

Abstract
Hydroamination of alkenes catalyzed by transition-metal complexes is an atom-economical method for the synthesis of amines, but reactions of unactivated alkenes remain inefficient. Additions of N–H bonds to such alkenes catalyzed by iridium, gold, and lanthanide catalysts are known, but they have required a large excess of the alkene. New mechanisms for such processes involving metals rarely used previously for hydroamination could enable these reactions to occur with greater efficiency. We report ruthenium-catalyzed intermolecular hydroaminations of a variety of unactivated terminal alkenes without the need for an excess of alkene and with 2-aminopyridine as an ammonia surrogate to give the Markovnikov addition product. Ruthenium complexes have rarely been used for hydroaminations and have not previously catalyzed such reactions with unactivated alkenes. Identification of the catalyst resting state, kinetic measurements, deuterium labeling studies, and DFT computations were conducted and, together, strongly suggest that this process occurs by a new mechanism for hydroamination occurring by oxidative amination in concert with reduction of the resulting imine.
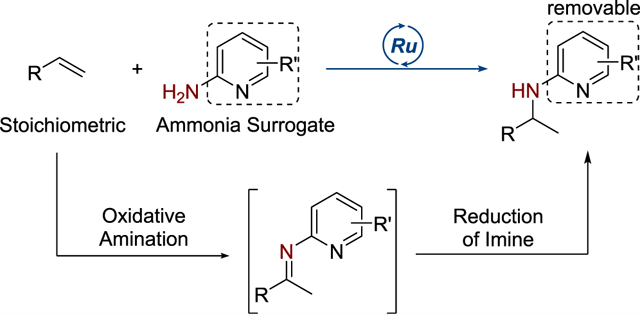
Graphical Abstract
INTRODUCTION
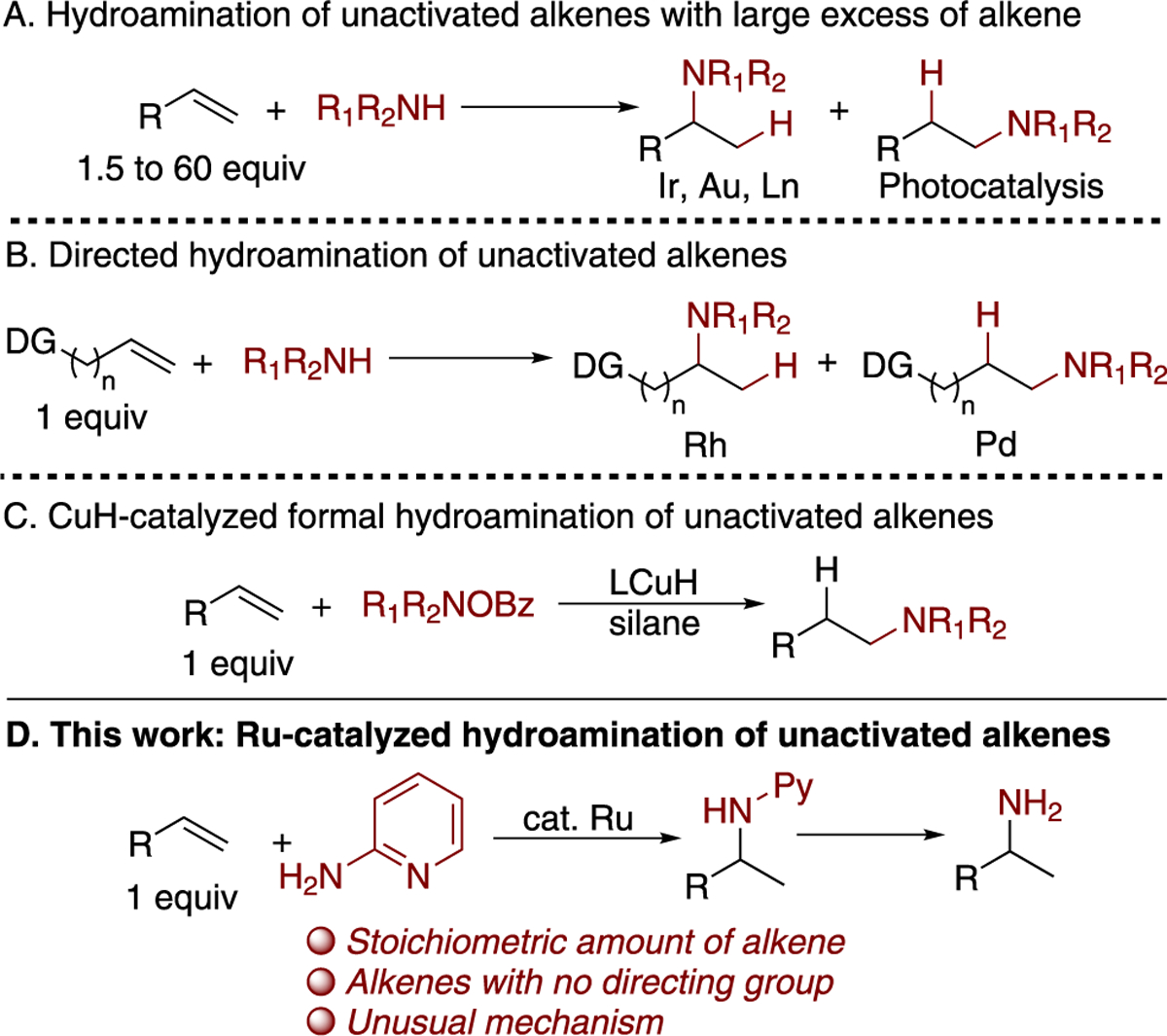
Amines and their derivatives are important as both pharmaceuticals and agrochemicals.1–3 Traditional methods to synthesize amines include nucleophilic substitution of organic halides,4 reductive amination of carbonyl compounds,5 and reduction of amides, nitriles, and azides.6 The hydroamination of alkenes catalyzed by transition-metal complexes is an attractive alternative to these methods because it occurs directly with alkenes and could be applied to the functionalization of both simple alkenes and complex molecules containing alkene units.7–9 Despite the potential utility of hydroamination, examples of intermolecular hydroaminations are often limited to conjugated and strained alkenes, such as dienes,10,11 vinylarenes,12,13 norbornenes,14,15 and cyclopropenes.16,17 Hydroaminations of unactivated alkenes are rare and generally require a large excess of alkene (Scheme 1a).18–26
