

Abstract
HIV nucleotide sequence data can identify clusters of persons with genetically similar strains suggesting transmission. We simulated the effect of lowered data completeness, defined by the percent of persons with diagnosed HIV with a reported sequence, on transmission patterns and detection of growing HIV transmission clusters. We analyzed HIV surveillance data for persons with HIV diagnosed during 2008–2014 who resided in Michigan or Washington. We calculated genetic distances, constructed the inferred transmission network for each jurisdiction, and compared transmission network characteristics and detection of growing transmission clusters in the full dataset with artificially reduced datasets. Simulating lower levels of completeness resulted in decreased percentages of persons linked to a cluster from high completeness (full dataset) to low completeness (5%) (Michigan: 54%–18%; Washington, 46%–16%). Patterns of transmission between certain populations remained robust as data completeness level was reduced. As data completeness was artificially decreased, sensitivity of cluster detection substantially diminished in both states. In Michigan, sensitivity decreased from 100% with the full dataset, to 62% at 50% completeness and 21% at 25% completeness. In Washington, sensitivity decreased from 100% with the full dataset, to 71% at 50% completeness and 29% at 25% completeness. Lower sequence data completeness limits the ability to detect clusters that may benefit from investigation; however, inferences can be made about transmission patterns even with low data completeness, given sufficient numbers. Data completeness should be prioritized, as lack of or delays in detection of transmission clusters could result in additional infections.
Keywords: HIV, transmission cluster, sequence, detection, completeness, sensitivity
Introduction
Because HIV rapidly evolves over time, molecular HIV sequences can be compared to one another to determine relatedness and can be used to identify clusters of recent and rapid transmission.1–6 Molecular sequences generated through drug resistance testing, which the U.S. Department of Health and Human Services recommends be done at entry to HIV care,7 are reported to CDC through the National HIV Surveillance System (NHSS). These data have previously been used to describe HIV transmission patterns overall and within and between certain populations.8
CDC has recently expanded use of these data to identify growing molecular clusters that warrant public health investigation.9 Since 2016, CDC has routinely identified growing clusters, informing jurisdictions affected by these clusters, and offering assistance with cluster investigations as needed.10–12 To date, more than 60 such clusters have been identified by CDC and reviewed or investigated by state and local health departments.13 It is critical to identify these clusters in a timely manner to focus effective public health interventions aimed at stopping transmission and engaging persons with diagnosed infection in medical care.
Despite these advances in using molecular data to identify and halt transmission, completeness of reported HIV sequence data in the United States, defined by the proportion of HIV diagnoses with an associated sequence, varies by jurisdiction. A recent analysis showed that although drug resistance testing is considered part of standard of care in the United States, only 66% of persons with HIV diagnosed in 2013 who were in medical care received resistance testing.14 Reduced completeness of HIV sequence data limits detection of HIV transmission network patterns and growing clusters, although it is unclear by how much. There has been limited work in exploring the effect of data completeness on HIV transmission network patterns, and this has not been previously explored in the United States.15 Quantifying the effect of reduced data completeness on detection of network patterns and growing clusters is critical to understand the minimum threshold for completeness needed to provide informative results. Thus, in this analysis, we examined the effect of missing sequence data on observed HIV transmission network patterns and detection of growing HIV molecular clusters that warrant public health investigation.
Materials and Methods
Population
NHSS collects demographic, clinical, and risk information on all persons with diagnosed HIV infection in the United States. A subset of jurisdictions conducted molecular HIV surveillance during 2008–2014, collecting HIV sequence data generated through routine drug resistance testing, which requires nucleotide sequencing of a person’s HIV strain. Sequence data are reported routinely to NHSS with HIV case information. For this analysis, data reported through December 2015 were restricted to persons who received an HIV diagnosis during 2008–2014 and resided in either Michigan or Washington, which had the highest completeness of reported HIV sequence data during the analysis period (73% and 66% respectively). Data from each state were analyzed separately.
Analytic methods
Briefly, using molecular sequences submitted for persons residing in Michigan and Washington, we constructed the inferred transmission network for each jurisdiction separately, reported characteristics of the observed network, and calculated the number of concerning clusters warranting public health investigation. Subsequently, we simulated the effect of lowered data completeness on key characteristics of the observed network by comparing characteristics using the full dataset to observed characteristics in artificially reduced datasets for each jurisdiction using random selection. Finally, we calculated the sensitivity of detecting growing HIV molecular clusters at decreased levels of simulated completeness, again, based on random selection. Molecular clusters were constructed using a local installation of HIV-TRACE (HIV Transmission Cluster Engine; www.hivtrace.org) based on methods previously described.8,10,16,17 All other analyses were conducted using R. More details on analytic methodology follows.
