

Summary
Thrombocytopenia-absent radius (TAR) syndrome is caused by RBM8A insufficiency. We generated megakaryocyte-specific Rbm8a knockout (Rbm8aKOMK) mice that exhibited marked thrombocytopenia, internal hemorrhage, and splenomegaly, providing evidence that genetic deficiency of Rbm8a causes a disorder of platelet production. Rbm8aKOMK mice accumulated low-ploidy immature megakaryocytes in the bone marrow and exhibited defective platelet activation and aggregation. Accordingly, depletion of Y14 (RBM8A) in human erythroleukemia (HEL) cells compromised phorbol-ester-induced polyploidization. Notably, Y14/RBM8A deficiency induced both p53 and p21 in megakaryocytes and HEL cells. Treatment with a p53 inhibitor restored ex vivo differentiation of Rbm8aKOMK megakaryocytes and unexpectedly activated Y14 expression in HEL cells. Trp53 knockout partially restored megakaryocyte differentiation by reversing cell-cycle arrest and increased platelet counts of Rbm8aKOMK, indicating that excess p53 in part accounts for thrombocytopenia in TAR syndrome. This study provides evidence for the role of the Y14-p53 circuit in platelet production and a potential therapeutic strategy.
Subject areas: Biological sciences, Cell biology, Functional aspects of cell biology
Graphical abstract
Highlights
-
•
Rbm8a (Y14) knockout in megakaryocyte drastically lowers the platelet count
-
•
Rbm8a knockout activates the p53-p21 axis and disrupts cell cycle in megakaryocytes
-
•
Y14 deficiency impairs the expression of cell-cycle factors in megakaryocytic cells
-
•
Trp53 knockout partially restores platelet levels in megakaryocytic Rbm8a knockout
Biological sciences; Cell biology; Functional aspects of cell biology
Introduction
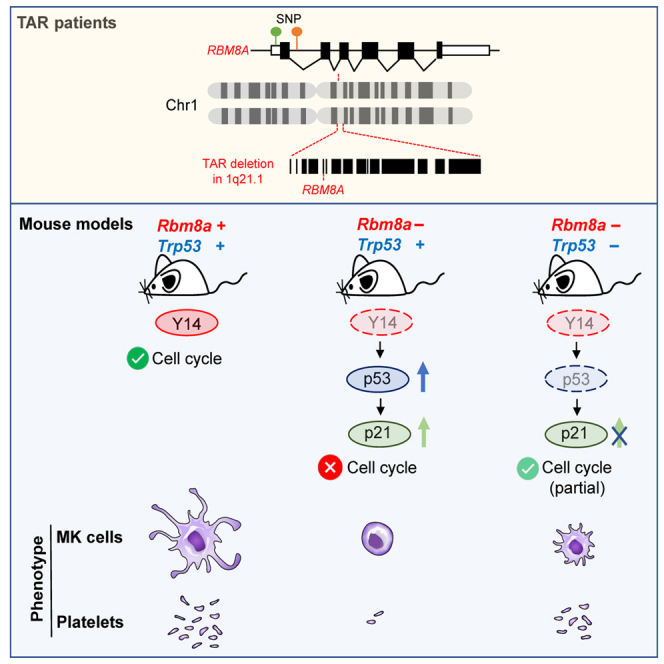
Thrombocytopenia-absent radii (TAR) syndrome is a rare congenital disorder characterized by bilateral radial aplasia and a reduced platelet count (Hall, 1987). Thrombocytopenia of the TAR syndrome results from defective differentiation of megakaryocyte precursors (Letestu et al., 2000). TAR is caused by heterozygosity for a deletion within chromosome 1q21.1 encompassing RBM8A and concomitant noncoding single nucleotide polymorphisms in another RBM8A allele that downregulate RBM8A expression (Albers et al., 2012). The deletion and mutations together result in diminished RBM8A expression in platelets from TAR syndrome patients (Albers et al., 2012), suggesting that RBM8A deficiency impacts bone morphogenesis and platelet production.
RBM8A encodes the RNA processing factor Y14/RBM8A (hereafter referred to as Y14) that participates in multiple steps of mRNA metabolism. Y14 is a constituent of the exon junction complex, which marks the position of the splice junction in mature mRNAs and initiates mRNA surveillance during the pioneer round of translation (Maquat et al., 2010). Depletion of Y14, however, alters mRNA splicing patterns, indicating that Y14 also modulates alternative splicing (Fukumura et al., 2016; Michelle et al., 2012). Moreover, Y14 is involved in DNA damage repair through its direct interaction with the nonhomologous end-joining machinery (Chuang et al., 2019). Y14 deficiency leads to accumulation of DNA damage and cell-cycle arrest at G2/M phase (Ishigaki et al., 2016; Lu et al., 2017). Rbm8a haploinsufficiency impairs mouse cortical development due to reduced numbers of progenitors and neurons and the perturbation of cortical lamination (Mao et al., 2015). Morpholino-mediated knockdown of rbm8a in Danio rerio results in disorganized myofibers and motor axon outgrowth defects (Gangras et al., 2020).
Platelets arise from cytoplasmic blebbing and fragmentation of mature megakaryocytes. Megakaryopoiesis is a multi-step process consisting of the commitment of hematopoietic stem cells to the megakaryocytic lineage, megakaryocyte maturation, and terminal differentiation (Machlus and Italiano, 2013; Mazzi et al., 2018). Promegakaryoblasts undergo endomitosis to increase their DNA content ranging from 2N to 128N (64N in mice). The endomitotic process is an abortive mitosis without telophase and cytokinesis, giving rise to a polylobulated nucleus. A number of cell-cycle regulators have been implicated in endomitosis, such as the cyclin-dependent kinase inhibitors p21CIP1/WAF1 and p19INK4D and the anaphase-promoting complex regulator Cdc20 (Raslova et al., 2007; Taniguchi et al., 1999; Trakala et al., 2015). Ablation of Cdc20 in megakaryocytes through Pf4-Cre (platelet factor 4-driven Cre recombinase) reduces ploidy level and impairs platelet production in mice (Trakala et al., 2015). The tumor suppressor p53 controls both cell-cycle progression and apoptosis. In contrast to Cdc20 knockout, however, TP53-knockout megakaryocytes exhibit increased ploidy and differentiation potential in vitro (Apostolidis et al., 2012b). Overexpression or pharmacological activation of p53 leads to decreased polyploidization of megakaryocytes (Apostolidis et al., 2012a; Mahfoudhi et al., 2016). Therefore, Cdc20 and p53 play opposite roles in polyploidization of megakaryocytes.
In this study, we generated a conditional knockout mouse model to investigate the role of RBM8A in megakaryocyte differentiation and platelet production.
Results
Megakaryocyte-specific Rbm8a knockout causes thrombocytopenia
To ablate Rbm8a in the megakaryocyte lineage, we generated a loxP-flanked Rbm8a allele using a CRISPR/Cas9-mediated insertion system (Figure S1A). Mice carrying homologous loxP-flanked Rbm8a alleles (Figure S1B) were mated to Pf4-Cre mice (i.e. Pf4Cre/+;Rbm8a+/+, wild-type) that express Cre primarily in the megakaryocytic lineage (Tiedt et al., 2007) to generate Pf4Cre/+;Rbm8af/+ (Con) and Pf4Cre/+;Rbm8af/f (Rbm8aKOMK) (Figure 1A and Table S1). Immunohistochemical (IHC) staining revealed that Y14 was expressed in Con megakaryocytes but was almost undetectable in Rbm8aKOMK megakaryocytes (Figures 1B and S1C), indicating efficient depletion of Y14 in megakaryocytes. The vast majority of male Rbm8aKOMK mice died within 2–8 weeks after birth (Figure 1C). Subcutaneous and gastrointestinal bleeding was frequently observed in 2-week-old male mice, indicating platelet defects and inflammation (Figure 1D). All female Rbm8aKOMK mice survived until the end of the study (up to 3 months) while showing moderate subdermal hemorrhage and internal hemorrhage near lymph nodes (Figure S1D). Moreover, Rbm8aKOMK mice of both genders exhibited splenomegaly (Figure 1E), suggesting sequestration of immature megakaryocytes in the spleen (see below).
Figure 1.

Megakaryocyte-specific Rbm8a knockout impairs platelet production
(A) Schematic diagram illustrating the generation of megakaryocyte-specific Rbm8a knockout mice.
(B) Immunohistochemical (IHC) staining for Y14 in the sections of bone marrow and spleen from 9-week-old Con and Rbm8aKOMK mice, magnified images (from Figure S1C) of representative cells are shown. Bar graph with overlapping dots shows the relative intensity unit (RIU; mean ± SEM) of Y14 in megakaryocytes from Con and Rbm8aKOMK bone marrow and spleen measured by using ImageJ. N, sample number; ∗∗∗∗p<0.0001.
(C) Death rate of 8-week-old male and female Rbm8aKOMK mice.
(D) Subcutaneous tissue (skin) and gastrointestinal tract of 14-day-old male Rbm8aKOMK mice.
