

Summary
Oriented enzyme immobilization on electrodes is crucial for interfacial electrical coupling of direct electron transfer (DET)-based enzyme-electrode systems. As inorganic-binding peptides are introduced as molecular binders and enzyme-orienting agents, inorganic-binding peptide-fused enzymes should be designed and constructed to achieve efficient DET. In this study, it is aimed to compare the effects of various gold-binding peptides (GBPs) fused to enzymes on electrocatalytic activity, bioactivity, and material-binding behaviors. Here, GBPs with identical gold-binding properties but different amino acid sequences were fused to the FAD-dependent glucose dehydrogenase gamma-alpha complex (GDHγα) to generate four GDHγα variants. The structural, biochemical, mechanical, and bioelectrochemical properties of these GDHγα variants immobilized on electrode were determined by their fused GBPs. Our results confirmed that the GBP type is vital in the design, construction, and optimization of GBP-fused enzyme-modified electrodes for facile interfacial DET and practical DET-based enzyme-electrode systems.
Subject areas: Electrochemistry, Bio-electrochemistry, Materials science, Materials chemistry
Graphical abstract
Highlights
-
•
The four GBP sequences are genetically fused to catalytic subunit of GDHγα complex
-
•
The cofactor-surface interface was investigated with 3D models of fusion enzymes
-
•
The four systems exhibit diverse electrochemical results depending on GBP type
Electrochemistry; Bio-electrochemistry; Materials science; Materials chemistry
Introduction
There is growing interest in direct electron transfer (DET) between enzyme active sites and electrode surfaces for the development of enzyme-based biodevices such as biofuel cells, biosensors, photosynthetic systems, and enzymatic electrosynthesis systems, among others (Guo et al., 1991; Ikeda et al., 1993; Wollenberger, 2005; Hatada et al., 2018). DET-based enzyme electrodes do not require mediators to shuttle electrons during redox reaction of surface-immobilized enzyme, which is, namely, “mediator-less” ET system. Thereby, the ET agent (i.e. FAD, Moco, heme, Fe-S cluster, etc.) fixed within the enzyme molecule should be the last electron donor or first electron acceptor while oxidation or reduction is taking place. Hence, DET between redox enzymes and electrode surfaces can avoid the drawbacks associated with redox mediators such as toxicity, high costs, and instability caused by mediator leaching among others (Holland et al., 2011; Martins et al., 2014).
The establishment and efficiency of DET at the enzyme-electrode interface are determined by the method of enzyme immobilization on the electrode surface. Immobilization strategies have been evaluated in terms of the selection of enzymes, protein carriers, and linking agents among others. To achieve intimate direct electrical coupling between enzymes and electrodes, the following parameters related to DET must be carefully considered during the selection of immobilization strategies: (1) Enzyme catalytic activity must be preserved and unfavorable conformational changes must be avoided to ensure functional active site and prevent inhibition of enzymatic activity (Jia et al., 2014). (2) The enzymes must be stabilized by attaching them to solid supports that determine the number and packing density of enzyme molecules on the electrode surface (Küchler et al., 2016). (3) The redox cofactor (final electron donor) and the electrode surface must be in close proximity for efficient electrical communication. It has been reported that the ET must be within 14 Å for direct “electron tunneling” to occur since ET kinetics decrease logarithmically with increasing distance (Page et al., 1999; Vazquez-Duhalt et al., 2014; Yates et al., 2018).
For designing enzyme-electrode systems, establishing a short ET distance is particularly challenging. The redox active center is often deeply buried in the proteinaceous shell. Consequently, the electrically accessible area in the protein structure is extremely narrow (Hess et al., 2003; Lee et al., 2019). Thus, development of an immobilization technology that precisely controls the enzyme orientation is essential for enhanced direct electrical communication at the enzyme-electrode interface (Hitaishi et al., 2018a, 2018b; Sorrentino et al., 2021).
Table 1 lists a wide range of enzyme orientation technologies including electrostatic adsorption (Das et al., 2014; Liu et al., 2018), chemical cross-linking (Gutierrez-Sanchez et al., 2012; Lalaoui et al., 2016; Lee et al., 2018a; Ma et al., 2019), DNA-directed hybridization (Fu et al., 2012; Chakraborty et al., 2015), and affinity binding (Lee et al., 2018b, 2019). They were developed to increase the DET rate in DET-based enzyme-electrode systems (Table 1). However, the first two strategies may destabilize binding (electrostatic adsorption) or cause structural deformation that shields the enzyme active sites (chemical cross-linking). Moreover, the orientation efficiency of both methods is low because the surface attachment points in the enzymes are not specifically designated. DNA hybridization-based enzyme immobilization using DNA tethered onto the electrode surface could effectively regulate enzyme orientation via site-specific attachment. As DNA sequences are long (∼33 Å/10 bp), they may widen cofactor-surface spacing, increase overpotential, and eventually reduce DET efficiency.
Table 1.
Immobilization technologies for orientation-controlled enzyme on electrode and their advantages and disadvantages
| Orientation methods | Platform | Enzyme (cofactor) | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Electrostatic adsorption | Reduced graphene oxide-gold nanoparticles composites | Glucose oxidase (FAD) |
|
|
Das et al. (2014) |
| Carbon nanotubes | Glucose oxidase (FAD) | Liu et al. (2018) | |||
| Chemical covalent linking | Functionalized gold nanoparticles on porous graphite | Laccase (multi-copper) |
|
|
Gutierrez-Sanchez et al. (2012) |
| Gold nanoparticles-modified carbon nanotubes | Laccase (multi-copper) | Lalaoui et al. (2016) | |||
| Self-assembly monolayer-modified gold disk electrode | Glucose dehydrogenase (FAD) | Lee et al., 2018a, 2018b | |||
| Maleimide-modified gold electrodes | Cellobiose dehydrogenase (FAD) | Ma et al. (2019) | |||
| DNA-directed hybridization | Single-walled carbon nanotubes | Bilirubin oxidase (multi-copper) |
|
|
Chakraborty et al. (2015) |
| DNA origami tiles | Glucose oxidase (FAD)/horseradish peroxidase (Heme) | Fu et al. (2012) | |||
| Genetic fusion of inorganic binding peptide | Plane gold surface | Glucose dehydrogenase (FAD) |
|
|
Lee et al. (2018a, 2018b, 2019), this article |
Affinity binding technology comprises the genetic fusion of inorganic-binding peptides to enzyme structures and could satisfy the foregoing DET-determining parameters. Inorganic-binding peptides are short amino acid sequences with binding affinity for the surfaces of their solid material targets (Tamerler et al., 2006; Vallee et al., 2010; Adams et al., 2015). This simple, versatile bioconjugation method can directly immobilize and orient biomolecular entities on solid supports without impeding enzyme function in principle (Kacar et al., 2009).
Several gold-binding peptides (GBPs) and other types of inorganic-binding peptides have been identified through phage display and selection-based studies (Hnilova et al., 2012; Care et al., 2015). Although different inorganic-binding peptides may have identical material specificity, there may be wide variation in the structural conformations of the peptides immobilized on solid surfaces. The peptide side chains differ in terms of their orientation preferences (Seker et al., 2007; Palafox-Hernandez et al., 2014). Moreover, the mechanisms by which inorganic-binding peptides recognize solid materials are poorly understood. The peptide-material interfaces are complex and are affected by buffer type, pH, temperature, and other factors. Hence, inorganic-binding peptides differing in amino acid content and sequence would bind in various conformations and have diverse physicochemical properties (Hughes et al., 2017). As various inorganic-binding peptides are fused to the same sites in an enzyme, the binding orientation and cofactor-surface distance in the fusion enzyme-immobilized electrode would differ. Interfacial DET efficiency in the enzyme-electrode could be substantially altered even by an infinitesimal change (Å units) in the cofactor-surface distance (Wardlaw and Marcus, 1985; Vazquez-Duhalt et al., 2014).
In the construction of enzyme-electrode using GBP-fused enzymes, thereby, precise selection of the inorganic-binding peptide, with optimal amino acid content, length, and sequence, is important as well as fusion site of GBP. We recently developed a DET-capable enzyme-electrode system using the genetic expression of a GBP consisting of 12 amino acids (LKAHLPPSRLPS, 1.3 kDa) to a thermostable FAD-dependent glucose dehydrogenase (FAD-GDH) gamma-alpha complex (GDHγα) consisting of FAD-GDH catalytic subunits (α subunits) complexed with small subunits (γ subunits) (Lee et al., 2018a, 2018b). Here, The GDH α subunit contains an FAD cofactor to oxidize glucose substrates, as well as a [3Fe-4S] cluster that accepts the generated electrons from the FAD cofactor and transfers the electrons toward external electron acceptor (Yoshida et al., 2019). In the previous study, the effect of GBP fusion site on the DET capability was investigated, and it was found that the GBP fusion at the C terminus of GDH α subunit established optimum DET condition at the enzyme-electrode interface. Furthermore, the GBP-enzyme fusion technology effectively orients the enzyme and enables DET communication between the enzyme and the electrode while maintaining enzyme activity (Lee et al., 2018b, 2019, 2020).
