

Abstract
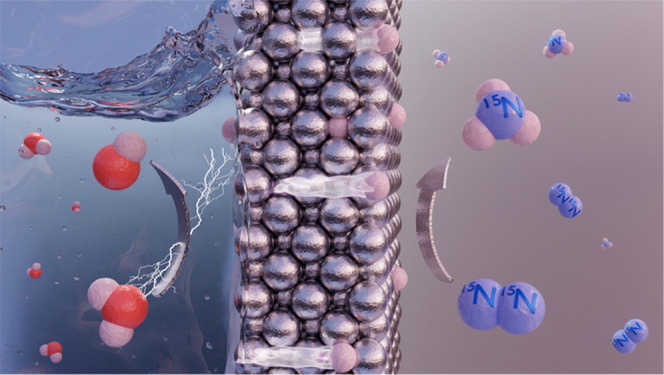
Direct electrochemical nitrogen reduction holds the promise of enabling the production of carbon emission-free ammonia, which is an important intermediate in the fertilizer industry and a potential green energy carrier. Here we show a strategy for ambient condition ammonia synthesis using a hydrogen permeable nickel membrane/electrode that spatially separates the electrolyte and hydrogen reduction side from the dinitrogen activation and hydrogenation sites. Gaseous ammonia is produced catalytically in the absence of electrolyte via hydrogenation of adsorbed nitrogen by electrochemically permeating atomic hydrogen from water reduction. Dinitrogen activation at the polycrystalline nickel surface is confirmed with 15N2 isotope labeling experiments, and it is attributed to a Mars–van Krevelen mechanism enabled by the formation of N-vacancies upon hydrogenation of surface nitrides. We further show that gaseous hydrogen does not hydrogenate the adsorbed nitrogen, strengthening the benefit of having an atomic hydrogen permeable electrode. The proposed approach opens new directions toward green ammonia.
The synthesis of ammonia (NH3) from nitrogen (N2) and hydrogen (H2) is widely considered one of the most important discoveries of the 20th century1 as ammonia plays an essential role as intermediate in nitrogen-based fertilizer production, ultimately sustaining the exponential growth in human population.2 Despite being abundant in Earth’s atmosphere, dinitrogen is stable and relatively inert, requiring high temperature and pressure for its activation and conversion to ammonia. The current Haber–Bosch process, with an estimated annual global production of 150 million metric tons of NH3,3 consumes roughly 1–2% of the global energy demand4 and 5% of the yearly extracted methane as hydrogen source5 via steam-methane re-forming. As a consequence, this process alone is responsible for about 1.4% of the worldwide CO2 emissions.6 In a world aiming to become CO2 neutral and renewable energy based by 2050, such fossil derived ammonia must be replaced by an alternative renewable option.6 In addition to the current primary use as fertilizer intermediate, liquid ammonia is also regarded as a potential high-density energy carrier (22.5 MJ kg–1, liquid at 8 bar and room temperature (RT), or 1 bar and −33 °C),7,8 yet its production with the current state of the art fossil-based technology would be largely unsustainable.
Electrochemical ammonia synthesis is an attractive solution as it can produce carbon-free NH3 in a flexible and scalable manner from the intermittent surplus of electricity generated by decentralized renewables.9 However, despite considerable growing interest and recent developments,10−13 electrochemical ammonia production at near ambient conditions remains unpractical and multiple challenges have to be tackled to further enable direct electrolytic ammonia synthesis. First, the nitrogen reduction reaction to ammonia is in competition with the relatively easier hydrogen evolution reaction (HER). The activation of dinitrogen is an arduous process due to the stable N≡N bond (941 kJ mol–1) and due to the absence of a permanent dipole in the N2 molecule, thus involving only neutral species. In contrast, hydrogen evolution proceeds according to the Volmer mechanism, involving charged hydrogen species (protons or polar water molecules). Conventional electrochemistry is carried out in the presence of an aqueous electrolyte, which contains an excess of hydrogen species (as H2O, H+, or OH–), and therefore, in an aqueous solution, at negative potential, hydrogen reduction and adsorption on the catalyst’s active sites easily prevail over nitrogen adsorption and activation.14−17 The adsorbed H readily recombines on the catalyst surface to form H2, rather than being employed in the production of ammonia; consequently, the electrochemical process is characterized by low faradaic efficiency. Second, the solubility of dinitrogen in aqueous electrolytes is rather low (0.7 mM at ambient conditions15); thus, the availability of nitrogen at the catalyst surface becomes limiting. Third, NH3 is produced in contact with the electrolyte and partitioned between gas phase and liquid phase, demanding appropriate product separation. Fourth, the catalyst surface preferentially adsorbs oxygen traces from the electrolyte; this poisons nitrogen activation sites and deactivates the catalyst.17,18
Substantial efforts have been recently made to develop new strategies to overcome the mentioned challenges in direct electrochemical nitrogen reduction to ammonia at ambient conditions. In particular, studies suggested that NH3 selectivity could be improved by engineering the catalyst–electrolyte interface14,15,19−22 or using nonaqueous electrolytes14,21,23−25 to increase nitrogen concentration and limit the proton and electron transfer at the catalyst interface. However, both approaches do not ensure a complete separation from the electrolyte nor do they prevent the competition of different adsorbate species at the catalyst surface, as previously discussed.