Construction of inferred transmission network.
We analyzed protease and reverse transcriptase sequences, excluding sequences shorter than 500 nucleotides and sequences that were unexpectedly similar to the HXB2 reference sequence (i.e., potential laboratory contaminants), and limited to the earliest sequence per person. Briefly, to identify molecular links, we aligned all sequences to the HXB2 reference sequence and quantified Tamura-Nei 93 genetic distance between all possible sequence pairs.18 Subsequently, we identified molecular clusters of HIV transmission, which consisted of connected network components, and subsequently, inferred observed network patterns.
Characteristics of observed network.
For each jurisdiction, we identified molecular clusters using a genetic distance threshold of 1.5% and inferred the observed network. A genetic distance threshold between 1% and 2% can indicate possible relatedness in HIV sequences.17 The methodology for evaluating characteristics of the observed network mirrored a previous analysis of molecular cluster data.8 We described characteristics of the observed network using the entire dataset, including the number of links and clusters, and pairings (partnerships) by race/ethnicity and transmission category. Race/ethnicity categories included black/African American (henceforth referred to as black), Hispanic/Latino (henceforth referred to as Hispanic), white, and other (American Indian/Alaska Native, Asian, Native Hawaiian/other Pacific Islander, multiple races, and other groups). We combined sex and transmission category with the following categories: male-to-male sexual contact [men who have sex with men (MSM)], MSM who inject drugs, male persons who inject drugs (PWID), female PWID, heterosexual men, heterosexual women, and other/unknown.
We examined how the characteristics of the observed network were affected by simulating decreasing levels of data completeness through random selection. To do this, we took 100 random samples of the data at each predetermined completeness level, in increments of 5%, from 5% to 70% for Michigan and from 5% to 65% for Washington. All persons were eligible for selection for each random sample; however, once a person was selected, they were no longer eligible for selection within that sample. Based on these simulations, we estimated the median and 5% and 95% quantiles for each network characteristic at every completeness level, and presented results for high (using the full analytic dataset) and low (5%) completeness.
Detection of priority clusters.
To focus on how data completeness affects detection of the most concerning HIV molecular clusters that should be prioritized for investigation, we limited the data to persons with HIV diagnosed during 2012–2014 and used a more stringent genetic distance threshold of ≤0.5% to identify molecular clusters, as is CDC’s standard approach for detecting such clusters.11,12 We calculated the number of HIV molecular clusters identified that should be prioritized for public health investigation (henceforth known as priority clusters), defined as those with ≥2 new HIV diagnoses in the last 12 months of the analysis period (2014).
Using this definition of priority clusters as the gold standard, we calculated the sensitivity of priority cluster detection at decreased levels of simulated completeness, again, based on random selection. To do this, we artificially decreased sequence data completeness by taking 100 random samples of the full dataset, ranging from 5% up to the completeness level of the full dataset for each jurisdiction, in increments of 5%. All persons were eligible for selection for each random sample; however, once a person was selected, they were no longer eligible for selection within that sample. We calculated the sensitivity of cluster detection at each level of data completeness, defined as the median number of priority clusters identified at each completeness level divided by the total number of priority clusters detected using the full dataset.
Results
Population
Overall, 215,692 persons were reported to have an HIV diagnosis in the United States during 2008–2014, of whom 5,509 resided in Michigan at the time of diagnosis and 3,520 resided in Washington at the time of diagnosis (Table 1). Of persons with diagnosed HIV in Michigan, 4,040 of 5,509 (73%) had an associated sequence; 30% were white, 61% were black, 5% were Hispanic, and 4% were of other race/ ethnicity. Fifty-seven percent were aged 13–34 years and 69% were MSM. Of persons with diagnosed HIV in Washington, 2,310 of 3,520 (66%) had an HIV sequence; 55% were white, 18% were black, 16% were Hispanic, and 11% were of other race/ethnicity. Forty-six percent were aged 13– 34 years, 56% were white, and 70% were MSM.
Table 1.