Scheme 1.
Catalytic Hydroamination of Unactivated Terminal Alkenes
Two main strategies have been followed to enable hydroamination to occur without an excess amount of alkene. The first involves catalytic reactions of alkenes possessing a directing group (Scheme 1b);27,28 the second involves a formal catalytic hydroamination achieved by combining a silane reducing agent and a nitrogen-based electrophile (Scheme 1c).29,30 These strategies require special alkenes or generate stoichiometric amounts of waste from the silane and aminating reagents and require synthesis of the aminating reagent. Thus, the development of a method that involves the direct addition of the N–H bond of an amine to unactivated alkenes is needed and would address these drawbacks.
Two general classes of mechanisms have been followed by most late, transition-metal catalysts for the hydroamination of alkenes. The first mechanism involves nucleophilic attack of a nitrogen nucleophile on a coordinated alkene, followed by protonation of the resulting amino-alkyl intermediate. The second involves oxidative addition of an N–H bond, followed by migratory insertion of the alkene into the metal–nitrogen bond and reductive elimination to form the C–H bond. Cationic rhodium31 and gold22 systems react by the first pathway, whereas neutral iridium complexes19 react by the second pathway. Ruthenium complexes, the subject of this work, catalyze the hydroamination of terminal alkynes32 and vinylarenes,13 but they have not been shown to catalyze the hydroamination of unconjugated alkenes. We considered that such complexes and a carefully designed amine could catalyze the hydroaminations of alkenes and do so by mechanisms that are distinct from those followed by complexes of other transition metals.