(E) The spleen/body weight ratio (mg/g; mean ± SEM, N = 12 in each group, ∗∗p < 0.01) of 10- to 12-week-old mice.
(F) Hematological analysis of blood from 6- to 8-week-old mice of the indicated genotypes (mean ± SEM). N, sample number; M, male mice; F, female mice; ns, not statistically significant; ∗∗∗p < 0.001.
(G) Tail bleeding time of 8- to 10-week-old Con and Rbm8aKOMK mice was recorded up to 6 min. N, sample number; ∗∗p < 0.01.
Knockout of one Rbm8a allele slightly reduced the count of platelets and white blood cells (Figure 1F, wild-type versus Con, and Table S2), the latter was possibly due to minimal expression of Pf4 in nonmegakaryocyte hematopoietic cells (Gollomp and Poncz, 2019). Nevertheless, a drastic reduction in the number of platelets was observed in Rbm8aKOMK mice at 6–8 weeks of age, including one male (Figure 1F, platelets), whereas the counts of other blood cells were nearly the same between Con and Rbm8aKOMK mice (red and white blood cells). Accordingly, Rbm8aKOMK mice showed a robust increase in tail bleeding compared with control mice (Figure 1G). The result that homozygous, but not heterozygous, Rbm8a deletion abolished platelet production echoed the scenario of TAR syndrome, i.e., thrombocytopenia resulted from biallelic disruption of RBM8A.
Rbm8a knockout impairs megakaryocyte differentiation
Next, we explored how Rbm8a participates in platelet production. Hematoxylin and eosin (H&E) staining revealed that Rbm8a knockout significantly reduced the average size of megakaryocytes in the bone marrow although the number of immature megakaryocytes was increased (Figure 2A). IHC staining revealed that the level of von Willebrand factor (vWF), a marker for mature megakaryocytes, was reduced by ∼60% (Figure 2B), suggesting that megakaryocyte differentiation was incomplete in Rbm8aKOMK mice. In the spleen, Rbm8aKOMK megakaryocytes formed clumps (Figure 2C, yellow dashed line), so that their number was greatly increased (bar graph), and, however, their size was reduced (dot graph). Accordingly, IHC staining revealed a substantial reduction of vWF in splenic megakaryocytes (Figure 2D), indicating impairment of their differentiation pathway. Moreover, the relatively high nuclear-to-cytoplasmic ratio for Rbm8aKOMK megakaryocytes reflected their immaturity. The observed accumulation of megakaryocytes in the spleen constituted evidence of splenomegaly in Rbm8aKOMK mice.
Figure 2.

Rbm8a knockout impairs megakaryopoiesis
(A and B) Hematoxylin and eosin (H&E) staining was performed using bone marrow sections from 9-week-old Con and Rbm8aKOMK mice. Yellow arrows indicate megakaryocytes. A selected megakaryocyte in the dashed-line square is magnified in the inset. The dot plots shows the size of megakaryocytes measured from H&E staining (1.0 = 500 μm2); for each, N indicates the number of cells from five mice of each genotype (mean ± SEM) that were measured. The bar graph shows the number of megakaryocytes in a 0.15 mm2 area; mean ± SEM was calculated from five areas each. ∗∗∗p < 0.001. MK, megakaryocyte. Scale bars in panels B, C, and D are the same with panel (A).
(B) IHC staining was performed using anti-vWF in the bone marrow as in panel (A). Bar graph with overlapping dots shows the level of vWF as measured in Figure 1B.
(C) H&E and IHC of the spleen sections from 9-week-old Con and Rbm8aKOMK mice were respectively performed as in panels A and (B). In panel C, yellow dashed lines indicate a cluster of megakaryocytes. The dot plots and bar graph were as in panel (A).
(D) IHC staining was performed using anti-vWF in the spleen as in panel (C). The level of vWF was measured as in Figure 2B.
(E) A representative DNA content histogram of bone marrow cells (megakaryocytes) from Con and Rbm8aKOMK mice. Quantitative analysis is shown in Figure S1E.
(F) Stacked-column bar shows the proportion (%) of each nuclear shape of Con and Rbm8aKOMK megakaryocytes. Representative images of the different nuclear shapes were obtained from Con mice. All images were collected from five mice for each genotype.
Because polyploidization is an important step during megakaryocyte maturation, we measured DNA content of Rbm8aKOMK megakaryocytes. Flow cytometry revealed an ∼1/3 reduction in the fraction of megakaryocytes with higher ploidy (≥8N) in Rbm8aKOMK mice, indicating that Rbm8a knockout interfered with megakaryocytic polyploidization (Figure 2E, a representative graph; Figure S1E, quantitative data). Along with an increase in ploidy, differentiated megakaryocytes display a single polylobulated nucleus (Mazzi et al., 2018). Therefore, we examined nuclear lobulation of megakaryocytes according to Trakala et al. (2015). H&E staining revealed that ∼50% of control megakaryocytes were polylobulated and nearly 20% bilobulated, whereas ∼50% of Rbm8aKOMK megakaryocytes were monolobulated (Figure 2F). Moreover, Rbm8a knockout likely prolonged mitosis and/or caused mitotic arrest (Figure 2F, ∼30% were condensed nuclei as indicated by red and blue columns). Together, these results indicated that Rbm8a is possibly essential for polyploidization during megakaryocyte differentiation.
Y14 depletion impairs cell-cycle progression and impairs endomitosis
Next, we attempted to evaluate the role of Y14 in megakaryocyte polyploidization and differentiation in vitro. Among three cell lines, i.e., human erythroleukemic K562 and HEL cells and megakaryoblastic leukemic MEG01 cells, tested, HEL cells exhibited a higher efficiency for phorbol 12-myristate 13-acetate (PMA)-induced polyploidization and short hairpin (sh) RNA-mediated gene knockdown and were then used in the following experiments. PMA treatment of HEL cells induced the expression of megakaryocyte markers, CD41/GPIIb and CD61/GPIIIa, in HEL cells (Figure 3A, cp. bars 1 and 3). Depletion of Y14 by lentiviral transduction of a Y14-targeting shRNA reduced their expression in PMA-treated HEL cells (Figure 3A, cp. bars 3 and 4), indicating that Y14 is essential for megakaryocyte differentiation. Flow cytometry revealed that PMA increased the 8N population from 7% to 23% in HEL cells (Figure 3B). Depletion of Y14 in PMA-treated cells substantially abolished the 4N/8N peaks and intriguingly induced a peak between 2N and 4N (Figure 3B, shY14_PMA, arrow), which was not observed without PMA treatment (shY14_mock). Ethynyl deoxyuridine (EdU) labeling revealed that Y14 depletion reduced DNA synthesis in PMA-treated HEL cells (Figure S2A). Although Y14 depletion slightly increased the level of cleaved caspase 3, a marker of apoptosis, PMA treatment substantially suppressed apoptosis (Figure S2B). Therefore, the peak between 2N and 4N neither represented the sub-G1 population nor resulted from prolonged S phase. Nevertheless, an increased level of phosphorylated histone 3 (phospho-H3) in Y14-depleted HEL cells and bone-marrow megakaryocytes (Figures 3C and S1F) indicated cell-cycle arrest, which likely impeded endomitosis. Because the features of Rbm8aKOMK megakaryocytes, including reduced ploidy and nuclear lobe number and increased phospho-H3 and γH2AX (Figures 2E, 2F, and S1F), were reminiscent of Cdc20 null megakaryocytes (Trakala et al., 2015), we examined Cdc20 in Y14-depleted HEL cells. Immunoblotting revealed that Y14 knockdown reduced the level of Cdc20 by ∼60% in PMA-treated HEL cells (Figure 3C), further supporting a critical role for Y14 in endomitosis.
Figure 3.

Depletion of Y14 impairs polyploidization
(A) HEL cells were transduced with lentivirus expressing shY14 (+) or control shRNA (−) for 48 h, followed by mock-treated or PMA (5 nM) treatment for 3 days. RT-qPCR of CD41 and CD61 and immunoblotting (IB) of Y14 and GAPDH were performed; experiments were performed in triplicate (mean ± SEM; ∗p < 0.05, ∗∗p < 0.01).
(B) DNA histogram analysis of HEL cells as in panel A was performed. The stacked-column bar shows ploidy distribution; a peak between 2N and 4N (arrow) was quantified as 2N.
(C) HEL cells were transduced and treated as in panel (A). Immunoblotting was performed using antibodies as indicated. The relative expression level of phospho-H3 (pH3) and CDC20 was indicated; average was obtained from 3 times of the experiments.
