characterized the functional consequences of noncanonical PIK3CA mutations by various platforms, including a subset of the 32 mutations found in HNSCC. Ten of 11 noncanonical PIK3CA mutations identified in various human cancers were found to induce transformation in primary chicken embryo fibroblasts, 4 of which occur in HNSCC (K111N, N345K, C420R, and M1043V; ref. 3). Five colorectal cancer–associated noncanonical PIK3CA mutations, 1 of which is also found in HNSCC (C420R), showed increased in vitro lipid kinase activity relative to WT p110α and promoted loss of contact inhibition and anchorage-independent growth in NIH3T3 cells (6). We previously assessed the functional impact of 21 HNSCC-associated noncanonical PIK3CA mutations in a murine pro-B cell line (Ba/F3) and a human breast epithelial cell line (MCF10A; refs. 4, 5). Using a growth factor/cytokine withdrawal assay, 19 of the 21 mutants showed a gain of function in either Ba/F3 or MCF10A or both.
Clinical trials have tested the possibility that tumors harboring canonical PIK3CA mutations are sensitive to PI3Kα-targeted agents. A recent phase III trial found that alpelisib (BYL719), a selective p110α inhibitor, in combination with fulvestrant, a hormone receptor antagonist, prolonged progression-free survival among patients with hormone receptor–positive/human epidermal growth factor receptor 2–negative (HER2-negative) breast cancer. Clinical benefit was found in patients whose tumors contained canonical PIK3CA mutations, or 1 of 3 frequently occurring noncanonical mutations (C420R, Q546E, and Q546K; ref. 7) that exhibited oncogenic activity in preclinical models (8). These results led to FDA approval of alpelisib in 2019. PI3Kα-targeted inhibitors are under clinical investigation in HNSCC as monotherapy or combinational therapy. Most ongoing trials are enrolling unselected patient populations, but one phase II trial requires genetic alterations in the PI3K pathway for enrollment (ClinicalTrials.gov NCT03292250), and a preoperative window study is selecting patients with HPV-positive HNSCC, a naturally biomarker-enriched population where genomic activation of PI3K due to mutation or amplification has been reported in approximately half the patients with HNSCC who are HPV positive (ClinicalTrials.gov NCT03601507; ref. 1).
Here, we report the remarkable response of a patient with recurrent and metastatic HNSCC to alpelisib. This patient’s tumor contained a noncanonical PIK3CA mutation, Q75E, which has not been previously evaluated in any preclinical model, to our knowledge. We profiled the functional and phenotypic impact of all 32 noncanonical PIK3CA mutations found in human HNSCC, including Q75E, using a serum-dependent HNSCC model, which we previously reported as an approach to identify oncogenic “driver” mutations (9). We determined that the activating or nonactivating properties of individual PIK3CA mutations were independent of cell lineage by assessing each mutation in multiple cell line models. The oncogenic properties of each mutation were also assessed using colony formation and cell migration assays. To further determine the translational significance of our findings, we tested the antitumor efficacy of alpelisib in relevant HNSCC preclinical models, including a patient-derived xenograft (PDX) harboring a representative activating noncanonical PIK3CA mutation. Our collective findings suggest that the majority of noncanonical PIK3CA mutations encode oncogenic activated proteins, which may serve as predictive biomarkers to PI3Kα-targeted therapies.
Results
Clinical response to alpelisib of a patient with HNSCC harboring the noncanonical PIK3CA mutation Q75E.
FDA approval of alpelisib in breast cancer is restricted to patients whose tumors harbor specified activating PIK3CA mutations. In recurrent or metastatic HNSCC, alpelisib is currently being evaluated in one arm of the Translational biomarker driven UMbrella Project (TRIUMPH, ClinicalTrials.gov NCT03292250). This phase II umbrella trial assigns subjects to 1 of 5 molecularly defined therapeutic regimens based on the results of comprehensive genomic profiling of the tumor (10, 11). A 55-year-old man with a history of smoking 40 packs a year presented with a right hard palate mass in 2017, which, upon biopsy, was revealed to be an HPV-negative (p16 negative) squamous cell carcinoma. Although the patient responded to an initial treatment of definitive concurrent chemoradiation therapy (CRT) with cisplatin, he experienced a local recurrence 6 months after completing CRT and lung metastasis shortly thereafter. His recurrent/metastatic HNSCC was first treated with 5-fluorouracil (5-FU) (1200 mg/m2 from day 1 to day 4) and cisplatin (80 mg/m2 every 3 weeks), but he progressed after 2 cycles. Next-generation sequencing of his recurrent tumor for 244 genes known to be somatically altered in HNSCC revealed a noncanonical PIK3CA mutation, Q75E, which had not been previously reported or characterized in any cellular platform, to our knowledge (Figure 1A and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI150335DS1). In addition to this PIK3CA mutation, 3 single nucleotide variants (SNVs) were also detected (FBXW7, TGFBR2, and ATR). Notably, mutations in NOTCH1, which could affect the response to alpelisib (12, 13), were not detected. The patient was enrolled in the TRIUMPH trial and assigned to receive alpelisib monotherapy (350 mg once daily) based on detection of the PIK3CA mutation in his tumor. He experienced a partial response with 73% tumor shrinkage based on Response Evaluation Criteria in Solid Tumors (RECIST) criteria after 2 months on treatment (Figure 1, B and C). The patient was treated for a total of 12 months with single-agent alpelisib until his disease progressed, and he was switched to durvalumab, an anti–PD-L1 antibody that is approved by the FDA for certain types of bladder cancer and lung cancer (14, 15). He is currently alive with stable disease on durvalumab, approximately 2 years after completing alpelisib monotherapy.
Figure 1. Clinical response to alpelisib in a patient with HNSCC harboring the noncanonical PIK3CA mutation Q75E.

(A) Clinical history of the alpelisib-treated patient with HNSCC. (B) Head and neck MRI showing the lesions (red arrows) in oral cavity. The representative images were obtained prior to treatment (left) and 2 months later (right). (C) Chest CT images were captured prior to treatment (left) and 2 months later (right). The well-defined pulmonary metastatic nodules (red arrows) were found diminished in diameter from 11 to 7 mm. The patient exhibited a partial response with 73% tumor shrinkage, based on RECIST criteria.
The Q75E PIK3CA mutation exhibits an activating phenotype in an HNSCC platform.
Given the potent clinical utility of alpelisib in this patient with HNSCC harboring a previously uncharacterized noncanonical PIK3CA mutation, we evaluated the activity of this mutant using an HNSCC platform that we previously developed to assess the ability of mutant proteins to sustain cell growth in low serum (Supplemental Table 2 and ref. 9). We engineered this HPV-negative HNSCC cell line platform (PCI-52 serum-dependent cells; hereafter PCI-52-SD1) for doxycycline-inducible exogenous overexpression of FLAG-tagged WT PIK3CA, a canonical PIK3CA mutant (E545K), the Q75E noncanonical PIK3CA mutant, or the negative control LUC. Immunoblotting with anti-FLAG was performed to confirm expression of the WT or mutant (MT) proteins (Figure 2A; see complete unedited blots in the supplemental material), and FLAG-expressing cells were cultured in normal FBS (10%) or low FBS (1%–2%) to assess serum-dependent cell growth (Figure 2B).
Figure 2. The Q75E PIK3CA mutation exhibits an activating phenotype in an HNSCC platform.

(A) Generation of isogenic models in PCI-52-SD1 cells. Immunoblot analysis of the indicated proteins using extracts from cells expressing LUC, WT PIK3CA, canonical E545K mutant, or noncanonical Q75E mutant, following growth in the presence or absence of doxycycline (Dox, 1 μg/mL) for 24 hours. GAPDH, loading control. The fold-changes of the ratio of pAKT/(total AKT) were quantified by densitometry 3 times independently and normalized to no Dox treatment (n = 3). Data are shown as mean ± SD. (B) Serum-dependence assays. Cells were cultured for 72 hours in medium containing Dox with either normal FBS (10%) or low FBS (1%–2%) followed by crystal violet staining. The cell growth rate in low FBS was normalized to the individual control of normal FBS (n = 6). Data are shown as the mean ± SD. The experiment was repeated 3 times with similar results. (C) Colony formation assays. Cells were cultured in the absence or presence of Dox for 3 weeks followed by crystal violet staining. Colonies were quantified using ImageJ and the relative colony counts were normalized to no Dox treatment (n = 3). Data are shown as the mean ± SD. (D) Cell migration assays. Cells were applied to Boyden chambers and incubated for 48 hours in the absence or presence of Dox followed by crystal violet staining. Images were taken from the bottom side of transmembrane chambers. Scale bar: 100 μm. The migrated cells were quantified using ImageJ and the relative cell migration level was normalized to no Dox treatment (n = 3). Data are shown as the mean ± SD. The experiments in C and D were repeated twice with similar results. In all bar graphs, *P < 0.05, **P < 0.01, ***P < 0.001, NS ≥ 0.05 for 1-tailed Student’s pairwise t test.