Multidentate coordination of an amine that is tethered to a Lewis basic group, rather than an alkene tethered to such a group, can accelerate hydroaminations due to a series of effects. First, the Lewis basic group can stabilize the intermediates resulting from oxidative addition of the N–H bond and products from insertion. Second, such an amine can serve as an ammonia equivalent by removal of the Lewis basic group. Third, an appropriate heteroaryl substituent can alter the thermodynamics of this nearly thermoneutral process33 to favor addition by rendering the N–H bond of the ammonia surrogate weaker than the N–H bond of ammonia.
We report ruthenium-catalyzed Markovnikov hydroaminations of unactivated, terminal alkenes with 2-aminopyridine as an ammonia surrogate (Scheme 1d). Terminal alkenes containing a series of functional groups undergo hydroamination in the presence of this catalyst to afford the corresponding amine products without excess alkene. Detailed experimental and computational mechanistic studies provide strong evidence that this reaction occurs by a new pathway for hydroamination that comprises oxidative amination of the alkene and reduction of the corresponding imine intermediate.
RESULTS AND DISCUSSION
Reaction Development.
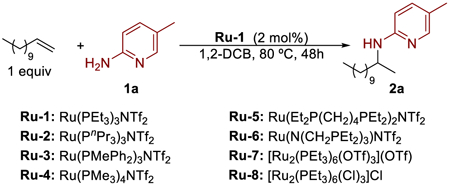
Our studies on ruthenium-catalyzed hydroamination of unactivated terminal alkenes began by examining the reaction between 1-dodecene (1.0 equiv) and 2-aminopyridine 1a (1.0 equiv) in the presence of a variety of electrophilic ruthenium complexes, a class of catalyst previously reported by our group to catalyze the oxidation of alcohols34 (Table 1). 2-Aminopyridine was investigated as the amine component because it could coordinate to the cationic ruthenium complex in a bidentate fashion to form a four-membered ruthenacycle that would retain reactivity at the Ru–N bond, due to the ring strain.
Table 1.
Evaluation of the Conditions for the Hydroamination of 1-Dodecene with 1aa
| ||
|---|---|---|
| entry | conditions | yield (%)b |
| 1 | standard | 67% |
| 2 | Ru-2 as catalyst | 43% |
| 3 | Ru-3 as catalyst | 18% |
| 4 | Ru-4 as catalyst | <1% |
| 5 | Ru-5 as catalyst | <1% |
| 6 | Ru-6 as catalyst | <1% |
| 7 | Ru-7 as catalyst | 62% |
| 8 | Ru-8 as catalyst | <1% |
| 9 | 1,2-DCE as solvent | <1% |
| 10 | toluene as solvent | 29% |
| 11 | PhCl as solvent | 48% |
| 12 | CH3CN as solvent | <1% |
| 13 | THF as solvent | 56% |
| 14 | dioxane as solvent | 61% |
| 15 | Ru-1 (2 mol %) | 45% |
| 16 | no Ru-1 | <1% |
| 17 | HNTf2 (5 mol %) | <1% |
Standard condition: 1-dodecene (0.2 mmol), Ru-1 (0.01 mmol), 1a (0.2 mmol), 1,2-DCB (50 μL), 80 °C, 48 h.
Determined by 1H NMR spectroscopy of the crude reaction mixture with 1,3,5-trimethoxybenzene as the internal standard.