Characteristics of the Inferred HIV Transmission Network Identified Using the 1.5% Genetic Distance Threshold and Potential Transmission Partners, Michigan and Washington, 2008–2014
| Michigan | Washington | |
|---|---|---|
| No. of HIV diagnoses total | 5,509 | 3,520 |
| No. of sequences total | 4,040 | 2,310 |
| Completeness of reported HIV sequences (%) | 73 | 66 |
| Attributes of links | ||
| Total no. of links | 9,076 | 5,589 |
| No. of sequences linked to ≥1 other sequence | 2,175 | 1,062 |
| Percentage of sequences linked to ≥1 other sequence | 54 | 46 |
| No. of links involving at least one sequence with ≥4 links | 8,415 | 5,228 |
| Percentage of links involving at least one sequence with ≥4 links | 93 | 94 |
| No. of links (among those with any links) | ||
| Median | 4 | 4 |
| Interquartile range | 1–10 | 1–10 |
| Maximum | 79 | 77 |
| Attributes of clusters | ||
| Total no. of distinct clusters | 419 | 234 |
| Cluster size | ||
| Median | 3 | 2 |
| Interquartile range | 2–5 | 2–4 |
| Maximum | 134 | 82 |
| Characteristics of potential transmission partners | ||
| Percentage of whites linked to other whites | 423/600 = 71 | 513/597 = 74 |
| Percentage of blacks linked to other blacks | 1,194/1,393 = 86 | 45/107 = 42 |
| Percentage of Hispanics/Latinos linked to other Hispanics/Latinos | 17/100 = 17 | 53/153 = 35 |
| Percentage of MSM linked to other MSM | 1,186/1,433 = 83 | 654/786 = 83 |
| Percentage of MSM linked to MSM or MSM who inject drugs | 1,215/1,433 = 85 | 724/786 = 92 |
| Percentage of MSM linked to heterosexual women | 22/1,433 = 2 | 4/786 = 1 |
| Percentage of heterosexual women linked to MSM | 24/121 = 20 | 5/25 = 21 |
| Percentage of heterosexual women linked to heterosexual men | 16/121 = 13 | 4/25 = 14 |
MSM, men who have sex with men.
In both jurisdictions, persons <13 years of age, male PWID, and those with other or unknown transmission risk were less likely to have a reported sequence (Supplementary Table S1). In Washington, female PWID and heterosexuals were also less likely to have a reported sequence.
Characteristics of observed network
Michigan.
Among 4,040 persons in Michigan for whom sequences were available, 2,175 (54%) were linked to ≥1 other person at the 1.5% genetic distance threshold (Table 1). A total of 9,076 links in 419 distinct clusters were detected at the 1.5% genetic distance threshold. The median number of persons in a cluster was 3 [interquartile range (IQR): 2–5], with a maximum of 134 persons in a cluster.
When we simulated lower levels of completeness, the percent of persons linked to ≥1 other person decreased from 54% at high completeness (full dataset) to 18% (5%–95%: 13%–22%) at low completeness (5%) (Fig. 1). The number of clusters decreased from 419 at high completeness to 18 (5%– 95%: 13–23) at low completeness. Assortative pairing among blacks was high and consistent (median 86%–87%) regardless of completeness, with narrower ranges of estimates with higher completeness (Fig. 2). Among Hispanics/Latinos, the percentage of Hispanic/Hispanic pairings was much lower and ranged from 17% at high completeness to 0% (5%–95%: 0%–2%) at low completeness. Pairings of MSM with other MSM remained consistent (median 82%–83%), with narrower ranges of estimates with higher completeness. The proportion of women with HIV infection attributable to heterosexual contact who were linked to MSM ranged from 20% at high completeness to a median of 14% (0%–100%) at low completeness. Additional information on simulated effects of lower data completeness on attributes of links and clusters and characteristics of potential transmission partners can be seen in Supplementary Figure S1.
FIG. 1.