Comparison of results from HNSCC cells engineered to overexpress WT PIK3CA with the LUC control was used to assess the impact of PIK3CA gene amplification. Overexpression of WT PIK3CA led to a 1.59-fold increase in cell growth (Figure 2B) compared with LUC control cells. This finding supports an oncogenic role of PIK3CA gene amplification in patients with HNSCC tumors, as previously reported (16). Comparison of HNSCC cells overexpressing MT with WT PIK3CA was then used to determine the functional impact of PIK3CA mutations on serum-dependent cell growth. Because the exogenous MT and WT proteins were expressed at similar levels (Figure 2A), comparison of MT and WT engineered cells removed overexpression (amplification) as a confounding variable and allowed for direct assessment of the impact of specific mutations. Under low serum conditions, cells expressing the E545K canonical mutant demonstrated a 1.34-fold increase in cell growth relative to cells expressing WT (Figure 2B), similar to what was previously reported in Ba/F3 and MCF10A models (3). The Q75E mutant exhibited a 1.30-fold increase in cell growth in comparison with WT, comparable to the E545K canonical PIK3CA mutation.
Canonical PIK3CA mutations have been reported to activate the PI3K signaling pathway (5). Biochemical investigation of the PI3K/AKT signaling pathway showed that PCI-52-SD1 HNSCC cells overexpressing WT PIK3CA exhibited a 5.20-fold increase in the relative ratio of phospho-AKT (pAKT)/(total AKT) compared with LUC control cells, verifying that PIK3CA overexpression stimulated the PI3K signaling cascade (Figure 2A). Cells overexpressing the E545K canonical mutant showed a 2.73-fold increase, and cells expressing the Q75E noncanonical mutant showed a 2.34-fold increase in relative pAKT/(total AKT) level when compared with cells engineered to overexpress WT PIK3CA (Figure 2A), confirming that the Q75E mutant drives activation of the PI3K signaling pathway more strongly than WT PIK3CA.
We next assessed the impact of WT or MT PIK3CA on colony formation in the PCI-52-SD1 HNSCC cell line model. Doxycycline-induced overexpression of WT PIK3CA led to a 1.59-fold increase in colony formation relative to untreated cells, whereas induction of the E545K canonical mutant promoted a 4.52-fold increase in colony formation, confirming the oncogenic phenotype of this known canonical mutation (Figure 2C). The Q75E mutant promoted a 3.30-fold increase in colony formation, similar to E545K (Figure 2C). We also evaluated the ability of WT or MT PIK3CA to promote cell migration using Boyden chamber assays. Treatment with doxycycline resulted in only a negligible increase in migration of cells engineered to overexpress WT PIK3CA, whereas induction of the E545K and Q75E mutants led to 3.30-fold and 3.15-fold increases in migration, respectively (Figure 2D). To ensure that the impact on cell migration was independent of the effect of the activating mutants on proliferation, we repeated the experiment at an earlier time point (24 hours) to minimize the potential impact of the activating mutation on proliferation (Supplemental Figure 1, A and B). These collective findings indicate the noncanonical Q75E PIK3CA mutation detected in the HNSCC tumor of the patient with a clinical response to alpelisib has the properties of a “driver” oncogene.
Functional characterization of all 32 noncanonical PIK3CA mutations in the HNSCC serum-dependent model.
We identified 97 tumors with somatic PIK3CA mutations from the set of 530 HNSCC tumors in TCGA (Table 1). Canonical mutations (E542K, E545K, H1047R/L/Q) comprised 63% (61/97) of the PIK3CA mutations in this HNSCC cohort. The remaining 37% consisted of 32 distinct noncanonical mutations. Most noncanonical PIK3CA mutations were found in a single tumor (28/32, 87.5%), and 4 noncanonical PIK3CA mutations (E81K, E726K, Q546R, and M1043V) were detected in 2 cases each.
Table 1. The PIK3CA mutational landscape in HNSCC.
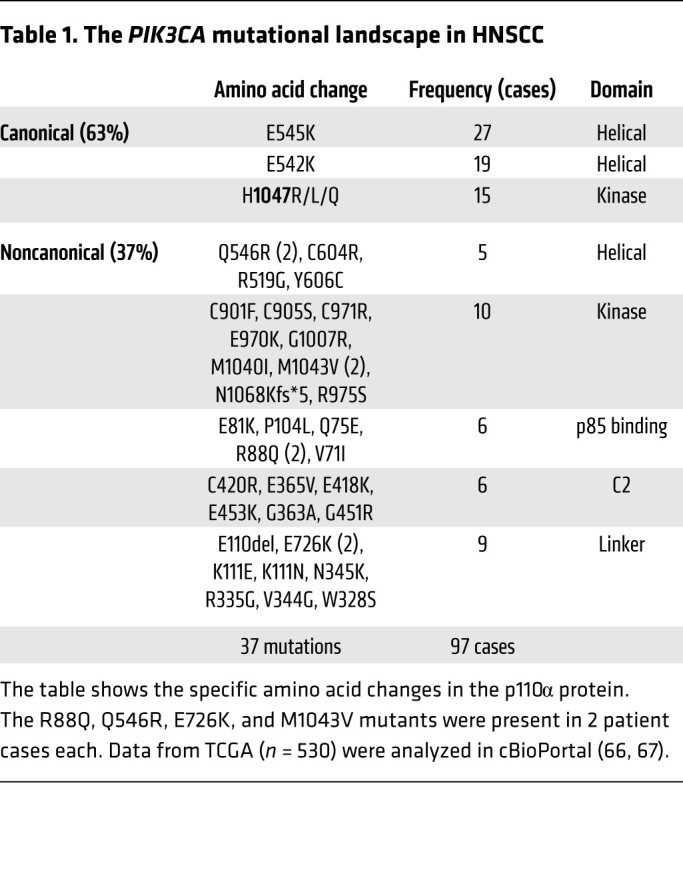
We next characterized the functional properties of each noncanonical PIK3CA mutation in our HNSCC serum-dependent platform to determine which, like Q75E, conferred a gain of function. Exogenous expression of each mutant protein in PCI-52-SD1 cells was confirmed by immunoblotting with anti-FLAG (Figure 3A; see complete unedited blots in the supplemental material), and cells were incubated in medium containing doxycycline with either normal FBS (10%) or low FBS (1%–2%) to assess serum-dependent cell growth. We observed that 22 of the 32 noncanonical mutants promoted enhanced cell growth in low serum ranging from 1.1-fold to 1.4-fold change relative to the WT PIK3CA (Figure 3B). The remaining 10 mutants promoted cell growth in low serum to an equal or lesser degree than WT PIK3CA (Figure 3B). Mutants were classified as activating if they exhibited significantly enhanced cell growth in low serum compared with WT PIK3CA; otherwise, they were annotated as nonactivating.
Figure 3. Functional characterization of all 32 noncanonical PIK3CA mutations in the HNSCC serum-dependent model.

(A) Generation of isogenic PCI-52-SD1 cells expressing the 32 noncanonical PIK3CA mutations. Immunoblotting with anti-FLAG was used to confirm expression of LUC, WT p110α, or the indicated p110α mutants following growth in the presence of Dox (1 ug/mL) for 24 hours; β-actin, loading control. (B) Serum-dependency assays. Cells were cultured for 72 hours in medium containing Dox (1 μg/mL) with either normal FBS (10%) or low FBS (1%–2%) followed by crystal violet assays. Shown is the growth of cells cultured in the medium containing low FBS relative to the individual control of normal FBS (n = 6) followed by normalization to WT PIK3CA control. The dashed line represents the significance limit set as the value equal to WT PIK3CA. Mutants were classified as activating if they exhibited statistically enhanced cell growth in low serum compared with WT PIK3CA. Data are represented as box and whiskers. The whiskers go down to the minimum and up to the maximum value and plot each individual value as a point superimposed on the graph. The box extends from the 25th to 75th percentiles. The lines within the boxes represent the median value. *P < 0.05, **P < 0.01, ***P < 0.001, NS ≥ 0.05 for 1-tailed Student’s pairwise t test. The experiment was repeated 3 times with similar results. (C) Heatmap indicating the concordance of functionality conferred by HNSCC-associated noncanonical mutations tested in different cell line models. The mutations were assigned as “activating” if they exhibited an activity significantly higher than WT; otherwise, the mutations were annotated as “nonactivating.”
