

Abstract
The mechanisms by which environmental exposures contribute to the pathogenesis of lung fibrosis are unclear. Here, we demonstrate an increase in cadmium (Cd) and carbon black (CB), common components of cigarette smoke (CS) and environmental particulate matter (PM), in lung tissue from subjects with idiopathic pulmonary fibrosis (IPF). Cd concentrations were directly proportional to citrullinated vimentin (Cit-Vim) amounts in lung tissue of subjects with IPF. Cit-Vim amounts were higher in subjects with IPF, especially smokers, which correlated with lung function and were associated with disease manifestations. Cd/CB induced the secretion of Cit-Vim in an Akt1- and peptidylarginine deiminase 2 (PAD2)–dependent manner. Cit-Vim mediated fibroblast invasion in a 3D ex vivo model of human pulmospheres that resulted in higher expression of CD26, collagen, and α-SMA. Cit-Vim activated NF-κB in a TLR4-dependent fashion and induced the production of active TGF-β1, CTGF, and IL-8 along with higher surface expression of TLR4 in lung fibroblasts. To corroborate ex vivo findings, mice treated with Cit-Vim, but not Vim, independently developed a similar pattern of fibrotic tissue remodeling, which was TLR4 dependent. Moreover, wild-type mice, but not PAD2−/− and TLR4 mutant (MUT) mice, exposed to Cd/CB generated high amounts of Cit-Vim, in both plasma and bronchoalveolar lavage fluid, and developed lung fibrosis in a stereotypic manner. Together, these studies support a role for Cit-Vim as a damage-associated molecular pattern molecule (DAMP) that is generated by lung macrophages in response to environmental Cd/CB exposure. Furthermore, PAD2 might represent a promising target to attenuate Cd/CB-induced fibrosis.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic disease of the lung (1, 2) whose etiology remains unclear. Although underlying genetic factors increase susceptibility (3), the trigger(s) that initiates the pathologic expression of the disease remains unknown. The possible role of environmental factors, especially cigarette smoke (CS) inhalation or exposure to particulate matter (PM) in ambient air pollution in lung fibrosis, needs further clarification. Cadmium (Cd), a heavy metal and a recognized cause of lung fibrosis in humans (4) and in rodents including hamster (5), rat (6), and mouse (7), can adsorb onto carbon nanomaterials (8). Ultrafine PM (<2.5 μm in diameter) from both CS and ambient air pollution from biomass fuels and coal furnaces consists of carbon black (CB) with other toxicants, such as Cd, adsorbed onto its surface (9–11). Adsorption onto the surface of carbon nanoparticles is intensified by combustion in lighted cigarettes and emissions from coal-fired power plants (12).
Up to two-thirds of subjects with IPF have a history of smoking, and CS is considered a potential factor that contributes to the pathogenesis of lung fibrosis (2, 13–15). Each cigarette contains 2 to 3 μg of Cd (7), and Cd accumulates in the lung because of its long half-life of 10 to 30 years (16). Similar to Cd, CB is also a universal constituent in CS (17).
Lung macrophages can phagocytose Cd particles, and both Cd and metallothionein (MT), a cysteine-rich Cd-binding protein, can accumulate within the lung macrophages (18, 19). Macrophages secrete many factors that modify extracellular matrix (ECM) deposition and collagen metabolism (20), highlighting the critical role of cell-cell interaction in lung fibrosis (21). We have demonstrated that Cd induces α-smooth muscle actin (α-SMA) activation, ECM protein accumulation, and collagen secretion, suggesting that Cd is fibrogenic, in which fibroblast differentiation and ECM deposition play a critical role (7). However, whether Cd/CB can trigger the development of lung fibrosis and whether they play an essential role in the pathogenesis of IPF has not been completely elucidated.
Innate immune responses are prominent in subjects with IPF, suggesting the potential role of Toll-like receptors (TLRs) in the pathogenesis of IPF (22, 23). Damage-associated molecular pattern molecules (DAMPs) serve as TLR ligands, and growing evidence implicates DAMP-triggered TLR mediated in fibrotic disorders (23, 24). Citrullination, specially citrullinated vimentin (Cit-Vim), has been linked to the pathogenesis of CCl4-induced liver fibrosis (25), rheumatoid arthritis (RA) (26), and interstitial lung disease associated with RA (RA-ILD) (27). Moreover, smoking induces airway inflammation by triggering protein citrullination through up-regulating peptidylarginine deiminase 2 (PAD2), a citrullination catalyzing enzyme (28, 29). PAD2 can also be induced by silica nanoparticles (30) and is important in inflammation and autoimmune diseases (31–33).
In the current study, we showed that secretion of Cit-Vim from lung macrophages is augmented by Cd/CB, and this action is Akt1 and PAD2 dependent. Cit-Vim drives an invasive subtype of fibroblasts and activates lung fibroblasts. Furthermore, Cit-Vim induced the production of active transforming growth factor–β1 (TGF-β1), connective tissue growth factor (CTGF), and interleukin-8 (IL-8). We confirmed that wild-type (WT), but not TLR4 mutant (MUT), mice developed Cit-Vim–induced, but not Vim-induced, fibrosis. In addition, we demonstrated greater amounts of citrullinated proteins/Cit-Vim in the lung tissue and plasma from subjects with IPF and a positive correlation between Cd and Cit-Vim. These data suggest that Cd/CB-induced Cit-Vim acts as a DAMP to activate fibroblasts through TLR4 signaling and contributes to the pathogenesis of IPF.
RESULTS
Both Cd and CB substantially accumulate in lung tissue from subjects with IPF
As smoking is known to be associated with IPF (2, 13, 14), and both Cd and CB are present in CS, we first investigated the accumulation of Cd and CB in subjects with IPF. MT has been considered as an indicator of metal exposure, including Cd (34, 35). Both MT-1E and MT-2A mRNA were increased in lung macrophages from subjects with IPF (fig. S1, A to D), and these macrophages also showed markedly increased MT-1/2 protein (fig. S1, E and F), and mRNA with greater expression in those cells from subjects with IPF that smoked (fig. S1, B and D). In addition, the concentrations of Cd in IPF lung tissues were significantly higher (P < 0.001) than in control lungs and were notably higher in smokers (Fig. 1, A and B). The clinical characteristics of the lung tissue from subjects with IPF for cohort 1 and a detailed analysis by smoking status are shown in tables S1 and S2. To identify the existence of CB in subjects with IPF, bronchoalveolar lavage fluid (BALF) samples were analyzed together with lung tissues (control BALF, n = 5; IPF BALF, n = 10; Cohort 1, table S1). A quantitative analysis of BALF cells stained with Hema 3 showed higher deposition of CB in macrophages from subjects with IPF, especially smokers (fig. S2, A and B, and Fig. 1, C and D). Transmission electron microscopy (TEM) was further used to demonstrate the presence of CB in lung macrophages of BALF from subjects with IPF (fig. S2, C to E).
Fig. 1. Cd and CB accumulate highly in lung tissue from subjects with IPF, especially smokers.
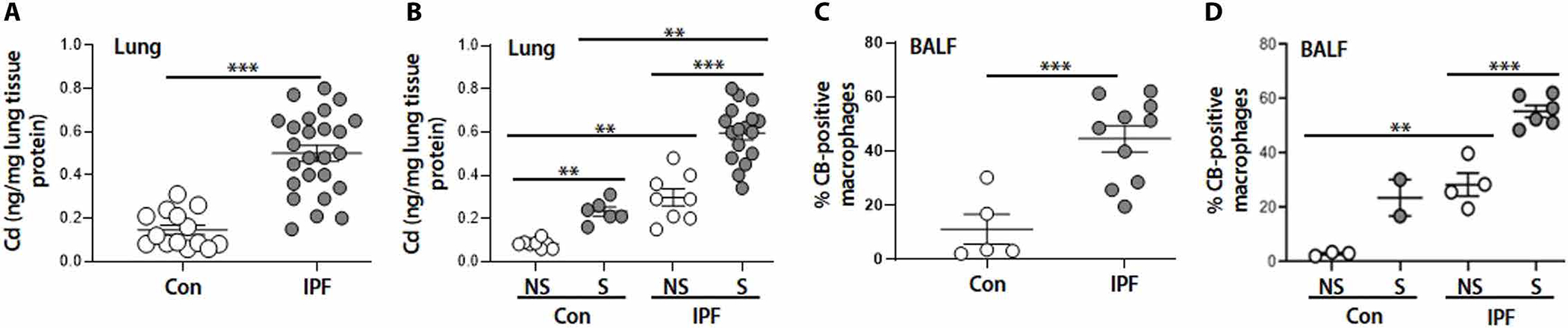
(A and B) Cd contents by ICP-MS from lung tissues for (A) control (Con) (n = 14) and IPF (n = 25) and (B) control never-smoker (NS) (n = 8), control smoker (S) (n = 6), IPF never-smoker (n = 8), and IPF smoker (n = 17). (C and D) Quantification of CB-positive macrophages from Hema 3–stained BALF for (C) control (n = 5) and IPF (n = 10) and (D) control never-smoker (n = 3), control smoker (n = 2), IPF never-smoker (n = 4), and IPF smoker (n = 6). **P < 0.01 and ***P < 0.001 using two-tailed t test for (A) and (C) and one-way ANOVA followed by Tukey’s post hoc analysis for (B) and (D).
Cd in subjects with IPF correlates with Cit-Vim, and Cit-Vim correlates with lung function and transplant-free survival
Initial studies showed an increase of citrullinated proteins in the lung tissue from subjects with IPF compared to controls, whereas these proteins were not detectable in normal lung parenchyma except few lung macrophages (Fig. 2, A to C). Citrullinated proteins were found predominantly in cytoplasm of cells. We further found that Cit-Vim and Vim protein expression was significantly increased (P < 0.001) in lung macrophages from subjects with IPF compared to controls (Fig. 2, D and E). Furthermore, Cit-Vim amounts were significantly higher (P < 0.001) in lung from subjects with IPF than in normal control lung (Fig. 2F). Similar to Cd (Fig. 1B), Cit-Vim amounts were also notably higher in smokers compared to nonsmokers (Fig. 2G). A significant correlation (r = 0.76, P = 0.005) was also observed between Cd and Cit-Vim in IPF lungs (Fig. 2H), suggesting that Cd may have an important role in the pathogenesis of IPF by increasing Cit-Vim. In subjects with IPF, the Cit-Vim amounts were also higher in plasma (Fig. 2, I and J; Cohort 2, plasma samples, table S1) and correlated inversely with the predicted percentage of forced vital capacity (FVC) (r = −0.69, P < 0.001; Fig. 2K) and diffusing capacity for carbon monoxide (DLCO) (r = −0.46, P < 0.001; Fig. 2L). Survival analysis using our previous strategy (36) showed that the subjects with higher Cit-Vim had significantly decreased [hazard ratio (HR) = 2.18, P = 0.04] transplant-free survival than subjects with lower Cit-Vim (Fig. 2M), suggesting that Cit-Vim might serve as a prognostic factor in IPF.
Fig. 2. Cd positively correlates with Cit-Vim, and Cit-Vim correlates with lung function parameters and transplant-free survival.