Y14 deficiency compromises megakaryocyte maturation and disrupts platelet function
Next, we attempted to gain a more comprehensive insight into how Y14 may affect gene expression during megakaryocyte differentiation. Using HEL cells, we performed RNA-seq (NCBI accession number: PRJNA684792) and observed that PMA treatment induced a significant fraction of megakaryocyte-specific transcripts reported previously (Chen et al., 2014) (Figure 4A, diagram for the procedure, and S3A). This observation was supported by gene set enrichment analysis (GSEA) (Figure S3B). Gene Ontology (GO) enrichment analysis revealed that PMA-induced genes are involved in cell adhesion, endomembrane and cytoskeleton organization, and cellular signaling (Figure S3C). This analysis in addition indicated a role for Y14 in PMA-induced ER stress response and Fc receptor signaling (Figure S3D, upper). Reverse transcription and quantitative PCR (RT-qPCR) confirmed that the expression of several genes in these two functional groups was indeed PMA inducible as well as Y14 dependent (Figure 4B). Among the factors analyzed, interleukin-8 (IL-8/CXCL8), transcription factor NFATC1, platelet immunoglobulin G Fc receptor FcγRIIa (FCGR2A), and protein kinase C have been implicated in megakaryopoiesis and platelet activation (Bournazos et al., 2016; Kim et al., 2019; Lumelsky and Schwartz, 1997; Zhu et al., 2018).
Figure 4.

Y14 deficiency compromises megakaryocyte-maturation-related gene expression and impairs platelet function
(A) The diagram shows the procedure of RNA-seq analysis in control or Y14-depleted HEL cells that were treated or untreated with PMA (see panel B).
(B) HEL cells were transduced with lentivirus expressing shY14 (+) or control shRNA (−) for 48 h at a multiplicity of infection of 5, followed by mock-treated or PMA (5 nM) treatment for 3 days. RT-qPCR analysis of the indicated genes in HEL cells that were transfected with shRNA and then mock-treated or treated with PMA as in panel (A). The experiments were performed in triplicate (mean ± SEM; ∗p < 0.05, ∗∗p < 0.01).
(C) Whole blood of Con and Rbm8aKOMK mice was collected and stained for IgG, GP-VI, GP-IX, α2bβ3, and CD41 (Con: blue; Rbm8aKOMK: red). Platelets were gated by forward scattering/side scattering characteristics. Representative flow cytometry profiles showing the indicated glycoproteins or integrins.
(D) Blood was mock-treated (resting; dashed lines) or treated with PAR4 agonist (solid lines) and subsequently stained with an antibody against JON/A (phycoerythrin), fibrinogen, vWF, or P-selectin. Flow cytometry profiles of platelet glycoproteins are shown as in panel (C).
The above observation prompted us to evaluate whether Rbm8a knockout influences platelet activity. Although the count of platelets was drastically reduced in Rbm8aKOMK, the remaining was sufficient for flow cytometric analysis. Using immunoglobulin G (IgG) as control, we observed a substantial reduction in the expression of the glycoproteins GP-VI and GP-IX and integrins α2bβ3 and CD41, which are important for platelet function, in Rbm8aKOMK platelets (Figure 4C). Furthermore, we treated whole blood with the protease-activated receptor 4 (PAR4) agonist peptides, which induce platelet activation (Kahn et al., 1998). Flow cytometry revealed that Rbm8aKOMK platelets exhibited a severe defect in inside-out activation of β3 integrin (JON/A) and α-granule secretion of fibrinogen, vWF, and P-selectin after treatment with a PAR4 agonist (Figure 4D). Therefore, Rbm8a knockout impaired not only megakaryocyte differentiation but also platelet activity.
Inhibition of p53 reverses differentiation defects of Rbm8a knockout megakaryocytes
Previous reports have indicated that Y14 deficiency causes cell-cycle arrest and inappropriately activates p53 protein (Ishigaki et al., 2016; Lu et al., 2017; Mao et al., 2016). A recent RNA-seq analysis has revealed that cell-cycle genes and p53 pathway are respectively upregulated and downregulated during the commitment of megakaryocytic-erythroid progenitors to the megakaryocyte lineage (Lu et al., 2018). GSEA of the above RNA-seq data showed that Y14 depletion downregulated cell-cycle genes and upregulated p53 pathway (Figure S3E). This result coincided with previous reports (Ishigaki et al., 2016; Lu et al., 2017; Mao et al., 2016) and supported the role of Y14 in megakaryocyte differentiation. Moreover, in light of this observation, we suspected that Rbm8a knockout impedes megakaryocyte differentiation in part through aberrant induction of p53. IHC staining indeed revealed a higher level of p53 in Rbm8aKOMK megakaryocytes than control samples (Figure 5A, p53). Next, we explored how the possible Y14-p53 regulatory axis influences megakaryocyte differentiation. Interestingly, we observed that PMA treatment increased Y14 and reduced p53 protein levels in HEL cells, indicating inverse expression of Y14 and p53 during megakaryocyte differentiation (Figure 5B). Knockdown of Y14 increased the level of p53 in PMA-treated HEL cells (Figure 5C), consistent with that observed in bone-marrow megakaryocytes (Figure 5A).
Figure 5.

p53 inhibition reverses the differentiation defects of Rbm8aKOMK megakaryocytes
(A) IHC staining of Con and Rbm8aKOMK bone-marrow sections using antibodies against p53 and p21; their relative level was measured as in Figure 1B.
(B) HEL cells were mock- or PMA-treated for 3 days. Immunoblotting was performed to examine Y14 and p53 proteins.
(C) HEL cells were transduced with control or shY14-lentivirus, followed by PMA induction. Immunoblotting was performed as in panel (B).
(D) HEL cells were treated as in panel C; immunoblotting was performed using antibodies against indicated proteins. A diagram shows that Y14 depletion increases the expression of p53 and 21 and decreases the expression of four cell-cycle regulators examined.
(E) HEL cells were transduced as in panel C followed by mock (−) or PFT-α (1 or 5 μM) treatment. The relative levels of p21, Cdc20, and phospho-H3 (pH3) are indicated below the respective blots.
(F) HEL cells were transduced with shY14-lentivirus and treated with PMA followed by mock or PFT-α treatment. DNA histogram analysis was performed as in Figure 2E.
(G) The diagram depicts isolation and culture of megakaryocytes from E14.5 fetal liver. Isolated fetal liver stromal cells were cultured for 3 days in the presence of thrombopoietin (TPO). Megakaryocytes were enriched and cultured for another 3 days in the presence of TPO and then subjected to different analyses (panels H and I).
(H) Giemsa staining of an enriched fraction containing megakaryocytes (yellow arrows). Bar graphs show the average size (μm2) of >350 cells (middle panel) and the percentage of CD41+ (right panel); experiments were performed in triplicate (mean ± SEM; ∗∗p < 0.01, ∗∗∗p < 0.001).
(I) Immunoblotting of the megakaryocyte-containing fraction was performed using antibodies against indicated proteins; residual Y14 signal was from nonmegakaryocytes in the coculture.
(J) As in panel H, the megakaryocyte-containing fraction was mock- or PFT-α treated, followed by Giemsa staining or flow cytometry using anti-CD41. Bar graphs are shown as in panel F (mean ± SEM; ∗p < 0.05, ∗∗p < 0.01). Scale bars in panel G are the same as that in panel (F).
(K) Immunoblotting was performed in HEL cells that were transfected with the empty (vec) or p53-expressing vector (lanes 1–2) or transduced with control (shC) or p53-targeting shRNA (shp53) lentivirus (lanes 3–4). A diagram shows that p53 suppresses Y14 expression.
Next, we evaluated whether accumulated p53 dysregulates cell-cycle factors. Upregulation of the p53-cyclin-dependent kinase inhibitor 1A (p21) pathway has been observed in a number of congenital hematopoietic disorders such as Fanconi anemia and Diamond-Blackfan anemia (Ceccaldi et al., 2012; Schneider et al., 2016). IHC staining showed that p21 signal increased in Rbm8aKOMK bone marrow samples (Figure 5A, p21). Immunoblotting also showed that the level of p21 increased in Y14-depleted HEL cells (Figure 5D). Therefore, it is possible that p53 activated p21, leading to cell-cycle arrest. In addition, we wondered whether Y14 depletion also alters the expression of additional cell-cycle regulators. GSEA revealed a set of cell-cycle factors as potential targets of Y14 (Figure S3F). We selected several for verification, including CCNB1, CDK1 and CDC20 that are negatively regulated by p53 and have been implicated in megakaryocyte endomitosis (Bassini et al., 1999; Apostolidis et al., 2012a; Banerjee et al., 2009). Both RT-qPCR and immunoblotting confirmed downregulation of all selected genes/proteins (Figures S3G and 5D) in Y14-depleted HEL cells. Therefore, Y14 depletion causes cell-cycle arrest possibly via p53 activation. To assess this possibility, we treated Y14-depleted HEL cells with the p53 inhibitor pifithrin-α (PFT-α). PFT-α reduced the level of p21 and phospho-H3 and restored the level of CDC20 in a dose-dependent manner (Figure 5E, lanes 2–4) and dissolved the promiscuous peak between 2N and 4N and induced 2N and minorly 4N (Figure 5F). This result suggested that p53 inactivation at least partially restored the cell cycle.