The activation status of 21 of the 32 noncanonical, HNSCC-associated mutants was previously tested in cytokine/growth factor–dependent Ba/F3 and MCF10A cell line models (4, 5). Thus, we evaluated whether the functional impact of noncanonical mutations in our HNSCC model was concordant with the findings in these 2 models by completing assessment of the 11 previously uncharacterized noncanonical PIK3CA mutations in Ba/F3 and MCF10A cells as well as our HNSCC platform (Supplemental Table 3). All 22 of the activating noncanonical mutants we identified as activating in HNSCC cells (Figure 3B) were also activating in the MCF10A model, and 20 were activating in the Ba/F3 model (Figure 3C). The C971R and E365V PIK3CA mutants were activating in both the HNSCC and MCF10A models but nonactivating in Ba/F3 cells.
HNSCC can arise from infection with HPV or from tobacco carcinogen exposure (HPV-negative HNSCC). Our primary HNSCC functional genomics platform was derived from an HPV-negative cell line. We tested the functional properties of a subset (n = 16) of noncanonical mutants, including the 4 HNSCC-associated, noncanonical PIK3CA mutations (E81K, M1043V, R88Q, and Q546R) recorded by the TCGA in HPV-positive patients, using the cervical cancer–derived HPV-positive cell line HeLa in a different functional platform. In this platform, we measured fluorescently tagged FOXO1 that shuttles from the nucleus to the cytoplasm upon activation of the PI3K/AKT pathway (17). Although this model only screened 16 of the noncanonical PIK3CA mutations, we found a high concordance of the functional properties of 15 out of 16 noncanonical mutants tested in this platform with the results obtained in the other 3 models (HPV-negative HNSCC, Ba/F3, and MCF10A) (Figure 3C and Supplemental Figure 2A). Taken together, our results suggest that the activating properties of noncanonical PIK3CA mutations are generally cell lineage independent (Supplemental Figure 2B). Thus, these findings may have broad implications for any cancer harboring an activating noncanonical PIK3CA mutation, including HPV-negative and HPV-positive cancers.
Colony formation and migratory phenotypes of activating and nonactivating PIK3CA noncanonical mutations.
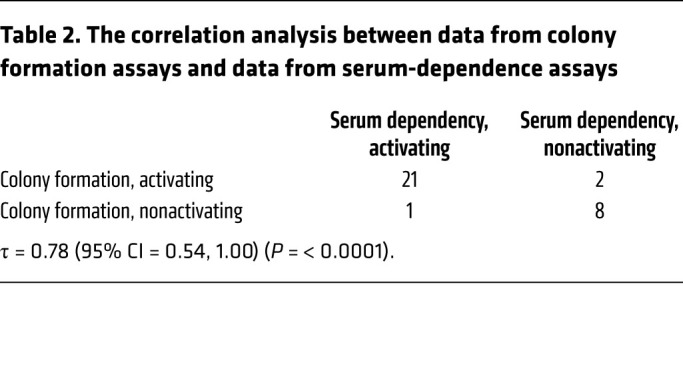
We next assessed the oncogenic properties of all 32 noncanonical PIK3CA mutations on cell transformation by assessing colony formation in standard serum condition (10% FBS). In total, 23 of the 32 noncanonical mutants increased colony formation compared with the WT control (Figure 4A and Supplemental Figure 3A). Of the 22 mutations that were classified as activating by our serum-dependent assay, 21 also demonstrated an activating phenotype in colony formation assay, showing that the results of the serum-dependent assay and the colony formation assay were highly concordant (Kendall’s correlation coefficient [τ] = 0.78 [95% CI = 0.54, 1], P < 0.0001; Table 2).
Figure 4. Colony formation and migratory phenotypes of activating and nonactivating noncanonical PIK3CA mutations.

(A) Cumulative results of colony formation assays of noncanonical PIK3CA mutations. The colonies were quantified using ImageJ, and the relative geometric means of colony counts in the presence of Dox were normalized to no Dox treatment (n = 3). Data were analyzed on a log-transformed scale and a 2-way ANOVA was performed to compute the fold-change conferred by an individual PIK3CA mutation (Dox versus no Dox). The dashed line represents the limit set by the upper 95% CI of the fold-changes for WT PIK3CA. The experiment was repeated twice with similar results. (B) Cumulative from the cell migration assays. Cell migration was quantified as described in A (n = 3). Mutants were classified as activating or nonactivating as in A. The dashed line represents the significance limit set at the upper 95% CI of the fold-changes for WT PIK3CA. The experiment was repeated twice with similar results. (C) The alignment between the serum-dependent assay and 2 phenotypic assays in HNSCC model.
Table 2. The correlation analysis between data from colony formation assays and data from serum-dependence assays.
Several studies have reported a role for the canonical PIK3CA mutations in promoting epithelial-mesenchymal transition (EMT) and cell invasion, an obligate step during tumor metastasis, through activation of the PI3K pathway (18, 19). Thus, we next evaluated the ability of the 32 noncanonical HNSCC mutants to promote cell migration using Boyden chamber assays. All 22 noncanonical mutants identified as activating in the serum-dependent assay demonstrated increased cell migration compared with controls (Figure 4B and Supplemental Figure 2B). Similarly, 8/10 noncanonical mutants identified as nonactivating in the serum-dependent assay promoted cell migration at levels comparable to that promoted by WT PIK3CA (Figure 4B and Supplemental Figure 3B). Results from 30 of the 32 noncanonical mutants concurred with results from the serum-dependent assay (Table 3). All the mutations classified as activating by the serum-dependent assay also increased cell migration (τ = 0.86 [95% CI = 0.68, 1], P < 0.0001) (Table 3). The significant alignment between the serum-dependent assay and the other 2 phenotypic assays reflects the broad oncogenic properties of activating noncanonical PIK3CA mutations (Figure 4C).
Table 3. The correlation analysis between data from cell migration assays and data from serum-dependence assays.

To investigate the biochemical impact of the noncanonical PIK3CA mutations, we evaluated activation of PI3K/AKT pathway after doxycycline induction of all 32 noncanonical mutants (Supplemental Figure 3C). Most mutants showing an activating phenotype in the serum-dependent assay also increased levels of pAKT relative to WT PIK3CA (τ = 0.63 [95% CI = 0.44, 0.83], P < 0.0001) (Supplemental Figure 3D). These findings suggest that activating noncanonical p110α mutants stimulate signaling through the PI3K pathway.
In summary, our systematic functional/phenotypic and biochemical profiling across a variety of cellular platforms distinguished activating from nonactivating noncanonical PIK3CA mutations (Supplemental Figure 3E).
Response to alpelisib in HNSCC preclinical models expressing noncanonical PIK3CA mutations.
We and others have reported that tumors harboring canonical PIK3CA mutations are more sensitive to PI3K pathway inhibition in preclinical models (20–22). Such observations led to the design of “basket” clinical trials and FDA approval of alpelisib in breast cancer (7). We previously determined that HNSCC PDXs exhibit greater similarity than cell line–derived xenografts to primary HNSCC tumors (23, 24). We also reported that a PDX derived from a patient with HNSCC with a canonical PIK3CA mutation (E542K) was sensitive to PI3K inhibitor therapy (20). To determine whether activating noncanonical PIK3CA mutations might serve as predictive biomarkers for treatment with PI3Kα inhibitors, mice bearing HNSCC PDX tumors harboring either WT PIK3CA or a representative, activating noncanonical PIK3CA mutation (M1043V) were treated with vehicle or alpelisib (Supplemental Table 4). In vehicle-treated mice, the pAKT/(total AKT) ratio was significantly higher in the mutant PDX compared with the WT PDX, indicating baseline in vivo activation of PI3K signaling by the M1043V noncanonical mutant protein (Supplemental Figure 4A; see complete unedited blots in the supplemental material). We selected the dosage of 25 mg/kg for the in vivo studies based on the published pharmacokinetic and pharmacodynamics properties of alpelisib in a PIK3CA-dependent mouse model (22). Alpelisib treatment resulted in modest, albeit statistically significant, inhibition of tumor growth in the M1043V PDX model (Figure 5A). Moreover, immunoblotting revealed significant downregulation of pAKT levels in the alpelisib-treated M1043V mutant tumors (Figure 5B; see complete unedited blots in the supplemental material). By contrast, tumor growth in the WT PDX model was not significantly affected by alpelisib treatment when compared with vehicle-treated controls (Figure 5C). Alpelisib treatment modestly decreased pAKT levels in WT PDX tumors, but to a lesser degree compared with the M1043V mutant PDX tumors (Figure 5D; see complete unedited blots in the supplemental material).