To the best of our knowledge, there are no reports on the effects of inorganic-binding peptide types on ET efficiency or material-binding behavior in fusion enzyme-incorporated electrode systems. Herein, we used a series of GBPs with identical gold-binding function but different amino acid sequences to clarify the mechanical and bioelectrochemical properties of the interface between the GBP-fused enzyme and the electrode surface. The aim of this work was to establish whether GBP type with different amino acid content and sequence affects interfacial DET capability. The GBP sequences were selected based on their amino acid content, sequence, and length and physical properties such as total charge and hydroxyl richness (Table S1).
The following steps were used to determine the influences of variations in the fused GBPs on enzyme-binding orientation and charge transfer resistance at the enzyme-electrode interface, as well as material-binding behaviors. (1) Each of selected GBPs was fused to the C terminus of the GDH α subunit in GDHγα to prepare GDHγα variants (GDHγα-XGBP, where X = L, M, T, or V) with different GBPs tagged. (2) Next, we quantitatively and qualitatively evaluated the binding affinity and selectivity of the GDHγα variants on the Au surface. (3) The interfacial DET rates in the enzyme-electrode systems were analyzed after immobilizing wild-type GDHγα and GDHγα variants on screen-printed Au electrodes.
The objective of this study was to ascertain whether the type of inorganic-binding peptide, fused genetically as a molecular linker to the enzyme, is a determinant of the interfacial DET property. Our results provide evidence that linker peptide selection must be optimized for the design and construction of enzyme-electrode systems incorporating inorganic-binding peptide-based enzymes.
Results and discussion
Preparation of wild-type GDHγα and GDHγα variants using different gold-binding peptides
By comparing the sequences of the various GBPs that were used as fusion tags in the enzyme electrodes, LGBP (LKAHLPPSRLPS) (Nam et al., 2006), MGBP (MHGKTQATSGTIQS) (Brown, 1997), TGBP (TGTSVLIATPGV) (Kim et al., 2010), and VGBP (VSGSSPDS) (Huang et al., 2005) were selected (Table S1). They differed in terms of amino acid content and physical properties such as total charge and hydroxyl richness. However, all peptides showed gold-binding capability. Each GBP sequence was genetically fused to the C terminus of the catalytic α subunit of the FAD-dependent glucose dehydrogenase gamma-alpha complex (GDHγα). The fusion site was selected based on the results of our previous study (Lee et al., 2018a, 2018b); (Gly)5 was inserted as a flexible linker to provide spatial separation between the catalytic domain and the GBPs and maintain the independent function of each domain (Figure 1).
Figure 1.

Construction of native and fusion GDHγα
(A) Schematic diagram showing the recombinant GDH proteins with GBP candidates described as XGBP (X = L, M, T, and V). 6X-His: 6X his-tag, 5xGly: five consecutive glycine, GDHγ: GDH γ subunit, GDHα: GDH α subunit.
(B) Structural analysis of GBP sequences. Best models generated by PEP-FOLD 3.5 are shown.
Plasmids encoding wild-type GDHγα and four different GDHγα variants (GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP) were expressed in E. coli Rosetta (DE3) cells and purified by nickel affinity chromatography. The enzymes were analyzed by SDS-PAGE to verify their purity and determine their molecular weight (Figure S1). The protein bands of the wild-type and recombinant enzymes were observed at ∼60 kDa (α subunit) and ∼23.5 kDa (γ subunit). The α subunit of the GBP-fused proteins shifted by ∼1 kDa relative to the native α subunit. These results showed that the GBPs were successfully tagged.
High retention of catalytic activity is a major advantage of using solid-binding peptides as linkers for immobilizing enzymes on electrodes. We compared the activity of mutant enzymes bearing the GBP tag with that of the unmodified enzyme. Wild-type GDHγα and GDHγα variants were evaluated using glucose as the substrate. Figure S2 shows that the activity levels of GDHγα-LGBP, GDHγα-MGBP, and GDHγα-TGBP were nearly conserved. However, that of GDHγα-VGBP decreased by ∼50% due to the occurrence of unfavorable conformational changes in the GDHγα after non-native amino acids were introduced. Hence, careful GBP selection is necessary to prevent any negative impact on enzyme activity.
Structural prediction of catalytic domains of wild-type and fusion GDHγα via molecular simulations
We analyzed the interfacial bioelectrochemistry and inorganic binding behavior of proteins using computational modeling and catalytic domain simulation of the GDH α subunit fused to various GBPs. The I-TASSER tool was used to generate 3D models of the α subunits of the fusion constructs and the native protein. The best model was generated using protein structures previously reported in the Protein Data Bank (PDB). The closest protein structure template was FAD-dependent glucose dehydrogenase from Burkholderia cepacia (PDB ID: 6A2U). The template modeling (TM)-scores were 0.979, 0.949, 0.941, 0.950, and 0.956 for wild-type GDHα, GDHα-LGBP, GDHα-MGBP, GDHα-TGBP, and GDHα-VGBP, respectively. The sequence identities between all these GDH proteins and the template were ∼94%. Hence, the predicted structures were nearly analogous to the model (PDB #6A2U) and were, therefore, reliable (Figure 2). The models were visualized in a PyMOL Molecular Graphics System, and the protein dimensions were determined. The analysis showed that the overall dimensions of the GBP-fused GDH α subunits were similar to those of the wild-type GDH α subunit. However, the z axes were extended by 2.9 Å and 5 Å in GDHα-LGBP and GDHα-MGBP, respectively (Figure 2). Further modeling studies were done to find out the location of cofactors, FAD and the iron-sulfur (3Fe-4S) cluster, as well as predict complexed form with GDH γ subunit, by comparative alignment of GDH α mutants with crystal structure of FAD-dependent glucose dehydrogenase from Burkholderia cepacia (PDB ID: 6A2U), using PyMOL (Figure S3A).
Figure 2.

Prediction models of native and fusion GDHα using homology modeling
(A–E) Structural representation of (A) native GDHα and (B–E) fusion GDHα of different GBP types tagged at C terminus. GDHα is displayed in light gray, and LGBP, MGBP, TGBP, and VGBP are depicted in yellow, red, blue, and green, respectively. Each protein was modeled using the iterative threading assembly refinement (I-TASSER) method on the online server: http://zhanglab.ccmb.med.umich.edu/I-TASSER/. The models were visualized using PyMOL, and protein dimensions were analyzed.
The conformation of GBP fused to the C terminus of the GDH α subunit was the most critical parameter determining the enzyme-electrode interface because it established the orientation of the enzyme immobilized on the electrode surface. The model simulations predicted that the LGBP and MGBP tagged to the GDH α subunit showed highly coiled conformation, whereas TGBP and VGBP tagged to the GDH α subunit had relatively extended conformation (Figure 2).
Quantitative and qualitative characterization of binding affinity of wild-type and fusion proteins toward gold substrate
Before investigating the interfacial properties of the enzyme-electrode, we quantitatively and qualitatively examined the variations in the binding behaviors of the different fusion constructs toward the Au substrate.
Surface plasmon resonance (SPR) analysis was performed to determine the changes in the Au substrate-binding kinetics of the GDHγα variants relative to those of the wild-type GDHγα. Kinetic titration methods involving sequential binding and sample washing were used. Figures 3A–3E show SPR sensorgrams obtained with 6.25, 12.5, 25, 50, and 100 nM enzyme. The steady-state SPR response units (RU) of five samples per concentration were plotted for direct comparison (Figure 3F). The RU profiles revealed significantly stronger binding properties of all fusion proteins compared with the wild-type, mostly attributed to the material-specific binding affinity of the GBPs. Next, we determined the association (ka) and dissociation (kd) rate constants by nonlinear curve fitting of the SPR sensorgram. We also calculated the equilibrium constant (Keq) indicating binding affinity as follows:
| Keq = kd/ka | (Equation 1) |
Figure 3.

Surface plasmon resonance (SPR) characterization of native and fusion GDHγα
(A–E) Single cycle kinetics analysis by SPR of the direct binding of (A) native GDHγα, (B) GDH γα-LGBP, (C) GDHγα-MGBP, (D) GDHγα-TGBP, or (E) GDHγα-VGBP to gold substrate. (A–E) Experimental data are shown in black; calculated fits using a 1:1 binding model with a global fitting on all injected concentrations are shown in red. Injections of proteins for the association phase are indicated by the solid arrows, and injection of pure PBS buffer for dissociation phase are indicated by dashed arrows. Protein concentrations injected were 6.25, 12.5, 25, 50, and 100 nM for all samples.
(F) Comparison of titration curve obtained by plotting binding responses against protein concentration.
The kinetic parameters ka, kd, and Keq are summarized in Table 2. Comparison of the Keq of the fusion constructs and the native enzyme indicated that the binding affinities of GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP were 2.29-, 1.61-, 2.72-, and 2.23-fold higher, respectively, than that of the wild-type GDHγα. The relative increase in binding affinity of the fusion constructs could be attributed to the enhanced adsorption rates of GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP. The relative decrease in the desorption rate of GDHγα-LGBP enhanced its binding affinity at the Au surface (Table 2).