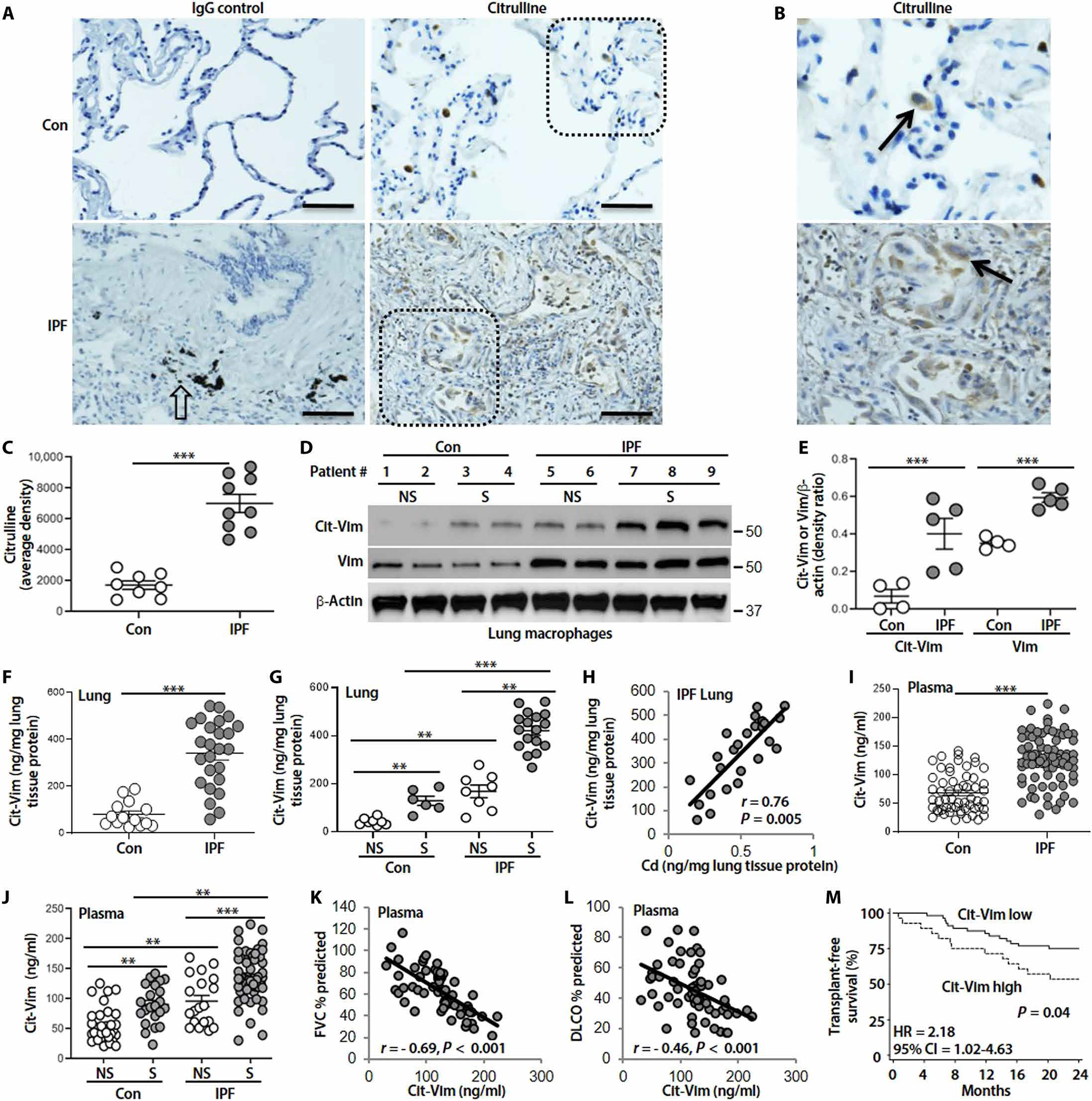
(A) Representative human lung histology with citrulline staining (arrows: stained brown) from control (n = 8) and subjects with IPF (n = 9). (B) Higher magnification of inset from (A). Open arrow indicates CB. Scale bars, 100 μm. (C) DAB staining density from (A) was quantified. (D) Immunoblot analysis of Cit-Vim and Vim in lung macrophage. (E) Quantification of Cit-Vim and Vim expression from (D). (F) Cit-Vim amounts by ELISA in lung tissues and (G) analyzed by smoking status. (H) Correlation analysis of Cd with Cit-Vim in lung tissue from subjects with IPF (n = 25). (I) Cit-Vim amounts by ELISA in plasma and (J) analyzed by smoking status. Correlation analysis of plasma Cit-Vim with predicted percentage of (K) FVC and (L) DLCO. P and r values were determined using Spearman rank correlations. (M) Transplant-free survival analysis during the next 2 years between the subjects with higher Cit-Vim and subjects with lower Cit-Vim. HR and CI values were established by Cox proportional hazard regression. **P < 0.01 and ***P < 0.001 using two-tailed t test for (C) and (F) and one-way ANOVA followed by Tukey’s post hoc analysis for (E), (G), and (J).
Cd/CB-induced Vim citrullination and Cit-Vim secretion are dependent on Akt1 and PAD2 activation
Lung macrophages are the first defensive cells that phagocytize environmental Cd and CB particles (19). We explored the mechanism of Vim citrullination using lung macrophages isolated from subjects with IPF. To evaluate the role of oxidative stress after Cd/CB exposure, intracellular reactive oxygen species (ROS) were analyzed. Treatment with Mito-TEMPO, an inhibitor of mitochondria ROS production, blocked Cd- and/or CB-induced ROS production (fig. S3, A and B). We have found that CdCl2 induced ECM protein deposition and phosphorylation of Akt and Vim, especially at Ser39 and Ser55, but caused less phosphorylation of extracellular signal–regulated kinase (ERK), protein kinase C (PKC), and PKA (7). Here, we demonstrated that Cd and CB in combination caused a noticeable increase in phosphorylation of Akt1 as well as expression of PAD2 (but not PAD4) and Cit-Vim over control or either alone (fig. S3, C and D). These actions were inhibited in the presence of Mito-TEMPO, suggesting that Akt1 activation and expression of PAD2 and Cit-Vim are redox dependent.
We further demonstrated that induction of both PAD2 and Cit-Vim by Cd/CB was blocked by both calcium inhibitor, cyclosporine A (CsA), and Akt inhibitor, LY (Fig. 3, A and B), indicating that both Akt1 and calcium signaling pathways are important for the induction of PAD2 and Cit-Vim, and Akt1 activation is also essential for the activation of PAD2. Vim can be secreted in response to Cd stimulation (7). Here, by using a similar strategy, we found that Cd and CB substantially induced the secretion of Cit-Vim compared to Cd or CB alone (Fig. 3, C and D). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a secreted protein (37), was used as a loading control. Secretion of Cit-Vim occurred earlier than that for Vim (Fig. 3, E and F). Moreover, pretreating with either a PAD2 (AFM-30a) or Akt (LY) inhibitor could effectively reverse Cd/CB-induced Cit-Vim secretion (Fig. 3, G and H). Dose-response viability analysis indicated that the dose we used (CdCl2, 1.8 μg/ml plus CB, 10 μg/ml) did not affect viability in human macrophages (fig. S4, A to C), and the combination of Cd/CB did not induce apoptosis (fig. S4D), suggesting that Vim/Cit-Vim is secreted from non-apoptotic lung macrophages.
Fig. 3. The secretion of Cit-Vim is dependent on the activation of Akt1 and PAD2, and citrullinated arginine (R) is identified by IP-MS.
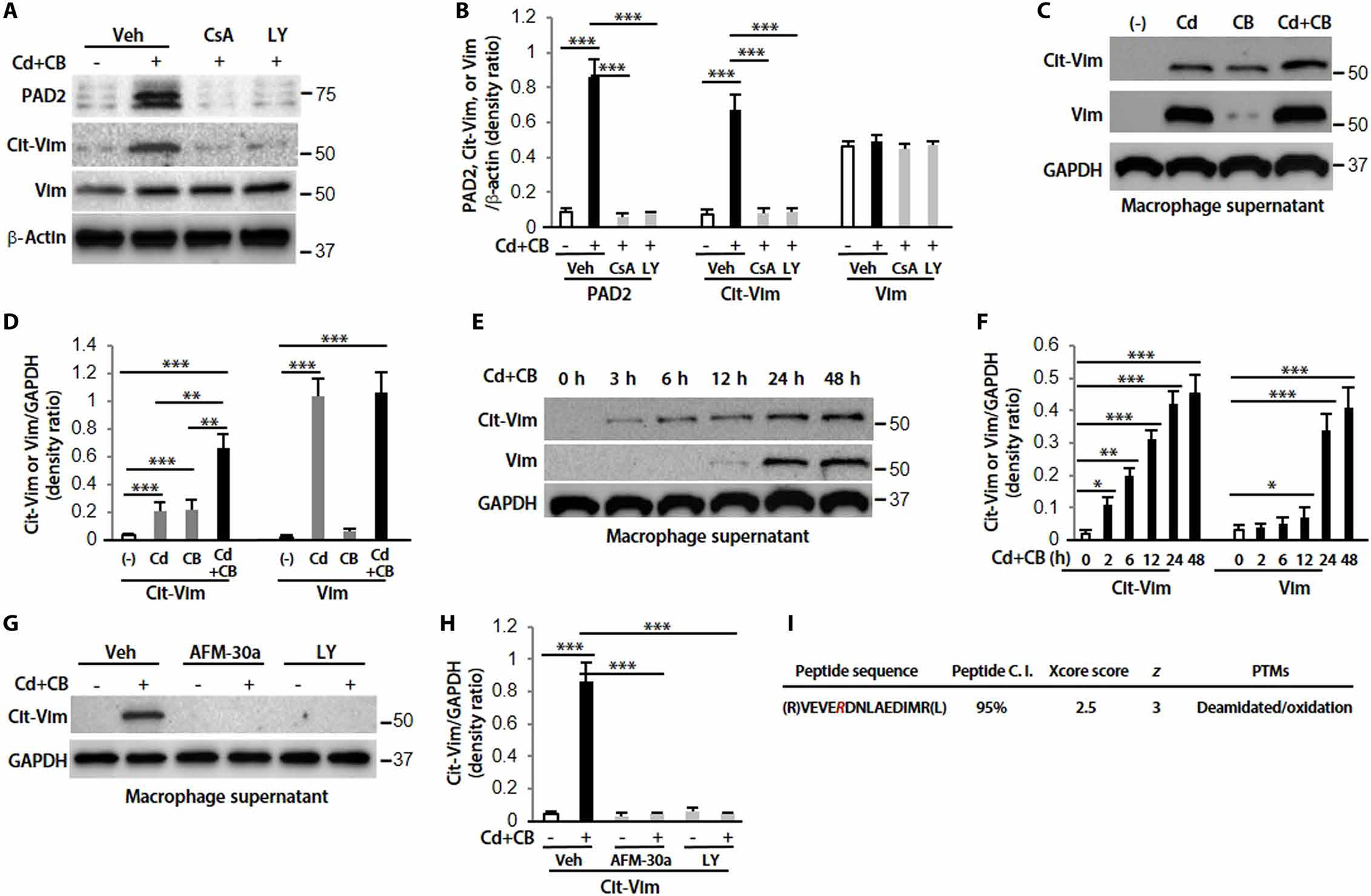
(A) Immunoblot analysis in lung macrophages treated with Sal or CdCl2 (1.8 μg/ml) plus CB (10 μg/ml) for 2 hours. Cells were pretreated with vehicle (Veh), CsA (20 μM), or LY (20 μM) for 2 hours. (B) Quantification of PAD2, Cit-Vim, and Vim expression from (A) (n = 3). (C) Supernatants were concentrated and analyzed by immunoblot analysis at 48 hours. (D) Quantification of Cit-Vim and Vim expression from (C) (n = 3). (E) Different time points by CdCl2 plus CB. (F) Quantification of Cit-Vim and Vim expression from (E) (n = 3). (G) Pretreatment with AFM-30a (5 μM) overnight or LY for 2 hours, followed by CdCl2 plus CB for 48 hours. (H) Quantification of Cit-Vim expression from (G) (n = 3). (I) Immunoprecipitated samples were performed with MS analysis for the citrullinated cites. The deamination (also termed citrullination) site was indicated by R (as highlighted in magenta; the residue number is AA175 based on the UniProtKB sequence). *P < 0.05, **P < 0.01, and ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
Immunoprecipitation coupled with mass spectrometry (IP-MS) was also conducted on these samples. Vim was the most abundant protein isolated with very few potentially nonspecific proteins observed (fig. S5A). The Vim peptide with the citrullination (R) was identified (Fig. 3I) and characterized by tandem MS2 (fig. S5B). The accompanying fragmentation was also shown in table S3. These data suggest that Cd/CB-induced activation of Akt1 and PAD2 in lung macrophages is required for Vim citrullination and Cit-Vim secretion (fig. S6).