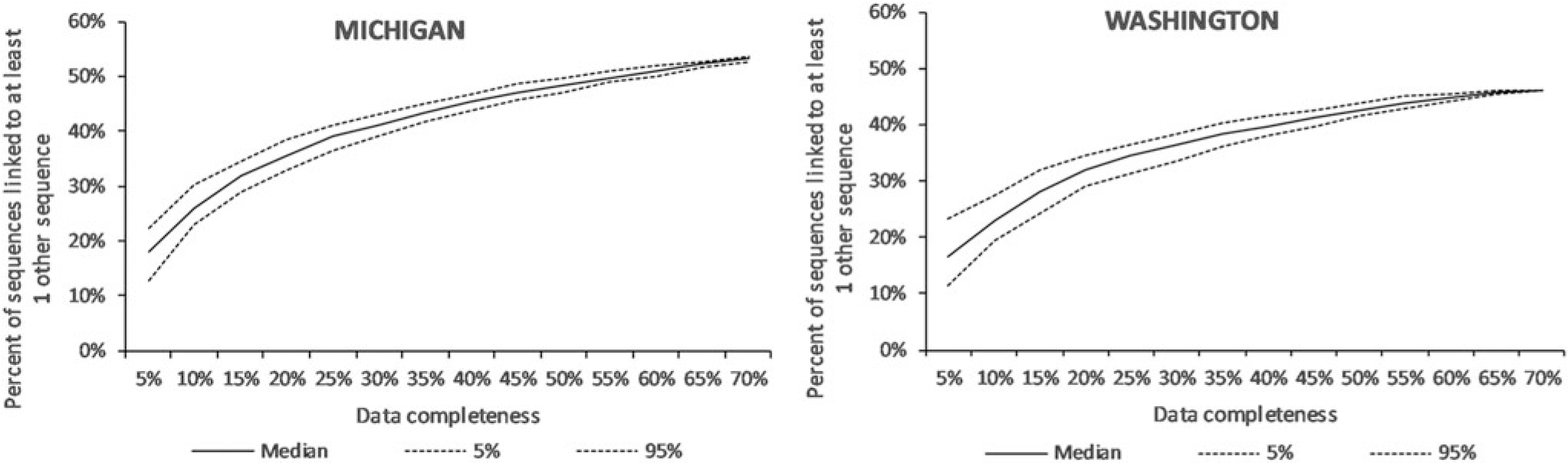
Effect of lower levels of completeness of HIV sequencing data on the percentage of sequences linked to at least one other sequence identified at the 1.5% genetic distance threshold in Michigan (n = 4,040 sequences at 73% completeness) and Washington (n = 2,310 sequences at 66%), 2008–2014.
FIG. 2.
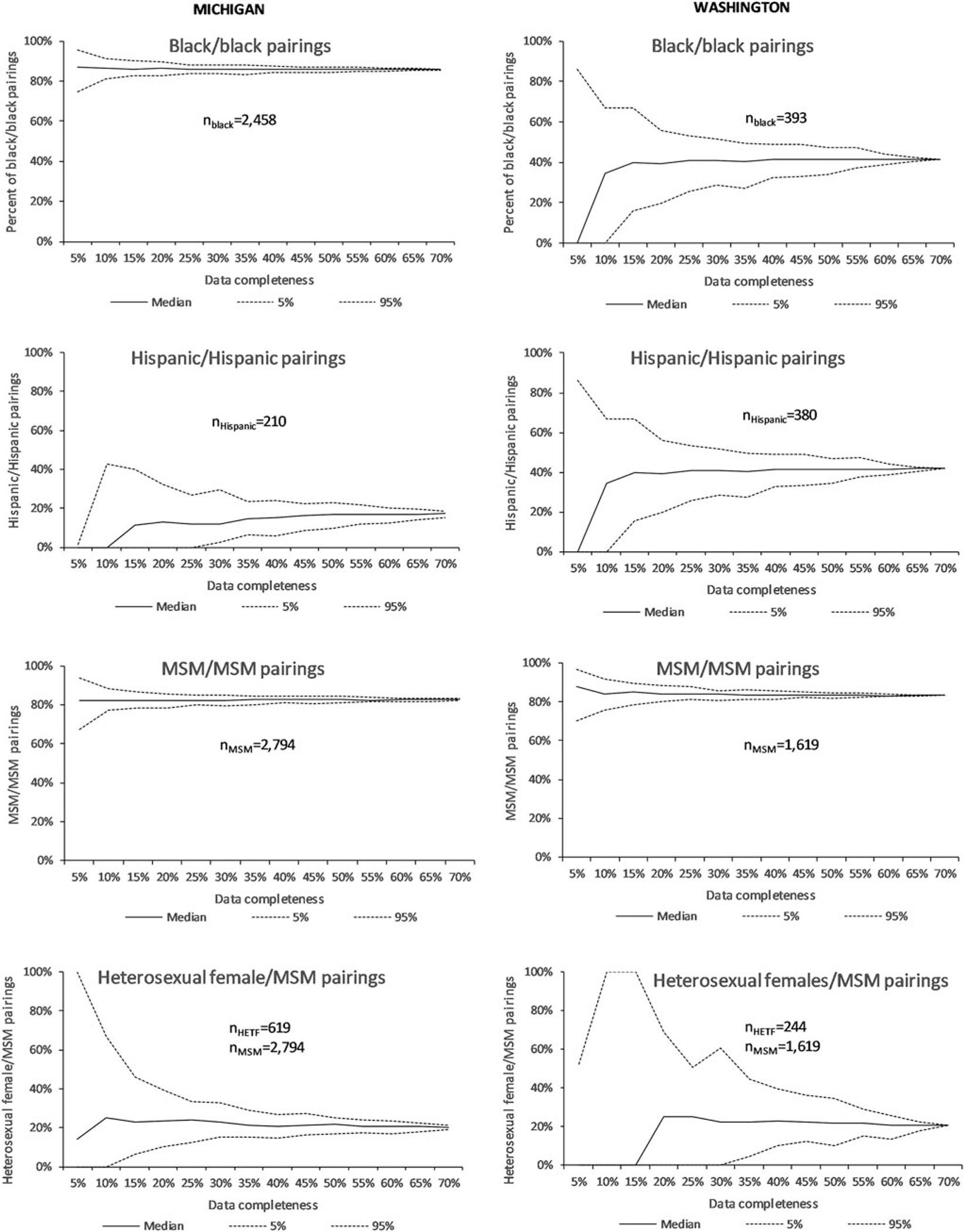
Effect of lower levels of completeness of HIV sequencing data on transmission network patterns (characteristics of potential transmission partners) in Michigan (n = 4,040 sequences at 73% completeness) and Washington (n = 2,310 sequences at 66%), 2008–2014.