Table 2.
Kinetic constants obtained from the kinetic fit of a 1:1 binding model to the SPR single-cycle kinetics sensorgrams
| Analyte | Kinetic constants |
||
|---|---|---|---|
| ka (M−1 s−1) | kd (10−5 s−1) | KD (10−9 M) | |
| GDHγα | 31,112 | 4.93 | 1.58 |
| GDHγα-LGBP | 34,635 | 2.40 | 0.69 |
| GDHγα-MGBP | 50,671 | 4.98 | 0.98 |
| GDHγα-TGBP | 49,563 | 4.10 | 0.58 |
| GDHγα-VGBP | 58,976 | 4.20 | 0.71 |
We examined the morphologies of the wild-type GDHγα and GDHγα variants bound to the Au surface using atomic force microscopy (AFM) in non-contact mode. The enzymes were first immobilized on the Au surface by immersing the Au chip for 2 h in PBS containing 0.1 μM GDH proteins. Weakly bound proteins were washed out by sonication in pure PBS.
The binding distribution and thickness of the protein biofilm on the Au surface were analyzed by AFM. The bare Au surface used for the enzyme-binding studies was shown to be atomically flat with a root-mean-square (rms) roughness as low as 0.286 nm, thus enabling the examination of thickness of enzymatic film on Au surface (Figure S4). The surface morphology showed that the GDHγα variants fused with GBPs were densely packed on the surfaces (Figure 4). In contrast, the wild-type GDHγα was sparsely populated on the same surface. The GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP biofilms were uniform, and their thicknesses were 5.96 ± 0.82 nm, 5.90 ± 0.74 nm, 4.96 ± 0.78 nm, and 4.49 ± 0.72 nm, respectively. The thickness of the wild-type GDHγα biofilm was 3.92 ± 1.34 nm in average, from the cross-sectional analysis. The average height of cross-section of wild-type GDHγα biofilm seems to be lower than the actual dimension of GDHγα molecule since they have not occupied the Au surface compactly, due to the non-specificity of wild-type protein toward the Au surface. In addition, the disparity in the biofilm thickness from the AFM result and the estimated GDHγα molecule dimension can be due to the non-specific interaction of wild-type GDHγα with Au surface that may possibly cause undesirable conformational change upon surface adsorption of enzyme. Also, the standard deviation of wild-type GDHγα thickness was approximately 2-fold that of each GDHγα variant film, since the height variation of wild-type GDHγα is relatively greater compared with GDHγα variants that are bound on Au surface with more compactly packed formation.
Figure 4.

Surface morphology of enzyme-immobilized Au surfaces
(A–E) AFM images for (A) native GDHγα, (B) GDHγα-LGBP, (C) GDHγα-MGBP, (D) GDHγα-TGBP, and (E) GDHγα-VGBP immobilized on gold substrates. 5 mm × 5 mm gold substrate was immersed in 0.1 μM of each protein for 2 h, for formation of enzymatic biofilm; scanned area is 1 μm × 1 μm; upper panel, 3D topography; middle panel, 2D topography; lower panel, cross-sectional profile along the arrow in middle panel.
It was observed that the average thickness of GDHγα-LGBP and GDHγα-MGBP was ∼10 Å greater than that of GDHγα-TGBP and GDHγα-VGBP. The z axes for GDHα-LGBP, GDHα-MGBP, GDHα-TGBP, and GDHα-VGBP, predicted by the molecular modeling study, is 54.5 Å, 56.6 Å, 51.6 Å, and 51.2 Å, respectively, in which the z axis length of GDHα-LGBP, GDHα-MGBP is slightly higher compared with the wild-type GDHα, GDHα-TGBP, or GDHα-VGBP. However, it is worth to note that this prediction is not sufficient for explanation of differences in biofilm thickness, and further in-depth analysis is required. The differential binding orientation among the fusion proteins may underlie the observed variations in enzyme topographies because each GBP forms a unique conformation on the Au surface. As the shape of GDHγα protein is oval, its thickness may change depending on the binding direction or conformation of the peptide linkers on the Au surface.
Influences of the GBP sequences fused to GDHγα on DET at enzyme-electrode interface
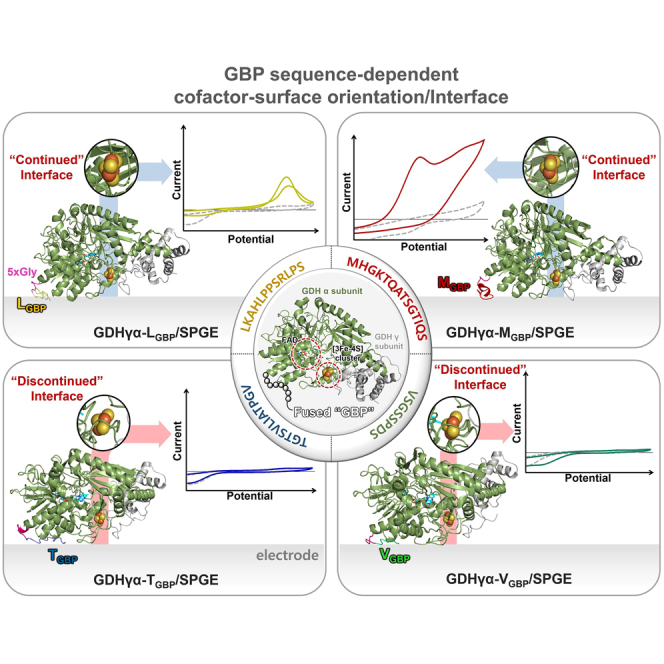
The metal recognition and binding conformation of GBP vary with amino acid content and sequence. The interfacial DET rates differ among the GDHγα-XGBP (X = L, M, T, and V)-immobilized electrodes because of the diverse binding conformations of the candidate GBPs and the different binding orientations of the model enzyme.
Before the bioelectrochemical analysis of the interfacial DET, the ET distance between the enzyme cofactor and the electrode surface was estimated according to the structure prediction shown in Figure S3. Depending on the binding direction of the native GDHγα on the electrode surface, the cofactor-to-surface distance could be in the range of 4.80–49.50 Å (Figure S5). For more in-depth estimation of cofactor-surface distance in each case of GDHγα-XGBP (X = L, M, T, and V)-immobilized Au electrode surface, it was hypothesized that the fused GBPs are closely lying on the electrode surface and the hydroxyl-containing amino acids would have a role as the anchoring point during surface immobilization of fusion enzymes. Previous studies have indicated that amino acids containing side chains with hydroxyl group, such as serine, threonine, and tyrosine, have the intrinsic ability to break through hydration layers and form anchor points on the Au surface (Yu et al., 2012; Adams et al., 2015). Therefore, serine and threonine molecules are believed to form direct contact points between the peptide and the electrode surface during docking of the simulated GBP-fused GDHγα. Since each fused GBP consists of more than one the hydroxyl-containing group (i.e., serine [S] or threonine [T]), the corresponding amino acid was selected for each and the vertical distance between [3Fe-4S] cluster and underlying surface in each case of anchoring points was averaged as shown in Figure 5. It was estimated that the ET distances in the enzyme-electrodes comprising GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP were 10.84 ± 0.16 Å, 8.88 ± 0.80 Å, 10.36 ± 0.65 Å, and 11.20 ± 0.66 Å, respectively. Page et al. (1999) reported that an electron tunneling distance of <14 Å between the electron donor and the electron acceptor enables efficient interfacial DET. Similarly, Holland et al. (2011) demonstrated that DET occurred when the orientation of the redox enzyme was regulated such that the spacing between the active site and the Au nanoparticle surface was <14 Å. Furthermore, during the positioning the GBP tag to be close to the electrode surface, the height of enzyme molecules on the electrode surface shown to be varied. The predicted height of GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP upon immobilization on the Au surface was 50.3 Å, 49.6 Å, 40.6 Å, and 40.4 Å, respectively, in which the latter two cases showed to be lower by approximately 10 Å in height. These predictions are highly in agreement with the AFM results where the average thickness of GDHγα-LGBP and GDHγα-MGBP on Au surface was ∼10 Å greater than that of GDHγα-TGBP or GDHγα-VGBP on Au surface, supporting the accuracy of prediction analysis. Thereby, it could be expected that DET may occur in four of GDHγα mutant-modified electrodes since the estimated ET distances are within the DET-capable range.
Figure 5.

Depiction of predicted orientation and estimated cofactor-to-surface distance of GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP on Au surface
The amino acid sequence of each GBP is notated right below the GBP site in fusion enzyme. The hydroxyl-containing amino acids (serine, S, or threonine, T) are colored differently (dark green). The estimation of distance between [3Fe-4S] cluster and Au surface is estimated under the assumption that hydroxyl-containing amino acid would serve as anchoring site on the Au surface. Since more than one hydroxyl-containing amino acid is composed in the GBPs, each case was averaged and marked in the scheme.