Figure 5. Response to alpelisib in HNSCC preclinical models expressing noncanonical PIK3CA mutations.

(A) Tumor growth curve of an HNSCC PDX (HN6851) harboring the noncanonical M1043V PIK3CA mutation. Mice were dosed with alpelisib (25 mg/kg, p.o.) daily for 21 days (n = 12 tumors per group). Data are shown as the mean ± SEM. For statistical assessment of tumor growth, the tumor volumes were compared between vehicle- and alpelisib-treated groups at multiple time points. *P < 0.05, **P < 0.01, ***P < 0.001 for 1-tailed Student’s pairwise t test. (B) Immunoblot analysis of AKT activation in the HN6851 PDX tumors. Tumor samples were collected 3 hours after the last dosing. AKT activation was detected by phosphorylation of S473; β-actin, loading control. The fold-change in the ratio of pAKT/(total AKT) expression in vehicle- and alpelisib-treated tumors was quantified by densitometry and normalized to the mean value in vehicle-treated tumors. ***P < 0.001 for 1-tailed Student’s pairwise t test. (C) Tumor growth curve of an HNSCC PDX (HN6431) with WT PIK3CA. Mice were dosed with alpelisib (25 mg/kg, p.o.) daily for 21 days (n = 12 tumors per group). Data were presented as indicated in A. (D) Immunoblot analysis of AKT activation in the HN6431 PDX tumors. Tumor samples were collected and analyzed as indicated in B. *P < 0.05 for 1-tailed Student’s pairwise t test. (E). Alpelisib sensitivity in colony formation assay. Isogenic PCI-52-SD1 expressing the indicated proteins were treated with vehicle (DMSO) or alpelisib (1, 3, 10 μM) for 3 weeks followed by crystal violet staining. The colonies were quantified using ImageJ (n = 3). Data are shown as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, NS ≥ 0.05 for 1-tailed Student’s pairwise t test. The experiment was repeated twice with similar results.
We also evaluated alpelisib sensitivity in an additional panel of representative activating or nonactivating noncanonical PIK3CA mutations using our engineered cell line models. Alpelisib demonstrated a dose-dependent abrogation of pAKT expression (Supplemental Figure 4B; see complete unedited blots in the supplemental material). Alpelisib treatment of HNSCC cells engineered to express the patient case–derived, activating noncanonical Q75E mutant or the activating noncanonical mutants M1043V or N345K resulted in modest induction of apoptosis (Supplemental Figure 4C) and dose-dependent inhibition of colony formation (Figure 5E). In contrast, colony formation in cells expressing the representative nonactivating PIK3CA mutant E81K were not significantly inhibited by alpelisib. Nonactivating PIK3CA mutants W328S and Y606C responded to alpelisib but to a lesser degree compared with activating noncanonical mutants (Figure 5E). These collective findings demonstrated antitumor activity of the FDA-approved PI3Kα inhibitor alpelisib in HNSCC in in vivo and in vitro models harboring activating noncanonical PIK3CA mutations. In contrast, this agent was ineffective in tumors containing WT PIK3CA or nonactivating noncanonical mutations, highlighting the implications for precision medicine.
Structural analysis of activating and nonactivating noncanonical PIK3CA mutations.
To understand whether the functional properties of different mutations could be predicted by the existing structures of PIK3CA, we mapped all 32 HNSCC-associated noncanonical mutations onto autoinhibited structures of the WT PI3Kα heterodimer that represent a spectrum of inactive conformational states captured collectively in crystal structures. This group includes 2 apo structures (Protein Data Bank [PDB] ID: 4OVU, 4L1B), 1 lipid substrate-bound (PDB ID: 4OVV), and 3 inhibitor-bound (PDB ID: 4L23, 4L2Y, 5XGI) structures. PI3Kα is an obligate heterodimer comprised of a p85-type regulatory subunit and the p110α catalytic subunit encoded by the PIK3R1-3 and PIK3CA genes, respectively. p110α contains 5 domains, including an adaptor binding domain (ABD), a Ras-binding domain (RBD), a C2 domain, a helical domain (HD), and the catalytic kinase domain (Figure 6A and ref. 25). The lipid kinase activity of PI3Kα is tightly controlled by the inter-SH2 (iSH2) and N-terminal SH2 (nSH2) domains of its regulatory subunit (Figure 6A and refs. 25–27). In the autoinhibited enzyme, the nSH2 domain forms additional interactions with p110α not present in the active conformation, while the iSH2 domain is more withdrawn from the membrane interface (28). Furthermore, polar and hydrophobic interactions between the activation loop of p110α and the nSH2/iSH2 domains of p85 are predicted to maintain the activation loop in an inactive conformation (28). This inhibition is released when PI3Kα binds to phosphorylated YxxM motifs on other proteins via its nSH2 domain, dislodging these inhibitory contacts, leading to global conformational changes that activate the kinase domain (25–27, 29). These conformational changes include movement of the ABD relative to the catalytic subunit, exposure of the iSH2/C2 interface, and exposure of the nSH2/HD interface, all of which happen in concert with binding of PI3Kα to membranes (30). Activating disease mutations in PI3Kα have been shown to facilitate at least one of these conformational changes or enhance interaction with membrane lipids (30–34).
Figure 6. Structural analysis of activating and nonactivating noncanonical PIK3CA mutations.

(A) Domain architecture of p85α and p110α with PIK3CA mutations mapped (activating mutations in red; nonactivating mutations in blue). (B) Activating non–kinase domain mutations, shown as red spheres, mapped onto an autoinhibited structure of PI3Kα (PDB ID: 4l23). (C) Schematic of PI3Kα domains with activating non–kinase domain mutations. (D–G) Representative activating mutations from mutation clusters. (H) Nonactivating non–kinase domain mutations, shown as blue spheres, mapped onto PDB ID: 4l23. (I) Schematic of PI3Kα domains with nonactivating non–kinase domain mutations. (J and K) Representative nonactivating mutations from mutation clusters. (L) Activating and nonactivating kinase domain mutations, shown as red and blue spheres, respectively, mapped onto PDB ID: 4l23, relative to the predicted membrane binding interface (red dashed line represents extension of the C-terminus as a function of N1068fs mutation). (M) Schematic of the kinase domain with activating and nonactivating kinase domain mutations. (N–Q) Representative activating and nonactivating kinase domain mutations from mutation clusters.
We mapped activating and nonactivating mutations to 4 of the 5 PIK3CA domains, including linkers between the ABD/RBD (linker 1), RBD/C2 (linker 2), and C2/HD (linker 3) and excluding the RBD itself (Figure 6A). Our initial analysis focused on mutations located outside of the kinase domain, which are distant from the lipid binding interface and the catalytic pocket. Activating non–kinase domain mutations largely clustered at the 3 interfaces previously shown to be compromised by mutations: ABD and linker 1/KD; iSH2 and C2; and nSH2 and HD (Figure 6, B and C). Mutations in the first cluster broke electrostatic interactions between the ABD and kinase domain (R88Q) or between the ABD and linker 1 (K111E/N) and disrupted hydrogen bonding between linkers 1 and 2 (E110del), destabilizing the ABD (Figure 6D and Supplemental Table 5). The patient with HNSCC–associated mutation Q75E falls within this cluster and forms only one direct polar contact with the backbone carbonyl of glycine 8, the first resolved N-terminal residue in p110α (PDB ID: 4L23, 4L2Y, 4L1B, 5XGI, 4OVU; Figure 6D and Supplemental Table 5). The preceding 7 N-terminal residues of the ABD are disordered in most of the PI3Kα crystal structures and it is possible that the loss of glutamine at position 75 further increases dynamics in this region by releasing glycine 8. The adjacent loop of the ABD, which interfaces with the kinase domain, may be a direct sensor of these dynamics, leading to increased kinase activity in the Q75E mutant. Interestingly, in 4OVV, Q75 is within hydrogen bonding distance of S7, which lies in a loop that forms a noncanonical second lipid binding site in PI3Kα (Figure 6E and Supplemental Table 5). This site has been hypothesized to anchor PI3K to the membrane, and increased dynamics in this region may further facilitate membrane binding, increasing kinase activity (28). Mutations in the second cluster eliminated electrostatic interactions (E418K, E453K), disrupted polar contacts and introduced charge repulsion (N345K, G451R), and destabilized a vast hydrophobic network within the C2 domain (V344G) at the interface of iSH2 and C2 domains (Figure 6F and Supplemental Table 5). Cluster 2 mutations weaken inhibitory contacts between p85α and p110α and may additionally position the iSH2 domain closer to the membrane to facilitate membrane binding, thereby activating PI3Kα. Interestingly, there seems to be a link between the ABD/linker 1 region and the iSH2/C2 interface as both G106V, located at the C-terminus of the ABD, and N345K, sandwiched between iSH2/C2 domains, simultaneously induce movement of ABD/linker 1 and disrupt the iSH2/C2 interface (30). Only one mutation mapped to the nSH2/HD interface, Q546R (Figure 6G). Q546R mimics frequently occurring mutations within the helical domain, E542K and E545K, by introducing a positive charge within the highly basic surface of the nSH2 domain. Q546R likely destabilizes this critical inhibitory interface while breaking inhibitory contacts between the nSH2 domain and the activation loop (28).