Washington.
Among 2,310 persons for whom sequences were available, 1,062 (46%) were linked to ≥1 other person at the 1.5% genetic distance threshold (Table 1). In total, 5,589 links were detected in 234 distinct molecular clusters. The median number of persons in a cluster was 2 (IQR: 2–4), with a maximum of 82 persons in a single cluster.
When data completeness was artificially reduced, the percent of persons linked to ≥1 other person decreased from 46% at high completeness to a median of 16% (5%–95%: 11%–23%) at low completeness (Fig. 1). The number of clusters decreased from 82 at high completeness to 7 (5%– 95%: 4–11) at low completeness (Supplementary Fig. S1). Assortative pairing among blacks ranged from 42% at high completeness to a median of 0 (5%–95%: 0%–86%) (Fig. 2). Among Hispanics, Hispanic/Hispanic pairings ranged from 35% at high completeness to a median of 0% (5%–95%: 0%– 39%) at low completeness. Pairings of MSM with other MSM remained consistent (median 83%–87%), with narrower ranges of estimates with higher completeness. The proportion of heterosexual women linked to MSM ranged from 21% at high completeness to 0% (5%–95%: 0%–53%) at low completeness. Additional information on simulated effects of lower data completeness on attributes of links and clusters and characteristics of potential transmission partners can be seen in Supplementary Figure S1.
Detection of priority clusters
Michigan.
Among 1,614 persons with HIV diagnosed during 2012–2014 and an associated sequence in Michigan, 1,061 (66%) were linked to ≥1 other person at the threshold used for priority cluster analysis, and a total of 130 distinct clusters were detected. The median number of persons in a cluster was 2 (IQR: 2–3), with a maximum of 17 persons in a cluster. Of the 130 clusters, 34 (26%) were considered to be priority clusters. The median size of a priority cluster was 3 (IQR: 2–6), with a maximum size of 17.
As data completeness was artificially decreased, sensitivity of priority cluster detection drastically diminished, from 34 clusters detected with the full analytic dataset to a median of 21 (5%–95%: 18–25) at 50% completeness and 7 (5%– 95%: 4–12) at 25% completeness. This resulted in a decrease in sensitivity from 100% with the full dataset, to 62% at 50% completeness, and to 21% at 25% completeness (Fig. 3).
FIG. 3.