To verify the predictions, the wild-type GDHγα and GDHγα variants were immobilized on the working electrode surfaces of the screen-printed gold electrode (SPGE) to characterize the DET. Cyclic voltammetry (CV) was conducted on the wild-type GDHγα- or GDHγα variants-modified SPGE (GDHγα/SPGE and GDHγα-XGBP/SPGE; X = L, M, T, or V) to evaluate the oxidative current response, ET reaction mechanisms, and overpotential during electron passage (Figure 6).
Figure 6.

Cyclic voltammetry (CV) of enzyme-modified SPGEs
(A–E) CV profiles of glucose oxidation at (A) native GDHγα/SPGE, (B) GDHγα-LGBP/SPGE, (C) GDHγα-MGBP/SPGE, (D) GDHγα-TGBP/SPGE, and (E) GDHγα-VGBP/SPGE in the absence and presence of 100 mM glucose in PBS buffer (pH 7.4) (scan rate: 100 mV s−1); inserted graph in (A), (D), and (E): CV graphs of the selected region in the figures.
After glucose addition, weak signal of oxidative current appeared at the GDHγα/SPGE interface as unfavorable ET conditions were created by the uncontrolled orientation of the native enzyme on the electrode surface due to non-specific interaction of enzyme with inorganic surface. The onset potential (Eon) of GDHγα/SPGE (0.364 ± 0.006 V (versus Ag/AgCl) shifted to the far positive direction, considering that the standard potential of FAD cofactor is −460 mV (versus Ag/AgCl) at pH 7.9 (Holland et al., 2011) and the common standard potential (E°') of the [3Fe-4S] cluster is generally in the range of −250 mV to −650 mV (versus Ag/AgCl) (Liu et al., 2014). CV of the GDHγα-XGBP/SPGE (X = L, M, T, or V) was conducted to determine the variation in the DET capability of fused GBPs. We observed a wide range of peak currents, catalytic potentials, and voltammogram shapes. The CV of GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE indicated a clear oxidation wave and a negative shift in the starting potential of current generation, compared with that of GDHγα/SPGE. Almost nil current was observed for GDHγα-TGBP/SPGE. Slight increases in oxidative current and negative shifts in onset potential were observed for GDHγα-VGBP/SPGE (Figure 6; Table S2). It should be noted that the observed anodic current is originated by transferred electrons from [3Fe-4S] cluster toward electrode followed by the glucose oxidation at FAD cofactor. The difference in the onset potential as well as the oxidative current size might be due to the physical condition at the enzyme-electrode interface such as ET distance, structural inhibitory condition in between cofactor and electrode surface, etc.
The CV results of GDHγα-TGBP/SPGE and GDHγα-VGBP/SPGE were remarkable because the previously estimated cofactor-surface distance was within DET-capable range. Subsequently, it was investigated whether the difference in oxidative current of each enzyme-electrode in CV results was due to the catalytic activity of the immobilized enzymes. For this, the mediated ET (MET) systems were constructed by injecting 1 mM hydroquinone (HQ) as mediator in the previously constructed enzyme-electrode systems. In the MET conditions, the oxidative peaks were observed at two regions (c.a. +200 mV and +420 mV versus Ag/AgCl) in the presence of 100 mM glucose substrate. When comparing the redox wave in the absence and presence of glucose, the formal peak at c.a. +200 mV is primarily from redox reaction of HQox/HQred molecules directly with Au substrate, and the later peak around +420 mV (versus Ag/AgCl) is attributed to redox reaction of HQox/HQred led by enzymatic glucose oxidation. The generation of distinct anodic waves, followed by glucose addition, was observed in all enzyme-electrode constructs, indicating that the surface-immobilized wild-type GDHγα, GDHγα-TGBP, and GDHγα-VGBP are still capable of glucose oxidation and their catalytic activities are barely deteriorated. In addition, it was examined that different GBP sequences fused to enzyme could have impact on varied electrocatalytic phenomenon observed during CV measurement. The oligomers corresponding to each GBP (LGBP, MGBP, TGBP, VGBP) were synthesized and immobilized on electrode surface by immersing the SPGEs in the 50 μM peptide solution contained in PBS buffer. The CV of each GBP-modified SPGE was examined under identical condition with electrochemical assessment of enzyme-electrodes, both in the absence and presence of glucose. As shown in Figure S7, four GBP-modified SPGEs have presented almost no current response following glucose injection, providing evidence supporting the fact that the differed fusion peptide sequences did not influence the CV results of enzyme-electrodes. Thereby, this implies that the diminished DET capacities at GDHγα-TGBP/SPGE and GDHγα-VGBP/SPGE, as well as GDHγα/SPGE, mainly originate from the undesirable interfacial condition at those enzyme-electrodes.
Next, we performed electrochemical impedance spectroscopy (EIS) to assess the interfacial charge transfer resistance (Rct) of the GDHγα variants immobilized on the Au surface (Figure 7). It was performed to prove whether the variation of CV results is mainly attributed to interfacial ET condition, excluding the external influences. From the measurements, EIS revealed that Rct values were 131.44 ± 0.55, 24.092 ± 0.972, 659.33 ± 23.93, and 324.49 ± 46.05 kΩ for GDHγα-LGBP/SPGE, GDHγα-MGBP/SPGE, GDHγα-TGBP/SPGE, and GDHγα-VGBP/SPGE, respectively. Among the enzyme-electrode systems, the GDHγα-MGBP/SPGE exhibited the lowest Rct value, indicating that overpotential during ET process is relatively low and facile ET condition was built at the enzyme-electrode interface, in terms of enzyme-binding orientation, ET distance, etc. In the GDHγα-LGBP/SPGE, its Rct was relatively higher than GDHγα-MGBP/SPGE. Furthermore, the interfacial Rct of the GDHγα-TGBP/SPGE and GDHγα-VGBP/SPGE showed significant increase compared with GDHγα-LGBP/SPGE or GDHγα-MGBP/SPGE. The measured interfacial resistances at those fusion enzymes-incorporated electrodes are highly correspondent with the CV results in which the order of the oxidative current size was GDHγα-MGBP/SPGE > GDHγα-LGBP/SPGE > GDHγα-VGBP/SPGE > GDHγα-TGBP/SPGE. This result implies that the variation in CV result (i.e., anodic current, onset potential) is mainly attributed to the difference in interfacial condition during DET process between enzymatic cofactor and electrode surface.
Figure 7.

Electrochemical impedance spectroscopy (EIS) of enzyme-modified SPGEs
(A) Nyquist plots of GDHγα-LGBP/SPGE, GDHγα-MGBP/SPGE, GDHγα-TGBP/SPGE, and GDHγα-VGBP/SPGE obtained from EIS characterization.
(B) Comparison of charge transfer resistance (Rct) of fusion enzyme-electrodes incorporating different types of gold-binding peptide.
Although the predicted ET distance at GDHγα-LGBP/Au, GDHγα-MGBP/Au, GDHγα-TGBP/Au, and GDHγα-VGBP/Au was estimated to be within the electron tunneling capable distance (i.e., <14 Å), all enzyme-electrode systems showed significant differences in their interfacial ET capability. To comprehend such bioelectrochemical phenomena of fusion enzyme-immobilized electrode, the enzyme-electrode interfaces were closely inspected in the structural aspects. In Figure 8, each GBP fused to GDHγα was oriented to face toward and contact closely with electrode surface, as described earlier. In this state, the bottom view of surface-immobilized fusion enzyme molecules was examined. It is interesting to note that the [3Fe-4S] clusters of GDHγα-TGBP and GDHγα-VGBP on Au seemed to be blocked in vertical direction by partial polypeptides that constitute the corresponding fusion enzymes, whereas [3Fe-4S] clusters of GDHγα-LGBP and GDHγα-MGBP on Au were not sterically shielded, but rather highly exposed toward Au surface. From the predicted adsorption structures of fusion enzymes on Au surface, it could be demonstrated that the GDHγα-LGBP/Au and GDHγα-MGBP/Au have built electrically “continued interface,” whereas GDHγα-TGBP/Au and GDHγα-VGBP/Au have formed electrically “discontinued interface” that electron tunneling between cofactor to surface is inhibited. This analysis could explain the CV and EIS results, in which GDHγα-TGBP/Au and GDHγα-VGBP/Au showed comparably low oxidative currents and high interfacial Rct. Since the ET cleft where [3Fe-4S] cluster is bound is quite narrow, the cofactor could not completely be exposed toward electrode surface even by slight shift of enzyme-binding orientation. To predict the ET capability of enzyme-electrode construction, thereby, the cofactor-surface interface should be closely inspected to determine whether the electron-donating cofactor and electron-accepting electrode are electrically continued or conductive, before the estimation of ET distance.
Figure 8.