Our analysis revealed that nonactivating non–kinase domain mutations in general did not cluster and were dispersed throughout the C2 and helical domains, along with linkers 2 and 3 (W328S, R335G, G363A, R519G, Y606C; Figure 6, H and I), with the 2 most nonconservative mutations (R335G and R519G) mapping to the protein surface (Figure 6J and Supplemental Table 5). Two nonactivating mutations, V71I and E81K, did map to the ABD/linker 1 region (Figure 6, H and I). V71 is part of a hydrophobic network involving I69 and I102 of the ABD, and it is likely that the protein is able to accommodate the additional methyl group from isoleucine without disrupting the inactive structure (Figure 6K and Supplemental Table 5). It is somewhat surprising that E81K is also nonactivating because it spans the ABD/linker 1 interface with E81, forming a salt-bridge with K111 in 4 out of 6 structures analyzed (PDB ID: 4L23, 4L2Y, 4L1B 5XGI; Figure 6D and Supplemental Table 5). The K111E mutant, which we determined to be activating (Figure 3C), has also been shown to be activating in in vitro kinase activity and lipid binding assays (30), as well as cell-based signaling assays monitoring Akt phosphorylation (35).
Several activating kinase domain mutants gained a positive charge (E726K, E970K, C971R, N1068fs) and were positioned at the membrane binding interface (Figure 6, L and M, and Supplemental Table 5). E726K resides on a loop, which binds the secondary PIP2 molecule via E722 in 4OVV (Figure 6O and refs. 21, 36). Thus, not surprisingly, E726K is predicted to enhance interaction of the kinase domain with anionic membrane phospholipids (32), and it is likely that E970K and C971R act through a similar mechanism. N1068fs, a frameshift mutation that results in the insertion of basic residues at the C-terminus (37), likely enhances the interaction with lipid substrate at the membrane binding interface in the similar way as E726K (Figure 6N and Supplemental Table 5). The nonactivating effect of mutations such as C905S and R975S could be explained by a conservative side-chain substitution or localization to the protein surface, away from the membrane binding interface, respectively (Figure 6, O and P, and Supplemental Table 5). Interestingly, M1043V and M1040I, which are similar in amino acid substitution and localize to the kα11 helix of the regulatory arch, have opposing effects on PI3Kα activity in our assays (Figure 3D and Figure 6Q). M1043V reduces the bulkiness of the hydrophobic side chain and is believed to activate the enzyme by allowing the activation loop to adopt an extended conformation for catalysis (38). Although the side-chain of M1043 is oriented toward the activation loop, buried deep within the kinase domain, M1040 faces outward, and its mutation to isoleucine is less likely to affect the conformation of the activation loop (Figure 6Q and Supplemental Table 5).
Collectively, our structural analysis showed that, although less prevalent than canonical mutations, noncanonical PIK3CA mutations impinge on similar molecular mechanisms to activate PI3Kα signaling in cancer by targeting the autoinhibitory structural locks that under normal conditions restrain the basal activity of the PI3Kα holoenzyme. By breaking these locks, noncanonical mutations elevate PI3Kα signaling, consequently sensitizing patients to PI3Kα-targeted therapies. Thus, the noncanonical mutations we describe here constitute a potentially new predictor for the success of PI3Kα-targeted therapies in HNSCC.
Discussion
Studies with PI3K inhibitors in HNSCC have, to date, yielded conflicting results. The majority of clinical trials have enrolled patients independent of the PIK3CA status of their tumors. Class I pan-PI3K inhibitors were designed to inhibit all class PI3K isoforms (α/β/δ/γ). PX-866 was developed as an analog to wortmannin to limit adverse effects and increase potency. However, 2 phase II studies of PX-866 have reported limited clinical efficacy in HNSCC in combination with docetaxel (ClinicalTrials.gov NCT01204099) or cetuximab (ClinicalTrials.gov NCT01252628; refs. 39, 40). The BERIL-I phase II trial (ClinicalTrials.gov NCT01852292) showed modest improvement of progression-free survival after treatment with class I pan-PI3K inhibitor buparlisib (BKM120) plus paclitaxel in patients with HNSCC previously treated with platinum compared with paclitaxel plus placebo (41). A comparable proportion of patients in each treatment arm harbored PIK3CA mutations (11% in buparlisib plus paclitaxel vs. 13% in placebo plus paclitaxel). Thus, the impact of PI3K inhibition in HNSCC remains incompletely understood. The encouraging response of the patient reported here underscores the need for additional studies that incorporate the PIK3CA mutational status of the patient’s tumor.
Recent efforts have largely shifted toward the development and testing of isoform-selective inhibitors. A number of ongoing clinical trials are testing the possibility that tumors harboring PIK3CA mutations are sensitive to PI3Kα-targeted agents. A few studies have looked at PI3K inhibitors as single agents. A phase II trial (ClinicalTrials.gov NCT01737450) reported that buparlisib had limited antitumor activity independent of PIK3CA mutational status in heavily pretreated patients with HNSCC (42). GDC-0077 is a potent PI3Kα inhibitor and a mutant PI3Kα degrader and is being evaluated as a single agent in patients with locally advanced or metastatic PIK3CA-mutant solid tumors (ClinicalTrials.gov NCT03006172). A phase II MATCH trial is evaluating targeted therapy directed by genetic testing in patients with solid tumors, which includes investigating the PI3Kα, δ inhibitor copanlisib; the PI3Kβ inhibitor GSK2636771; or the PI3Kα, δ, γ inhibitor taselisib as single agents in HNSCC (ClinicalTrials.gov NCT02465060). Alpelisib, a PI3Kα-selective inhibitor, demonstrated encouraging preliminary activity as monotherapy in patients with PIK3CA-altered solid tumors in a first-in-human phase Ia study (ClinicalTrials.gov NCT01219699; ref. 43). The disease control rate (DCR) in PIK3CA-altered patients with HNSCC was 68.4% (13 of 19). Of the 17 patients with HNSCC with tumor volume assessments, 7 patients showed tumor shrinkage. Alpelisib was approved by the FDA for patients with breast cancer whose tumors harbored mutations at PIK3CA canonical sites or specific noncanonical mutations (C420R, Q546E, Q546K). In addition, a PIK3CA C2 domain mutation (P447_L455 deletion), which was reported in a patient with estrogen receptor–positive (ER-positive) breast cancer, resulted in hyperactivation of p110α and was associated with an excellent clinical response to alpelisib (44). The evolving use of PI3Kα inhibitors in breast cancer demonstrates that the clinical benefit of these agents can be extended to patients whose tumors contain noncanonical PIK3CA mutations that are shown to be activating in relevant preclinical models.
In HNSCC, where PIK3CA is the most commonly altered oncogene, alpelisib and other PI3Kα-targeted drugs are being tested as monotherapy or in combination. In a phase Ib study of alpelisib and cetuximab with concurrent radiation therapy in stage III-IVb HNSCC (ClinicalTrials.gov NCT02282371), 1 patient, whose tumor harbored a canonical PIK3CA mutation, experienced a rapid response to the treatment, suggesting that an activating PIK3CA mutation may serve as a biomarker for therapy that includes a PI3K inhibitor (45). Overall, the success of PI3Kα inhibitors as monotherapy or in combination with other agents in HNSCC has been limited to date. One potential reason is the lack of biomarkers to reliably predict response to these agents. The FDA approval of alpelisib in breast cancer supports the utility of activating PIK3CA mutations as predictive biomarkers.