A survey of a series of phosphine-ligated ruthenium triflimide and triflate complexes showed that the triethylphosphine-ligated ruthenium complex Ru-1 containing triflimide as the anionic ligand catalyzes the hydroamination of 1-dodecene to afford the amine product 2a in 67% yield (Table 1, entry 1). Entries 2–6 show the effect of the phosphine ligand. Ruthenium complexes that contain more sterically demanding phosphine ligands (entry 2) and less electron-rich phosphine ligands (entry 3) catalyzed the reactions to form the addition product in lower yields. Complexes of ruthenium ligated by trimethylphosphine, 1,2-bis(diethylphosphino)-butane, and a tripodal phosphine ligand, N(CH2PEt2)3, did not catalyze the hydroamination reaction (entries 4–6). Entries 7 and 8 show the effect of the anionic ligand. Hydroamination conducted with the PEt3-ligated ruthenium complex containing triflate counteranion (Ru-7) afforded the product in a slightly lower 62% yield (entry 7) than that with Ru-1, and the PEt3-ligated ruthenium complex containing chlorides (Ru-8) did not catalyze the hydroamination reaction (entry 8). This hydroamination proceeded in a variety of polar, noncoordinating solvents, and the reaction in 1,2-DCB (entry 1) occurred in a higher yield than did reactions in other solvents of this class (entries 9–14). The reaction performed with 2 mol % of Ru-1 instead of 5 mol % gave a lower yield of product (entry 15).
Hydroaminations of alkenes catalyzed by electrophilic metal complexes, in some cases, have been suspected to occur by acid generated from the amine and the complex.35 Although the basicity of the 2-aminopyridine would render such a pathway unlikely, we conducted control experiments to test this potential. To assess the role of the ruthenium complex in this reaction, we conducted the reaction in the absence of Ru-1. Without Ru-1, no detectable product formed, indicating that the ruthenium complex is necessary for this transformation (entry 16). The hydroamination of 1-dodecene with substoichiometric amounts of strong acid also did not form detectable amounts of product (entry 17), providing evidence against a pathway for hydroamination with 2-aminopyridine catalyzed by a Brønsted acid.