In this contribution, we present an unconventional electrochemical design to perform catalytic nitrogen reduction to ammonia by separating the electrolytic hydrogen activation from the catalytic dinitrogen activation and hydrogenation at the two opposite sides of a dense metallic hydrogen permeable electrode. The working principle is demonstrated using a thin nickel foil as hydrogen permeable electrode. Ammonia is produced catalytically at ambient conditions via the unprecedented reaction between electrochemically permeating atomic hydrogen and adsorbed nitrogen, brought together in the absence of the electrolyte through this Ni-electrode. The formation of N-vacancies, enabled by the hydrogenation of surface nitrogen atoms, shows a clear effect on the catalytic cycle, indicating the occurrence of a Mars–van Krevelen mechanism for N2 activation. We also show that the presence of active nitrogen on the Ni-surface hinders the activation of gaseous H2 that is typically needed for NH3 synthesis. This finding corroborates the benefit of applying permeating atomic hydrogen for the hydrogenation of adsorbed N. The demonstrated reaction pathway represents a novel mechanism for direct electrolytic ammonia production at ambient conditions, which has the potential to be an effective strategy to overcome most of the challenges in electrochemical ammonia synthesis.
System Design
To limit the competition between nitrogen and hydrogen, a dense hydrogen permeable electrode is used to separate two independent compartments dedicated to the spatially decoupled hydrogen activation and nitrogen adsorption respectively, as shown in Figure 1.
Figure 1.
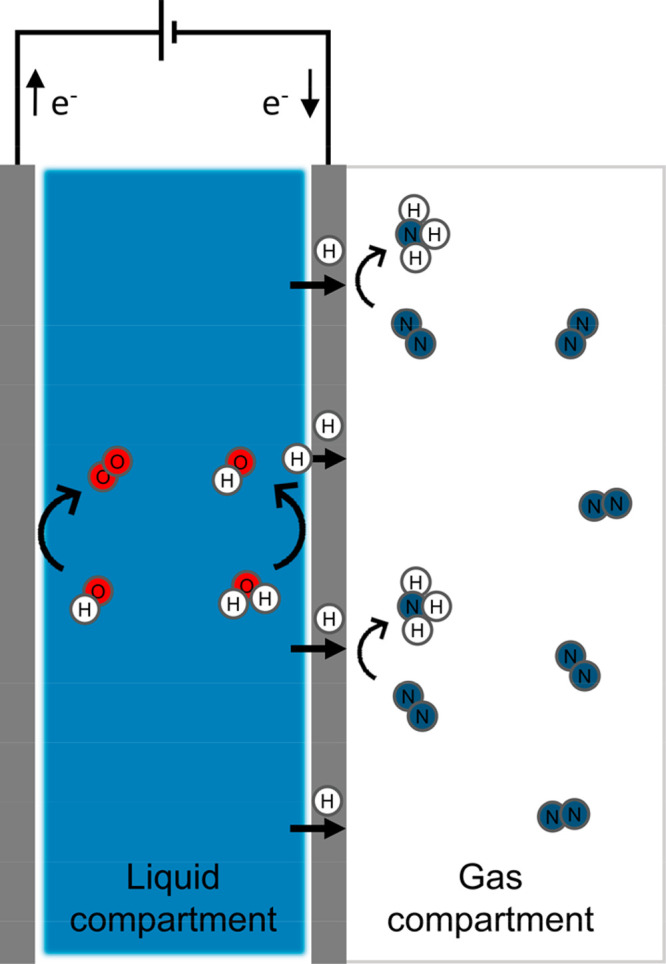
Schematic representation of the proposed system for direct electrolytic ammonia synthesis via electrochemical generated and permeated atomic hydrogen. The cathode is a thin dense metallic electrode permeable to atomic hydrogen and characterized by two active interfaces: an electrode–electrolyte interface dedicated to the electrochemical hydrogen activation (liquid compartment side) and an electrode–gas interface dedicated to the dinitrogen activation and hydrogenation (gas compartment side). Ammonia is generated directly in the gas compartment from the interaction between adsorbed nitrogen and permeating atomic hydrogen.
The liquid compartment contains the aqueous electrolyte required for the coupled electrochemical water oxidation and reduction reactions, driven by the applied potential. Here, the hydrogen is electrochemically inserted in the lattice of the negatively charged thin metallic electrode, permeable only to atomic hydrogen via a solution–diffusion mechanism.26 Importantly, the hydrogen atoms flux can be tuned, by a large extent, by changing the cathodic charging and water reduction driving force, which is a potential advantage of a hydrogen permeable electrode.27 Thus, the dense hydrogen permeable electrode provides a controlled access of protons and electrons, delivered as atomic hydrogen to the nitrogen active sites, preventing at the same time the poisoning of the catalyst by blocking the access of O2, H2O and electrolyte.
In the gas compartment, the electrode is only in contact with gaseous nitrogen, which can chemisorb onto the catalyst surface in the absence of competing adsorbate molecules from the electrolyte. Despite the stable N≡N bond, dissociative chemisorption of dinitrogen at room temperature has been reported to happen spontaneously on several clean transition metal surfaces.28−33 However, one of the major limitations of ambient condition (catalytic) ammonia synthesis is the formation of stable intermediates at the catalyst surface that hinder the advancement of the reaction, due to the lack of available active sites for both H2 and N2 dissociation.34,35 To overcome such an issue, we propose an electrochemical cell configuration where the hydrogenation of the adsorbed nitrogen (and its intermediates) to ammonia proceeds via hydrogen atoms emerging from the bulk of a thin metallic electrode. Therefore, the feeding of reactive hydrogen atoms from the bulk circumvents the necessity of available active sites for H2 activation on the catalyst surface. In this case, the catalytic hydrogenation reactions can continue upon NH3 formation and desorption; hence a free active site becomes available exclusively for further dinitrogen adsorption. Moreover, NH3 is produced directly in the gas phase in a separate compartment, facilitating product separation and preventing NH3 back-diffusion to the anode surface and subsequent NH3 oxidation. The following sections will address in detail each step: nitrogen adsorption, hydrogen permeation, and nitrogen hydrogenation to ammonia.