Structural analysis of cofactor-surface interface
(A–D) Predicted adsorption structure of (A) GDHγα-LGBP, (B) GDHγα-MGBP, (C) GDHγα-TGBP, and (D) GDHγα-VGBP on Au surface, shown as ribbon diagrams in two orientations. Most left: side view showing binding formation of fusion proteins on Au surface. Middle: bottom view representing whether the cofactor-surface interface is open or sterically blocked. Most right: simplified depiction of the interfacial spaces between [3Fe-4S] cluster and electrode surface is open or blocked in the vertical direction, forming “continued interface” or “discontinued interface.”
Next, the GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE, in which distinct anodic currents were observed, were used to examine changeable aspects of DET signal dependent on glucose concentration. For this, Ipeak obtained from CV was plotted against glucose concentrations (0–100 mM) and the apparent kinetic parameters of GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE were estimated (Figure S8). The linear portion of the graph and the Lineweaver-Burk transformation were used to calculate the electrochemical KMapp and Imax values. The KMapp were 21.00 ± 7.57 and 9.74 ± 0.10 mM for GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE, respectively. At the GDHγα-MGBP-electrode interface, the electrons moved toward the electrode. Moreover, Imax was 3.48-fold higher for GDHγα-MGBP/SPGE than GDHγα-LGBP/SPGE (741.76 ± 27.47 μA cm−2 versus 213.39 ± 24.71 μA cm−2). Hence, controlling the orientation of GDHγα-MGBP on the Au surface may be conducive for DET at the enzyme-electrode surface because it can shorten the ET distance.
GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE had identical GBP fusion sites. However, differences in terms of the types of GBPs fused to their GDHγα could account for the Angstrom-scale differences in their ET distances and significantly change their ET kinetic mechanisms and rates.
Although the GBPs differ in terms of their physical properties and structural conformations on inorganic surfaces, they have identical material specificity. Thus, interfacial ET rates may vary depending on the type of GBP used as the molecular binder. Therefore, the GBP sequence is an important parameter to consider during the design, construction, and optimization of GBP-fused, enzyme-modified electrodes for efficient interfacial DET.
Comparative analysis of fusion protein binding on various inorganic surfaces
Solid binding peptides exhibit unique material selectivity and affinity. Thus, they are promising biolinkers for targeted biomolecule assembly at the micro- to even nano-scale. The genetic linking of solid binding peptides with enzymes may confer material selectivity to the engineered enzymes. We assessed whether GBP selectivity could be imparted to fusion constructs wherein the GBPs were genetically tagged as linkers. To this end, we used a quartz crystal microbalance (QCM) to evaluate the Au and Si surface-binding properties of wild-type GDHγα, GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP.
The binding characteristics between the wild-type GDHγα or GDHγα variants and the Au or Si surfaces are shown in Figure S9. The adsorption phases were compared based on the types of protein and surface materials. GDH binding was represented by the change in frequency (Δf) that occurs during the mass change in the molecules adhering to the quartz crystal surfaces.
The frequency changes and binding properties of the wild-type GDHγα were comparable on the Au and Si surfaces (Figure 9) as the native enzyme bound nonspecifically to the inorganic surface. By contrast, the adsorption kinetics of the fusion constructs GDHγα-LGBP, GDHγα-MGBP, GDHγα-TGBP, and GDHγα-VGBP were enhanced on the Au surface relative to the Si surface. These findings correlated with the SPR data. However, the binding affinities of the fusion constructs on the Si surface were lower than that of the wild-type GDHγα. Moreover, unlike wild-type GDHγα, the GDHγα variants preferentially bound to the Au surface rather than the Si surface. The modified binding property of the GDHγα variants could be attributed to the distinct material selectivity of their fused GBPs.
Figure 9.

Quartz crystal microbalance (QCM) analysis
Comparison of frequency shift (Δf) of wild-type GDHγα and GDHγα-XGBP (X = L, M, T, and V) flowed on the Au and Si surface, measured at 800 s after sample injection.
Selective immobilization of the GDHγα variants on the Au surfaces was visualized by fluorescence imaging (Figure 10). Wild-type GDHγα and GDHγα variants were immobilized on a micropatterned array consisting of the Si background and square-shaped Au micropatterns (100 μm × 100 μm; 50-μm intervals). The immobilized proteins were then biotinylated and labeled with streptavidin-conjugated quantum dots. Next, they were washed with pure PBS and subjected to fluorescence micrography. The fluorescence intensity lines were measured as shown in Figure 10. All GDHγα variants specifically bound to Au regions, whereas only faint binding signatures were obtained on the Si surfaces. On the Au surfaces, the fluorescence intensity of the wild-type GDHγα was much lower than those of the fusion constructs. On the wild-type GDHγα-immobilized microtemplate, no significant differences in fluorescence intensity were observed between the Au and Si regions. Hence, the binding specificity of the mutant proteins was determined by the GBP tags. Even after they were washed in PBS for 12 h, the fluorescence intensity of the GDHγα variant-immobilized microtemplates remained the same. Thus, the mutants exhibited a high binding stability by virtue of their GBPs (data not shown). These results indicate that GBP incorporation is a highly effective strategy for regulating the binding behavior of biomolecules. Significantly, the fusion enzyme binding experiments were conducted in aqueous solutions, at physiological pH, and under biologically viable conditions. Therefore, our results provide convincing evidence that solid binding peptide fusion technology may have wide applicability in biological environments.
Figure 10.

Comparison of fluorescence images of native and fusion GDHγα proteins
(A) Scanning electron microscopic image of the microfabricated template composed of gold region and Si substrate.
(B–F) The fluorescent images of (B) native GDHγα, (C) GDH γα-LGBP, (D) GDHγα-MGBP, (E) GDHγα-TGBP, and (F) GDHγα-VGBP, revealing QD525-SA immobilization on self-assembled GDHγα-XGBP (X = L, M, T, and V). (G) Comparison of fluorescence intensities of GDHγα proteins along the line profile.
In conclusion, we explored the biochemical, mechanical, and bioelectrochemical properties of enzymes fused with GBPs having identical gold-binding properties and different amino acid sequences. After fusion of the GBPs (LGBP, MGBP, TGBP, and VGBP) at the C termini of the α subunits of GDHγα, the catalytic activity was found to be conserved for GDHγα-LGBP, GDHγα-MGBP, and GDHγα-TGBP. For GDHγα-VGBP, however, it decreased by ∼50%. The gold-binding strengths of all GDHγα variants were greater than that of the wild-type GDHγα. All GDHγα variants formed densely packed, monolayered enzyme films on the Au surfaces. On surfaces with both Au and Si regions, the GDHγα variants were highly selective for Au. Moreover, the GDHγα variants exhibited enhanced binding strength for the Au surface and significant lower ability to recognize the Si surface.
We predicted the cofactor-surface distances in the enzyme-electrode constructs using a fusion enzyme model predicted by homology modeling and molecular alignments. Based on the analysis of adsorption structure of fusion enzymes on Au electrode surface, the electrochemical results were comprehended. The GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE showed agreeable oxidative current on Au electrode, whereas GDHγα-TGBP/SPGE and GDHγα-VGBP/SPGE presented unexpected CV trends such as low or no oxidation currents. Upon a close inspection of interfacial space at enzyme-electrode, it was revealed that GDHγα-TGBP and GDHγα-VGBP forms electrically discontinued interface between cofactor and electrode surface, with electron tunneling blocked. Hence, an in-depth comparison of GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE demonstrated remarkable catalytic currents. GDHγα-MGBP/SPGE had a negatively shifted onset potential and a significantly elevated oxidation current. A change in the GBP fused to the GDHγα could lead to Angstrom-scale alterations in the ET distance and modulate the thermodynamics and kinetics of interfacial ET.
Thereby, the present study demonstrated that the GBP sequence is a vital parameter in the design, construction, and optimization of GBP-fused, enzyme-modified electrodes with efficient interfacial DET. Moreover, it influences the applicability of DET-based enzyme-electrodes in biological systems.