Next, we evaluated whether p53 inhibition could promote ex vivo differentiation of Y14-deficient megakaryocytes. The fetal liver provides a source of megakaryocyte progenitors. After TPO treatment and enrichment (Figure 5G), we observed that Rbm8aKOMK megakaryocytes were smaller than the control and exhibited a reduced expression level of CD41 (Figure 5H). Rbm8aKOMK megakaryocytes in fetal liver exhibited a higher level of p53, p21, and γH2AX (Figure 5I for immunoblotting and Figure S1G for immunofluorescence staining), as observed in bone marrow (Figures 5A and S1F). PFT-α treatment restored their average size and CD41 level by 32% and 20%, respectively (Figure 5J). Together, our results indicated that excessive p53-induced p21 accounts for cell-cycle dysregulation and hence impeded differentiation of Rbm8aKOMK megakaryocytes.
The above result unexpectedly revealed that a high dose of PFT-α increased the cellular level of Y14 (Figure 5E, lane 4). Overexpression or knockdown of p53 indeed decreased or increased the expression of Y14, respectively (Figure 5K). p53 may restrict Y14 expression in megakaryoblasts at the transcriptional level (Figure S4). Upon induction of differentiation, the consequent downregulation of p53 allows an increase of Y14, which is essential for polyploidization and maturation of megakaryocytes.
p53 knockout rescues platelet production in Rbm8aKOMK mice
In light of the in vitro observation that p53 inhibition could in part reverse the differentiation defects of Rbm8aKOMK megakaryocytes, we had attempted to evaluate the effect of PFT-α in mice. Following daily intravenous injection of PFT-α for several days, mice unfortunately died of excessive bleeding. Meanwhile, we also genetically ablated p53 in Rbm8aKOMK mice. Pf4Cre/+;Rbm8af/+ mice were crossed with Trp53f/f. The resulting Rbm8af/+;Trp53f/+ mice were then crossed with Trp53f/f to generate Rbm8af/+;Trp53f/f. Subsequently, the use of Pf4Cre/+;Rbm8af/+;Trp53f/+ and Rbm8af/+;Trp53f/f mice generated all genotypes needed for subsequent analysis (Figure 6A; a detailed diagram in Figure S5A). PCR and Sanger sequencing confirmed Cre-mediated excision of floxed Trp53 sequences from bone marrow cells of floxed allele-bearing mice (Figures S5B–S5D).
Figure 6.

Trp53 knockout partially restores platelet production in Rbm8aKOMK mice
(A) Schematic diagram showing the generation of megakaryocyte-specific Rbm8a/Trp53 double-knockout mice (for the detail, see Figure S5A).
(B) Bar graph shows platelet counts of 6- to 8-week-old mice of the indicated genotypes (mean ± SEM). N, sample number. Because most of Rbm8aKOMK-p53Homo mice died at 3–5 weeks of age, data were acquired from only three mice.
(C) H&E staining of bone marrow sections from mice of indicated genotypes; magnified images of representative cells are shown.
(D) Stacked-column bar graph shows the proportion (%) of megakaryocytes in bone marrow sections with different morphological features. All images were collected from three to five mice for each genotype.
(E) Dot plots show the size of megakaryocytes measured from H&E staining (1.0 = 500 μm2); for each, 100–200 megakaryocytes from three to five mice of each genotype (mean ± SEM; ns, not statistically significant, ∗∗∗∗p < 0.0001) that were measured.
(F) IHC staining of Rbm8aKOMK-p53Het/p53Homo bone-marrow sections using anti-p21. Yellow arrow indicates megakaryocytes. The level of p21 was measured as in Figure 1B; ∗∗∗∗p < 0.0001.
(G) IHC was performed as in panel F using anti-phospho-H3. Red and black arrowheads indicate phospho-H3-positive and -negative megakaryocytes, respectively. The percentage of phospho-H3-positive cells was measured from three mice for each genotype (∼100 megakaryocytes); ∗∗p < 0.01.
Knockout of one Trp53 allele in Rbm8a+/+ or Rbm8af/+ mice did not affect platelet production, as compared with control samples (Figure 6B, cp. lanes 4 and 5 to lanes 1 and 2). Complete ablation of Trp53 minimally elevated platelet counts in Rbm8a wild-type mice (lane 7), which may result from enhanced megakaryocyte differentiation as reported previously (Apostolidis et al., 2012b). Pf4Cre/+;Rbm8af/f;Trp53f/f (Rbm8aKOMK-p53Homo), however, acquired tumors in lymph nodes, and most of them died at 3–5 weeks old regardless of gender (Figure S5E and Table S3). Nevertheless, blood analysis of the three surviving mice showed a partial restoration of the platelet count (Figure 6B, lane 9), supporting our hypothesis that p53 inactivation/deficiency could in part rescue the defects caused by Rbm8a knockout. These three mice also bore tumors and died after blood collection. Knockout of one Trp53 allele was unable to restore platelet production of Rbm8aKOMK (Figure 6B, lane 6, Pf4Cre/+;Rbm8af/f;Trp53f/+; Rbm8aKOMK-p53Het). These mice were in general alive >2 months, although two out of eight still developed tumors in lymph nodes. H&E staining of bone marrow revealed that the control Trp53f/+ or Trp53f/f megakaryocytes showed normal morphological features (Figure S5F for a representative field of cells; Figure 6C for a magnified image). The majority of Rbm8aKOMK-p53Het megakaryocytes had a hypolobated or condensed nucleus, indicating cell-cycle arrest and impaired polyploidization (Figure 6C, image). Homozygous knockout of p53 substantially reduced the population of mitotic cells, and hence Rbm8aKOMK-p53Homo megakaryocytes exhibited monolobular and even polylobular nuclei (Figure 6D). Moreover, the size of these megakaryocytes was moderately increased (Figure 6E), but their small cytoplasmic volume and accumulation in the bone marrow indicated immaturity.
The aforementioned result showed that PFT-α could partially rescue differentiation of fetal megakaryocytes by relieving cell-cycle block (Figures 5E, 5F, and 5J). Finally, we attempted to evaluate whether Trp53 knockout could reduce p21 accumulation in Rbm8aKOMK megakaryocytes (Figure 5A). IHC staining showed that Rbm8aKOMK-p53Het megakaryocytes exhibited a high level of p21 (Figure 6F), consistent with the observation in Rbm8aKOMK (Figure 5A). Homozygous Trp53 knockout largely reduced p21 signals (Figure 6F) and also reduced the percentage of phospho-H3-positive megakaryocytes (Figure 6G). This observation was in line with increased cell size and nuclear complexity of megakaryocytes (Figures 6C–6E). In conclusion, we provided evidence supporting that p53 hyperactivation in part accounts for Rbm8a knockout-induced blockage of megakaryocyte differentiation. Knockout of p53 in Rbm8a-deficient megakaryocytes could partially restore megakaryopoiesis and platelet counts.
Discussion
In this study, we generated Rbm8aKOMK using Pf4-Cre driven recombination and demonstrated that Rbm8a deficiency drastically reduced platelet counts, which phenocopies TAR syndrome. It is noteworthy that only complete loss of Rbm8a abolished platelet production, whereas heterozygous Rbm8a knockout had no effect (Figure 1). This result echoes the scenario of TAR syndrome, in which one RBM8A allele is deleted in concert with mutations (and consequent downregulation) of another allele (Albers et al., 2013). Therefore, only drastic reduction of Y14 impairs megakaryocyte differentiation. This is unlike the scenario that Rbm8a haploinsufficiency is sufficient to cause microcephaly (Mao et al., 2015). However, in contrast to megakaryocytic hypoplasia in TAR patients, Rbm8aKOMK mice had increased numbers of immature megakaryocytes in the bone marrow and spleen (Figure 2). Such a phenotypic difference has been observed in human congenital amegakaryocytic thrombocytopenia (CAMT) and mouse models. CAMT caused by thrombopoietin receptor MPL mutations shows a reduced number of megakaryocytes, whereas Pf4-Cre-driven Mpl knockout results in megakaryocyte accumulation (Ng et al., 2014). Analogously, we reasoned that germline RBM8A deficiency may impair propagation of hematopoietic stem cells in TAR patients, whereas Rbm8a knockout in late megakaryocyte precursors essentially blocked subsequent differentiation, leading to accumulation of immature megakaryocytes in the bone marrow (Figure 1). The common microdeletion region on chromosome 1q21.1 in TAR patients is conserved in mouse. Of note, within this region, PIAS3 has a role in thrombopoietin signaling (Fiedler et al., 2012). Therefore, co-deletion of Rbm8a and Pias3 and/or other neighboring genes may address whether those genes contribute to megakaryocyte differentiation. More intriguingly, although both genders exhibited internal hemorrhage and splenomegaly, male Rbm8aKOMK mice had a high risk of preweaning mortality (Figure 1). The possibilities including whether Rbm8a knockout in nonmegakaryocytes or Rbm8a-knockout-induced epigenetic alteration results in a life-threatening lesion in male mice and whether female mice are protected by sex hormones remain to be determined.