Nitrogen Adsorption
Dissociative nitrogen chemisorption occurs at room temperature on Ni-surfaces.28,30−32 Moreover, the formation of “bulk-like” surface nitrides subsequent to nitrogen adsorption has been suggested for Ni,32 similarly to the one proposed for Fe.36
X-ray photoelectron spectroscopy (XPS) was used to characterize nitrogen adsorption on a rigorously Ar/H2 plasma cleaned Ni-foil (Supporting Information Figure S1a). The N 1s spectra before and after the exposure to 1 bar N2(g) at room temperature (Figure 2a,b) were compared to the spectrum obtained for a Ni-foil exposed to a low-pressure plasma nitriding process (Figure 2c; the nitriding treatment is described in Materials and Methods of the Supporting Information). The appearance of a main peak centered at 397.8 eV, ascribed to atomic nitrogen (Nad),37,38 confirms the formation of surface nitrides and thus that N2(g) is activated on a sufficiently clean Ni-surface. However, the Ar/H2 cleaned nickel foil rapidly oxidizes to form surface hydroxides when exposed to air (Figure S1c), while the formed surface nitrides provide a protective layer against nickel oxidation from molecular oxygen and moisture (Figure S1b). Moreover, during the electrochemical ammonia production experiments, the hydrogenation of surface nitrides generates in situ highly active sites on the Ni-surface without the necessity of an ultrahigh vacuum (UHV) and oxygen-free controlled environment, used for nitrogen adsorption. As such, in this contribution we deliberately use nitrided, protected nickel surfaces with the aim of preventing unwanted nickel oxidation that would hamper the possibility of demonstrating that nitrogen can be catalytically hydrogenated to ammonia by electrochemically permeating hydrogen at ambient conditions.
Figure 2.

Comparison between the N 1s XPS spectra of an Ar/H2 cleaned polycrystalline nickel surface (a) before and (b) after the exposure to 1 bar gaseous molecular nitrogen and (c) an Ar/H2 cleaned polycrystalline nickel surface after the exposure to low-pressure nitrogen plasma. The difference in peak intensity can be attributed to the additional contribution of nitrogen found in the subsurface of the plasma treated sample. The open circles are the measured values. The fitting is shown as a continuous black line, LA(1.3, 2.4, 69). Pass energy: 50 eV.
Hydrogen Permeation and Nitrogen Reduction to Ammonia
Nickel has a good hydrogen
permeability (Figure S2) and does not form very stable hydrides due to its only
marginally negative enthalpy of formation.39 The 12.5 μm thin dense Ni-electrode (2.5 cm2) was
placed in the cell with the nitrided surface oriented toward the gas
side of the electrochemical cell (Figure 1). A simple two-electrode setup was used,
in which the counter electrode was a Ni-wire of 6.5 cm2. All of the experiments were carried out at ambient conditions and
with a constant N2 flow of 1 mL min–1 supplied at the cathodic gas compartment, unless otherwise stated.
The inlet gas was sufficiently purified with an in-line filter before entering the cell to remove possible contaminations40 (Table S1). The outlet
gases were constantly monitored with in-line TRACE 1300 gas chromatography
(GC). Details of the in-line detection method have been reported elsewhere.41 The insertion of atomic hydrogen into the metal
lattice of the electrode was achieved with a constant cathodic charging
current density of 5 mA cm–2 in 1 M potassium hydroxide
aqueous solution, resulting in a cell potential between 1.9 and 2.0
V (Figure S3) and corresponding to an overall
energy input of about 9.2 kWh 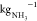 . The theoretical minimum energy
investment
for the presented hydrogen-permeation methodology would be between
5.5 and 5.8 kWh
. The theoretical minimum energy
investment
for the presented hydrogen-permeation methodology would be between
5.5 and 5.8 kWh 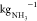 (energetic calculations are reported
in
the Supporting Information). It is acknowledged
that electrolytic ammonia synthesis at reduced cell potentials represents
a craved energetic requirement, sometimes difficult to achieve even
for some of the most promising studies.21,42,43 We investigated the ammonia production in response
to the electrochemical hydrogen permeation, by alternating open circuit
and cathodic charging conditions, while continuously analyzing the
composition of the gas compartment (Figure 3a). No detectable amounts of ammonia were
found during open circuit conditions, indicating that no contaminations
nor other sources of ammonia were present in the cathodic gas compartment
of the electrolytic cell. Upon electrochemical charging, hydrogen
is generated at the electrode–electrolyte interface and hydrogen
atoms permeate through the nickel lattice. Gaseous ammonia is then
produced by the reaction between active adsorbed nitrogen and the
permeating lattice hydrogen, as revealed from the appearance of a
distinct peak in the gas chromatograms. When interrupting the charging
current, the resulting decay of the permeating hydrogen flux follows
a characteristic diffusion profile as a function of time.44 Consequently, the synthesis of ammonia ceases
until a successive cathodic charging current is applied, when both
hydrogen permeation and ammonia production are restored. Therefore,
this result shows the hydrogenation of Nad to ammonia via
electrochemical atomic hydrogen permeation at ambient conditions.
(energetic calculations are reported
in
the Supporting Information). It is acknowledged
that electrolytic ammonia synthesis at reduced cell potentials represents
a craved energetic requirement, sometimes difficult to achieve even
for some of the most promising studies.21,42,43 We investigated the ammonia production in response
to the electrochemical hydrogen permeation, by alternating open circuit
and cathodic charging conditions, while continuously analyzing the
composition of the gas compartment (Figure 3a). No detectable amounts of ammonia were
found during open circuit conditions, indicating that no contaminations
nor other sources of ammonia were present in the cathodic gas compartment
of the electrolytic cell. Upon electrochemical charging, hydrogen
is generated at the electrode–electrolyte interface and hydrogen
atoms permeate through the nickel lattice. Gaseous ammonia is then
produced by the reaction between active adsorbed nitrogen and the
permeating lattice hydrogen, as revealed from the appearance of a
distinct peak in the gas chromatograms. When interrupting the charging
current, the resulting decay of the permeating hydrogen flux follows
a characteristic diffusion profile as a function of time.44 Consequently, the synthesis of ammonia ceases
until a successive cathodic charging current is applied, when both
hydrogen permeation and ammonia production are restored. Therefore,
this result shows the hydrogenation of Nad to ammonia via
electrochemical atomic hydrogen permeation at ambient conditions.