We are currently conducting a preoperative window of opportunity trial evaluating biomarker modulation and tumor volume change in patients with HPV-positive HNSCC scheduled for transoral robotic surgery, a context naturally enriched for genomic PI3K activation by amplification or mutation (ClinicalTrials.gov NCT03601507) (1). Among other ongoing clinical trials evaluating alpelisib in HNSCC, only the study that our patient case was enrolled in (ClinicalTrials.gov NCT03292250) specifically requires the presence of genetic alterations of the PI3K pathway be present in the tumor for enrollment. In this trial, we identified a patient with HNSCC whose tumor contained the noncanonical PIK3CA mutation Q75E and who had recurrent and metastatic disease after cisplatin-based chemoradiation. In addition to the noncanonical PIK3CA mutation (Q75E), there were 3 single nucleotide variants (SNVs) found in the index patient’s tumor (FBXW7, TGFBR2, and ATR). Of note, FBXW7 is considered as a potent tumor suppressor gene responsible for the degradation of several proto-oncogenes, including mTOR, and loss of function of FBXW7 has been reported to increase levels of total and activated mTOR (46–48). The index patient tumor in our study contained 3 missense mutations of FBXW7 (R361Q, R399Q, R479Q). R479Q has been reported to confer a loss of FBXW7-substrate interaction (49, 50). Thus, it possible that these SNV alterations contributed to the patient’s clinical response to alpelisib.
To our knowledge, this is the first report of this noncanonical PIK3CA mutation in a patient with cancer. To determine the functional impact of all noncanonical PIK3CA mutations in HNSCC, we systematically characterized the oncogenic properties of the 32 noncanonical mutants found in the TCGA HNSCC cohort. The majority of these noncanonical mutants (22/32, 68.8%) demonstrated an activating phenotype, comparable to the more common canonical mutants, when compared with the WT p110α. Notably, the Q75E mutation found in our patient also demonstrated an activating phenotype in our serum dependency, colony formation, and cell migration assays. It is possible that the method we used, exogenous overexpression via a doxycycline-inducible promoter, may overestimate the potency of the constructs tested, although this limitation would be true of all mutations, including noncanonical PIK3CA mutants found to be nonactivating (51, 52). In addition, 2 other noncanonical PIK3CA mutations (E722K, R808Q) were reported in a larger cohort of HPV-positive HNSCC (53). The functional impact of these 2 mutations has not been elucidated, thus representing a limitation of our study.
Comparison of the impact of noncanonical PIK3CA mutants in 3 other cell line models confirmed that the functional consequences of noncanonical PIK3CA mutations were largely lineage independent. Although the results across the models showed strong agreement, the C971R and E365V mutants demonstrated discordant behaviors. These mutants were found to be nonactivating in the Ba/F3 model, but activating in the HNSCC and MCF10A cells (and were not tested in the HeLa model). We previously reported discrepant results between the Ba/F3 and MCF10A cell models in the functional properties of mutations in other genes, including ERBB2, BRAF, and EGFR (5). It may be important to note that Ba/F3 is murine in origin, whereas PCI-52-SD1, MCF10A, and HeLa are all human cell lines. In a platform using the HPV-positive cell line HeLa, 16 of the 32 HNSCC-associated noncanonical PIK3CA mutations were tested and displayed significant concordance with the other 3 models, except for E81K, which was nonactivating in the HNSCC, Ba/F3, and MCF10A models, yet activating in the HeLa model. Others have reported that the E81K mutant induces AKT phosphorylation, activation of mTOR signaling, and enhanced growth in patient-derived fibroblasts in vitro (54). Discordant results with E81K, E365V, and C971R suggest that these are neomorphs activating only a subset of PIK3CA responses or new responses that activate only some processes. Alternatively, particular mutations may collaborate and have context-dependent effects in some models or lineages. These collective findings highlight the challenges of restricting studies to any single preclinical platform to predict functionality and hence, clinical translation.
Our broad functional profiling demonstrated that activating noncanonical p110α mutants in HNSCC were more sensitive than WT p110α to PI3Kα inhibition with alpelisib in PDX tumors and cell line models. This supports our contention that patients with noncanonical mutants defined as activating in the HNSCC platform may benefit from treatment with PI3Kα-targeting agents. However, several studies have reported that not all patients respond to alpelisib despite the presence of canonical PIK3CA mutations in their tumors. In a phase Ib trial, patients with ER-positive/HER2-negative metastatic breast cancer with PIK3CA mutations, including canonical and noncanonical (P447_L455del, Q546K/P, I273V, C420R, D939G, E78K, E726K) mutations, and concurrent alterations in KRAS, TP53, or FGFR1 did not benefit from alpelisib (55). Additionally, Elkabets et al. found that persistent mTORC1 activation mediated resistance to alpelisib in breast cancer cell lines with canonical or noncanonical (N345K, C420R, K111N) PIK3CA mutations (56). They also reported that persistent activation of mTOR signaling in HNSCC cell lines bearing either mutated (H1047R) or amplified PIK3CA confers resistance to alpelisib, and that the activation is mediated via AXL interaction with EGFR (57, 58). Another study reported that acquired resistance to alpelisib in a PIK3CA-mutant (H1047R) HNSCC cell line was associated with increased expression of the TAM family receptors TYRO3 and AXL (59). Therefore, as with many other targeted therapies, dissecting the underlying mechanisms driving resistance may lead to improved treatment approaches.
Mapping of the noncanonical PIK3CA mutations onto inactive apo, lipid-bound, and inhibitor-bound structures of the PI3Kα heterodimer outlined potential mechanisms behind their activating effects. A breadth of biochemical and biophysical studies on canonical mutations as well as a number of noncanonical mutations, including in vitro lipid binding and kinase assays (29, 30), hydrogen-deuterium exchange mass spectrometry (30), and molecular dynamics simulations (32, 60–62), have demonstrated several mechanisms by which PI3Kα autoinhibition is subverted in cancer. These include (a) disruption of critical interdomain inhibitory contacts, (b) conformational rearrangement of the regulatory arch and subsequent reorientation of the activation loop for catalysis, and (c) enhanced affinity of the enzyme for anionic membrane lipids due to an increase in positive charge at the membrane-binding interface. Our analysis suggests that many noncanonical mutations tap into these mechanisms, frequently mimicking those of canonical mutations. For example, canonical helical domain mutations E542K and E545K activate the enzyme by breaking the nSH2/HD interface (33), and we showed that the noncanonical mutant, Q546R, is positioned to do the same. In another example, noncanonical mutation M1043V is likely to affect the conformation of the regulatory arch, and noncanonical mutations E726K, E970K, C971R, and N1068fs are likely to enhance membrane association, similar to the canonical kinase domain mutant H1047R (34). More in-depth biophysical and structural studies are needed to fully understand the molecular mechanisms behind the effect noncanonical mutations have on PI3Kα activity and signaling, but we showed that although less prevalent, noncanonical mutations can be used as predictors of patients’ responsiveness to PI3Kα-targeted therapies.
The collective results of the present study reveal the clinical significance of noncanonical PIK3CA mutations in HNSCC and possibly other cancers that harbor these mutations. In 2018, there were 890,000 new HNSCC cases and 450,000 deaths worldwide from this malignancy. The incidence of HNSCC, particularly HPV-positive HNSCC, continues to rise and is anticipated to increase by 30% by 2030 (63, 64). In 2021, an estimated 66,630 people will be diagnosed with HNSCC in the United States (65). Nearly 20% of patients with HNSCC whose tumors were characterized in TCGA harbor a PIK3CA mutation. Our study suggests that inclusion criteria should be broadened to encompass all activating PIK3CA mutations, including the 22 activating noncanonical PIK3CA mutations identified in our preclinical models. It is worth noting that one of the HNSCC-associated activating noncanonical mutants, R88Q, is the most common noncanonical PIK3CA mutation across 32 cancer types in TCGA, underscoring the broad significance of our findings to non-HNSCC cancers. Additional investigation is needed to determine the role of activating noncanonical PIK3CA mutations in predicting responses to PI3K inhibitors in cancer, including HNSCC.
In summary, our systematic functional profiling of HNSCC-associated noncanonical PIK3CA mutations across a variety of cellular platforms identified activating mutations. These mutations were further associated with antitumor responses to alpelisib in a patient with HNSCC treated in a phase II trial and in several HNSCC preclinical models, including PDXs. These findings suggest that noncanonical PIK3CA mutations identified as activating in preclinical models should be considered as predictive biomarkers for treatment with PI3K pathway inhibitors. The high level of concordance of the activating phenotype across 4 distinct cellular platforms highlights the significance of noncanonical PIK3CA mutations to a broad spectrum of cancers.