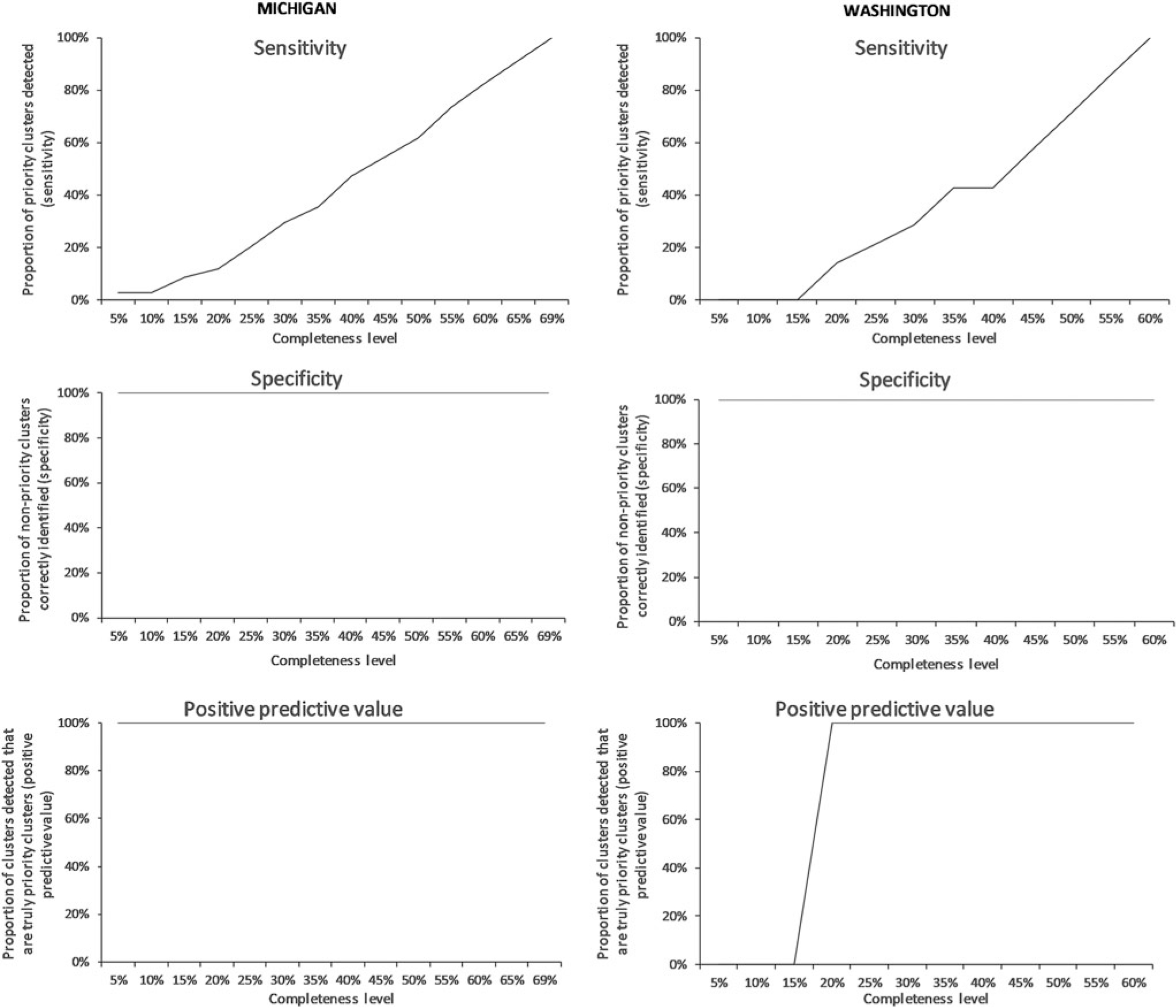
Effect of lower levels of completeness of HIV sequencing data and changing the threshold for detection on the identification of priority clusters, Michigan (n = 4,040 sequences at 73% completeness) and Washington (n = 2,310 sequences at 66%), 2008–2014.
Washington.
Of 817 persons with diagnosed HIV infection during 2012–2014 who had an associated sequence, 542 (66%) were linked to ≥1 other person at the threshold used for priority cluster analysis, and a total of 52 distinct clusters were detected. The median number of persons in a cluster was 2 (IQR: 2–3), with a maximum of 17 persons in a cluster. Of these 52 clusters, 7 (13%) were considered to be priority clusters. The median size of priority clusters was 2 (IQR: 2– 4), with a maximum size of 17.
Similar to Michigan, as Washington’s data completeness was artificially decreased, sensitivity of priority cluster detection decreased substantially, from seven clusters detected with the full analytic dataset to a median of 5 (5%–95%: 3–7) at 50% completeness and 2 (5%–95%: 0–3) at 25% completeness. This resulted in a decrease in sensitivity from 100% with the full dataset, to 71% at 50% completeness, and to 29% at 25% completeness (Fig. 3).
Discussion
Although it is expected that reduced completeness of reported HIV sequence data limits detection of HIV transmission network patterns and growing clusters, this analysis was the first in the United States to quantify these effects. Despite some variability in transmission network patterns observed at lower levels of completeness, consistency in patterns largely depended on availability of sequence data for selected populations. Nevertheless, inferences about transmission patterns were still informative, even at lower data completeness. However, the sensitivity of priority cluster detection substantially decreased with even modest reductions in sequence completeness, which may affect the ability to intervene on clusters contributing to a considerable amount of transmission.