Cit-Vim induces fibroblast invasion in lung pulmospheres and enhances the expression of collagen and α-SMA
The presence of Cit-Vim in lungs from subjects with IPF prompted us to investigate the pathologic and immunogenic effects of these soluble proteins. Vim and Cit-Vim were purified from cell culture supernatants of human lung macrophages treated with Cd/CB that was free of endotoxin contamination (fig. S7).
Three-dimensional (3D) lung pulmospheres can determine the invasiveness of fibroblasts (38). We questioned the effect of Cit-Vim and found a noticeable increase in the invasive phenotype of lung pulmospheres compared to treatment with the noncitrullinated protein, Vim (Fig. 4, A and B). CD26 (+) fibroblasts are highly proliferative and exhibit more invasive potential compared to CD26 (−) fibroblasts (39). We found that CD26 was markedly up-regulated in response to Cit-Vim in lung fibroblasts (Fig. 4, C and D). The ability of Cit-Vim to initiate invasion was also confirmed by using transwell assay (fig. S8). Furthermore, Cit-Vim, but not Vim, induced a significant increase (P < 0.001) in col1a1 and Col3a1 mRNA expression (Fig. 4, E and F), and Cit-Vim treatment mediated differentiation to myofibroblasts as indicated by the increased expression of α-SMA (Fig. 4, G and H). These data suggest that Cit-Vim regulates the phenotype of fibroblasts and enhances the expression of collagen. We further demonstrated that the effects of Cit-Vim on the expression of collagen are more evident when fibroblasts isolated from subjects with IPF were used (fig. S9), suggesting the possible pathogenic role of Cit-Vim in IPF fibroblasts.
Fig. 4. Cit-Vim exposure induces a more invasive subtype of fibroblast and enhances the expression of collagen and α-SMA.
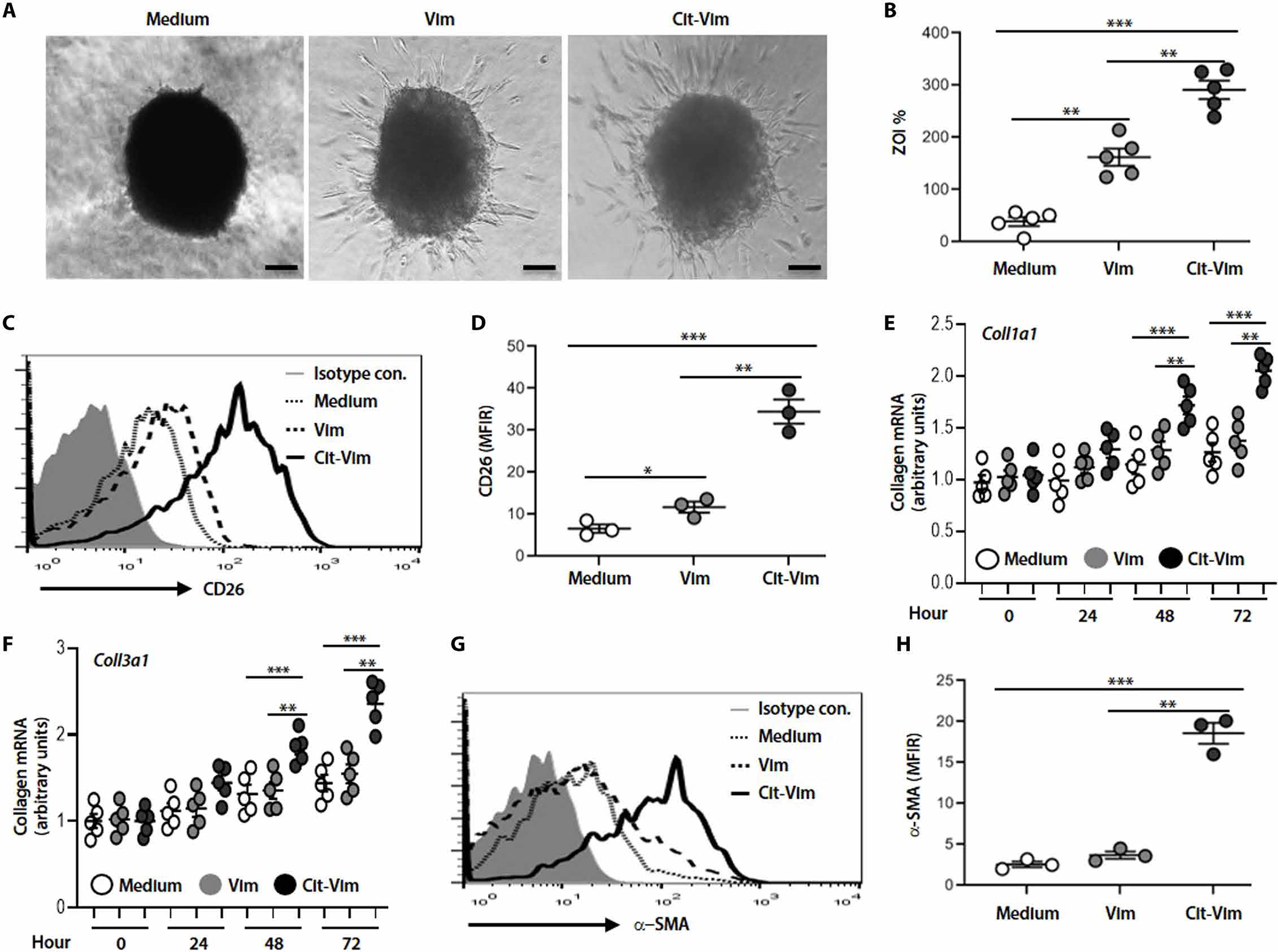
Primary human lung fibroblasts (5 × 105/ml) isolated from normal subjects were incubated with Vim or Cit-Vim (2 μg/ml). (A) Representative phase-contrast images of pulmospheres from normal human lung (n = 5) with Vim or Cit-Vim for 12 hours. Scale bars, 250 μm. (B) Invasiveness of pulmospheres evaluated using the percentage of ZOI. Each dot represents each subject indicated by the mean value of ZOI from six lung pulmospheres. (C) Representative histograms on flow cytometry showing expression of CD26. (D) Quantification of CD26 expression in (C) using the mean fluorescence intensity ratio (MFIR) calculated by dividing the MFI of medium (dot line), Vim (dashed line), or Cit-Vim (solid line) by the MFI of isotype control (gray-filled histogram). (E) Col1α1 and (F) Col3α1 mRNA expression in fibroblasts stimulated with Vim or Cit-Vim. Data are representative from three individual subjects. (G) Representative histogram on flow cytometry showing expression of α-SMA. (H) Quantification of α-SMA expression in (G) using MFIR as shown in (D). *P < 0.05, **P < 0.01, and ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
Cit-Vim activates TLR4/NF-κB signaling pathway and induces profibrotic cytokine production
Next, we explored the potential of Cit-Vim to function as a DAMP. We initially found that exposure of human lung fibroblasts to Cit-Vim increased the surface expression of TLR4 but not TLR1, TLR2, or intracellular TLR9 (Fig. 5, A and B). We further assessed the critical role of TLR4 in Cit-Vim–induced fibroblast activation. Cit-Vim treatment promoted the phosphorylation of inhibitor of nuclear factor κB (IκB) and nuclear localization of p65 in fibroblasts, and incubation together with an anti-TLR4 antibody or nuclear factor κB (NF-κB) inhibitor remarkably blocked Cit-Vim–induced NF-κB activation (Fig. 5, C to F).
Fig. 5. Cit-Vim exposure induces TLR4/NF-κB-mediated cytokine/chemokine production.