Scope of Terminal Alkenes and 2-Aminopyridines for Catalytic Hydroamination.
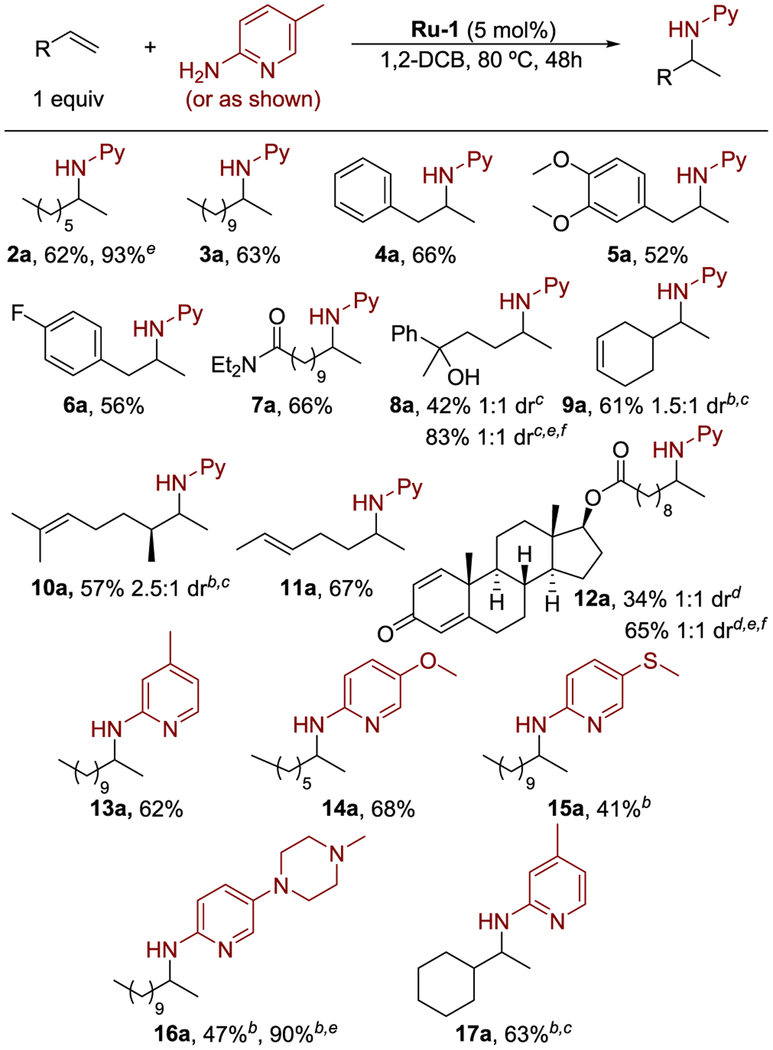
Studies on the scope of the ruthenium-catalyzed hydroamination under the developed conditions are summarized in Table 2. The hydroamination of terminal alkenes with alkyl and aryl substituents occurred to afford products in good yields (2a–4a). The hydroamination of a series of alkenes bearing ether, fluoride, amide, and tertiary alcohol functional groups also formed the hydroamination products in good to moderate yields (5a–8a). Alkenes containing multiple double bonds underwent hydroamination exclusively at the terminal alkene (9a–11a). Even the hydroamination of boldenone undecylenate, an anabolic steroid, occurred, showing the potential of this methodology for late-stage functionalization of architecturally complex molecules containing potential competing functionality (12a). An investigation of the scope of 2-aminopyridines for this reaction showed that a variety of 2-aminopyridines containing alkyl, ether, sulfide, and methylpiperazine substituents reacted to give the hydroamination products (13a–17a).
Table 2.
|
Isolated yields.
100 °C.
Ru-1 10 mol %.
Ru-1 15 mol %.
2 equiv of alkene.
72 h.
In addition to unconjugated, terminal alkenes, vinylarenes underwent this hydroamination reaction (Table 3). Vinylarenes bearing a variety of electron-donating and electron-withdrawing para-substituents underwent hydroamination to afford products in good yields (18a–24a). However, higher temperatures were required for the hydroaminations of electron-deficient vinylarenes catalyzed by Ru-1 (20a, 22–24a). A lower yield was observed from the reaction of a vinylarene bearing an ortho-substituent (25a), indicating that the reaction is sensitive to steric hindrance. The hydroamination of vinylnaphthalene and vinylferrocene also proceeded to afford hydroamination products (27a–28a). The major side reaction observed during the hydroamination was the isomerization of the terminal alkene to form internal alkenes, which did not undergo hydroamination under our reaction conditions. High yields of the amine products 2a, 8a, 12a, 16a, and 28a were obtained for reactions with just 2 equiv of alkene with respect to the aminopyridine.