Maximizing sequence data completeness is critical for detection of transmission clusters that would benefit from public health intervention. However, there are many challenges involving sequence reporting, including that multiple steps are required for a sequence to be reported to CDC and included in sequence analyses. Specifically, a person needs to receive an HIV diagnosis, be linked to medical care, and have drug resistance testing ordered and a specimen drawn. Subsequently, the laboratory conducting the test has to report the results to the health department, and the health department needs to process and report the sequence to CDC. There may be a delay or a potential for reduced data completeness associated with each of these steps.
Further delays may occur due to complexity of data processing. CDC currently only analyzes sequence data to identify transmission clusters quarterly, and there may be limited capacity at state and local health departments to conduct analyses to identify HIV transmission. However, a user friendly point-and-click molecular analytic tool called Secure HIV-Trace (https://secure.hivtrace.org; University of California, San Diego) was recently released, allowing state and local health departments to securely analyze molecular HIV sequence data locally as often as needed, which is improving timeliness of cluster detection.
Identifying and addressing reasons for low sequence reporting are key to improving low data completeness. Activities planned for improving data completeness will vary based on these reasons. For instance, if data completeness is low because persons are not seeking medical care or getting a specimen drawn for testing, patient navigation and linkage to care services could help bridge this gap. Specifically, strategies for improving linkage to, and retention in, HIV care could focus on populations that may be less likely to be engaged in HIV care, and thus, may be contributing disproportionately to HIV transmission.19,20 If providers are ordering drug resistance tests differentially with respect to demographic characteristics or risk behaviors, then provider education on the importance of drug resistance testing may be helpful. Addressing any legal barriers to reporting, such as lack of requirements for reporting molecular HIV sequences to the health department, can improve completeness. Delays in laboratory reporting to the health department or to NHSS can be investigated. Administering a laboratory survey to identify laboratories that report drug resistance testing results may help in regular monitoring of laboratory testing, and thus, it can help in identifying laboratories with delayed reporting and addressing reasons for gaps in reporting. A laboratory survey can also be used to assess capacity for electronic reporting. Lack of robust infrastructure for electronic laboratory reporting, and limited ability to efficiently process these sequences at the health department, could lead to additional delays.
Even with 100% sequence data completeness, transmission clusters may not be fully represented, as data on persons with undiagnosed infection would not be included. During 2014, 15% of persons with HIV were living with undiagnosed infection in the United States, but this proportion varied by state and was as high as 24%.21 Not having a complete picture of HIV transmission may limit effectiveness of interventions during investigation of transmission clusters. Examining social and sexual network data (through contact tracing) alongside molecular cluster data may provide insight into the true size of an HIV transmission cluster, and could help identify persons potentially linked to a cluster who may not have otherwise been identified using molecular sequence data alone because of undiagnosed infection or lack of sequence data.22 If further investigation uncovers that information on a cluster was missing because of undiagnosed infections, increasing HIV testing initiatives may be helpful in quantifying the true size of the cluster, increasing linkage to care, and mitigating further transmission.
These findings are subject to some limitations. This analysis assumed that sequence data were missing at random, but certain providers or laboratories may be more likely to report testing results or sequencing may be more likely to be ordered for certain groups of people. Future analyses would inform how systematic bias could affect cluster detection. Results are based only on two jurisdictions and may not be representative of patterns in all U.S. jurisdictions. Effects of data completeness on observed transmission patterns or cluster detection should not be extrapolated beyond observed sequence data completeness levels, as this requires assuming a specific relationship beyond what was observed, which may or may not be correct.
These findings demonstrate that lower data completeness substantially limits the ability to detect clusters that should be prioritized for public health investigation; however, inferences can be made on transmission patterns even with low data completeness, given there are sufficient numbers. The results highlight how essential data completeness is in obtaining a complete picture of HIV transmission and preventing infections. Absence of, or delay in, detection and investigation of transmission clusters could result in additional infections and potentially fewer people engaged in the medical care needed to reduce the risk of transmission. These results emphasize the need for ordering HIV drug resistance testing for all persons with diagnosed HIV, not just for identifying drug resistance but to understand HIV transmission, and will help inform national guidance on data completeness standards for reporting HIV sequences.
Supplementary Material
Acknowledgments
We acknowledge the work of state and local health department staff in collecting and reporting HIV sequence data to CDC, and M. Cheryl Bañez Ocfemia for her support and expertise.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Sequence Data
States and U.S. territories collect and report demographic, behavioral, and clinical characteristics (including HIV sequences) on persons with diagnosed HIV infection according to local reporting requirements. Local jurisdictions voluntarily share these data with CDC. Each jurisdiction has the authority to determine whether their laws and regulations allow for the submission of sequences to open databases, such as Genbank, EMBL, or DDBJ, and thus, CDC is not able to submit sequences to these databases.