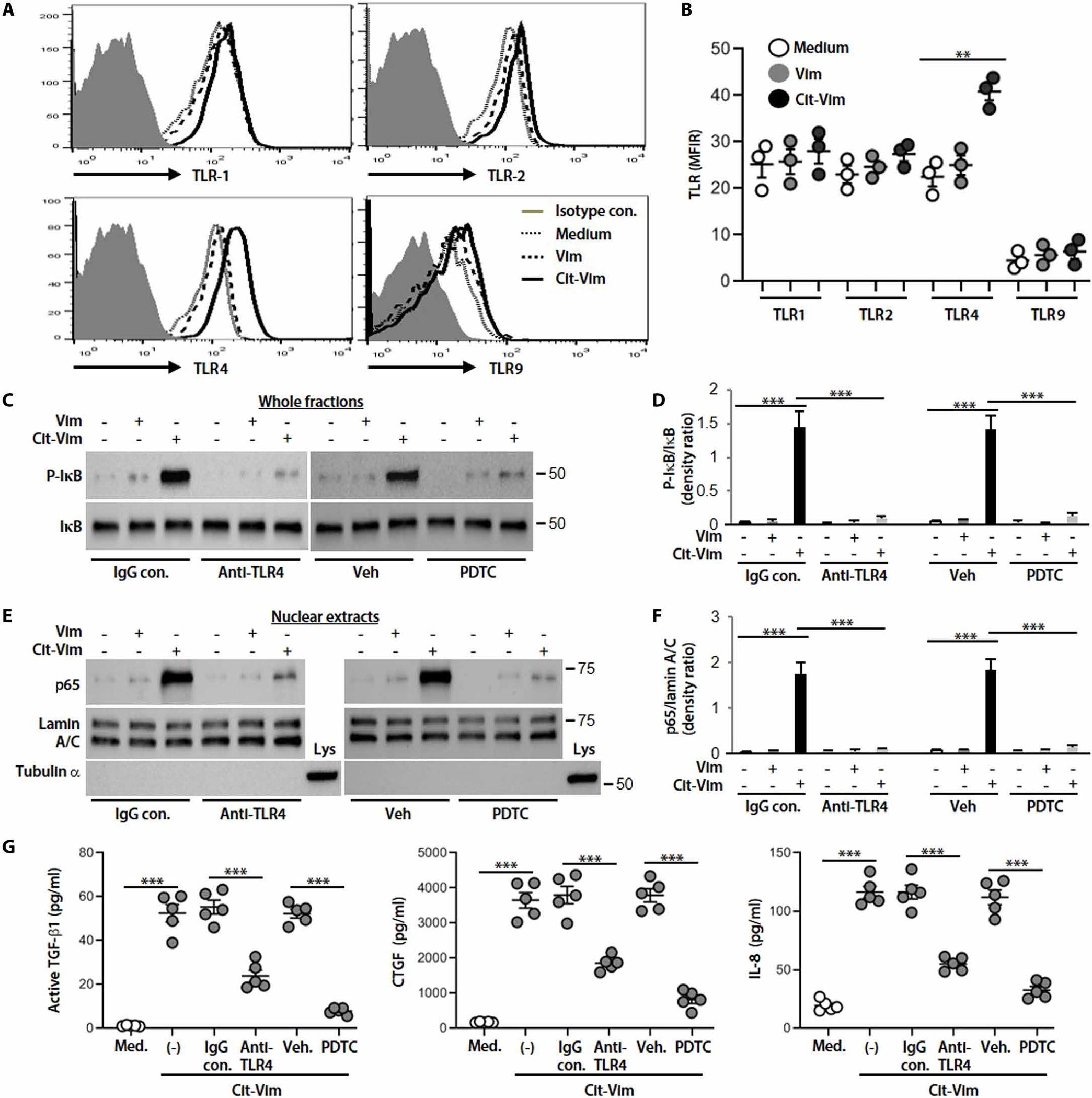
(A) Representative histograms on flow cytometry showing surface expression of TLR1, TLR2, TLR4, and intracellular TLR9. (B) Quantification TLR expression using the MFIR as shown in Fig. 4D. Lung fibroblasts isolated from normal subjects were incubated for 48 hours, and each dot represents each subject. (C) Immunoblot analyses of p-IκB and IκB expression in whole fractions. (D) Quantification of p-IκB expression from (C) (n = 3). (E) p65 and lamin A/C expression in nuclear extracts. (F) Quantification of p65 expression from (E) (n = 3). Lung fibroblasts were preincubated with IgG control, anti-TLR4 antibody (10 μg/ml), vehicle, or NF-κB inhibitor (pyrrolidine dithiocarbamate, PDTC, 100 μM) for 1 hour and then stimulated with Vim or Cit-Vim (2 μg/ml) for 2 hours. Lamin A/C was used as a nuclear protein loading control, and α-tubulin was used as a cytoplasm protein control. (G) Cytokine/chemokine concentrations by ELISA in fibroblast supernatants stimulated with Cit-Vim for 24 hours in the presence of IgG control, anti-TLR4 antibody, vehicle, or PDTC. **P < 0.01 and ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
Cytokines/chemokines, such as TGF-β1 (40), CTGF (41), IL-8 (42), and monocyte chemoattractant protein 1 (MCP-1) (43), produced by fibroblasts (for active TGF-β1, CTGF, and IL-8) and monocytes/macrophages (active TGF-β1, IL-8, and MCP-1) have previously been shown to be increased in lung tissues, serum, and/or BALF of subjects with IPF and to correlate with signs of lung fibrosis, indicating an important role of these cytokines/chemokines in the pathogenesis of IPF. Therefore, we evaluated whether the ability of Cit-Vim to induce cytokine/chemokine production in primary lung fibroblasts was dependent on the presence of TLR4. We observed that active TGF-β1, CTGF, and IL-8 secretion increased substantially in response to Cit-Vim stimulation but not Vim, and noticeable production occurred in 12 hours and peaked at 24 hours after stimulation (fig. S10). These events occur earlier relative to the production of collagen (48 hours; Fig. 4, E and F). As expected, human fibroblast cytokine/chemokine production in Cit-Vim–treated cultures was diminished remarkably by pretreatment with the anti-TLR4 antibody or NF-κB inhibitor (Fig. 5G). These data confirmed that TLR4 likely serves as a functional receptor linking fibroblast activation with exposure to Cit-Vim.
Cit-Vim independently induces lung fibrosis
To evaluate whether Cit-Vim could independently induce lung fibrosis in vivo, TLR4 WT versus MUT mice were administered intratracheal Cit-Vim purified from the Cd/CB-treated lung macrophages. We demonstrate that administration of Cit-Vim induced greater architectural destruction and increased collagen deposition in TLR WT mice, whereas the lungs from TLR4 MUT mice had normal architecture and markedly less collagen (Fig. 6, A and B). Cit-Vim, but not Vim, challenge significantly increased (P < 0.001) hydroxyproline content, validating biochemically the histological assessment (Fig. 6C). Cit-Vim also substantially increased active TGF-β1, CTGF, and KC (keratinocyte-derived chemokine) (Fig. 6, D to F) in TLR WT mice only, and the concentrations peaked at day 14 (fig. S11). These data suggest that Cit-Vim induces lung fibrosis in mice in a TLR4-dependent fashion (fig. S12).
Fig. 6. Cit-Vim, but not Vim, can effectively induce cytokine/chemokine production and lung fibrosis.
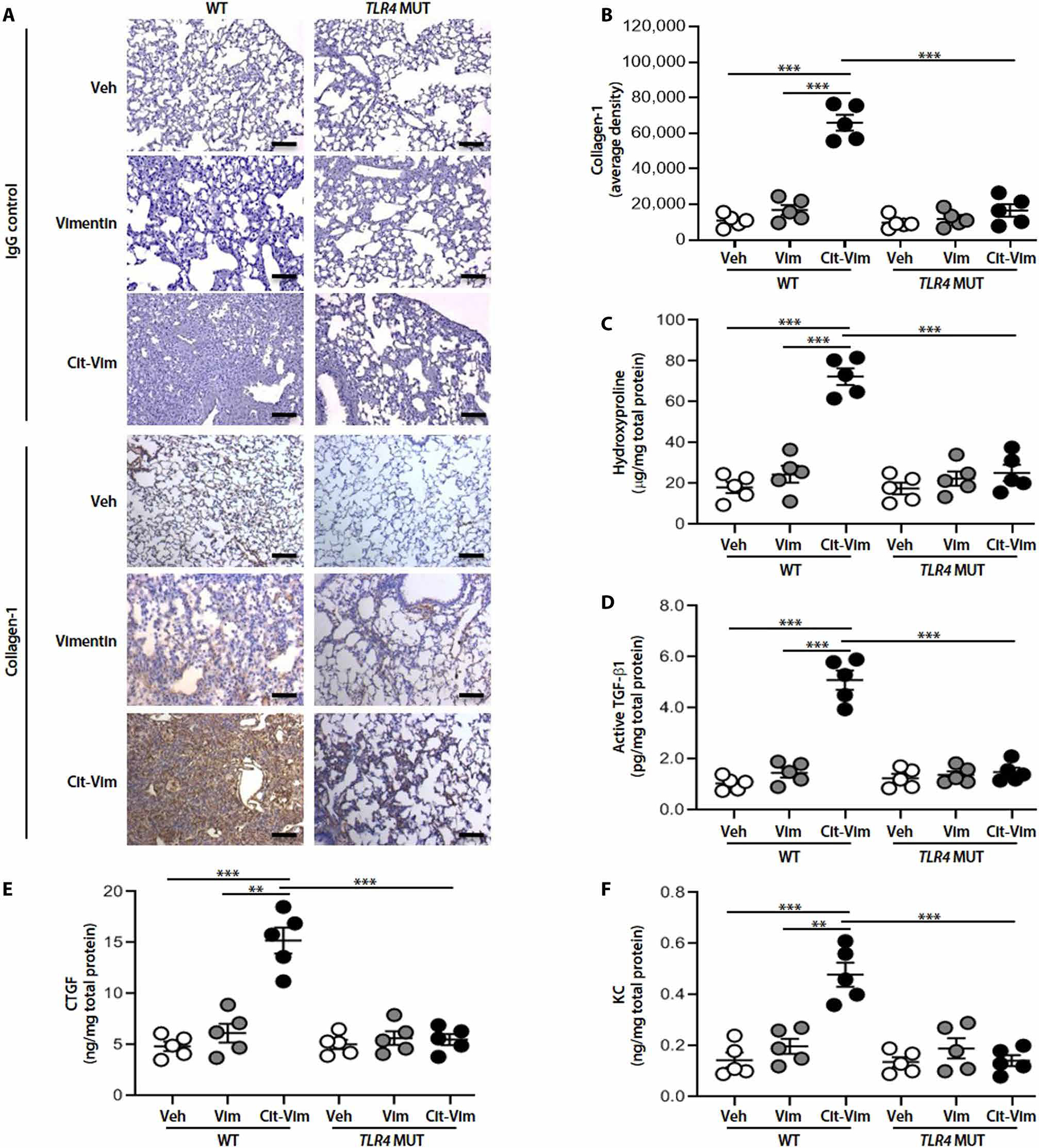
TLR4 WT or TLR4 MUT mice (n = 5 per group) were treated intratracheally with Vim or Cit-Vim (2 mg/kg). (A) Representative lung histology with collagen-1 staining at day 14. (B) DAB staining density from (A) was quantified. Scale bars, 100 μm. (C) Hydroxyproline content at day 14. Cytokine/chemokine concentrations by ELISA at day 14 in lung tissue suspensions for (D) active TGF-β1, (E) CTGF, and (F) KC. Each dot represents individual mouse. **P < 0.01 and ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
Cd and CB induce pulmonary fibrosis in mice
Next, we examined whether airway delivery of Cd/CB was able to induce fibrogenesis in mice lungs. To achieve Cd concentrations in mice similar to those in subjects with IPF, the concentrations of Cd in mice lung tissues after exposure to different doses of CdCl2 were measured. After 2 weeks of exposure, Cd concentration in CdCl2-treated mice (0.16 mg/kg) (fig. S13A) was similar as human tissue from subjects with IPF (Fig. 1A). The data indicate that we were able to achieve a similar concentration of Cd in our mouse model as those found in the lung parenchyma of subjects with IPF.
We found that a single dose of Cd could induce signs of interstitial lung fibrosis in mice. Cd increased collagen deposition in the lungs as evaluated by immunohistochemistry (IHC) staining (fig. S13, B and C). The pulmonary morphological fibrotic changes were assessed by the semiquantitative morphological index (SMI) (fig. S13D). With exposure to Cd, hydroxyproline concentrations were increased in a concentration-dependent manner and peaked at day 14 (fig. S13, E and F). Whether CB alone can induce fibrosis is debated (17, 44, 45). Our data showed that CB alone caused minimal hydroxyproline expression, but the combination of Cd/CB increased hydroxyproline compared to Cd or CB alone (fig. S13, G and H). Cd and CB exposure also induced inflammatory cell infiltration indicated by an increase in the total cell count (fig. S13I), with the dominant cell type being macrophages (fig. S13J). TEM images also demonstrated the presence of CB within the cytoplasm of lung macrophages (fig. S13, K to M). BALF analysis of Cd/CB-treated mice was characterized by higher active TGF-β1, CTGF, KC (the murine homolog of human IL-8), and MCP-1 concentrations compared with those of saline (Sal)–treated mice (fig. S14).