Limitations of the study
The ET (cofactor-surface) distances in GDHγα-XGBP (X = L, M, T, and V) were predicted by setting structural models of the fusion GDHγα on imaginary electrode surfaces. However, the peptide-surface interactions and the peptide conformations on the Au surfaces must also be computationally simulated. Furthermore, detailed analysis of differed voltammogram shape from the GDHγα-LGBP/SPGE and GDHγα-MGBP/SPGE should be supplemented to provide the in-depth explanation of interfacial electrocatalytic process in the enzyme-electrodes. This approach could facilitate the in-depth interpretation of the electrochemical results. Also, this approach would be helpful in designing the GBP-fused enzymes for construction of highly efficient, DET-based enzyme-electrode systems.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Rosetta (DE3) | (Enzynomics, Daejeon, Republic of Korea) | CP1010 |
| Chemicals, peptides, and recombinant proteins | ||
| 2,6-dichloroindophenol | Sigma-Aldrich (St. Louis, MO | D1878 |
| phenazine methosulfate | Sigma-Aldrich (St. Louis, MO | P9625 |
| TEV protease | New England Biolabs (Ipswich, MA, USA) | N.A. |
| Ni-NTA Agarose | Qiagen (Hilden, Germany) | 30210 |
| NHS-PEG4-Biotin | Thermo Fisher Scientific (Waltham, MA, USA) | A39259 |
| Qdot™ 525 Streptavidin Conjugate | Thermo Fisher Scientific (Waltham, MA, USA) | Q10141MP |
| Peptide (LKAHLPPSRLPS) | Cosmo Genetech Co., Ltd. (Seoul, Korea) | N.A. |
| Peptide (MHGKTQATSGTIQS) | Cosmo Genetech Co., Ltd. (Seoul, Korea) | N.A. |
| Peptide (TGTSVLIATPGV) | Cosmo Genetech Co., Ltd. (Seoul, Korea) | N.A. |
| Peptide (VSGSSPDS) | Cosmo Genetech Co., Ltd. (Seoul, Korea) | N.A. |
| Software and algorithms | ||
| Scrubber2 | BioLogic | http://www.biologic.com.au/scrubber.html |
| winEchem | Seiko EG&G | https://www.sii.co.jp/en/corp/base-domestic/map-segg/ |
| XEI software | Park Systems | https://www.parksystems.com/kr/ |
| I-TASSER | Zhang (2008) | http://zhanglab.ccmb.med.umich.edu/I-TASSER/ |
| PyMOL v. 2.5.0 | Schrödinger | https://pymol.org/2/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the In Seop Chang (ischang@gist.ac.kr).
Material availability
This study did not generate new materials.
Methods details
Plasmid construction
Genes encoding GDHγα were cloned into a pET21a(+) plasmid. The GDH α subunit and γ subunit of FAD-GDH from Burkholderia lata (GenBank ID; α subunit of FAD-GDH: Bcep18194_B1293, γ subunit of FAD-GDH: Bcep18194_B1292) was used. The GDHγα-pET21a(+) plasmid product included a 6× His tag and a tobacco etch virus (TEV) protease cleavage site at the N-terminus of the GDHγ subunit for purification and affinity tag removal, respectively. The GDH γ subunit consisted of 216 amino acids (23.5 kDa). The GDHα subunit included 5× Gly at the C-terminus for the spacer consisting of 544 amino acids (60 kDa). The plasmid encoding GDHγα variants (GDHγα-XGBP, X = L, M, T, and V) was produced by site-directed mutagenesis. Sequences encoding LGBP, MGBP, TGBP, and VGBP were tagged at the C-terminus of the GDH α subunit. The clones were sequenced and confirmed before transformation and expression in the host E. coli BL21-Rosetta (DE3) (Table S1).
Cell growth and purification
Escherichia coli BL21-Rosetta (DE3) expressing the GDHγα and GDHγα variants were cultured in Terrific Broth (TB) medium containing 100 mg/mL ampicillin at 37°C and 180 rpm until OD600 = 0.6 was reached. IPTG was added to a final concentration of 0.2 mM, and the cell culture was further incubated at 16°C and 180 rpm for 18–20 h. The cells were harvested by centrifugation at 4,000 rpm and 4°C for 20 min. The harvested cells were resuspended in 20 mM sodium phosphate buffer (pH 7.4) supplemented with 50 mM imidazole and 500 mM NaCl. The resuspended cells were disrupted by sonication. The lysates were centrifuged at 10,000 rpm and 4°C for 30 min, and the soluble fractions were filtered to remove cell debris and inclusion bodies. The target proteins were purified by nickel affinity chromatography. The crude extracts of the native and variant GDHγα were equilibrated in nickel affinity agarose resin (Qiagen, Hilden, Germany) and washed with 20 mM sodium phosphate buffer (pH 7.4) containing 50 mM imidazole and 500 mM NaCl. Each protein was eluted with two column volumes of a stepwise imidazole gradient (70-, 350-, and 500-mM imidazole in 20 mM sodium phosphate buffer (pH 7.4) and 500 mM NaCl) at 1 mL min−1. The protein-containing buffers were exchanged with phosphate-buffered saline (PBS; pH 7.4) using Amicon Ultra-15 (nominal MW limit, 30,000; Merck Millipore, Carrigtwohill, Ireland). Protein concentrations were estimated by the Bradford assay (Bradford, 1976). The His-tag was removed by cleaving the TEV protease recognition site via incubation with TEV protease (New England Biolabs, Ipswich, MA, USA) at 30°C for 1 h. After the TEV protease treatment, the products were observed on 10% SDS-PAGE gel. All proteins were used only after cleavage of the 6× His tag.
Enzyme activity assay
Purified wild type and engineered GDHγα activity levels were assayed by the 2,6-dichloroindophenol (DCIP) method. Briefly, the enzyme samples were incubated at room temperature (25°C) with 10 mM potassium phosphate buffer containing 6 mM phenazine methosulfate (PMS), 0.5 mM DCIP, and 100 mM glucose. Glucose oxidation was recorded at 600 nm using a microplate spectrophotometer (Epoch Microplate Spectrophotometer; BioTek Instruments, Winooski, VT, USA). To measure the decrease in adsorption, enzyme activity was calculated by the Beer-Lambert Law (Swinehart, 1962). The molar extinction coefficient (ԑ) of DCIP was 16.3 mM−1 cm−1 (Inose et al., 2003).
Surface plasmon resonance (SPR) assay
SPR binding experiments were performed on a Reichert SR7500DC dual channel SPR system (Reichert Technologies, Buffalo, NY, USA) at 25°C in PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl) and a flow rate of 30 μL min−1. To establish the baseline, the reaction buffer was injected until the response signal stabilized. Direct binding of protein sample to the gold surface was then estimated. The kinetic parameters were derived from datasets acquired in single-cycle mode (Kushwaha et al., 2019). Each run comprised five consecutive analyte injections at 6.25, 12.5, 25, 50, and 100 nM. The duration of each analyte injection was 180 s, and the injected analytes were separated by 180-s dissociation periods in which pure PBS was injected. Binding was calculated in resonance units per unit time. The data were evaluated with Scrubber2 software (BioLogic Software Pty. Ltd., Campbell, ACT, Australia).
Quartz crystal microscopy (QCM) characterization
Quartz crystal microbalance (QCM) characterization was conducted to compare protein binding strengths of GBPs fused to GDHγα on Au and Si surfaces. Piezoelectric quartz crystals in Au or Si electrode mode (QA-A9M-AU and QA-A9M-SI; Seiko EG&G, Matsudo, Japan) were used. The electrodes were mounted on a QCM Teflon flow cell (QA-CL6; SEIKO EG&G), and the adsorption signals were measured with an analyzer (QCM922A; SEIKO EG&G).
Prior to the analyte injection, PBS (pH 7.4) was run through the system until a stable baseline signal was established. The adsorption behavior of the proteins was measured by injecting PBS containing 0.1 μM target analyte. The duration of each analyte injection was 800 s. The frequency shift (Δf) on the protein adsorption was monitored with winEchem software (Seiko EG&G). The adsorption phase and Δf at the 800-s point were compared between the variant and wild type GDHγα.
Atomic force microscopy
Before enzyme immobilization, Au chips were cleaned with 1:3 (v/v) of 30% H2O2/H2SO4 (piranha solution) at room temperature for 10 min, rinsed with deionized water, and dried under a nitrogen gas stream to remove organic contaminants. The Au chips were immersed in 0.5 μM wild type or variant GDHγα in PBS and immobilized with gentle shaking for 120 min. Weakly bound proteins on the Au surface were removed by sonication in pure PBS for 30 s, washing with deionized water, and drying with nitrogen gas.
Morphology of the prepared samples was visualized by AFM in non-contact mode (XE-100; Park Systems, Langen, Germany). Measurements were conducted using 125-μm Si/Al-coated cantilevers (PPP-NCHR 10 M; Park Systems) with 200–400-kHz resonance frequency and 42 N/m spring constant. Images of 1 μm × 1 μm scan fields were acquired. Surface topography and cross-sectional analysis were performed with XEI software (Park Systems).
Fluorescence imaging by confocal laser scanning microscopy (CLSM)
Micro-patterns (100-μm resolution) of Au on Si substrate were obtained by Ti (5 nm) and Au (20 nm) deposition with an e-beam evaporator through shadow masks toward the Si substrate. The micro-patterned substrate was immersed for 2 h in PBS containing 0.1 μM wild type or variant GDHγα. After a gentle PBS rinse, the enzyme-immobilized substrate was incubated in 1 mM NHS-PEG4-Biotin (Thermo Fisher Scientific, Waltham, MA, USA) for biotinylating the immobilized proteins. The products were labeled by incubation with 2 nM streptavidin-conjugated quantum dots (QD525) for 1 h. The quantum dots self-assembled with the biotinylated proteins via strong biotin-streptavidin binding. The prepared samples were visualized by fluorescence confocal laser-scanning microscopy (LSM 880; Carl Zeiss AG, Oberkochen, Germany) using a 488-nm laser for excitation and a 495–550-nm emission filter.
Electrochemical analysis
A screen-printed gold electrode (SPGE) integrated with three electrodes was purchased from Metrohm DropSens Corp (Oviedo, Spain). The SPGE consisted of Au working electrode 4 mm in diameter, Pt auxiliary electrode, and Ag pseudo-reference electrode.