Methods
Targeted sequencing of patient tumor. Genomic DNA of the patient’s tumor was isolated from FFPE samples using the QIAamp DNA FFPE Tissue Kit (Qiagen) and subjected to targeted sequencing of 244 genes known to be somatically altered in HNSCC. Sequencing was performed using the customized SureSelectXT Target Enrichment library generation kit (Agilent), and then sequenced by the Illumina HiSeq 2500 platform with a depth of coverage of more than 1000×. The data set for next-generation sequencing was deposited in NCBI’s Sequence Read Archive (PRJNA764203). Copy number alterations were called using CNVkit (https://cnvkit.readthedocs.io/en/stable/). To reduce ambiguity from individual variations, all normal samples were pooled and used as a control. Of the initial copy number alteration calls, genes with 4 or more and 0 measured copy numbers were considered amplified and deleted, respectively, to secure high confidence (11).
Plasmids
pHAGE-PIK3CA WT has been previously described (5). The PIK3CA WT gene (full-length coding sequence) was cloned into pENTR-vector using BP Clonase (Invitrogen, 11789100). For generation of individual PIK3CA mutants, site-directed mutagenesis of the pENTR-PIK3CA WT gene was performed with designated primers and Phusion High-Fidelity DNA Polymerase (New England Biolabs, M0530L). The site-specific recombination reaction was then performed using Gateway LR Clonase (Invitrogen, 11791019) to generate the mutants in the doxycycline-inducible pLVX-3X-FLAG (N-terminus) vector (Clontech). All constructs were sequence-verified in the final pLVX destination vector (Quintarabio).
Lentiviral infection of HNSCC cells.
HEK293T cells (4 × 106 cells in 10 cm dish) were transfected with 5 μg of lentiviral vector carrying the gene of interest (pLVX-LUC as control, pLVX-PIK3CA WT, and pLVX-PIK3CA mutants). Two days after transfection, fresh lentiviral particles (in the supernatant of the HEK293T cells) were collected, followed by filtering through a 0.45 μm PVDF syringe to remove debris. Fresh lentiviral particles were used for infection of PCI-52-SD1 cells. Cells were plated at 40% to 50% confluency (4 × 106 cells in 10 cm dish) 1 day before infection. Infection was performed by adding lentivirus to the cells containing complete culture media, with polybrene (Sigma-Aldrich, TR-1003-G) at a final concentration of 8 μg/mL. Cells were then incubated for an additional 48 hours, and the infection medium was replaced with fresh complete medium containing puromycin (1 μg/mL) for 3-day selection.
Animal experiments.
PDXs from patients with HNSCC were established in 5- to 6-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, catalog 005557) as described previously (24). Tumor-bearing mice were randomized into alpelisib and vehicle treatments (n = 12 tumors per group) when the average tumor volume reached 100 to 200 mm3. Alpelisib was administered at 25 mg/kg daily for 5 days/week by oral gavage for 21 days. Alpelisib was solubilized in 1% (w/v) carboxymethylcellulose containing 0.5% (w/v) Tween 80. Tumors were harvested 3 hours after the last treatment. Tumor growth was determined by measurement with calipers every 3 days and calculated using the formula (length × width2)/2. The individual relative tumor volume (RTV) was calculated as follows: RTV = Vt/V0, where Vt is the volume on each day and V0 is the volume at the beginning of treatment.
Structural analysis.
To visualize the putative effect of activating and nonactivating noncanonical mutations, mutations were mapped onto autoinhibited structures of PI3Kα in the PyMOL Molecular Graphics System (version 1.8.2.3, Schrödinger, LLC) using the “find contacts” tool and the structure of the inactive PI3Kα (PDB ID: 4l23) as a template. The find tool was used to identify polar and backbone contacts between residues that were mutated and their surrounding residues and to demonstrate how these contacts are disrupted upon mutagenesis. All figures illustrating the location of the mutations and their PI3Kα activation mechanisms were prepared in PyMOL.
Statistics.
Data are shown as SD, except in tumor growth curves (Figure 5, A and C), in which data are shown as SEM. P values of less than 0.05 were considered significant. A 1-tailed Student’s t test was used for comparison of any 2 groups and if needed to meet the normality assumption of the test, data were analyzed on a log-transformed scale. For comparison of more than 2 groups determined by more than 1 experimental condition, ANOVA was used with 2-group comparisons of interest conducted via contrasts. The agreement in the activation status between 2 assays was measured by Kendall’s tau correlation coefficient (τ).
Study approval.
The TRIUMPH trial was conducted according to the guidelines of the Declaration of Helsinki and approved by the IRB of Yonsei University Severance Hospital. All animal procedures and maintenance were conducted in accordance with protocols approved by the IACUC of the University of California, San Francisco.
Author contributions
N Jin, DEJ, and JRG conceived the study. DEJ and JRG supervised the study. N Jin, JC, MJL, NKV, PKSN, ZB, and GT were involved in data acquisition and analysis. BK and HRK provided essential data about the patient case. HT and N Jura performed the structural analysis. N Jin, HL, and YZ contributed to animal studies. N Jin and MOK were involved in data statistical analysis. GBM, NJK, JEB, DEJ, and JRG provided research resources. N Jin and MJL wrote the manuscript with input from all authors. GBM, HK, JEB, DLS, NJK, DEJ, and JRG edited and reviewed the manuscript. All authors approved the manuscript.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 DE023685 (to JRG and DEJ), R35 CA231998 (to JRG), U54CA209891 (to NJK), and U01 CA217842 (to GBM) and the Howard Hughes Medical Institute Medical Research Fellows Program (to JC), and a translational grant from the V Foundation for Cancer Research (to JEB and JRG). The TRIUMPH trial was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (HA16C0015).
Version 1. 11/15/2021
Electronic publication
Funding Statement
JRG and DEJ
JRG
NK
GBM
JC
Translational bIomarker Driven UMbrella Project for Head and Neck (TRIUMPH trial)
JEB and JRG
Footnotes
Conflict of interest: JRG and DEJ are coinventors of cyclic STAT3 decoy and have financial interests in STAT3 Therapeutics, Inc. STAT3 Therapeutics, Inc. holds an interest in the cyclic STAT3 decoy oligonucleotide. ZB and GT are full-time employees of NovellusDx. The Krogan Laboratory received research support from Vir Biotechnology and F. Hoffmann-La Roche. NJK has consulting agreements with Maze Therapeutics and Interline Therapeutics. GBM is a member of the consultant/scientific advisory boards of Amphista Therapeutics, AstraZeneca, Chrysalis Biotechnology, GSK, ImmunoMet, Ionis, Eli Lilly, PDX Pharmaceuticals, SignalChem, Symphogen, Tarveda Therapeutics, Turbine, and Zentalis Pharmaceuticals. GBM has financial interests in Catena Pharmaceuticals, ImmunoMet, SignalChem, and Tarveda. The Mills Laboratory received foundation support from Adelson Medical Research Foundation; research support from NanoString Center of Excellence and Ionis (provision of tool compounds); and clinical trial support from AstraZeneca, Genentech, GSK, and Eli Lilly. The Mills Laboratory has licensed a homologous recombination deficiency assay to Myriad Genetics and digital spatial profiler to NanoString. JEB received clinical research grants from AstraZeneca, Aveo, Bristol Myers Squibb, Celldex, CUE Biopharma, Eli Lilly, and Novartis.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(22):e150335.https://doi.org/10.1172/JCI150335.
Contributor Information
Nan Jin, Email: jinnan199006@163.com.
Bhumsuk Keam, Email: bhumsuk@gmail.com.
Janice Cho, Email: janicecho89@gmail.com.
Michelle J. Lee, Email: michelle.lee3@ucsf.edu.
Hye Ryun Kim, Email: nobelg@yuhs.ac.
Hayarpi Torosyan, Email: hayarpi.torosyan@ucsf.edu.
Natalia Jura, Email: natalia.jura@ucsf.edu.
Patrick K.S. Ng, Email: patrick.ng@jax.org.
Gordon B. Mills, Email: millsg@ohsu.edu.
Hua Li, Email: hua.li2@ucsf.edu.
Yan Zeng, Email: yan.zeng@ucsf.edu.
Zohar Barbash, Email: zohar@novellus.bio.
Gabi Tarcic, Email: gabi@novellus.bio.
Hyunseok Kang, Email: Hyunseok.Kang@ucsf.edu.
Julie E. Bauman, Email: jebauman@arizona.edu.
Mi-Ok Kim, Email: miok.kim@ucsf.edu.
Nathan K. VanLandingham, Email: Nathan.vanlandingham@ucsf.edu.
Danielle L. Swaney, Email: danielle.swaney@gladstone.ucsf.edu.
Nevan J. Krogan, Email: Nevan.Krogan@ucsf.edu.