Cd/CB-induced pulmonary fibrosis is PAD2 dependent
To explore the role of PAD2 in Cd/CB-induced Cit-Vim secretion and lung fibrosis, we used PAD2−/− mice. PAD2 expression in lung macrophages was absent in PAD2−/− mice, and PAD2 expression was increased after exposure to Cd plus CB only in WT mice (Fig. 7, A and B). The production of Cit-Vim in plasma and BALF was markedly inhibited in PAD2−/− mice compared to WT mice (Fig. 7, C and D). Furthermore, WT mice exposed to Cd/CB had lung architectural destruction and collagen deposition, whereas the lung from PAD2−/− mice showed less collagen (Fig. 7, E to G). To investigate the role of macrophage Akt1 in the production of Cit-Vim, we generated mice harboring a conditional deletion of Akt1 (Akt1−/−Lyz2-cre) in macrophages (46). The expression of p-Akt1 and Akt1 in type II lung epithelial cells was found in both strains of mice (46). Similar results were also obtained by using Akt1−/−Lyz2-cre mice (fig. S15). As expected, decreases in lung compliance (Fig. 7, H and I) and inspiratory capacity (Fig. 7J) and increases in whole lung airway resistance (Fig. 7K), central airway resistance (Fig. 7L), alveolar tissue resistance (Fig. 7M), and tissue stiffness (Fig. 7N) were observed in CdCl2/CB-exposed mice using respiratory system mechanics after increasing methacholine challenge. However, the decreased compliances or increased resistance was markedly recovered in PAD2−/− mice compared to WT mice after CdCl2/CB administration. These data suggest that PAD2 likely plays a critical role in Cd/CB-induced Vim citrullination and might be linked to the pathogenesis of lung fibrosis.
Fig. 7. Cd/CB-induced Vim citrullination and lung fibrosis are dependent on PAD2 expression, and respiratory mechanics analysis demonstrates decreased disease susceptibility in PAD2−/− mice.
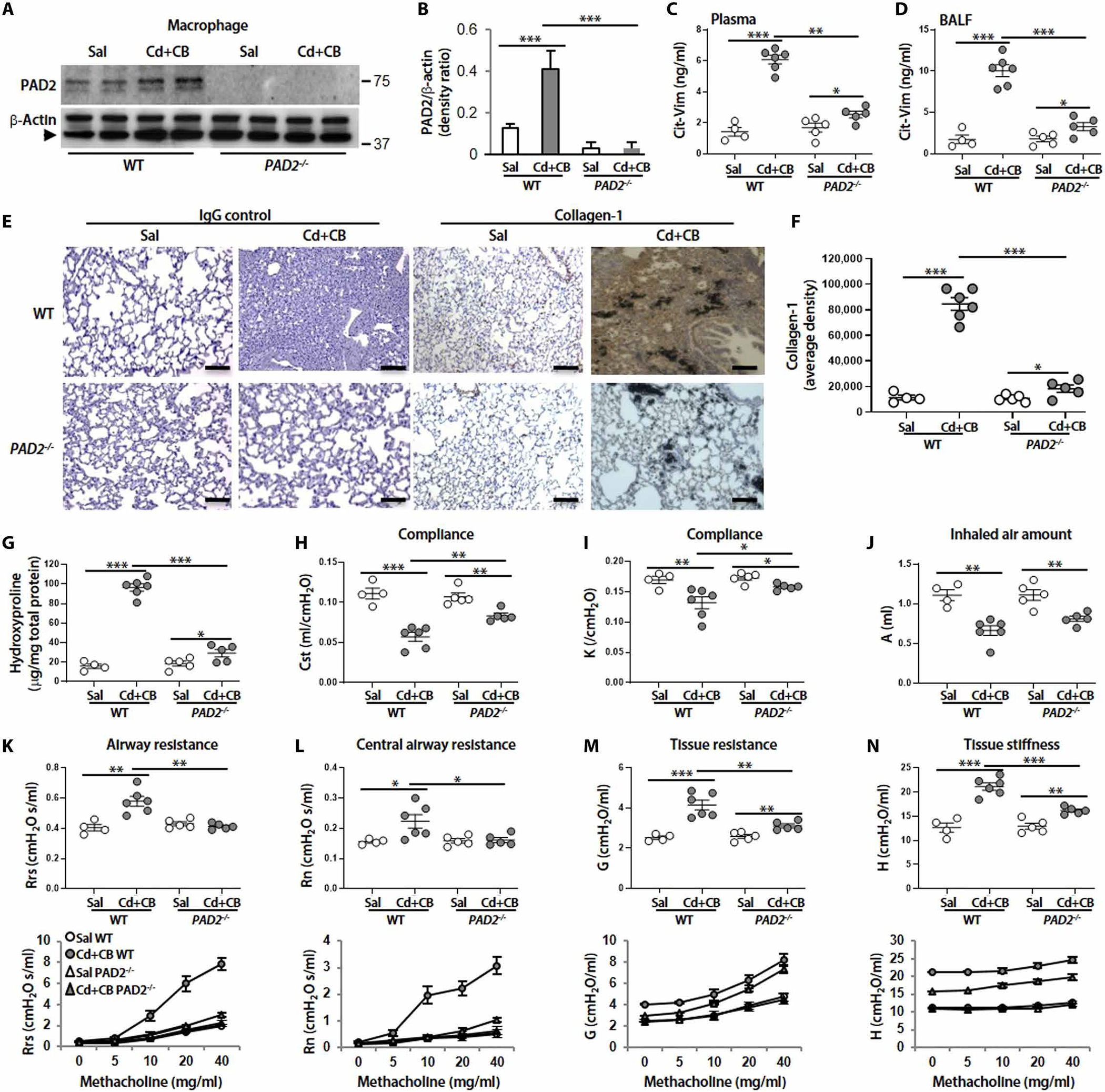
(A) Immunoblot analysis in mice lung macrophages from WT (Sal, n = 4; Cd + CB, n = 6) and PAD2−/− mice (Sal, n = 5; Cd + CB, n = 5) treated with Sal or CdCl2 (0.16 mg/kg) plus CB (5.0 mg/kg). Arrow represents specific band. (B) Quantification of PAD2 expression from (A) (n = 3). Cit-Vim amounts by ELISA in (C) plasma and (D) BALF at day 14. (E) Representative lung histology with collagen-1 staining at day 14. (F) DAB staining density from (E) was quantified. Scale bars, 100 μm. (G) Hydroxyproline content at day 14. Respiratory mechanics analysis was performed in paralyzed and mechanically ventilated mice 14 days after Sal or CdCl2 plus CB administration. The parameters (H) Cst, (I) K, and (J) A were assessed using the baseline values. (K) Rrs, (L) Rn, (M) G, and (N) H were evaluated using both baseline values (top) and average values from 12 measurements at different concentrations of methacholine (bottom). Each dot represents individual mouse. *P < 0.05, **P < 0.01, and ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
Cd/CB-induced pulmonary fibrosis is TLR4 dependent
To link Cd/CB-mediated Cit-Vim production to the pathogenesis of fibrosis, we examined whether Cd/CB-induced fibrogenesis is dependent on TLR4. Our data showed that the combination of Cd/CB induced extensive fibrosis in TLR4 WT mice, but not in TLR4 MUT mice, as assessed by the histological observations (Fig. 8, A and B) and biochemical confirmation (Fig. 8D). Citrullinated proteins were decreased in TLR4 MUT mice, and there was a greater increase in WT mice after Cd/CB exposure (Fig. 8, A and C). Furthermore, Cd/CB significantly increased (P < 0.001) Cit-Vim in plasma and BALF collected from WT mice, but not from TLR4 MUT mice (Fig. 8, E and F). TLR4 expression in lung fibroblasts was clearly expressed in WT mice but absent in TLR4 MUT mice (Fig. 8, G and H). Fibroblasts isolated from TLR4 WT mice showed an increase in both collagen-1 and α-SMA expression when they were incubated with BALF from Cd/CB-treated mice (Fig. 8, I to L). However, the collagen-1 and α-SMA expression was essentially less in fibroblasts isolated from TLR4 MUT mice, suggesting that TLR4 is essential for Cd/CB-induced collagen synthesis and fibroblast differentiation. These observations indicate that the environmental toxins, Cd and CB, might mediate lung fibrosis through a distinct macrophage-fibroblast interaction via the production of Cit-Vim.
Fig. 8. Cd/CB-induced interstitial fibrosis in mice is TLR4 dependent.
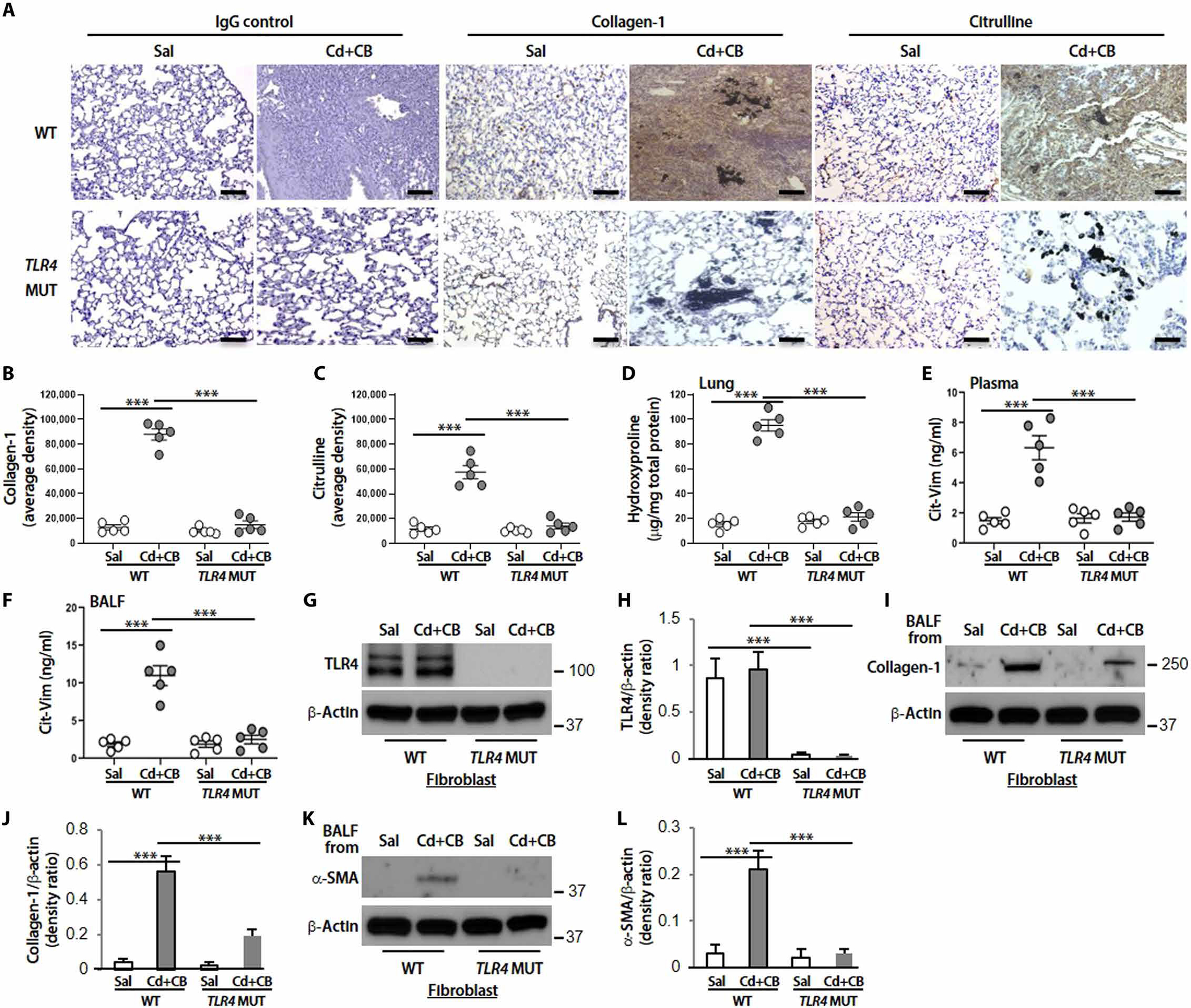
TLR4 WT or TLR4 MUT mice (n = 5 per group) were treated intratracheally with Sal or CdCl2 + CB (CdCl2, 0.16 mg/kg; CB, 5.0 mg/kg). (A) Representative lung histology with collagen-1 and citrulline staining at day 14. DAB staining densities from (A) for (B) collagen-1 and (C) citrulline were quantified. Scale bars, 100 μm. (D) Hydroxyproline content at day 14. Cit-Vim amounts by ELISA in (E) plasma and (F) BALF at day 14 after mice exposure. (G) Immunoblot analysis of TLR4. (H) Quantification of TLR4 expression from (G) (n = 3). (I) Collagen-1. (J) Quantification of collagen-1 expression from (I) (n = 3). (K) α-SMA. (L) Quantification of α-SMA expression from (K) (n = 3) in lung fibroblasts isolated from TLR4 WT or TLR4 MUT mice. For checking collagen-1 and α-SMA expression, cells were cultured with BALF collected from Sal- or CdCl2 + CB–treated mice. Each dot represents individual mouse. ***P < 0.001 using one-way ANOVA followed by Tukey’s post hoc analysis.