To construct the enzyme-electrode, the working electrode surface was immersed in enzyme solution containing 0.5 μM wild-type or variant GDHγα and incubated with gentle shaking for 120 min at room temperature. Excess proteins on Au surfaces were washed off with PBS. Then, enzyme-modified SPGEs were placed in reactors (working volume, 8 mL) connected to a potentiostat (Metrohm AutoLab, Utrecht, The Netherlands) fitted with a specific DropSens connector (Metrohm DropSens).
For electrochemical assessment in the GBP-only condition, the oligomers corresponding to LGBP (LKAHLPPSRLPS), MGBP (MHGKTQATSGTIQS), TGBP (TGTSVLIATPGV), and VGBP (VSGSSPDS) were commercially synthesized by Cosmo Genetech Co., Ltd. (Seoul, Korea). For the GBPs (LGBP, MGBP, TGBP, VGBP)-immobilized SPGEs, the SPGE was incubated in the 50 μM peptide solution in PBS buffer, for 2 h at room temperature, with mild shaking. The peptide-modified SPGEs were installed in the reactors and connected to the potentiostat, as mentioned earlier.
Electrochemical tests such as cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed with a potentiostat (Metrohm AutoLab) immersed in 10 mM potassium-based phosphate buffer (pH 7.4) containing various glucose concentrations (0–100 mM). The electrochemical properties of the protein (i.e., enzyme or peptide)-electrode were determined by CV over a −500 mV to 400 mV potential range (vs. Ag/Ag+) at a scan rate of 100 mV s−1, unless otherwise indicated. For the EIS measurements, an open circuit potential (OCP) was applied at 10 mV amplitude, and a Nyquist plot was obtained in the 100 kHz–0.1 Hz frequency range.
Modeling of 3D structure of wild type GDHα or GBP-fused GDHα
Full-length models of wild type GDHα or GDHα-XGBP (X = L, M, T, and V) were generated by the iterative threading assembly refinement (I-TASSER) method (Zhang, 2008; Shamriz and Ofoghi, 2016; Jamil et al., 2017). The full-length amino acid sequences of the GDHα proteins were submitted to the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). To predict the 3D structure by homology modeling, the protein with PDB ID: 6A2U (crystal structure of FAD-dependent glucose dehydrogenase gamma-alpha complex from Burkholderia cepacia) was used as the main template because it had the highest TM score (Yoshida et al., 2019). The models were imaged and analyzed with PyMOL v. 2.5.0 (Schrödinger, LLC, Cambridge, MA, USA). Further, the prediction models of wild type GDHα or GDHα-XGBP (X = L, M, T, and V) were aligned with crystal structure of FAD-dependent glucose dehydrogenase from Burkholderia cepacia (PDB ID: 6A2U) using PyMOL, to designate the location of FAD cofactor and [3Fe-4S] cluster within the GDH α subunit and to predict complexed form with GDH γ subunit.
Data and software availability
The detailed description of the methods and original data can be obtained from the authors upon a reasonable request.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (NRF), funded by the Korean Government (No. 2020R1A2C3009210 and No. 2021R1A5A1028138). The authors thank Prof. Jun-Ho Choi of the Department of Chemistry, Gwangju Institute of Science and Technology, Korea, for his insightful discussion. The authors also thank Prof. Jae Young Lee of the School of Materials Science and Engineering, Gwangju Institute of Science and Technology, Korea, for his technical support and providing the QCM instrument.
Author contributions
H.L.: Conceptualization, methodology, investigation, writing –original draft, writing – review & editing, visualization; E.M.L.: investigation; S.S.R.: writing – review & editing; I.S.C.: writing – review & editing, supervision, project administration, funding acquisition.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103373.
Supplemental information
Data and code availability
This study did not generate any unique code or data sets. All data supporting the finding of this study are available within the paper and its supplemental information files. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. All software's used in this study are commercially available.
References
- Adams B.L., Hurley M.M., Jahnke J.P., Stratis-Cullum D.N. Functional and selective bacterial interfaces using cross-scaffold gold binding peptides. JOM. 2015;67:2483–2493. [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brown S. Metal-recognition by repeating polypeptides. Nat. Biotechnol. 1997;15:269–272. doi: 10.1038/nbt0397-269. [DOI] [PubMed] [Google Scholar]
- Care A., Bergquist P.L., Sunna A. Solid-binding peptides: smart tools for nanobiotechnology. Trends Biotechnol. 2015;33:259–268. doi: 10.1016/j.tibtech.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Chakraborty S., Babanova S., Rocha R.C., Desireddy A., Artyushkova K., Boncella A.E., Atanassov P., Martinez J.S. A hybrid DNA-templated gold nanocluster for enhanced enzymatic reduction of oxygen. J. Am. Chem. Soc. 2015;137:11678–11687. doi: 10.1021/jacs.5b05338. [DOI] [PubMed] [Google Scholar]
- Das D., Ghosh S., Basumallick I. Electrochemical studies on glucose oxidation in an enzymatic fuel cell with enzyme immobilized on to reduced graphene Oxide surface. Electroanalysis. 2014;26:2408–2418. [Google Scholar]
- Fu J., Liu M., Liu Y., Woodbury N.W., Yan H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012;134:5516–5519. doi: 10.1021/ja300897h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.H., Allen H., Hill O. Direct electrochemistry of proteins and enzymes. Adv. Inorg. Chem. 1991;36:341–375. [Google Scholar]
- Gutierrez-Sanchez C., Pita M., Vaz-Dominguez C., Shleev S., De Lacey A.L. Gold nanoparticles as electronic bridges for laccase-based biocathodes. J. Am. Chem. Soc. 2012;134:17212–17220. doi: 10.1021/ja307308j. [DOI] [PubMed] [Google Scholar]
- Hatada M., Loew N., Inose-Takahashi Y., Okuda-Shimazaki J., Tsugawa W., Mulchandani A., Sode K. Development of a glucose sensor employing quick and easy modification method with mediator for altering electron acceptor preference. Bioelectrochemistry. 2018;121:185–190. doi: 10.1016/j.bioelechem.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Hess C.R., Juda G.A., Dooley D.M., Amii R.N., Hill M.G., Winkler J.R., Gray H.B. Gold electrodes wired for coupling with the deeply buried active site of Arthrobacter globiformis amine oxidase. J. Am. Chem. Soc. 2003;125:7156–7157. doi: 10.1021/ja029538q. [DOI] [PubMed] [Google Scholar]
- Hitaishi V.P., Clement R., Bourassin N., Baaden M., De Poulpiquet A., Sacquin-Mora, Ciaccafava A., Lojou E. Controlling redox enzyme orientation at planar electrodes. Catalysts. 2018;8:192. [Google Scholar]
- Hitaishi V.P., Mazurenko I., Harb M., Clément R., Taris M., Castano S., Duché D., Lecomte S., Ilbert M., Poulpiquet A., Lojou E. Electrostatic-driven activity, loading, dynamics, and stability of a redox enzyme on functionalized-gold electrodes for bioelectrocatalysis. ACS Catal. 2018;8:12004–12014. [Google Scholar]
- Hnilova M., So C.R., Oren E.E., Wilson B.R., Kacar T., Tamerler C., Sarikaya M. Peptide-directed co-assembly of nanoprobes on multimaterial patterned solid surfaces. Soft Matter. 2012;8:4327–4334. [Google Scholar]
- Holland J.T., Lau C., Brozik S., Atanassov P., Banta S. Engineering of glucose oxidase for direct electron transfer via site-specific gold nanoparticle conjugation. J. Am. Chem. Soc. 2011;133:19262–19265. doi: 10.1021/ja2071237. [DOI] [PubMed] [Google Scholar]
- Huang Y., Chiang C.Y., Lee S.K., Gao Y., Hu E.L., Yoreo J.D., Belcher A.M. Programmable assembly of nanoarchitectures using genetically engineered viruses. Nano Lett. 2005;5:1429–1434. doi: 10.1021/nl050795d. [DOI] [PubMed] [Google Scholar]
- Hughes Z.E., Nguyen M.A., Li Y., Swihart M.T., Walsh T.R., Knecht M.R. Elucidating the influence of materials-binding peptide sequence on Au surface interactions and colloidal stability of Au nanoparticles. Nanoscale. 2017;9:421–432. doi: 10.1039/c6nr07890g. [DOI] [PubMed] [Google Scholar]
- Ikeda T., Kobayashi D., Matsushita F., Sagara T., Niki K. Bioelectrocatalysis at electrodes coated with alcohol dehydrogenase, a quinohemoprotein with heme c serving as a built-in mediator. J. Electroanal. Chem. 1993;361:221–228. [Google Scholar]
- Inose K., Fujikawa M., Yamazaki T., Kojima K., Sode K. Cloning and expression of the gene encoding catalytic subunit of thermostable glucose dehydrogenase from Burkholderia cepacia in Escherichia coli. Bba-prot. Proteom. 2003;1645:133–138. doi: 10.1016/s1570-9639(02)00534-4. [DOI] [PubMed] [Google Scholar]