Daniel E. Johnson, Email: daniel.johnson@ucsf.edu.
Jennifer R. Grandis, Email: Jennifer.Grandis@ucsf.edu.
References
- 1.Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, et al. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 3.Gymnopoulos M, et al. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31(6):820–832. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng PK, et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell. 2018;33(3):450–462. doi: 10.1016/j.ccell.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikenoue T, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65(11):4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 7.Andre F, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–1940. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 8.Dogruluk T, et al. Identification of variant-specific functions of PIK3CA by rapid phenotyping of rare mutations. Cancer Res. 2015;75(24):5341–5354. doi: 10.1158/0008-5472.CAN-15-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lui VW, et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111(3):1114–1119. doi: 10.1073/pnas.1319551111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keam B, et al. TRIUMPH trial: one small step could become one giant leap for precision oncology in head and neck cancer. Cancer Res Treat. 2019;51(1):413–414. doi: 10.4143/crt.2018.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim SM, et al. Investigating the feasibility of targeted next-generation sequencing to guide the treatment of head and neck squamous cell carcinoma. Cancer Res Treat. 2019;51(1):300–312. doi: 10.4143/crt.2018.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keysar SB, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol. 2013;7(4):776–790. doi: 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janku F, et al. Abstract B109: Oral dual PI3K/mTOR inhibitor bimiralisib demonstrates tolerability and a signal of activity in head and neck squamous cell cancer with NOTCH1 loss-of-function mutation. Mol Cancer Ther. 2019;18(12):B109 [Google Scholar]
- 14.Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 15.Powles T, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020;21(12):1574–1588. doi: 10.1016/S1470-2045(20)30541-6. [DOI] [PubMed] [Google Scholar]
- 16.Du L, et al. Overexpression of PIK3CA in murine head and neck epithelium drives tumor invasion and metastasis through PDK1 and enhanced TGFβ signaling. Oncogene. 2016;35(35):4641–4652. doi: 10.1038/onc.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter ME, Brunet A. FOXO transcription factors. Curr Biol. 2007;17(4):R113–R114. doi: 10.1016/j.cub.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, et al. Distinct roles of PIK3CA in the enrichment and maintenance of cancer stem cells in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(1):139–158. doi: 10.1002/1878-0261.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonelli MA, et al. Inhibition of PI3K pathway reduces invasiveness and epithelial-to-mesenchymal transition in squamous lung cancer cell lines harboring PIK3CA gene alterations. Mol Cancer Ther. 2015;14(8):1916–1927. doi: 10.1158/1535-7163.MCT-14-0892. [DOI] [PubMed] [Google Scholar]
- 20.Lui VW, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazumdar T, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13(11):2738–2750. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritsch C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 23.Li H, et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Mol Cancer Res. 2014;12(4):571–582. doi: 10.1158/1541-7786.MCR-13-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, et al. Proteomic characterization of head and neck cancer patient-derived xenografts. Mol Cancer Res. 2016;14(3):278–286. doi: 10.1158/1541-7786.MCR-15-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 26.Yu J, et al. Regulation of the p85/p110alpha phosphatidylinositol 3’-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1998;273(46):30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 27.Yu J, et al. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18(3):1379–1387. doi: 10.1128/MCB.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller MS, et al. Structural basis of nSH2 regulation and lipid binding in PI3Kα. Oncotarget. 2014;5(14):5198–5208. doi: 10.18632/oncotarget.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hon WC, et al. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2012;31(32):3655–3666. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke JE, et al. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA) Proc Natl Acad Sci U S A. 2012;109(38):15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabelli SB, et al. Structural effects of oncogenic PI3Kalpha mutations. Curr Top Microbiol Immunol. 2010;347:43–53. doi: 10.1007/82_2010_53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang M, et al. The mechanism of PI3Kα activation at the atomic level. Chem Sci. 2019;10(12):3671–3680. doi: 10.1039/C8SC04498H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miled N, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317(5835):239–242. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 34.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106(40):16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudd ML, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17(6):1331–1340. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maheshwari S, et al. Kinetic and structural analyses reveal residues in phosphoinositide 3-kinase α that are critical for catalysis and substrate recognition. J Biol Chem. 2017;292(33):13541–13550. doi: 10.1074/jbc.M116.772426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spangle JM, et al. PIK3CA C-terminal frameshift mutations are novel oncogenic events that sensitize tumors to PI3K-α inhibition. Proc Natl Acad Sci U S A. 2020;117(39):24427–24433. doi: 10.1073/pnas.2000060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, et al. Structural features that distinguish inactive and active PI3K lipid kinases. J Mol Biol. 2020;432(22):5849–5859. doi: 10.1016/j.jmb.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jimeno A, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51(4):383–388. doi: 10.1016/j.oraloncology.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowles DW, et al. A multicenter phase 1 study of PX-866 and cetuximab in patients with metastatic colorectal carcinoma or recurrent/metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 2014;32(6):1197–1203. doi: 10.1007/s10637-014-0124-3. [DOI] [PubMed] [Google Scholar]
- 41.Soulieres D, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017;18(3):323–335. doi: 10.1016/S1470-2045(17)30064-5. [DOI] [PubMed] [Google Scholar]
- 42.Fayette J, et al. Buparlisib (BKM120) in refractory head and neck squamous cell carcinoma harbouring or not a PI3KCA mutation: a phase II multicenter trial. Ann Oncol. 2019;30(suppl_5):V455 [Google Scholar]
- 43.Juric D, et al. Phosphatidylinositol 3-kinase α-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36(13):1291–1299. doi: 10.1200/JCO.2017.72.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Croessmann S, et al. PIK3CA C2 domain deletions hyperactivate phosphoinositide 3-kinase (PI3K), generate oncogene dependence, and are exquisitely sensitive to PI3Kalpha inhibitors. Clin Cancer Res. 2018;24(6):1426–1435. doi: 10.1158/1078-0432.CCR-17-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunn LA, et al. A phase 1b study of cetuximab and BYL719 (Alpelisib) concurrent with intensity modulated radiation therapy in stage III-IVB head and neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2020;106(3):564–570. doi: 10.1016/j.ijrobp.2019.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao JH, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun. 2013;434(2):352–356. doi: 10.1016/j.bbrc.2013.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jardim DL, et al. FBXW7 mutations in patients with advanced cancers: clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS One. 2014;9(2):e89388. doi: 10.1371/journal.pone.0089388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang CC, et al. FBXW7 mutation analysis and its correlation with clinicopathological features and prognosis in colorectal cancer patients. Int J Biol Markers. 2015;30(1):e88–e95. doi: 10.5301/jbm.5000125. [DOI] [PubMed] [Google Scholar]
- 50.O’Neil J, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204(8):1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kinross KM, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122(2):553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gillison ML, et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019;29(1):1–17. doi: 10.1101/gr.241141.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loconte DC, et al. Molecular and functional characterization of three different postzygotic mutations in PIK3CA-related overgrowth spectrum (PROS) patients: effects on PI3K/AKT/mTOR signaling and sensitivity to PIK3 inhibitors. PLoS One. 2015;10(4):e0123092. doi: 10.1371/journal.pone.0123092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayer IA, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2- metastatic breast cancer. Clin Cancer Res. 2017;23(1):26–34. doi: 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elkabets M, et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5(196):196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elkabets M, et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27(4):533–546. doi: 10.1016/j.ccell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Badarni M, et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight. 2019;5:125341. doi: 10.1172/jci.insight.125341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruicci KM, et al. TAM family receptors in conjunction with MAPK signalling are involved in acquired resistance to PI3Kα inhibition in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2020;39(1):217. doi: 10.1186/s13046-020-01713-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghalamkari S, et al. A novel carcinogenic PI3Kα mutation suggesting the role of helical domain in transmitting nSH2 regulatory signals to kinase domain. Life Sci. 2021;269:118759:S0024-3205(20)31512-5. doi: 10.1016/j.lfs.2020.118759. [DOI] [PubMed] [Google Scholar]
- 61.Echeverria I, et al. Oncogenic mutations weaken the interactions that stabilize the p110α-p85α heterodimer in phosphatidylinositol 3-kinase α. FEBS J. 2015;282(18):3528–3542. doi: 10.1111/febs.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalsi N, et al. Biophysical aspect of phosphatidylinositol 3-kinase and role of oncogenic mutants (E542K & E545K) J Biomol Struct Dyn. 2016;34(12):2711–2721. doi: 10.1080/07391102.2015.1127774. [DOI] [PubMed] [Google Scholar]
- 63.Ferlay J, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 64.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 65.Siegel RL, et al. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 66.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.