DISCUSSION
Despite the identification of genetic variants that predispose individuals to the development of lung fibrosis, environmental influences on the overall risk of this disease cannot be overstated. Several reports have demonstrated that Cd/CB deposit within the lung macrophages and induce an increase in the percentage of lung macrophages (>90%) in mice BALF (19, 47). Here, we identify Cd and CB, known constituents of CS and environmental PM, as inducers of Cit-Vim production by lung macrophages, sentinel host defense cells of the mammalian lung. Our data demonstrate that Cit-Vim is present in plasma and lung tissue of subjects with IPF, especially smokers. Cit-Vim in lungs from subjects with IPF correlated positively with Cd in lungs. The presence of Cit-Vim in the plasma may be a quantitative marker of Cd exposure and directly correlates with lung function and transplant-free survival. We further show that Cit-Vim functions as a DAMP to activate TLR4-dependent fibrogenic pathways in lung fibroblasts. Our results indicate that CB/Cd mediate fibrotic effects in the lung. These effects are triggered by macrophage-derived Cit-Vim that activates TLR4-dependent NF-κB signaling in lung fibroblasts. We therefore speculate that Vim citrullination is a specific process that activates macrophages and has pathogenic effects in fibroblasts by binding TLR4, although other citrullinated proteins such as Cit-fibrinogen may also activate this pathway in RA (48). The relevance of this pathway to human disease is supported by the findings of higher amounts of Cit-Vim in association with Cd and CB in IPF lungs.
DAMPs trigger TLR signaling indicated by the activation of NF-κB and the secretion of profibrotic cytokines (23, 24). Our data indicate that Cit-Vim, but not Vim, functions as a DAMP to promote TLR4-dependent NF-κB activation. This finding is important because Cit-Vim is sufficient to provoke fibroblast activation in vitro and elicit profibrotic cytokine/chemokine production and TLR4-dependent lung fibrosis in vivo. TGF-β1 (40), CTGF (41), and IL-8 (42) are essential profibrotic cytokines/chemokines that play crucial roles in macrophage/fibroblast activation. Binding of TGF-β1 with both the TGF-β receptor type 1 (TβR-I) and TβR-II is required for the activation of TGF-β1 and their effects, such as synthesis of connective tissue proteins (49). Both receptors are present on all cells of normal lung. In IPF, however, only interstitial fibroblasts express both TβR-I and TβR-II, whereas TβR-I is markedly reduced in most cells of IPF (40), suggesting that active TGF-β1 is predominantly from interstitial fibroblasts. In the current study, exposure of fibroblasts to Cit-Vim induced the secretion of active TGF-β1, CTGF, and IL-8. Pathological lung fibrosis in IPF might be similarly elucidated through TLR4-dependent fibrosis driven by endogenous DAMPs induced by Cd/CB within the injured microenvironment. Endogenous production of Cit-Vim functions as a DAMP and activates an innate immune response in lung fibroblasts.
CS has been reported to be a risk factor in the pathogenesis of IPF (2, 13–15), yet little is known about the mechanisms underlying the fibrogenic actions of CS. Cd can be adsorbed spontaneously onto the surface of carbon particles in environmental PM, and this phenomenon may enhance the abundant accumulation of metals in human lungs (8). Our data demonstrate that Cd/CB is a risk factor not only in subjects with IPF that smoked but also in nonsmokers. Higher concentrations of Cd in patients that do not smoke may be caused by exposure to Cd through food and/or occupation/environment (50). So far, the precise mechanisms underlying the effects of both CB and Cd remain incompletely understood. The inhibition of Cit-Vim expression and secretion with specific PAD2 and Akt inhibitors as well as the use of PAD2−/− and Akt1−/−Lyz2-cre mice demonstrate that the production of Cit-Vim by macrophages requires Akt1 and PAD2 activation and emphasize the critical role of lung macrophages in the development of lung fibrosis.
In our studies, we provide evidence that Cit-Vim increased invasion by fibroblasts compared with native Vim in 3D lung pulmospheres, and confirmed by transwell analysis and increased CD26 expression, suggesting that, compared to native Vim, Cit-Vim might cause fibroblasts to invade; the binding sites and/or binding receptors of Cit-Vim on the fibroblast surface may be specific. Invasive fibroblasts in keloid skin lesions are characterized by accelerated proliferation and aberrant ECM deposition (39, 51). The fact that only Cit-Vim induces an increase in CD26 surface expression and the fact that CD26+ keloid fibroblasts have excess collagen and ECM deposition compared to CD26− fibroblasts (39) suggest that CD26 may be a surface marker to distinguish invasive activated subtypes of fibroblasts from the noninvasive subtype. In response to Cit-Vim stimulation, increased expression of a more differentiated type of myofibroblast is also recognized using α-SMA staining on flow cytometry analysis and induction of collagen expression by different fibroblasts derived from control or subjects with IPF strengthens the possible pathogenic role of Cit-Vim.
Protein citrullination is increasingly recognized as a critical pathway in mediating profibrotic effects in various clinical syndromes, in particular, connective tissue–associated RA-ILD (27). Cit-Vim peptides have been identified in the lung, plasma, and synovial tissue from subjects with RA (26). The presence of these peptides in plasma before the joint involvement suggests that initiation of the disease-specific immune response might occur at extra-articular sites, such as the lung, rather than at articular sites. Citrullinated proteins have been found highly expressed in lung sample from patients with chronic obstructive pulmonary disease (COPD) (52) and IPF (53). We demonstrated here that Cit-Vim was up-regulated in both lung and plasma from subjects with IPF in a large cohort, suggesting that Cit-Vim might play a role in lung fibrosis and can then be found in the circulation. The development of RA and the interaction of CS with the human leukocyte antigen (HLA)–DR shared epitope genes trigger immune response to citrulline-modified protein (54), suggesting the important role of gene-environment interaction in the etiology of RA. Recently, several cytokeratins have also been recognized as major citrullination targets for Cd exposure in lung epithelial cells (55). Together, these data suggest that Cd exposure may be a common etiological factor for RA-ILD, COPD, and IPF. A proposed working hypothesis for IPF is that exposure to Cd/CB through CS and/or other environmental PM may trigger mechanisms that induce Vim citrullination by activating both Akt1 and PAD2 in lung macrophages before the development of lung fibrosis. Our study also suggests that CdCl2-induced interstitial fibrosis in mice is a potential disease model for lung fibrosis and IPF. Compared to bleomycin-induced fibrosis (56, 57), CdCl2-induced interstitial fibrosis occurs early, lasts longer, and cannot be reversed even after 28 days of exposure (fig. S13, D and F) and therefore is more similar to IPF. This finding may be attributed to the long half-life of Cd.
PAD2 and PAD4 are the primary PADs expressed in macrophages (33). Here, we demonstrate that PAD2, but not PAD4, was induced by Cd/CB stimulation in macrophages isolated from subjects with IPF. The protective role of PAD inhibition has been observed in several diseases. One study reported that a pan-PAD inhibitor reduced disease severity in a collagen-induced arthritis model (58). PAD2 enzyme inhibition was also found to impair NLRP3 inflammasome assembly and IL-1β release in macrophages primed with lipopolysaccharide (LPS) and NLRP3 inflammasome activator (33). In vivo, PAD2 modulated autoimmunity and T cell function in TLR7-dependent lupus model indicated by lower amounts of autoantibodies and down-regulation of T helper 1 (TH1) and TH17 responses in PAD2−/− mice compared to WT mice (32). Our finding that PAD2−/− mice displayed protection from lung fibrosis with preservation of lung architecture, lower collagen deposition, and absence of the decreased lung compliance and other abnormal lung physiological parameters raises the possibility that PAD2 inhibition could function as a promising strategy for treating Cd/CB-induced lung fibrosis.
A two-strike model can be postulated for the pathogenesis of pulmonary fibrosis, where genetically susceptible individuals who are exposed to environmental Cd/CB through the induction of Cit-Vim subsequently develop lung fibrosis. Our results suggest a link between environmental Cd and Cit-Vim–induced lung fibrosis.
One of the limitations of this study is that although mice were treated with Cd to achieve similar concentrations in lung tissue to those in subjects with IPF, only a single dose of Cd cannot completely recapitulate the pathogenic course of IPF, a chronic, progressive, and irreversible lung disease. Another limitation of our study is the relatively small size of cohort 1 (lung tissue), especially when smoking status of the subjects was analyzed. Therefore, a larger cohort is necessary to evaluate the importance of smoking as a risk factor for IPF. Although Cit-Vim can be accurately measured in plasma and lung tissues, it may also be present in other body fluids, such as BALF and exhaled breath condensate. Given the existence of many toxins in CS and activation of multiple signaling pathways, it still remains difficult to determine quantitively the degree to which Cd/CB contribute to the pathogenesis of IPF.
MATERIALS AND METHODS
Study design
This observational study was designed to investigate the role of Cit-Vim and the pathogenesis of Cd/CB-induced Cit-Vim in lung fibrosis. To evaluate Cd and Cit-Vim accumulation in tissue, lung biopsies from 25 subjects with IPF (8 never-smokers and 17 smokers) and 14 control (8 never-smokers and 6 smokers) were processed in parallel for Cd using inductively coupled plasma mass spectrometry (ICP-MS) (7) and, for Cit-Vim, using IHC, immunoblot, and sandwich enzyme-linked immunosorbent assay (ELISA). IPF lung specimens were obtained from diseased parts removed from therapeutic transplantation or by video-assisted thoracic surgery (VATS). Control lung specimens were obtained from resections or lobectomy during surgery, or the uninvolved lobes of control subjects during VATS who did not have any lung parenchymal abnormality. Random selection of 10 BALF from subjects with IPF and 5 BALF from control subjects was obtained for evaluating the existence of CB using Hema 3 (Thermo Fisher Scientific) and TEM. Plasma contents of Cit-Vim were determined in 84 subjects with IPF (19 never-smokers and 65 smokers) and 57 healthy control (35 never-smokers and 22 smokers). The diagnosis was based on standard criteria (1), and samples were recruited from the University of Alabama at Birmingham (UAB) ILD Clinic after approval by the institutional review board. FVC and DLCO were performed for pulmonary function. Primary lung macrophages isolated from subjects with IPF were purified for evaluating the secretional mechanisms of Cit-Vim, and primary lung fibroblasts isolated from normal control or subjects with IPF were used to assess the roles of Cit-Vim ex vivo or in vitro, as described previously (7, 46). In vitro experiments were performed in triplicate unless specified. Three mice models, including TLR4 MUT, PAD2−/−, and Akt1−/−Lyz2-cre mice, were used to evaluate the role of TLR4, PAD2, and Akt1 in Cit-Vim– or Cd/CB-induced lung fibrosis, respectively. Fibrosis was evaluated by IHC and hydroxyproline assay. Cit-Vim amounts and Cd/CB-induced cytokine/chemokine production were evaluated by ELISA. Downstream protein expression was assessed by immunoblot analysis. Animal experiments were approved by the Institutional Committee of Animal Care at UAB. Mice were randomly assigned to experimental groups. Treatment groups were blinded, and no outliers were excluded from the datasets. An equal number of female and male mice (6 to 8 weeks old) were selected in this study. Mice were evaluated at the endpoints of days 3, 7, 14, 21, and 28.