Disclaimer
The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
References
- 1.Dennis AM, Herbeck JT, Brown AL, et al. : Phylogenetic studies of transmission dynamics in generalized HIV epidemics: An essential tool where the burden is greatest? J Acquir Immune Defic Syndr 2014;67:181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poon AF, Gustafson R, Daly P, et al. : Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016;3:e231–e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Little SJ, Kosakovsky Pond SL, Anderson CM, et al. : Using HIV networks to inform real time prevention interventions. PLoS One 2014;9:e98443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan PA, Hogan JW, Huang A, et al. : Phylogenetic investigation of a statewide HIV-1 epidemic reveals ongoing and active transmission networks among men who have sex with men. J Acquir Immune Defic Syndr 2015;70:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis AM, Pasquale DK, Billock R, et al. : Integration of Contact Tracing and Phylogenetics in an Investigation of Acute HIV Infection. Sex Transm Dis 2018;45:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ragonnet-Cronin M, Jackson C, Bradley-Stewart A, et al. : Recent and rapid transmission of HIV among people who inject drugs in scotland revealed through phylogenetic analysis. J Infect Dis 2018;217:1875–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents Department of Health and Human Services. 2018. https://aidsinfo.nih.gov/guidelines [Google Scholar]
- 8.Oster AM, Wertheim JO, Hernandez AL, Ocfemia MC, Saduvala N, Hall HI: Using molecular HIV surveillance data to understand transmission between subpopulations in the United States. J Acquir Immune Defic Syndr 2015;70: 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oster AM, France AM, Mermin J: Molecular epidemiology and the transformation of HIV prevention. JAMA 2018; 319:1657–1658. [DOI] [PubMed] [Google Scholar]
- 10.Oster AM, France AM, Panneer N, et al. : Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data. J Acquir Immune Defic Syndr 2018;79:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.France AM: Identifying growing clusters of recent, rapid HIV transmission to target prevention. International Workshop on HIV Transmission, Chicago, IL, 2016. [Google Scholar]
- 12.Oster AM, France AM, Panneer N, et al. : Analysis of U.S. HIV sequence data indicates that recent and rapid HIV transmission is focused among young Hispanic/Latino men who have sex with men. International AIDS Society Conference on HIV Science, Paris, France, 2017. [Google Scholar]
- 13.France AM, Panneer N, Ocfemia MCB, et al. : Rapidly growing HIV transmission clusters in the United States, 2013–2016. Conference on Retroviruses and Opportunistic Infections, Boston, MA, 2018. [Google Scholar]
- 14.Dasgupta S, Hall HI, Hernandez AL, Ocfemia MCB, Saduvala N, Oster AM: Receipt and timing of HIV drug resistance testing in six U.S. jurisdictions. AIDS Care 2017; 29:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novitsky V, Moyo S, Lei Q, DeGruttola V, Essex M: Impact of sampling density on the extent of HIV clustering. AIDS Res Hum Retroviruses 2014;30:1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiteside YO, Song R, Wertheim JO, Oster AM: Molecular analysis allows inference into HIV transmission among young men who have sex with men in the United States. AIDS 2015;29:2517–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pond S, Weaver S, Brown AJL, Wertheim JO: HIV-TRACE (Transmission Cluster Engine): A tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol Biol Evol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K: On the estimation of the rate of nucleotide substitution for the control region of human mitochondrial DNA. Gene 2000;259:189–197. [DOI] [PubMed] [Google Scholar]
- 19.Bradley H, Hall HI, Wolitski RJ, et al. : Vital signs: HIV diagnosis, care, and treatment among persons living with HIV—United States, 2011. MMWR Morb Mortal Wkly Rep 2014;63:1113–1117. [PMC free article] [PubMed] [Google Scholar]
- 20.CDC: Monitoring selected national HIV prevention and care objectives using HIV surveillance data: United States and 6 dependent areas, 2016. HIV Suppl Surveillance Rep 2018:23. [Google Scholar]
- 21.Satcher Johnson A, Song R, Hall HI: Estimated HIV incidence, prevalence, and undiagnosed infections in US States and Washington, DC, 2010–2014. J Acquir Immune Defic Syndr 2017;76:116–122. [DOI] [PubMed] [Google Scholar]
- 22.Monterosso A, Minnerly S, Goings S, et al. : Identifying and investigation a rapidly growing HIV transmission cluster in Texas. Conference on Retroviruses and Opportunistic Infections Seattle, Washington, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.