Figure 3.

(a) Rate of ammonia synthesis (solid green symbols) and electrochemical hydrogen permeation (open orange symbols) through a 0.0125 mm thick Ni-electrode as a function of time, while alternating open circuit condition (top horizontal axis: OFF) and cathodic charging (top horizontal axis: ON). Here, the permeating hydrogen is the sum of the detected H2, resulting from the recombinative desorption of atomic hydrogen at the gas compartment side and the hydrogen reacting with the nitrogen to form NH3. (b) Long-term ammonia production rate under N2 (blue) and He (black) atmosphere. Higher synthesis rate and up to 9 h longer NH3 production is measured under N2 atmosphere. The black dotted line indicates the limit of detection (LOD). In both cases, the experiments were conducted under galvanostatic charging of 5 mA cm–2, with cell potentials in Figure S3.
To elucidate the impact of atomic H on the hydrogenation pathway, the surface nickel nitride was exposed to two different concentrations of gaseous hydrogen in the absence of electrochemical hydrogen permeation, i.e., open circuit conditions. An Ar:H2 (98:2 (%)) mixture and pure H2 were sequentially fed with a flow rate of 1 mL min–1 in the cathodic gas compartment. In both cases, no ammonia was recorded at the GC. This is consistent with the reported stability of nickel nitrides under hydrogen gas up to a temperature of 430 K.45 Exclusively after switching to electrochemical hydrogen permeation, ammonia production was restored and detected with the in-line GC (Figure S4). This observation evidences that the formation of NH3(g) requires dissociated H2 to be present and that H2 dissociation does not occur from the gas phase on the nitrided Ni-surface. Hydrogenography measurements (Figure S5) confirmed that preadsorbed nitrogen on Ni hinders H2 spillover, therefore preventing the hydrogenation reaction to proceed and form NH3. However, the direct hydrogenation of Nad does become enabled when electrochemical inserted and permeated lattice hydrogen atoms are present.
Long-term experiments under a constant charging current show that the NH3 production decreases in time, yet when N2 gas is present in the gas compartment, NH3 production lasts up to 9 h longer and about three times more NH3 is produced compared to having He gas atmosphere in the gas compartment (Figure 3b and Figure 4). The three times larger production achieved with N2 atmosphere and the isotope labeled experiment indicate that gas phase N2 does participate in the catalysis, replenishing the surface nitride by dissociative adsorption (vide infra). The decay of NH3 synthesis with time, however, suggests that the available Nad for hydrogenation decreases with time. These results were obtained consistently for multiple experiments (Figure 4 and Figures S6 and S7).
Figure 4.
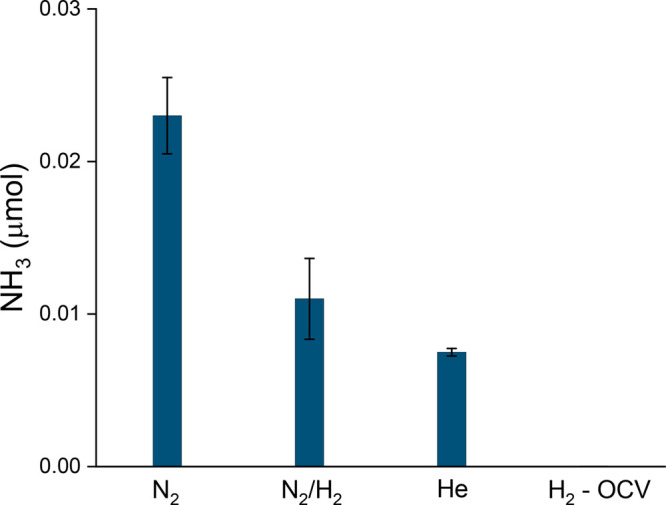
Amount of ammonia generated under N2, N2:H2 (96:4 (%)) or He after 12 h of electrochemical hydrogen permeation at ambient conditions. The NH3 amount under N2 atmosphere increases about 3-fold compared to He. No ammonia has been detected under pure H2 without electrochemical hydrogen permeation. Error bars correspond to the standard deviation of three or more measurements with fresh samples.
The activation of gaseous nitrogen, and its subsequent hydrogenation to ammonia, was confirmed using isotopically labeled 15N2 as feed gas. Prior to being introduced to the electrolytic cell, 15N2 gas was purified with an in-line filter to remove possible contaminations, as previously done for inlet 14N2 gas. 15NH3 was detected by coupling an in-line TRACE 1300 GC with an ISQ single quadrupole mass spectrometer (Figure S8). With this method we were capable of distinguishing isotopologues of NH3 via mass spectroscopy (14NH3m/z = 16, 17 and 15NH3m/z = 17, 18), thanks to the water/ammonia separation achieved with a suitable separation column.41 Once again, no NH3 (14N or 15N) was detected during open circuit conditions, i.e., in the absence of H-permeation (Figures S9–S12). Ammonia production was observed solely in response to electrochemical H-permeation and in close agreement with the amount of ammonia produced with 14N2 in Figure 3, proving the consistency of the experimental results, independently of the isotope used (Table S2). Under electrochemical hydrogen permeation, the appearance of a peak at the retention time corresponding to ammonia, at mass-to-charge (m/z) ratio equal to 18 (fragment, 100%), and a confirmation peak at m/z ratio equal to 17 (fragment, 80%), confirmed that 15NH3 was produced via the catalytic reaction between activated gaseous 15N2 and atomically permeating hydrogen.