Both Rbm8aKOMK megakaryocytes and Y14-depleted HEL cells showed an increased level of p21 and phospho-H3 (Figures 3, 5, and S1). According to RNA-seq analysis in HEL cells, we experimentally confirmed that Y14 depletion compromised the expression of several mitotic regulators (Cdk1, cyclin B1/B2, and Cdc20) in HEL cells (Figures 5 and S3′). Therefore, Rbm8a/Y14 deficiency in megakaryocytes resulted in cell-cycle blockage and even impaired polyploidization in vivo. Previous reports have indicated that Y14 deficiency results in cell-cycle arrest and apoptosis in various cell lines (Michelle et al., 2012; Ishigaki et al., 2016; Lu et al., 2017). However, Rbm8a/Y14 deficiency only minimally caused apoptosis in differentiating HEL cells and megakaryocytes (Figure S2). This may explain why Rbm8a knockout resulted in accumulation of immature megakaryocytes. Moreover, RNA-seq data also revealed that Y14 may contribute to the expression of a set of PMA-induced genes related to megakaryocyte maturation and platelet function; the result was confirmed by RT-qPCR (Figure 4). In addition, analysis of PMA-induced genes that are negatively regulated by Y14 identified a set of extracellular structure/matrix proteins, which contribute to bone marrow stiffness (Figure S3D, bottom). In general, a less stiff matrix favors platelet production (Abbonante et al., 2017). Therefore, we deduced that Y14 promotes polyploidization and the expression of differentiation-related gene and meanwhile suppresses platelet release from premature megakaryocytes. RNA-seq using HEL cells provided somewhat limited information; therefore, future investigation using primary murine megakaryocytes would be important for determining Y14 targets in vivo and elucidating cell populations in Rbm8a knockout.
The Y14-p53 regulatory circuit likely contributes to megakaryocyte differentiation. Using PMA-treated HEL cells, we observed that Y14 and p53 were respectively upregulated and downregulated upon cell differentiation and that p53 negatively regulates Y14 expression (Figures 5 and 7). Therefore, p53 may restrict Y14 expression in undifferentiated megakaryocytes. When the level of p53 is decreased upon differentiation induction, Y14 is upregulated and contributes to megakaryocyte differentiation and maturation (Figure 7). Rbm8a knockout impairs megakaryocyte differentiation likely through cell-cycle disruption and p53 activation (Figures S1 and 5). A similar result was observed in Y14-depleted HEL cells (Figures 3 and 5). Excessive p53 activation likely accounts for cell-cycle arrest and decreased ploidy in Y14-depleted HEL cells and Rbm8aKOMK megakaryocytes (Figures 2 and 3). It is possible that p53 inhibition by PFT-α or Trp53 knockout reactivates cell-cycle progression (Figures 5 and 6). The observations that PFT-α treatment partially restored Rbm8aKOMK megakaryocyte differentiation ex vivo and that Trp53 knockout moderately raised the platelet count in Rbm8a knockout mice (Figures 5 and 6) suggest p53 inactivation as a potential therapeutic strategy for TAR. Nevertheless, Y14-mediated gene regulation in a p53-independent manner may also contribute to megakaryocyte maturation and thrombopoiesis. Therefore, inactivation of p53 is not sufficient for full rescue of platelet counts. Finally, we observed that ablation of p53 induced tumors in lymph nodes of Rbm8a knockout mice, leading to early death (Figure S5 and Table S3). A high frequency of lymphomas has been observed in Trp53-null mice (Donehower et al., 1995). Because Y14 is also essential for maintaining genome stability (Chuang et al., 2019), concomitant loss of Rbm8a and p53 may thus increase the incidence of cancer.
Figure 7.

The Y14/p53 regulatory circuit in megakaryocyte differentiation and its dysregulation causes TAR syndrome
(A) Upper: p53 may suppress Y14 expression in megakaryoblasts. Upon induction of differentiation, p53 is downregulated, whereas Y14 is upregulated during megakaryocyte differentiation. Lower: Rbm8a knockout increases the expression of p53 and p21, leading to a blockade of megakaryopoiesis and subsequent platelet production. Rbm8a/Trp53 double knockout partially rescues megakaryocyte cell differentiation and platelet production.
(B) This study provides an explanation for how RBM8A deficiency causes TAR syndrome. Inactivation of p53 may be a therapeutic strategy for TAR.
Abnormal p53 activation has been observed in several congenital syndromes, such as Diamond-Blackfan anemia and Fanconi anemia, that are caused by mutations in ribosome biogenesis or DNA repair pathways. Mouse models of these disorders in general can recapitulate human traits, and disorder-related phenotypes can be alleviated by inactivation of p53 (Bowen and Attardi, 2019; Tsai et al., 2021). Therefore, our present result indicated that TAR syndrome belongs to a p53-activation-associated genetic disorder. Moreover, it is interesting to note that thrombopoietin can trigger the nonhomologous end-joining pathway in hematopoietic stem cells perhaps to ensure chromosomal integrity (de Laval et al., 2013). Knockout of Fanconi-anemia-associated Fanca results in defective megakaryopoiesis (Pawlikowska et al., 2014), further suggesting that the DNA damage response network is important for preventing DNA damage accumulation through endomitotic cycles. Thus, whether the role of Y14 in DNA damage repair and genome stability maintenance (Chuang et al., 2019) also contributes to megakaryocyte differentiation remains to be deciphered.
In conclusion, our results show that p53 hyperactivation accounts for impaired megakaryocyte differentiation via cell-cycle blockage in Rbm8aKOMK mice (Figure 7). Inhibition or knockout of p53 can partially restore megakaryocyte differentiation. Therefore, TAR syndrome represents a type of abnormal p53 activation that can cause developmental defects, and the use of p53 inhibitors may prevent hemorrhages of patients with TAR syndrome.
Limitations of the study
Using mouse models, the finding of this study indicates that p53 hyperactivation in Rbm8a-deficient megakaryocytes causes cell-cycle blockage, but how Y14 and p53 reciprocally regulate endomitosis and megakaryocyte differentiation warrants future investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Y14 | Abcam | Cat# ab5828; RRID: AB_2269494 |
| Mouse monoclonal anti-phospho-Histone H2A.X (gH2AX) | Sigma-Aldrich | Cat# 05-636 |
| Mouse monoclonal anti-beta actin | Proteintech | Cat# 60008-1-Ig; RRID: AB_2289225 |
| Mouse monoclonal anti-GAPDH | Proteintech | Cat# 60004-1-Ig; RRID: AB_2107436 |
| Mouse monoclonal anti-cyclin B1 | BD Pharmingen | Cat# 557149 |
| Rabbit monoclonal anti-phospho-Histone H3 | Cell Signaling Technology | Cat# 3377; RRID: AB_1549592 |
| Rabbit monoclonal anti-Histone H3 | Cell Signaling Technology | Cat# 4499; RRID: AB_10544537 |
| Rabbit polyclonal anti-cyclin B2 | ABclonal | Cat# A3351; RRID: AB_2765068 |
| Rabbit polyclonal anti-vWF | Proteintech | Cat# 11778-1-AP; RRID: AB_10642840 |
| Rabbit polyclonal anti-p53 | Proteintech | Cat# 10442-1-AP; RRID: AB_2206609 |
| Rabbit polyclonal anti-p21 | Proteintech | Cat# 10355-1-AP; RRID: AB_2077682 |
| Rabbit polyclonal anti-p21 (C-19) | Santa Cruz | Cat# sc-397; RRID: AB_632126 |
| Rabbit polyclonal anti-CDK1 | Proteintech | Cat# 19532-1-AP; RRID: AB_10638617 |
| Rabbit polyclonal anti-Cdc20 | Abcam | Cat# ab183479 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit polyclonal anti-Y14 | Bethyl | Cat# A301-033A; RRID: AB_2300943 |
| Rabbit polyclonal anti-gamma H2AX (p Ser 139) | Novus Biologicals | Cat# NB100-384; RRID: AB_10002815 |
| Rat Monoclonal anti-Mouse GP-VI | EMFRET Analytics | Cat# M011-1; RRID: AB_2827531 |
| Rat Monoclonal anti-GP-IX | EMFRET Analytics | Cat# M051-1; RRID: AB_2827529 |
| Rat anti-Mouse α2bβ3 | EMFRET Analytics | Cat# M021-1 |
| Rat Monoclonal anti-JON/A | EMFRET Analytics | Cat# M023-2; RRID: AB_2833084 |
| Rabbit polyclonal anti-Fibrinogen | EMFRET Analytics | Cat# P140-1 |
| Rabbit Polyclonal anti-vWF | EMFRET Analytics | Cat# P150-1 |
| Rat anti-Mouse P-selectin | EMFRET Analytics | Cat# M130-1; RRID: AB_2890922 |
| Rat anti-Mouse CD41 | BD Biosciences | Cat# 553848; RRID: AB_395085 |
| Rat anti-Mouse IgG | BD Biosciences | Cat# 554684; RRID: AB_395508 |
| Mouse anti-Human CD41 | BD Biosciences | Cat# 555466; RRID: AB_395858 |
| Anti-Mouse IgG HRP | GE Healthcare | Cat# NA931; RRID: AB_772210 |
| Anti-Rabbit IgG HRP | GE Healthcare | Cat# NA934; RRID: AB_772206 |
| Bacterial and virus strains | ||
| Luc-targeting shRNA-expressing lentivirus | RNAi Core Facility, Academia Sinica | N/A |
| Y14-targeting shRNA-expressing lentivirus | RNAi Core Facility, Academia Sinica | N/A |
| P53-targeting shRNA-expressing lentivirus | RNAi Core Facility, Academia Sinica | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PMA | Sigma-Aldrich | Cat# P1585 |
| Pifithrin-α | Sigma-Aldrich | Cat# 506132 |
| PAR4 activating peptide | Bachem | Cat# H6054 |
| Thrombopotein (TPO), Mouse | GenScript | Cat# Z03175 |
| Critical commercial assays | ||
| Click-iTTM EdU Alexa FluorTM 647 Flow Cytometry Assay kit | ThermoFisher Scientific | Cat# C10340 |
| Deposited data | ||
| RNA-seq data | This study | PRJNA684792 |
| Experimental models: Organisms/strains | ||
| Mouse: Pf4-Cre; Rbm8af/f (C57BL/6J strain) | This study | N/A |
| Mouse: Pf4-Cre; Rbm8af/f; Trp53f/f (C57BL/6J strain) | This study | N/A |
| Oligonucleotides | ||
| All primers used in this study were listed in Table S4. | This study | N/A |
| Recombinant DNA | ||
| Plasmid: pcDNA-FLAG-p53-expression vector | (Lu et al., 2017) | N/A |
| Plasmid: pcDNA-FLAG-p53 M133K mutant expression vector | This study | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2016 | http://imagej.nih.gov/ij/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Trimmomatic v.0.38 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| HISAT2 | Kim et al., 2015; Sahraeian et al., 2017 | http://daehwankimlab.github.io/hisat2/ |
| featureCounts | Liao et al., 2014 | N/A |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| clusterProfiler (3.10.1) | Subramanian et al., 2005 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Other | ||
| Mendeley data | This study | https://doi.org/10.17632/ycpm9nyfr2.1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Woan-Yuh Tarn (wtarn@ibms.sinica.edu.tw)
Materials availability
There are restrictions to the availability of new generated plasmids and mouse lines due to MTA.