Mice models of lung fibrosis
To evaluate the fibrogenetic role of Cit-Vim, C3H/HeOuJ TLR4 WT and C3H/HeJ TLR4 MUT (a spontaneous mutation in the TLR4 gene, Tlr4lps-d, The Jackson Laboratory) mice were treated by intratracheal instillation (IT) once to Sal, native Vim, or Cit-Vim (2 mg/kg). In other experiments, PAD2−/− mice (provided by S. A. Coonrod), Akt1−/−Lyz2-cre mice with a conditional deletion of Akt1 in macrophages (46), and TLR4 MUT and WT mice in parallel were given once with CdCl2 (0.16 mg/kg, Sigma-Aldrich) and/or CB (5.0 mg/kg, Sigma-Aldrich). For BALF, three instillations of 1 ml of phosphate-buffered saline (PBS) were performed for each mouse and the differential number of cells in BALF was counted according to standard hematological procedures. Lung tissues were dried and acid-hydrolyzed (6 N HCl at 95°C for 20 hours), and collagen deposition was assessed by checking hydroxyproline concentration as per the manufacturer’s protocols (Chondrex).
Citrulline and collagen-1 expression in lung biopsies
Immunostaining in formalin-fixed, paraffin-embedded lung sections (4 μm) was performed to assess expression of citrulline (1:400; MilliporeSigma) or collagen-1 (1:200; Rockland) in lung tissues using a diaminobenzidine (DAB) staining kit (Dako). Immunofluorescence was assessed and quantified using Bioquant Imaging Analysis software. The average positive area density in each subject was calculated from 10 random areas.
Evaluation of invasive capacity in ex vivo pulmospheres
3D pulmospheres from normal human lungs were prepared in Hema-coated (5 mg/ml in 95% ethanol) plates as described previously (38). To perform invasion assay, 96-well plates were coated with collagen–Dulbecco’s modified Eagle’s medium (DMEM) solution (3.98 mg/ml, pH 7 to 7.4) and single lung pulmospheres were seeded onto wells with 100 μl of the collagen-DMEM solution in the presence or absence of Vim or Cit-Vim (2 μg/ml) and cultured for 12 hours. Images were acquired and analyzed using an AxioCam camera and imaging software (Carl Zeiss). The invasiveness of pulmospheres was evaluated using the percentage of zone of invasion (ZOI) after the calculation was reported (38).
Measurements of cytokines/chemokines
ELISA was used to measure cytokine/chemokine concentrations in culture media, lung tissue suspensions, and BALF according to the manufacturer’s instructions. Mouse interferon-γ (IFN-γ), IL-6, IL-10, and MCP-1 were detected using the BD Cytometric Bead Array (BD Biosciences). ELISA kits for mouse IL-4, IL-13, KC, human IL-8, and CTGF were provided by R&D Systems, for active TGF-β1 by BioLegend, and for mouse CTGF by Lifespan Biosciences.
Flow cytometric analysis
For apoptosis analysis, lung macrophages were stained with annexin V-FITC (fluorescein isothiocyanate) and propidium iodide (PI) (Imgenex Corporation) together with Vim-PE (phycoerythrin) (BD). ROS production was evaluated by staining cells with 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA; Abcam) for 30 min and then cultured for additional 2 hours with CdCl2 and/or CB in the presence or absence of Mito-TEMPO (Sigma-Aldrich) before analysis by flow cytometry. Human fibroblasts were stained with CD26-PE (BioLegend), TLR1-FITC (Abcam), TLR2-FITC (Abcam), or TLR4-PE (Abcam) for measuring their surface expression in response to Vim or Cit-Vim (2 μg/ml) stimulation. Intracellular expression of α-SMA was detected by staining cells with α-SMA–APC (allophycocyanin) (R&D Systems), and TLR9 was characterized by staining unlabeled anti-TLR9 (Novus) followed by counterstaining with APC-conjugated anti-mouse immunoglobulin G (IgG). Data were acquired with LSR II (BD Biosciences) and analyzed using FlowJo software (version 10.6.1).
Statistical analysis
Two-tailed t test was performed for two group comparisons and one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for three or more group comparisons. Spearman rank correlations were used to analyze the relationships between variables. Product-limit estimation was used for survival analyses, and HR and 95% confidence intervals (CIs) were established using Cox proportional hazard regression. A P value of <0.05 was considered significant. Values are shown as means ± SEM unless specified.
Supplementary Material
Fig. S1. MT-1/2 mRNA and protein expression is higher in subjects with IPF, especially smokers.
Fig. S2. CB deposits are found in macrophages from subjects with IPF.
Fig. S3. Cd/CB-induced Akt1, PAD2 activation, and Vim citrullination are dependent on oxidative states.
Fig. S4. Cells that express Vim on their surface are not apoptotic.
Fig. S5. Proteins are identified and tandem MS2 spectrum is characterized by IP-MS.
Fig. S6. Schematic diagram of the Cit-Vim secretion pathway.
Fig. S7. Both Vim and Cit-Vim are purified and enriched from cell culture supernatants.
Fig. S8. Cit-Vim exposure induces more invasive subtypes of fibroblasts.
Fig. S9. Cit-Vim exposure enhances the expression of collagen in fibroblasts isolated from normal subjects and subjects with IPF.
Fig. S10. Cit-Vim, but not Vim, induces CTGF, IL-8, and active TGF-β1 production.
Fig. S11. Cit-Vim, but not Vim, can effectively induce cytokine/chemokine production only in TLR4 WT mice.
Fig. S12. Schematic diagram of the Cit-Vim–induced fibrosis pathway.
Fig. S13. Exposure of Cd/CB causes dose-dependent interstitial fibrosis in mice.
Fig. S14. Exposure of Cd/CB induces cytokine/chemokine secretion.
Fig. S15. Cd/CB-induced Vim citrullination and lung fibrosis are dependent on the activation of Akt1.
Table S1. Demographics and baseline characteristics of subjects with IPF.
Table S2. Smoking status analysis of subjects with IPF.
Table S3. Accompanying fragmentation for the same peptide identified by IP-MS.
Acknowledgments
Funding: This study was supported by NIEHS grant P42 ES027723 (V.B.A., A.B.C., and V.J.T.); NIH grants R01 ES029981 (V.B.A.), R01 ES015981–13 (A.B.C.), U54 ES030246 (M.A. and V.B.A.), P01 HL114470 (V.J.T. and V.B.A.), and R01 P30CA013148 (J.A.M.); NIGMS grant GM118112 (P.R.T.); and Department of Veteran Affairs grant 1 I01 CX001715–01 (A.B.C.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/585/eaba2927/DC1
Materials and Methods
Reference (59)
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr., Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis, An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh CK, Murray LA, Molfino NA, Smoking and idiopathic pulmonary fibrosis. Pulm. Med 2012, 808260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, Nakano Y, Okamoto T, Evans CM, Yang IV, Schwartz DA, Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 57, 91–99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith TJ, Petty TL, Reading JC, Lakshminarayan S, Pulmonary effects of chronic exposure to airborne cadmium. Am. Rev. Respir. Dis. 114, 161–169 (1976). [DOI] [PubMed] [Google Scholar]
- 5.Snider GL, Lucey EC, Faris B, Jung-Legg Y, Stone PJ, Franzblau C, Cadmium-chloride-induced air-space enlargement with interstitial pulmonary fibrosis is not associated with destruction of lung elastin. Implications for the pathogenesis of human emphysema. Am. Rev. Respir. Dis. 137, 918–923 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Damiano VV, Cherian PV, Frankel FR, Steeger JR, Sohn M, Oppenheim D, Weinbaum G, Intraluminal fibrosis induced unilaterally by lobar instillation of CdCl2 into the rat lung. Am. J. Pathol. 137, 883–894 (1990). [PMC free article] [PubMed] [Google Scholar]
- 7.Li FJ, Surolia R, Li H, Wang Z, Liu G, Liu RM, Mirov SB, Athar M, Thannickal VJ, Antony VB, Low-dose cadmium exposure induces peribronchiolar fibrosis through site-specific phosphorylation of vimentin. Am. J. Phys. Lung Cell. Mol. Phys. 313, L80–L91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morozesk M, Franqui LS, Mansano AS, Martinez DST, Fernandes MN, Interactions of oxidized multiwalled carbon nanotube with cadmium on zebrafish cell line: The influence of two co-exposure protocols on in vitro toxicity tests. Aquat. Toxicol. 200, 136–147 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Hulett LD Jr., Weinberger AJ, Northcutt KJ, Ferguson M, Chemical species in fly ash from coal-burning power plants. Science 210, 1356–1358 (1980). [DOI] [PubMed] [Google Scholar]
- 10.Dufka M, Docekal B, Characterization of urban particulate matter by diffusive gradients in thin film technique. J. Anal. Methods Chem. 2018, 9698710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang K, Fox J, DPF soot as an adsorbent for Cu(II), Cd(II), and Cr(VI) compared with commercial activated carbon. Environ. Sci. Pollut. Res. Int. 25, 8620–8635 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Wibowo N, Setyadhi L, Wibowo D, Setiawan J, Ismadji S, Adsorption of benzene and toluene from aqueous solutions onto activated carbon and its acid and heat treated forms: Influence of surface chemistry on adsorption. J. Hazard. Mater. 146, 237–242 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA, Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 155, 242–248 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Antoniou KM, Hansell DM, Rubens MB, Marten K, Desai SR, Siafakas NM, Nicholson AG, du Bois RM, Wells AU, Idiopathic pulmonary fibrosis: Outcome in relation to smoking status. Am. J. Respir. Crit. Care Med. 177, 190–194 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Ryu JH, Colby TV, Hartman TE, Vassallo R, Smoking-related interstitial lung diseases: A concise review. Eur. Respir. J. 17, 122–132 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Waalkes MP, Cadmium carcinogenesis. Mutat. Res. 533, 107–120 (2003). [DOI] [PubMed] [Google Scholar]
- 17.You R, Lu W, Shan M, Berlin JM, Samuel EL, Marcano DC, Sun Z, Sikkema WK, Yuan X, Song L, Hendrix AY, Tour JM, Corry DB, Kheradmand F, Nanoparticulate carbon black in cigarette smoke induces DNA cleavage and Th17-mediated emphysema. eLife 4, e09623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart BA, Gong Q, Eneman JD, Durieux-Lu CC, In vivo expression of metallothionein in rat alveolar macrophages and type II epithelial cells following repeated cadmium aerosol exposures. Toxicol. Appl. Pharmacol. 133, 82–90 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Grasseschi RM, Ramaswamy RB, Levine DJ, Klaassen CD, Wesselius LJ, Cadmium accumulation and detoxification by alveolar macrophages of cigarette smokers. Chest 124, 1924–1928 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Shekhter AB, Berchenko GN, Nikolaev AV, Macrophage-fibroblast interaction and its possible role in regulating collagen metabolism during wound healing. Biull. Eksp. Biol. Med. 83, 627–630 (1977). [PubMed] [Google Scholar]