On the other hand, the production of 14NH3 (Figure S12 and Table S2), in quantitative agreement with the experiments carried out under He, is attributed to the hydrogenation of preadsorbed nitrogen (14N), which also leads to the expected in situ generation of active N-vacancy sites (vide infra). The mechanism for the N2 activation depends on the nature and the abundance of available surface sites. The deactivation of the catalytic activity is related to the behavior of the N and N-vacancies on the surface and in the subsurface. The presence of surface N prevents poisoning (Figure S1b), while the NH3 desorption leaves behind a N-vacancy on which N2 can be adsorbed, still in competition with poisoning by impurities and vacancy migration to the subsurface. After the long-term experiment, the interruption of the galvanostatic charging and subsequent reactivation after a waiting period, brings back the NH3 production to a rate lower than the initial one. In view of the prolonged operation and activation of N2, the rate of N2 dissociative adsorption outweighs the other processes for an extended period of time. The contribution of N-vacancies to the NH3 synthesis reaction is discussed in the following section.
Nitrogen Mobility and Vacancies
As a consequence of the nitrogen plasma pretreatment, small traces of nitrogen were also found in the subsurface layer of the nickel electrode. The XPS N 1s depth profile in Figure 5 shows the nitrogen distribution over the sample thickness after the nitrogen plasma pretreatment and after electrochemical hydrogen permeation. The penetration depth of nitrogen was about 40 nm. Interestingly, upon electrochemical hydrogen permeation, we observed the consumption of the nitrogen from both the surface and the subsurface. Eventually, after 12 h operation nearly all nitrogen atoms in the bulk reacted to form NH3. No N2 was detected during the experiments under inert He carrier gas, suggesting that all of the implanted nitrogen was selectively converted to NH3.
Figure 5.
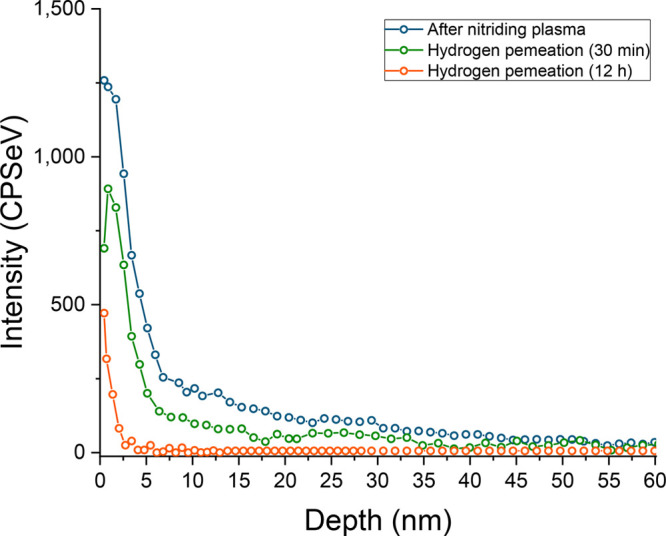
Intensity of the N 1s nitrogen signal as a function of the depth of the sample, resulting from X-ray photoelectron spectroscopy depth-profiling measurements: after N2-plasma exposure (blue) and after 30 min (green) and 12 h (orange) of electrochemical hydrogen permeation under N2.
The N 1s signal measured at the Ni-surface after prolonged H-permeation shows the formation of a small shoulder around 399.9 eV, indicating a distinct N-species, which could be ascribed to the presence of N-vacancies46 (Figure S13). The produced nickel nitride thin film is stable under ambient conditions; however, the hydrogenation of surface nitrogen atoms to ammonia results in the formation of nitrogen vacancies.47 These vacancies exhibit a low activation barrier for N-migration in nickel nitrides.17 Therefore, the created N-vacancies can be exchanged for nitrogen available from the bulk, accounting for the observed depletion of nitrogen atoms from the subsurface layers. Still, as stated above, N2 readily reacts with the vacancies. The presence of a pre-existing surface nitride layer and the role of N-vacancies point in the direction of molecular nitrogen activation via a Mars–van Krevelen mechanism.48 The N-vacancy, formed at the catalyst surface upon ammonia production from surface nitrogen atoms, represents an energetically more favorable active site for nitrogen adsorption and activation compared to a bare Ni-surface;17,47,49,50 although the latter also activates N2 when sufficiently clean as shown in Figure 2. However, in the long term of the experiment, poisoning apparently results in the reduction of available N-vacancy active sites. Migration of N from the subsurface also plays a role, but does not seem to deactivate the vacancy formation, but rather extends the NH3 formation from the plasma preadsorbed N. Thus, the prolonged reaction could benefit from strategies to increase N-vacancy stability on the surface.51,52 Nitrogen activation rates may be promoted by varying the operating temperature and pressure at the gas compartment, which can be realized with the presented electrochemical configuration. However, this falls outside the scope of the present investigation at near ambient conditions.
The surface sites can also interact with molecular or atomic hydrogen or other potential adsorbates that might preferentially occupy them over adsorbed N, preventing a persistent and more efficient N2(g) activation.18,35,50,53 To investigate whether the presence of H2 gas would help in creating a more reductive environment, or suppress oxide impurities, or interact with the active sites, we also carried out the NH3 synthesis using a mixture of 4% H2 in N2 in the gas compartment (Figure S14). In this case, the resulting produced ammonia (0.011 μmol) was less than obtained under pure nitrogen but higher when compared to He atmosphere (Figure 4); this is evidence that the competing action of H2 on the N-adsorption sites50 is more prominent than the potential reducing and surface protection action on, e.g., oxygen impurities. Therefore, it becomes relevant that, under reaction conditions, the surface is sufficiently contaminant free, as adsorbed N was observed with XPS in a well-controlled environment and after in situ Ar ion cleaning. This also suggests that the N2 activation on the polycrystalline Ni-surface controls the reaction rate.