Experimental model and subject details
Animal models
Experimental mice used in this study are C57BL/6J strain with both genders as indicated in Figure 1. The age of experimental mice has been indicated in Figure Legends. The influence of gender on survival rate of Rbm8aKOMK mice is shown in Figure 1 and described in the text. All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee, Academia Sinica (protocols 12-12-449 and 17-08-1104) and complied with the Ministry of Science and Technology, Taiwan.
Cell lines
HEL cells were grown in RPMI medium supplemented with penicillin-streptomycin and 10% fetal bovine serum (Gibco). HEK293 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with penicillin-streptomycin and 10% fetal bovine serum. All cell lines were incubated at 37°C and with 5% CO2.
Method details
Generation of megakaryocyte-specific knockout mice
Conditional allele of Rbm8a (Rbm8af/f) was created by insertion of loxP sites flanking its exon 2 and 6 using the CRISPR/Cas9 system. Rbm8af/f mice were then crossed with Pf4-Cre mice (Tiedt et al., 2007). We first generated heterozygous mice for Cre and floxed Rbm8a (Pf4Cre/+;Rbm8af/+, Con) that were then bred to Rbm8af/f to produce Rbm8a megakaryocyte/platelet-specific knockout (Pf4Cre/+;Rbm8af/f Rbm8aKOMK) mice (Figure 1A). These mice were born at the expected Mendelian frequency (Table S1). Genotyping was conducted by PCR of toe lysate (primers are listed in Table S4). To generate megakaryocyte/platelet-specific Rbm8a and Trp53 (p53) knockout, we crossed Rbm8aKOMK mice with Trp53F2-10/F2-10 (hereafter termed as Trp53f/f) (Jonkers et al., 2001). In brief, Pf4Cre/+;Rbm8af/+ were mated with Trp53f/f mice to generate Pf4Cre/+;Rbm8af/+;Trp53f/+. Meanwhile, Rbm8af/+;Trp53f/+ mice were bred with the Trp53f/f mice to generate Rbm8af/+;Trp53f/f. Next, Pf4Cre/+;Rbm8af/+;Trp53f/+ were bred with Rbm8af/+;Trp53f/f to generate Pf4Cre/+ mice with Rbm8a (+/+ , f/+ or f/f) and Trp53 (f/+ or f/f) (Figures 6A and S5A). All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee, Academia Sinica (protocols 12-12-449 and 17-08-1104) and complied with the Ministry of Science and Technology, Taiwan. In the text, Pf4Cre/+ is abbreviated as Pf4-Cre. Note that male Pf4-Cre;Rbm8af/f died before sexual maturity, and male Pf4-Cre; Trp53f/f had a high incidence of scrotal swelling possibly due to testicular teratoma, which restricted multiple mating.
Histochemistry and immunohistochemistry
Paraffin-embedded mouse spleen and femur (bone marrow) sections were prepared according to standard procedures. Deparaffinized and rehydrated sections were stained with HE for histology. For IHC, retrieved sections were sequentially incubated with monoclonal anti-Y14 (Abcam) or γH2AX (Sigma-Aldrich), polyclonal antibodies against vWF, p53 (Proteintech), p21 (Santa Cruz), cleaved caspase-3 (CC3) or phospho-histone-H3 (pH3) (Cell Signaling Technology) overnight at 4°C and horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, followed by staining with DAB Quanto (Thermo Fisher). The nuclei were counter-stained with hematoxylin. The size and number of megakaryocytes were analyzed and quantified by using ImageJ software.
Immunofluorescence staining of fetal liver
Fetal liver on embryonic day 13.5 was isolated, fixed with 4% paraformaldehyde and subjected to perform cryosectioning. The sections were co-stained with FITC-conjugated rat anti-mouse CD41 (BD Biosciences) and polyclonal antibodies against Y14 (Bethyl) or γH2AX (Novus) overnight at 4°C, and then were then incubated with appropriate secondary antibodies and stained with Hoechst 33258 (Sigma).
Hematological analysis
Fresh blood was collected in K2EDTA tubes (BD Biosciences) and analyzed using ProCyte Dx® Hematology Analyzer (IDEXX Laboratories) according to manufacturer’s instruction.
Platelet activation and flow cytometry
Blood samples were collected in heparinized tubes (365965, BD Biosciences) using submandibular bleeding into. Collected blood was diluted 1:20 with saline and subsequently stimulated with 250 μM PAR4 activating peptide (Bachem, Torrance, CA) for 5 min. Unstimulated or activated platelets were stained with fluorophore (FITC or PE)-conjugated antibodies against cell surface antigens for 15 min at room temperature, including monoclonal antibodies against GP-VI, GP-IX, α2bβ3, JON/A or P-selectin, and polyclonal antibodies against Fibrinogen or vWF (all above from EMFRET Analytics) or CD41 (BD Biosciences). Samples were analyzed by using an Attune NxT Flow Cytometer (Thermo Fisher).
DNA histogram analysis
Femoral and tibial bone marrow was isolated as described (Schulze, 2016). Bone-marrow cells were stained with FITC-conjugated rat anti-mouse CD41 or rat-IgG isotype control (both from BD Biosciences). Cells were washed in phosphate buffered saline (PBS) followed by fixation with 4% paraformaldehyde at room temperature for 1 h. After washing twice with PBS, cells were incubated with 5 μg/ml of DAPI and analyzed by flow cytometry (Trakala et al., 2015). HEL cells were stained with FITC-conjugated mouse anti-human CD41 (BD Biosciences), followed by flow cytometry analysis as above.
Tail bleeding assay
Mice were intraperitoneally anesthetized with a single dose of combined ketamine (100 mg/kg) and xylazine (15 mg/kg). Tail bleeding times were determined as described (Hoover-Plow et al., 2006). In brief, a distal 4-mm segment of the tail was amputated using a scalpel and immediately immersed in physiological saline maintained at 37°C. The time for complete cessation of bleeding was recorded up to 6 min. The experiment was subsequently terminated in any mice that failed to stop bleeding.
Cell culture and transfection
HEL cells were grown in RPMI medium supplemented with penicillin-streptomycin and 10% fetal bovine serum (Gibco). For Y14 knockdown, HEL cells were transduced for 48 h with a Y14-targeting shRNA-expressing lentivirus (the RNAi Core Facility, Academia Sinica) at a multiplicity of infection of 5; shLuc served as the control (Y14, 5’-CGAGAGCATTCACAAACTGAA and control, 5’-GCGGTTGCCAAGAGGTTCCAT, which targeted luciferase).
HEK293 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with penicillin-streptomycin and 10% fetal bovine serum. The pcDNA-FLAG-p53-expression vector was previously described (Lu et al., 2017). Transfection was performed using lipofectamine 2000 (Thermo Fisher Scientific) for 2 days.