- 21.Wynn TA, Barron L, Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, Simpson AJ, Millar AB, McGrath EE, Whyte MK, Hirani N, Hogaboam CM, Donnelly SC, The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 188, 1442–1450 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, Gross BH, Oak SR, Coelho AL, Evanoff H, Day E, Toews GB, Joshi AD, Schaller MA, Waters B, Jarai G, Westwick J, Kunkel SL, Martinez FJ, Hogaboam CM, TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci. Transl. Med 2, 57ra82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GR, Raparia K, Tamaki Z, Varga J, Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 7, 11703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vassiliadis E, Oliveira CP, Alvares-da-Silva MR, Zhang C, Carrilho FJ, Stefano JT, Rabelo F, Pereira L, Kappel CR, Henriksen K, Veidal SS, Vainer B, Duffin KL, Christiansen C, Leeming DJ, Karsdal M, Circulating levels of citrullinated and MMP-degraded vimentin (VICM) in liver fibrosis related pathology. Am. J. Transl. Res. 4, 403–414 (2012). [PMC free article] [PubMed] [Google Scholar]
- 26.Ytterberg AJ, Joshua V, Reynisdottir G, Tarasova NK, Rutishauser D, Ossipova E, Haj Hensvold A, Eklund A, Skold CM, Grunewald J, Malmstrom V, Jakobsson PJ, Ronnelid J, Padyukov L, Zubarev RA, Klareskog L, Catrina AI, Shared immunological targets in the lungs and joints of patients with rheumatoid arthritis: Identification and validation. Ann. Rheum. Dis. 74, 1772–1777 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Rocha-Munoz AD, Ponce-Guarneros M, Gamez-Nava JI, Olivas-Flores EM, Mejia M, Juarez-Contreras P, Martinez-Garcia EA, Corona-Sanchez EG, Rodriguez-Hernandez TM, Vazquez-del Mercado M, Salazar-Paramo M, Nava-Zavala AH, Cardona-Munoz EG, Celis A, Gonzalez-Lopez L, Anti-cyclic citrullinated peptide antibodies and severity of interstitial lung disease in women with rheumatoid arthritis. J. Immunol. Res. 2015, 151626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, Grunewald J, Skold CM, Klareskog L, Catrina AI, Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 67, 1488–1492 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Bidkar M, Vassallo R, Luckey D, Smart M, Mouapi K, Taneja V, Cigarette smoke induces immune responses to vimentin in both, arthritis-susceptible and -resistant humanized mice. PLOS ONE 11, e0162341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohamed BM, Verma NK, Davies AM, McGowan A, Crosbie-Staunton K, Prina-Mello A, Kelleher D, Botting CH, Causey CP, Thompson PR, Pruijn GJ, Kisin ER, Tkach AV, Shvedova AA, Volkov Y, Citrullination of proteins: A common post-translational modification pathway induced by different nanoparticles in vitro and in vivo. Nanomedicine 7, 1181–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vossenaar ER, van Venrooij WJ, Citrullinated proteins: Sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res. Ther. 6, 107–111 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Lightfoot YL, Seto N, Carmona-Rivera C, Moore E, Goel R, O’Neil L, Mistry P, Hoffmann V, Mondal S, Premnath PN, Gribbons K, Dell’Orso S, Jiang K, Thompson PR, Sun HW, Coonrod SA, Kaplan MJ, Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight 3, e124729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra N, Schwerdtner L, Sams K, Mondal S, Ahmad F, Schmidt RE, Coonrod SA, Thompson PR, Lerch MM, Bossaller L, Cutting edge: Protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome-dependent IL-1β maturation and ASC speck formation in macrophages. J. Immunol. 203, 795–800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh RK, Albrecht AL, Somji S, Sens MA, Sens DA, Garrett SH, Alterations in metal toxicity and metal-induced metallothionein gene expression elicited by growth medium calcium concentration. Cell Biol. Toxicol. 24, 273–281 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Hayes RA, Regondi S, Winter MJ, Butler PJ, Agradi E, Taylor EW, Kevin Chipman J, Cloning of a chub metallothionein cDNA and development of competitive RT-PCR of chub metallothionein mRNA as a potential biomarker of heavy metal exposure. Mar. Environ. Res. 58, 665–669 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Li FJ, Surolia R, Li H, Wang Z, Kulkarni T, Liu G, de Andrade JA, Kass DJ, Thannickal VJ, Duncan SR, Antony VB, Autoimmunity to vimentin Is associated with outcomes of patients with idiopathic pulmonary fibrosis. J. Immunol. 199, 1596–1605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaji R, Chatani E, Harada N, Sugimoto K, Inui H, Nakano Y, Glyceraldehyde-3-phosphate dehydrogenase in the extracellular space inhibits cell spreading. Biochim. Biophys. Acta 1726, 261–271 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Surolia R, Li FJ, Wang Z, Li H, Liu G, Zhou Y, Luckhardt T, Bae S, Liu RM, Rangarajan S, de Andrade J, Thannickal VJ, Antony VB, ` pulmospheres serve as a personalized and predictive multicellular model for assessment of antifibrotic drugs. JCI Insight 2, e91377 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xin Y, Wang X, Zhu M, Qu M, Bogari M, Lin L, Mar Aung Z, Chen W, Chen X, Chai G, Zhang Y, Expansion of CD26 positive fibroblast population promotes keloid progression. Exp. Cell Res. 356, 104–113 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Khalil N, Parekh TV, O’Connor R, Antman N, Kepron W, Yehaulaeshet T, Xu YD, Gold LI, Regulation of the effects of TGF-beta 1 by activation of latent TGF-beta 1 and differential expression of TGF-beta receptors (T beta R-I and T beta R-II) in idiopathic pulmonary fibrosis. Thorax 56, 907–915 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen JT, Knight RA, Bloor CA, Spiteri MA, Enhanced insulin-like growth factor binding protein-related protein 2 (Connective tissue growth factor) expression in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am. J. Respir. Cell Mol. Biol. 21, 693–700 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Muller-Quernheim J, Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am. J. Respir. Crit. Care Med. 157, 762–768 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM Jr., Hunter MG, Bringardner BD, Monick MM, Brigstock DR, Stromberg PC, Hunninghake GW, Marsh CB, Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 176, 78–89 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Driscoll KE, Carter JM, Howard BW, Hassenbein DG, Pepelko W, Baggs RB, Oberdorster G, Pulmonary inflammatory, chemokine, and mutagenic responses in rats after subchronic inhalation of carbon black. Toxicol. Appl. Pharmacol. 136, 372–380 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Cesta MF, Ryman-Rasmussen JP, Wallace DG, Masinde T, Hurlburt G, Taylor AJ, Bonner JC, Bacterial lipopolysaccharide enhances PDGF signaling and pulmonary fibrosis in rats exposed to carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 43, 142–151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB, Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44, 582–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frankel FR, Steeger JR, Damiano VV, Sohn M, Oppenheim D, Weinbaum G, Induction of unilateral pulmonary fibrosis in the rat by cadmium chloride. Am. J. Respir. Cell Mol. Biol. 5, 385–394 (1991). [DOI] [PubMed] [Google Scholar]
- 48.Sokolove J, Zhao X, Chandra PE, Robinson WH, Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 63, 53–62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Derynck R, Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 9, 274–279 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Ganguly K, Levänen B, Palmberg L, Åkesson A, Lindén A, Cadmium in tobacco smokers: A neglected link to lung disease? Eur. Respir. Rev 27, 170122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, Weissman IL, Longaker MT, Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lugli EB, Correia RE, Fischer R, Lundberg K, Bracke KR, Montgomery AB, Kessler BM, Brusselle GG, Venables PJ, Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: Implications for autoimmunity in rheumatoid arthritis. Arthritis Res. Ther. 17, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samara KD, Trachalaki A, Tsitoura E, Koutsopoulos AV, Lagoudaki ED, Lasithiotaki I, Margaritopoulos G, Pantelidis P, Bibaki E, Siafakas NM, Tzanakis N, Wells AU, Antoniou KM, Upregulation of citrullination pathway: From autoimmune to idiopathic lung fibrosis. Respir. Res. 18, 218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L, A. new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 54, 38–46 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Hutchinson D, Muller J, McCarthy JE, Gun’ko YK, Verma NK, Bi X, Di Cristo L, Kickham L, Movia D, Prina-Mello A, Volkov Y, Cadmium nanoparticles citrullinate cytokeratins within lung epithelial cells: Cadmium as a potential cause of citrullination in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 13, 441–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R, Time course of bleomycin-induced lung fibrosis. Int. J. Exp. Pathol. 83, 111–119 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M, The bleomycin animal model: A. useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, Luo Y, Levitt B, Glogowska M, Chandra P, Kulik L, Robinson WH, Arend WP, Thompson PR, Holers VM, N-α-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-l-Ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 186, 4396–4404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carnes RM, Mobley JA, Crossman DK, Liu H, Korf BR, Kesterson RA, Wallis D, Multi-omics profiling for NF1 target discovery in neurofibromin (NF1) deficient cells. Proteomics 19, e1800334 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. MT-1/2 mRNA and protein expression is higher in subjects with IPF, especially smokers.
Fig. S2. CB deposits are found in macrophages from subjects with IPF.
Fig. S3. Cd/CB-induced Akt1, PAD2 activation, and Vim citrullination are dependent on oxidative states.
Fig. S4. Cells that express Vim on their surface are not apoptotic.
Fig. S5. Proteins are identified and tandem MS2 spectrum is characterized by IP-MS.
Fig. S6. Schematic diagram of the Cit-Vim secretion pathway.
Fig. S7. Both Vim and Cit-Vim are purified and enriched from cell culture supernatants.
Fig. S8. Cit-Vim exposure induces more invasive subtypes of fibroblasts.
Fig. S9. Cit-Vim exposure enhances the expression of collagen in fibroblasts isolated from normal subjects and subjects with IPF.
Fig. S10. Cit-Vim, but not Vim, induces CTGF, IL-8, and active TGF-β1 production.
Fig. S11. Cit-Vim, but not Vim, can effectively induce cytokine/chemokine production only in TLR4 WT mice.
Fig. S12. Schematic diagram of the Cit-Vim–induced fibrosis pathway.
Fig. S13. Exposure of Cd/CB causes dose-dependent interstitial fibrosis in mice.
Fig. S14. Exposure of Cd/CB induces cytokine/chemokine secretion.
Fig. S15. Cd/CB-induced Vim citrullination and lung fibrosis are dependent on the activation of Akt1.
Table S1. Demographics and baseline characteristics of subjects with IPF.
Table S2. Smoking status analysis of subjects with IPF.
Table S3. Accompanying fragmentation for the same peptide identified by IP-MS.
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.