Through the adoption of a hydrogen permeable electrode, we have demonstrated a novel strategy for direct electrolytic ammonia synthesis from N2, H2O, and renewable electricity at room temperature and pressure. The proposed system uses cheap and abundant materials, and it is in line with the paramount need of innovative electrode designs for electrochemical ammonia synthesis.14,19 The dense metallic electrode ensures a complete separation of the nitrogen active sites from the electrolyte, while reactive atomic hydrogen is electrochemically fed from the bulk of the electrode. Hydrogenography and gas chromatography measurements revealed that adsorbed nitrogen on a polycrystalline Ni-surface does not react with gaseous H2, hindering the hydrogen spillover essential for the NH3 synthesis reaction. Nonetheless, NH3 synthesis was enabled upon atomic hydrogen permeation, showing that the hydrogenation of active nitrogen via permeating hydrogen circumvents the mutual competition between nitrogen and hydrogen activation. Our results indicate the occurrence of a Mars–van Krevelen dinitrogen activation mechanism, in which N-vacancies play a significant role in the catalytic process facilitating N2 adsorption.
This work highlights the role of the hydrogen permeable electrode and the electrochemical atomic hydrogen permeation in the catalytic hydrogenation of Nad to ammonia at ambient conditions, demonstrating the feasibility of this reaction. We envision that the presented work will encourage further developments toward a more efficient direct electrolytic ammonia synthesis.
Acknowledgments
This work is part of the Open Technology research program with Project No. 15234 which is (partly) financed by The Netherlands Organisation for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.1c01568.
Materials and methods, experimental details, additional XPS characterization, electrochemical hydrogen-permeation measurement, cell potential–time curve, hydrogenography data, long-term NH3 production rates, GC, GC-MS, and UV–vis calibration curves, 15N-isotope labeling experiment, NOx contaminations, and gas purification (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Smil V. Detonator of the population explosion. Nature 1999, 400 (6743), 415. 10.1038/22672. [DOI] [Google Scholar]
- Erisman J. W.; Sutton M. A.; Galloway J.; Klimont Z.; Winiwarter W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636. 10.1038/ngeo325. [DOI] [Google Scholar]
- Bernhardt D.; Reilly J. F.. Mineral Commodity Summaries 2020; U.S. Geological Survey, 2020; Vol. 1, pp 116–117. 10.3133/mcs2020. [DOI]
- Jiao F.; Xu B. Electrochemical Ammonia Synthesis and Ammonia Fuel Cells. Adv. Mater. 2019, 31 (31), 1805173. 10.1002/adma.201805173. [DOI] [PubMed] [Google Scholar]
- Rouwenhorst K. H. R.; Krzywda P. M.; Benes N. E.; Mul G.; Lefferts L.. Ammonia, 4. Green Ammonia Production. In Ullmann’s Encyclopedia of Industrial Chemistry; 2020; pp 1–20. 10.1002/14356007.w02_w02. [DOI] [Google Scholar]
- MacFarlane D. R.; Cherepanov P. V.; Choi J.; Suryanto B. H. R.; Hodgetts R. Y.; Bakker J. M.; Ferrero Vallana F. M.; Simonov A. N. A Roadmap to the Ammonia Economy. Joule 2020, 4 (6), 1186–1205. 10.1016/j.joule.2020.04.004. [DOI] [Google Scholar]
- Avery W. H. A role for ammonia in the hydrogen economy. Int. J. Hydrogen Energy 1988, 13 (12), 761–773. 10.1016/0360-3199(88)90037-7. [DOI] [Google Scholar]
- Lan R.; Irvine J. T. S.; Tao S. W. Ammonia and related chemicals as potential indirect hydrogen storage materials. Int. J. Hydrogen Energy 2012, 37 (2), 1482–1494. 10.1016/j.ijhydene.2011.10.004. [DOI] [Google Scholar]
- Mulder F. M. Implications of diurnal and seasonal variations in renewable energy generation for large scale energy storage. J. Renewable Sustainable Energy 2014, 6 (3), 033105. 10.1063/1.4874845. [DOI] [Google Scholar]
- Guo X.; Du H.; Qu F.; Li J. Recent progress in electrocatalytic nitrogen reduction. J. Mater. Chem. A 2019, 7 (8), 3531–3543. 10.1039/C8TA11201K. [DOI] [Google Scholar]
- Cao N.; Zheng G. Aqueous electrocatalytic N2 reduction under ambient conditions. Nano Res. 2018, 11 (6), 2992–3008. 10.1007/s12274-018-1987-y. [DOI] [Google Scholar]
- Soloveichik G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nature Catalysis 2019, 2 (5), 377–380. 10.1038/s41929-019-0280-0. [DOI] [Google Scholar]
- Qing G.; Ghazfar R.; Jackowski S. T.; Habibzadeh F.; Ashtiani M. M.; Chen C.-P.; Smith M. R.; Hamann T. W. Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020, 120 (12), 5437–4416. 10.1021/acs.chemrev.9b00659. [DOI] [PubMed] [Google Scholar]