Megakaryocytic differentiation and p53 inhibiton
Megakaryocytic differentiation of HEL cells was archived by treatment with 5 nM PMA (Sigma) for 3 days. For p53 inhibition, HEL cells were treated with 5 μM PFT-α (Millipore) for 4 h prior to PMA induction; DMSO was used as control. Primary cells isolated from embryonic day 14.5 mouse fetal liver were grown in Dulbecco’s modified Eagle’s medium (Gibco) supplemented as above and with 100 ng/ml murine thrombopoietin (GenScript) for 3 days. Megakaryocytes were enriched by 1.5% and 3% BSA density-gradient centrifugation, and then cultured in the presence of thrombopoietin for another 3 days; for p53 inhibition, 5 μM PFT-α was added. To express or knock down p53, HEL cells were transfected with the p53 expressing vector (Lu et al., 2017) or transduced with p53-targeting shRNA lentivirus for 2 days.
Giemsa staining
Primary cells from fetal liver stroma were cultured and PFT-α treated and subsequently by smeared and dried on slides, and then fixed in methanol for 5 min. The samples were stained with Giemsa stain (1:20 dilution) for 15 min, and air-dried after rinse.
Immunoblotting
Immunoblotting was performed as described (Lu et al., 2017), using monoclonal antibodies against beta actin, GAPDH (Proteintech), histone 3 (Cell Signaling Technology), cyclin B2 and cyclin B2 (BD Biosciences) and polyclonal antibodies against Y14 (Bethyl), p21, p53, cyclin-dependent kinase 1 (CDK1) (Proteintech), cleaved caspase-3 (CC3), phospho-histone H3 (Cell Signaling Technology), Cdc20 (Abcam).
RT-PCR and RT-qPCR
Total RNA was extracted by using TRIzol reagent (Thermo Fisher Scientific). RNA was treated with RQ1 DNase (Promega) followed by reverse-transcription using SuperScript III kit (Life Technologies). RT-PCR was performed using DreamTaq Green PCR Master Mix (Thermo Fisher Scientific OR Thermoscientific). RT-qPCR was performed using SYBR Green FastMix (Quantabio) in LightCycler480 (Roche). PCR primer sets were listed in Table S4.
EdU labeling
EdU labeling was performed using Click-iTTM EdU Alexa FluorTM 647 Flow Cytometry Assay kit (Thermo Fisher Scientific) according to manufacturer’s instructions. In brief, 10 μM EdU was added into the culture medium 1 h before cell harvest. The cells were stained with FITC-conjugated mouse anti-human CD41 (BD Biosciences) and fixed and permeabilized by 4% paraformaldehyde and saponin-based permeabilization/wash reagent for 15 min, respectively. The Click-iT reaction was performed using the Click-iT reaction buffer for 30 min followed by incubation with DAPI and analyzed by flow cytometry as described in the main text. PBS containing 3% BSA was used as the wash buffer between different steps.
RNA sequencing and bioinformatics analysis
Control shRNA or Y14-depleted HEL cells were mock- or PMA-treated for 3 days as described above. Two replicates of each treatment were subjected to RNA-seq. RNA was extracted by chloroform and selected by oligo-dT following the manufactory instructions. The libraries were constructed by KAPA mRNA HyperPrep Kit (KAPA Biosystems, Roche, Basel, Switzerland) and sequenced using Illumina Novaseq 6000 platform. A total of 53-81 million 150 bp paired-end reads were generated for each sample. Clean reads were generated by using Trimmomatic v.0.38 to filter low quality reads and remove adaptor sequences. Reads were aligned to GRCh38 database using HISAT2 (Kim et al., 2015; Sahraeian et al., 2017). Differential gene expression was analyzed using featureCounts (Liao et al., 2014). Differentially expressed coding genes were obtained using DESeq2 and defined as a fold-change of > 2 and a p-value of < 0.05 (Love et al., 2014). Gene Ontology Enrichment analysis was performed using clusterProfiler (3.10.1), showing top 10 differential expression categories of megakaryocytic differentiation-related (PMA-treated compared with mock-treated shLuc-HEL cells) or Y14-depletion-affected (shLuc- compared with Y14-depleted HEL cells after PMA treatment). A global expression profile of pre-defined curated gene sets and Gene Ontology gene sets from MSigDB were compared between control and Y14-depleted cells. Gene set enrichment analysis was performed as described (Subramanian et al., 2005) using clusterProfiler (3.10.1). The predefined gene sets were used from Molecular Signatures Database (MSigDB) v6.2 (Liberzon et al., 2015).
Quantification and statistical analysis
Statistical analyses were essentially performed using MedCalc statistic software (Ostend, Belgium) or Prism (GraphPad). Tail bleeding time data were analyzed by 2-tailed Mann-Whitney test. Additional statistical analyses were performed using Prism (GraphPad) including two-tailed, unpaired and paired Student’s t-test in this study. One-way ANOVA and Tukey’s multiple comparison test were used for statistical analysis in Figure 6E. Data were presented as mean ± SEM. In all Figures, p values are denoted by asterisks; ns: no statistical significance; ∗ <0.05; ∗∗ <0.01; ∗∗∗ <0.001; ∗∗∗∗<0.0001.
Acknowledgments
HEL cells and Trp53f/f mice were respectively obtained from Tien-Shun Yeh and Li-Ru You (National Yang-Ming University, Taipei, Taiwan). We thank Kuan-Yang Hung and Chang-Yi Lin for generation of Pf4Cre/+;Rbm8af/+ mice and initial characterization of HEL cells, respectively. We thank the Taiwan Animal Consortium funded by the Ministry of Science and Technology of Taiwan for technical support in hematological analysis. This study was supported by Academia Sinica Investigator Award (AS-IA-107-L04) to W.-Y.T.
Author contributions
C.-H.S. and W.-J.L. performed experiments (C.-H.S., Figures 2A, 2C, 2E, 2F, 3, 4B, 5, 6 and all supplementary Figures except 1C, 1D; W.-J.L., Figures 1, 2B, 2D, 2E, 4C, 4D, 6B, S1C and S1D), interpreted the data, and contributed to manuscript preparation. W.-C.K. raised and maintained mice and performed the experiment of Figure 6B. R.-B.Y. supervised the experiments. W.-Y.T. designed the study, supervised the experiments, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103368.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited in SRA and are publicly available. Accession numbers are listed in the key resources table. Original dataset or images have been deposited in Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required for analysis of the data reported in this paper is available from the lead contact upon request.
References
- Abbonante V., Di Buduo C.A., Gruppi C., De Maria C., Spedden E., De Acutis A., Staii C., Raspanti M., Vozzi G., Kaplan D.L., et al. A new path to platelet production through matrix sensing. Haematologica. 2017;102:1150–1160. doi: 10.3324/haematol.2016.161562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers C.A., Paul D.S., Schulze H., Freson K., Stephens J.C., Smethurst P.A., Jolley J.D., Cvejic A., Kostadima M., Bertone P., et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012;44:435–439. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers C.A., Newbury-Ecob R., Ouwehand W.H., Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr. Opin. Genet. Dev. 2013;23:316–323. doi: 10.1016/j.gde.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Apostolidis P.A., Lindsey S., Miller W.M., Papoutsakis E.T. Proposed megakaryocytic regulon of p53: the genes engaged to control cell cycle and apoptosis during megakaryocytic differentiation. Physiol. Genomics. 2012;44:638–650. doi: 10.1152/physiolgenomics.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolidis P.A., Woulfe D.S., Chavez M., Miller W.M., Papoutsakis E.T. Role of tumor suppressor p53 in megakaryopoiesis and platelet function. Exp. Hematol. 2012;40:131–142. doi: 10.1016/j.exphem.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee T., Nath S., Roychoudhury S. DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res. 2009;37:2688–2698. doi: 10.1093/nar/gkp110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassini A., Pierpaoli S., Falcieri E., Vitale M., Guidotti L., Capitani S., Zauli G. Selective modulation of the cyclin B/CDK1 and cyclin D/CDK4 complexes during in vitro human megakaryocyte development. Br. J. Haematol. 1999;104:820–828. doi: 10.1046/j.1365-2141.1999.01264.x. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S., Wang T.T., Ravetch J.V. The role and function of Fcγ receptors on myeloid cells. Microbiol. Spectr. 2016;4:6. doi: 10.1128/microbiolspec.MCHD-0045-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen M.E., Attardi L.D. The role of p53 in developmental syndromes. J. Mol. Cell. Biol. 2019;11:200–211. doi: 10.1093/jmcb/mjy087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R., Parmar K., Mouly E., Delord M., Kim J.M., Regairaz M., Pla M., Vasquez N., Zhang Q.S., Pondarre C., et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11:36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Kostadima M., Martens J., Canu G., Garcia S.P., Turro E., Downes K., Macaulay I.C., Bielczyk-Maczynska E., Coe S., et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014;345:1251033. doi: 10.1126/science.1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang T.-W., Lu C.-C., Su C.-H., Wu P.-Y., Easwvaran S., Lee C.-C., Kuo H.-C., Hung K.-Y., Lee K.-M., Tsai C.