- Jamil F., Ali A., Sehgal S.A. Comparative modeling, molecular docking, and revealing of potential binding pockets of RASSF2; a candidate cancer gene. Interdiscip. Sci. 2017;9:214–223. doi: 10.1007/s12539-016-0145-z. [DOI] [PubMed] [Google Scholar]
- Jia F., Narasimhan B., Mallapragada S. Materials-based strategies for multi-enzyme immobilization and co-localization: a review. Biotechnol. Bioeng. 2014;111:209–222. doi: 10.1002/bit.25136. [DOI] [PubMed] [Google Scholar]
- Kacar T., Zin M.T., So C., Wilson B., Ma H., Gul-Karaguler N., Jen A.K.Y., Sarikaya M., Tamerler C. Directed self-immobilization of alkaline phosphatase on micro-patterned substrates via genetically fused metal-binding peptide. Biotechnol. Bioeng. 2009;103:696–705. doi: 10.1002/bit.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Rheem Y., Yoo B., Chong Y., Bozhilov K.N., Kim D., Sadowsky M.J., Hur H.G., Myung N.V. Peptide-mediated shape-and size-tunable synthesis of gold nanostructures. Acta Biomater. 2010;6:2681–2689. doi: 10.1016/j.actbio.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Kushwaha A., Takamura Y., Nishigaki K., Biyani M. Competitive non-SELEX for the selective and rapid enrichment of DNA aptamers and its use in electrochemical aptasensor. Sci. Rep. 2019;9:1–11. doi: 10.1038/s41598-019-43187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küchler A., Yoshimoto M., Luginbühl S., Mavelli F., Walde P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016;11:409. doi: 10.1038/nnano.2016.54. [DOI] [PubMed] [Google Scholar]
- Lalaoui N., Rousselot-Pailley P., Robert V., Mekmouche Y., Villalonga R., Holzinger M., Cosnier S., Tron T., Le Goff A. Direct electron transfer between a site-specific pyrene-modified laccase and carbon nanotube/gold nanoparticle supramolecular assemblies for bioelectrocatalytic dioxygen reduction. ACS Catal. 2016;6:1894–1900. [Google Scholar]
- Lee H., Lee Y.S., Reginald S.S., Baek S., Lee E.M., Choi I.G., Chang I.S. Biosensing and electrochemical properties of flavin adenine dinucleotide (FAD)-dependent glucose dehydrogenase (GDH) fused to a gold binding peptide. Biosens. Bioelectron. 2020;165:112427. doi: 10.1016/j.bios.2020.112427. [DOI] [PubMed] [Google Scholar]
- Lee I., Loew N., Tsugawa W., Lin C.E., Probst D., La Belle J.T., Sode K. The electrochemical behavior of a FAD dependent glucose dehydrogenase with direct electron transfer subunit by immobilization on self-assembled monolayers. Bioelectrochemistry. 2018;121:1–6. doi: 10.1016/j.bioelechem.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Lee Y.S., Baek S., Lee H., Reginald S.S., Kim Y., Kang H., Choi I.G., Chang I.S. Construction of uniform monolayer-and orientation-tunable enzyme electrode by a synthetic glucose dehydrogenase without electron-transfer subunit via optimized site-specific gold-binding peptide capable of direct electron transfer. ACS Appl. Mater. Interfaces. 2018;10:28615–28626. doi: 10.1021/acsami.8b08876. [DOI] [PubMed] [Google Scholar]
- Lee H., Lee Y.S., Lee S.K., Baek S., Choi I.G., Jang J.H., Chang I.S. Significant enhancement of direct electric communication across enzyme-electrode interface via nano-patterning of synthetic glucose dehydrogenase on spatially tunable gold nanoparticle (AuNP)-modified electrode. Biosens. Bioelectron. 2019;126:170–177. doi: 10.1016/j.bios.2018.10.013. [DOI] [PubMed] [Google Scholar]
- Liu Y., Zhang J., Cheng Y., Jiang S.P. Effect of carbon nanotubes on direct electron transfer and electrocatalytic activity of immobilized glucose oxidase. ACS Omega. 2018;3:667–676. doi: 10.1021/acsomega.7b01633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Chakraborty S., Hosseinzadeh P., Yu Y., Tian S., Petrik I., Bhagi A., Lu Y. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev. 2014;114:4366–4469. doi: 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Laurent C.V., Meneghello M., Tuoriniemi J., Oostenbrink C., Gorton L., Bartlett P.N., Ludwig R. Direct electron-transfer anisotropy of a site-specifically immobilized cellobiose dehydrogenase. ACS Catal. 2019;9:7607–7615. [Google Scholar]
- Martins M.V., Pereira A.R., Luz R.A., Iost R.M., Crespilho F.N. Evidence of short-range electron transfer of a redox enzyme on graphene oxide electrodes. Phys. Chem. Chem. Phys. 2014;16:17426–17436. doi: 10.1039/c4cp00452c. [DOI] [PubMed] [Google Scholar]
- Nam K.T., Kim D.W., Yoo P.J., Chiang C.Y., Meethong N., Hammond P.T., Chiang Y.M., Belcher A.M. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312:885–888. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- Page C.C., Moser C.C., Chen X., Dutton P.L. Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Palafox-Hernandez J.P., Tang Z., Hughes Z.E., Li Y., Swihart M.T., Prasad P.N., Walsh T.R., Knecht M.R. Comparative study of materials-binding peptide interactions with gold and silver surfaces and nanostructures: a thermodynamic basis for biological selectivity of inorganic materials. Chem. Mater. 2014;26:4960–4969. [Google Scholar]
- Seker U.O.S., Wilson B., Dincer S., Kim I.W., Oren E.E., Evans J.S., Tamerler C., Sarikaya M. Adsorption behavior of linear and cyclic genetically engineered platinum binding peptides. Langmuir. 2007;23:7895–7900. doi: 10.1021/la700446g. [DOI] [PubMed] [Google Scholar]
- Shamriz S., Ofoghi H. Design, structure prediction and molecular dynamics simulation of a fusion construct containing malaria pre-erythrocytic vaccine candidate, Pf CelTOS, and human interleukin 2 as adjuvant. BMC Bioinform. 2016;17:1–15. doi: 10.1186/s12859-016-0918-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino I., Stanzione I., Piscitelli A., Giardina P., Le Goff A. Carbon-nanotube-supported POXA1b laccase and its hydrophobin chimera for oxygen reduction and picomolar phenol biosensing. Biosens. Bioelectron. 2021;8:100074. [Google Scholar]
- Swinehart D.F. The Beer-Lambert law. J. Chem. Educ. 1962;39:333. [Google Scholar]
- Tamerler C., Duman M., Oren E.E., Gungormus M., Xiong X., Kacar T., Parviz B.A., Sarikaya M. Materials specificity and directed assembly of a gold-binding peptide. Small. 2006;2:1372–1378. doi: 10.1002/smll.200600070. [DOI] [PubMed] [Google Scholar]
- Vallee A., Humblot V., Pradier C.M. Peptide interactions with metal and oxide surfaces. Acc. Chem. Res. 2010;43:1297–1306. doi: 10.1021/ar100017n. [DOI] [PubMed] [Google Scholar]
- Vazquez-Duhalt R., Aguila S.A., Arrocha A.A., Ayala M. QM/MM molecular modeling and Marcus theory in the molecular design of electrodes for enzymatic fuel cells. ChemElectroChem. 2014;1:496–513. [Google Scholar]
- Wardlaw D.M., Marcus R.A. Unimolecular reaction rate theory for transition states of partial looseness. II. Implementation and analysis with applications to NO2 and C2H6 dissociations. J. Chem. Phys. 1985;83:3462–3480. [Google Scholar]
- Wollenberger U. Third generation biosensors—integrating recognition and transduction in electrochemical sensors. Compr. Anal. Chem. 2005;44:65–130. [Google Scholar]
- Yates N.D., Fascione M.A., Parkin A. Methodologies for “wiring” redox proteins/enzymes to electrode surfaces. Chem. Eur. J. 2018;24:12164–12182. doi: 10.1002/chem.201800750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Kojima K., Shiota M., Yoshimatsu K., Yamazaki T., Ferri S., Tsugawa W., Kamitori S., Sode K. X-ray structure of the direct electron transfer-type FAD glucose dehydrogenase catalytic subunit complexed with a hitchhiker protein. Acta Crystallogr. D. 2019;75:841–851. doi: 10.1107/S2059798319010878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Becker M.L., Carri G.A. The influence of amino acid sequence and functionality on the binding process of peptides onto gold surfaces. Langmuir. 2012;28:1408–1417. doi: 10.1021/la204109r. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:1–8. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The detailed description of the methods and original data can be obtained from the authors upon a reasonable request.
This study did not generate any unique code or data sets. All data supporting the finding of this study are available within the paper and its supplemental information files. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. All software's used in this study are commercially available.