- Singh A. R.; Rohr B. A.; Schwalbe J. A.; Cargnello M.; Chan K.; Jaramillo T. F.; Chorkendorff I.; Nørskov J. K. Electrochemical Ammonia Synthesis—The Selectivity Challenge. ACS Catal. 2017, 7 (1), 706–709. 10.1021/acscatal.6b03035. [DOI] [Google Scholar]
- Hu L.; Xing Z.; Feng X. Understanding the Electrocatalytic Interface for Ambient Ammonia Synthesis. ACS Energy Letters 2020, 5 (2), 430–436. 10.1021/acsenergylett.9b02679. [DOI] [Google Scholar]
- Kibsgaard J.; Nørskov J. K.; Chorkendorff I. The Difficulty of Proving Electrochemical Ammonia Synthesis. ACS Energy Letters 2019, 4 (12), 2986–2988. 10.1021/acsenergylett.9b02286. [DOI] [Google Scholar]
- Abghoui Y.; Skúlason E. Computational Predictions of Catalytic Activity of Zincblende (110) Surfaces of Metal Nitrides for Electrochemical Ammonia Synthesis. J. Phys. Chem. C 2017, 121 (11), 6141–6151. 10.1021/acs.jpcc.7b00196. [DOI] [Google Scholar]
- Ertl G.; Huber M. Interaction of Nitrogen and Oxygen on Iron Surfaces. Z. Phys. Chem. 1980, 119 (1), 97. 10.1524/zpch.1980.119.1.097. [DOI] [Google Scholar]
- Ampelli C. Electrode design for ammonia synthesis. Nature Catalysis 2020, 3 (5), 420–421. 10.1038/s41929-020-0461-x. [DOI] [Google Scholar]
- Shi R.; Zhang X.; Waterhouse G. I. N.; Zhao Y.; Zhang T. The Journey toward Low Temperature, Low Pressure Catalytic Nitrogen Fixation. Adv. Energy Mater. 2020, 10 (19), 2000659. 10.1002/aenm.202000659. [DOI] [Google Scholar]
- Lazouski N.; Chung M.; Williams K.; Gala M. L.; Manthiram K. Non-aqueous gas diffusion electrodes for rapid ammonia synthesis from nitrogen and water-splitting-derived hydrogen. Nature Catalysis 2020, 3 (5), 463–469. 10.1038/s41929-020-0455-8. [DOI] [Google Scholar]
- Liu S.; Qian T.; Wang M.; Ji H.; Shen X.; Wang C.; Yan C. Proton-filtering covalent organic frameworks with superior nitrogen penetration flux promote ambient ammonia synthesis. Nature Catalysis 2021, 4 (4), 322–331. 10.1038/s41929-021-00599-w. [DOI] [Google Scholar]
- Suryanto B. H. R.; Du H.-L.; Wang D.; Chen J.; Simonov A. N.; MacFarlane D. R. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nature Catalysis 2019, 2 (4), 290–296. 10.1038/s41929-019-0252-4. [DOI] [Google Scholar]
- Singh A. R.; Rohr B. A.; Statt M. J.; Schwalbe J. A.; Cargnello M.; Nørskov J. K. Strategies toward Selective Electrochemical Ammonia Synthesis. ACS Catal. 2019, 9 (9), 8316–8324. 10.1021/acscatal.9b02245. [DOI] [Google Scholar]
- Zhou F.; Azofra L. M.; Ali M.; Kar M.; Simonov A. N.; McDonnell-Worth C.; Sun C.; Zhang X.; MacFarlane D. R. Electro-synthesis of ammonia from nitrogen at ambient temperature and pressure in ionic liquids. Energy Environ. Sci. 2017, 10 (12), 2516–2520. 10.1039/C7EE02716H. [DOI] [Google Scholar]
- Uemiya S. State-of-the-Art of Supported Metal Membranes for Gas Separation. Sep. Purif. Methods 1999, 28 (1), 51–85. 10.1080/03602549909351644. [DOI] [Google Scholar]
- Sherbo R. S.; Delima R. S.; Chiykowski V. A.; MacLeod B. P.; Berlinguette C. P. Complete electron economy by pairing electrolysis with hydrogenation. Nature Catalysis 2018, 1 (7), 501–507. 10.1038/s41929-018-0083-8. [DOI] [Google Scholar]
- Rao C. N. R.; Ranga Rao G. Nature of nitrogen adsorbed on transition metal surfaces as revealed by electron spectroscopy and cognate techniques. Surf. Sci. Rep. 1991, 13 (7), 223–263. 10.1016/0167-5729(91)90014-O. [DOI] [Google Scholar]
- Dietrich H.; Geng P.; Jacobi K.; Ertl G. Sticking coefficient for dissociative adsorption of N2 on Ru single-crystal surfaces. J. Chem. Phys. 1996, 104 (1), 375–381. 10.1063/1.470836. [DOI] [Google Scholar]
- Rao G. R.; Rao C. N. R. Adsorption of nitrogen on clean and modified single-crystal Ni surfaces. Appl. Surf. Sci. 1990, 45 (1), 65–69. 10.1016/0169-4332(90)90021-Q. [DOI] [Google Scholar]
- Wedler G.; Alshorachi G. Adsorption of Nitrogen on Polycrystalline Nickel Films between 77 and 333 K. Berichte der Bunsengesellschaft für physikalische Chemie 1980, 84 (3), 277–281. 10.1002/bbpc.19800840317. [DOI] [Google Scholar]
- Grunze M.; Driscoll R. K.; Burland G. N.; Cornish J. C. L.; Pritchard J. Molecular and dissociative chemisorption of N2 on Ni(110). Surf. Sci. 1979, 89 (1), 381–390. 10.1016/0039-6028(79)90624-1. [DOI] [Google Scholar]
- Ertl G.; Lee S. B.; Weiss M. Kinetics of nitrogen adsorption on Fe(111). Surf. Sci. 1982, 114 (2), 515–526. 10.1016/0039-6028(82)90702-6. [DOI] [Google Scholar]
- Vojvodic A.; Medford A. J.; Studt F.; Abild-Pedersen F.; Khan T. S.; Bligaard T.; Nørskov J. K. Exploring the limits: A low-pressure, low-temperature Haber–Bosch process. Chem. Phys. Lett. 2014, 598, 108–112. 10.1016/j.cplett.2014.03.003. [DOI] [Google Scholar]