-Y., et al. The RNA processing factor Y14 participates in DNA damage response and repair. iScience. 2019;13:402–415. doi: 10.1016/j.isci.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laval B., Pawlikowska P., Petit-Cocault L., Bilhou-Nabera C., Aubin-Houzelstein G., Souyri M., Pouzoulet F., Gaudry M., Porteu F. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell. 2013;12:37–48. doi: 10.1016/j.stem.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Donehower L.A., Harvey M., Vogel H., McArthur M.J., Montgomery C.A., Jr., Park S.H., Thompson T., Ford R.J., Bradley A. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol. Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- Fiedler J., Strauss G., Wannack M., Schwiebert S., Seidel K., Henning K., Klopocki E., Schmugge M., Gaedicke G., Schulze H. Two patterns of thrombopoietin signaling suggest no coupling between platelet production and thrombopoietin reactivity in thrombocytopenia-absent radii syndrome. Haematologica. 2012;97:73–81. doi: 10.3324/haematol.2011.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura K., Wakabayashi S., Kataoka N., Sakamoto H., Suzuki Y., Nakai K., Mayeda A., Inoue K. The exon junction complex controls the efficient and faithful splicing of a subset of transcripts involved in mitotic cell-cycle progression. Int. J. Mol. Sci. 2016;17:1153. doi: 10.3390/ijms17081153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangras P., Gallagher T.L., Parthun M.A., Yi Z., Patton R.D., Tietz K.T., Deans N.C., Bundschuh R., Amacher S.L., Singh G. Zebrafish rbm8a and magoh mutants reveal EJC developmental functions and new 3′UTR intron-containing NMD targets. Plos Genet. 2020;16:e1008830. doi: 10.1371/journal.pgen.1008830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollomp K., Poncz M. Gp1ba-Cre or Pf4-Cre: pick your poison. Blood. 2019;133:287–288. doi: 10.1182/blood-2018-11-887513. [DOI] [PubMed] [Google Scholar]
- Hall J.G. Thrombocytopenia and absent radius (TAR) syndrome. J. Med. Genet. 1987;24:79–83. doi: 10.1136/jmg.24.2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover-Plow J., Shchurin A., Hart E., Sha J., Hill A.E., Singer J.B., Nadeau J.H. Genetic background determines response to hemostasis and thrombosis. BMC Blood Disord. 2006;6:6. doi: 10.1186/1471-2326-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y., Nakamura Y., Tatsuno T., Hashimoto M., Shimasaki T., Iwabuchi K., Tomosugi N. Depletion of RNA-binding protein RBM8A (Y14) causes cell cycle deficiency and apoptosis in human cells. Exp. Biol. Med. (Maywood) 2016;238:889–897. doi: 10.1177/1535370213494646. [DOI] [PubMed] [Google Scholar]
- Jonkers J., Meuwissen R., van der Gulden H., Peterse H., van der Valk M., Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Kahn M.L., Zheng Y.W., Huang W., Bigornia V., Zeng D., Moff S., Farese R.V., Jr., Tam C., Coughlin S.R. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Lee M.-K., Kim H.R. Difference in megakaryocyte expression of GATA-1, IL-6, and IL-8 associated with maintenance of platelet counts in patients with plasma cell neoplasm with dysmegakaryopoiesis. Exp. Hematol. 2019;73:13–17. doi: 10.1016/j.exphem.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Letestu R., Vitrat N., Massé A., Le Couedic J.P., Lazar V., Rameau P., Wendling F., Vuillier J., Boutard P., Plouvier E., et al. Existence of a differentiation blockage at the stage of a megakaryocyte precursor in the thrombocytopenia and absent radii (TAR) syndrome. Blood. 2000;95:1633–1641. [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.-C., Lee C.-C., Tseng C.-T., Tarn W.-Y. Y14 governs p53 expression and modulates DNA damage sensitivity. Sci. Rep. 2017;7:45558. doi: 10.1038/srep45558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.C., Sanada C., Xavier-Ferrucio J., Wang L., Zhang P.X., Grimes H.L., Venkatasubramanian M., Chetal K., Aronow B., Salomonis N., et al. The molecular signature of megakaryocyte-erythroid progenitors reveals a role for the cell cycle in fate specification. Cell Rep. 2018;25:2083–2093.e4. doi: 10.1016/j.celrep.2018.10.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumelsky N.L., Schwartz B.S. Protein kinase C in erythroid and megakaryocytic differentiation: possible role in lineage determination. Biochim. Biophy. Acta. 1997;1358:79–92. doi: 10.1016/s0167-4889(97)00051-7. [DOI] [PubMed] [Google Scholar]
- Machlus K.R., Italiano J.E., Jr. The incredible journey: from megakaryocyte development to platelet formation. J. Cell. Biol. 2013;201:785–796. doi: 10.1083/jcb.201304054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahfoudhi E., Lordier L., Marty C., Pan J., Roy A., Roy L., Rameau P., Abbes S., Debili N., Raslova H., et al. P53 activation inhibits all types of hematopoietic progenitors and all stages of megakaryopoiesis. Oncotarget. 2016;7:31980–31992. doi: 10.18632/oncotarget.7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H., McMahon J.J., Tsai Y.-H., Wang Z., Silver D.L. Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. Plos Genet. 2016;12:e1006282. doi: 10.1371/journal.pgen.1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H., Pilaz L.-J., McMahon J.J., Golzio C., Wu D., Shi L., Katsanis N., Silver D.L. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 2015;35:7003–7018. doi: 10.1523/JNEUROSCI.0018-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat L.E., Tarn W.-Y., Isken O. The pioneer round of translation: features and functions. Cell. 2010;142:368–374. doi: 10.1016/j.cell.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzi S., Lordier L., Debili N., Raslova H., Vainchenker W. Megakaryocyte and polyploidization. Exp. Hematol. 2018;57:1–13. doi: 10.1016/j.exphem.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Michelle L., Cloutier A., Toutant J., Shkreta L., Thibault P., Durand M., Garneau D., Gendron D., Lapointe E., Couture S., et al. Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol. Cell. Biol. 2012;32:954–967. doi: 10.1128/MCB.06130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A.P., Kauppi M., Metcalf D., Hyland C.D., Josefsson E.C., Lebois M., Zhang J.G., Baldwin T.M., Di Rago L., Hilton D.J., et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc. Natl. Acad. Sci. U S A. 2014;111:5884–5889. doi: 10.1073/pnas.1404354111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowska P., Fouchet P., Vainchenker W., Rosselli F., Naim V. Defective endomitosis during megakaryopoiesis leads to thrombocytopenia in Fanca-/- mice. Blood. 2014;124:3613–3623. doi: 10.1182/blood-2014-01-551457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raslova H., Kauffmann A., Sekkaï D., Ripoche H., Larbret F., Robert T., Tronik Le Roux D., Kroemer G., Debili N., Dessen P., et al. Interrelation between polyploidization and megakaryocyte differentiation: a gene profiling approach. Blood. 2007;109:3225–3234. doi: 10.1182/blood-2006-07-037838. [DOI] [PubMed] [Google Scholar]
- Sahraeian S.M.E., Mohiyuddin M., Sebra R., Tilgner H., Afshar P.T., Au K.F., Bani Asadi N., Gerstein M.B., Wong W.H., Snyder M.P., et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat. Commun. 2017;8:59. doi: 10.1038/s41467-017-00050-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R.K., Schenone M., Ferreira M.V., Kramann R., Joyce C.E., Hartigan C., Beier F., Brümmendorf T.H., Germing U., Platzbecker U., et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 2016;22:288–297. doi: 10.1038/nm.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze H. Culture, expansion, and differentiation of murine megakaryocytes from fetal liver, bone marrow, and spleen. Curr. Protoc. Immunol. 2016;112:22F.6.1–22F.6.15. doi: 10.1002/0471142735.im22f06s112. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T., Endo H., Chikatsu N., Uchimaru K., Asano S., Fujita T., Nakahata T., Motokura T. Expression of p21(Cip1/Waf1/Sdi1) and p27(Kip1) cyclin-dependent kinase inhibitors during human hematopoiesis. Blood. 1999;93:4167–4178. [PubMed] [Google Scholar]
- Tiedt R., Schomber T., Hao-Shen H., Skoda R.C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109:1503–1506. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- Trakala M., Rodríguez-Acebes S., Maroto M., Symonds C.E., Santamaría D., Ortega S., Barbacid M., Méndez J., Malumbres M. Functional reprogramming of polyploidization in megakaryocytes. Dev. Cell. 2015;32:155–167. doi: 10.1016/j.devcel.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Tsai Y.-Y., Su C.-H., Tarn W.-Y. p53 activation in genetic disorders: different routes to the same destination. Int. J. Mol. Sci. 2021;22:9307. doi: 10.3390/ijms22179307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., Feng M., Sinha R., Seita J., Mori Y., Weissman I.L. Screening for genes that regulate the differentiation of human megakaryocytic lineage cells. Proc. Natl. Acad. Sci. U S A. 2018;115:E9308–E9316. doi: 10.1073/pnas.1805434115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data have been deposited in SRA and are publicly available. Accession numbers are listed in the key resources table. Original dataset or images have been deposited in Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required for analysis of the data reported in this paper is available from the lead contact upon request.