- Ertl G.; Huber M.; Lee S. B.; Paál Z.; Weiss M. Interactions of nitrogen and hydrogen on iron surfaces. Appl. Surf. Sci. 1981, 8 (4), 373–386. 10.1016/0378-5963(81)90092-1. [DOI] [Google Scholar]
- Bozso F.; Ertl G.; Grunze M.; Weiss M. Interaction of nitrogen with iron surfaces: I. Fe(100) and Fe(111). J. Catal. 1977, 49 (1), 18–41. 10.1016/0021-9517(77)90237-8. [DOI] [Google Scholar]
- Moulder J. F.; Stickle W. F.; Sobol P. E.; Bomben K. D.. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Perkin-Elmer: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Galtayries A.; Laksono E.; Siffre J.-M.; Argile C.; Marcus P. XPS study of the adsorption of NH3 on nickel oxide on Ni(111). Surf. Interface Anal. 2000, 30 (1), 140–144. . [DOI] [Google Scholar]
- Tkacz M. Enthalpies of formation and decomposition of nickel hydride and nickel deuteride derived from (p, c, T) relationships. J. Chem. Thermodyn. 2001, 33 (8), 891–897. 10.1006/jcht.2000.0797. [DOI] [Google Scholar]
- Hodgetts R. Y.; Du H.-L.; MacFarlane D. R.; Simonov A. N. Electrochemically Induced Generation of Extraneous Nitrite and Ammonia in Organic Electrolyte Solutions During Nitrogen Reduction Experiments. ChemElectroChem 2021, 8 (9), 1596–1604. 10.1002/celc.202100251. [DOI] [Google Scholar]
- Zaffaroni R.; Ripepi D.; Middelkoop J.; Mulder F. M. Gas Chromatographic Method for In Situ Ammonia Quantification at Parts per Billion Levels. ACS Energy Letters 2020, 5 (12), 3773–3777. 10.1021/acsenergylett.0c02219. [DOI] [Google Scholar]
- Suryanto B. H. R.; Matuszek K.; Choi J.; Hodgetts R. Y.; Du H.-L.; Bakker J. M.; Kang C. S. M.; Cherepanov P. V.; Simonov A. N.; MacFarlane D. R. Nitrogen reduction to ammonia at high efficiency and rates based on a phosphonium proton shuttle. Science 2021, 372 (6547), 1187–1191. 10.1126/science.abg2371. [DOI] [PubMed] [Google Scholar]
- Wang M.; Khan M. A.; Mohsin I.; Wicks J.; Ip A. H.; Sumon K. Z.; Dinh C.-T.; Sargent E. H.; Gates I. D.; Kibria M. G. Can sustainable ammonia synthesis pathways compete with fossil-fuel based Haber–Bosch processes?. Energy Environ. Sci. 2021, 14 (5), 2535–2548. 10.1039/D0EE03808C. [DOI] [Google Scholar]
- Devanathan M. A. V.; Stachurski Z. The adsorption and diffusion of electrolytic hydrogen in palladium. Proc. R. Soc. London, Ser. A 1962, 270 (1340), 90–102. 10.1098/rspa.1962.0205. [DOI] [Google Scholar]
- Baiker A.; Maciejewski M. Formation and thermal stability of copper and nickel nitrides. J. Chem. Soc., Faraday Trans. 1 1984, 80 (8), 2331–2341. 10.1039/f19848002331. [DOI] [Google Scholar]
- Liu B.; He B.; Peng H.-Q.; Zhao Y.; Cheng J.; Xia J.; Shen J.; Ng T.-W.; Meng X.; Lee C.-S.; Zhang W. Unconventional Nickel Nitride Enriched with Nitrogen Vacancies as a High-Efficiency Electrocatalyst for Hydrogen Evolution. Advanced Science 2018, 5 (8), 1800406. 10.1002/advs.201800406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T.-N.; Park S.-W.; Lu Y.; Li J.; Sasase M.; Kitano M.; Tada T.; Hosono H. Vacancy-enabled N2 activation for ammonia synthesis on an Ni-loaded catalyst. Nature 2020, 583 (7816), 391–395. 10.1038/s41586-020-2464-9. [DOI] [PubMed] [Google Scholar]
- Mars P.; van Krevelen D. W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. 10.1016/S0009-2509(54)80005-4. [DOI] [Google Scholar]
- Zeinalipour-Yazdi C. D.; Hargreaves J. S. J.; Catlow C. R. A. Nitrogen Activation in a Mars–van Krevelen Mechanism for Ammonia Synthesis on Co3Mo3N. J. Phys. Chem. C 2015, 119 (51), 28368–28376. 10.1021/acs.jpcc.5b06811. [DOI] [Google Scholar]
- Michalsky R.; Avram A. M.; Peterson B. A.; Pfromm P. H.; Peterson A. A. Chemical looping of metal nitride catalysts: low-pressure ammonia synthesis for energy storage. Chemical Science 2015, 6 (7), 3965–3974. 10.1039/C5SC00789E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.; Li L.; Liu X.; Tang C.; Xu W.; Chen S.; Song L.; Zheng Y.; Qiao S.-Z. Nitrogen Vacancies on 2D Layered W2N3: A Stable and Efficient Active Site for Nitrogen Reduction Reaction. Adv. Mater. 2019, 31 (32), 1902709. 10.1002/adma.201902709. [DOI] [PubMed] [Google Scholar]
- Abghoui Y.; Skúlasson E. Transition Metal Nitride Catalysts for Electrochemical Reduction of Nitrogen to Ammonia at Ambient Conditions. Procedia Computer Science 2015, 51, 1897–1906. 10.1016/j.procs.2015.05.433. [DOI] [Google Scholar]
- Abghoui Y.; Garden A. L.; Hlynsson V. F.; Björgvinsdóttir S.; Ólafsdóttir H.; Skúlason E. Enabling electrochemical reduction of nitrogen to ammonia at ambient conditions through rational catalyst design. Phys. Chem. Chem. Phys. 2015, 17 (7), 4909–4918. 10.1039/C4CP04838E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.