Table 3.
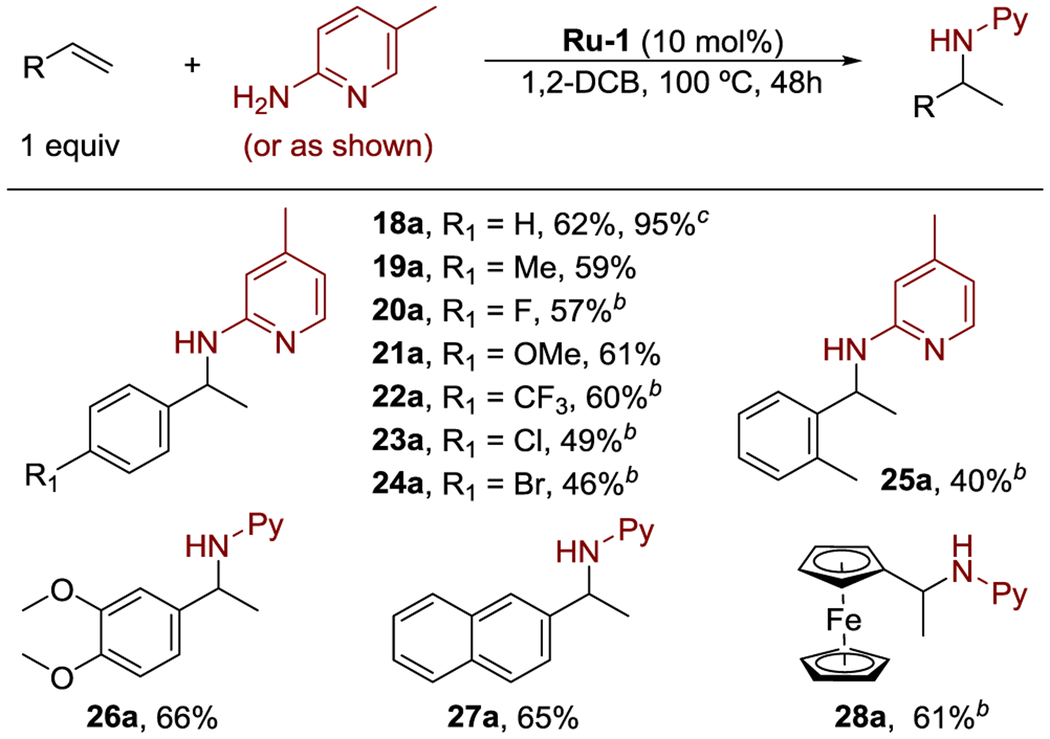
|
Isolated yields.
100 °C.
2 equiv of alkene.
Removal of Pyridyl Group from the Hydroamination Product.
The product from the hydroamination reaction was converted to a primary amine by a two-step sequence (Scheme 2).36–38 Compound 2a was first converted to an amidine, and the amidine was cleaved by NaBH4 to form primary amine 2b. For convenience on the laboratory scale, the Boc-protected amine 2c was isolated instead of the free amine 2b.
Scheme 2.
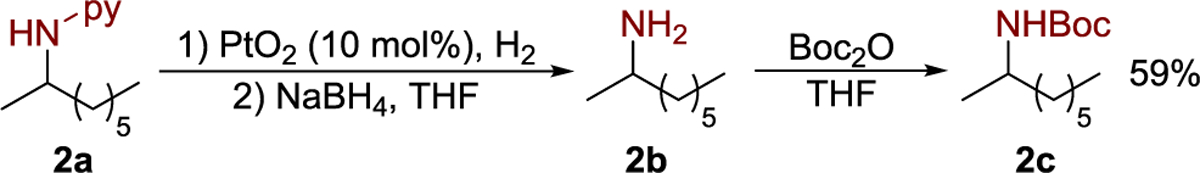
Removal of the Pyridyl Group from 2a
Mechanistic Studies of Alkene Hydroamination.
Because side reactions, such as alkene isomerization, alkene oligomerization, and oxidative amination, compete with alkene hydroamination,9,19,39 literature protocols for the hydroamination of unactivated alkenes require either the use of alkenes containing a directing group or excess alkene. Our hydroamination protocol represents a rare example of catalytic hydroamination of unactivated alkenes with a stoichiometric amount of alkene. Therefore, a detailed investigation of the mechanism of this ruthenium-catalyzed hydroamination reaction was conducted to provide insight into the features that promote the hydroamination reaction over other side reactions.
Determination of the Catalyst Resting State.
Our mechanistic investigation began with identifying the catalyst resting state. To obtain preliminary information about this species, we monitored the reaction of 2-amino-4-methylpyridine (1b) and 1-dodecene catalyzed by Ru-1 by 31P NMR spectroscopy at 80 °C (spectrum b of Figure 1). Aminopyridine 1b was used for these studies, instead of 1a, because ruthenium complexes containing 1b were more crystalline than those containing 1a (vide infra). A broad resonance at 50 ppm, corresponding to the resting state of the catalyst, was observed in the 31P NMR spectrum of the reaction mixture.
Figure 1.
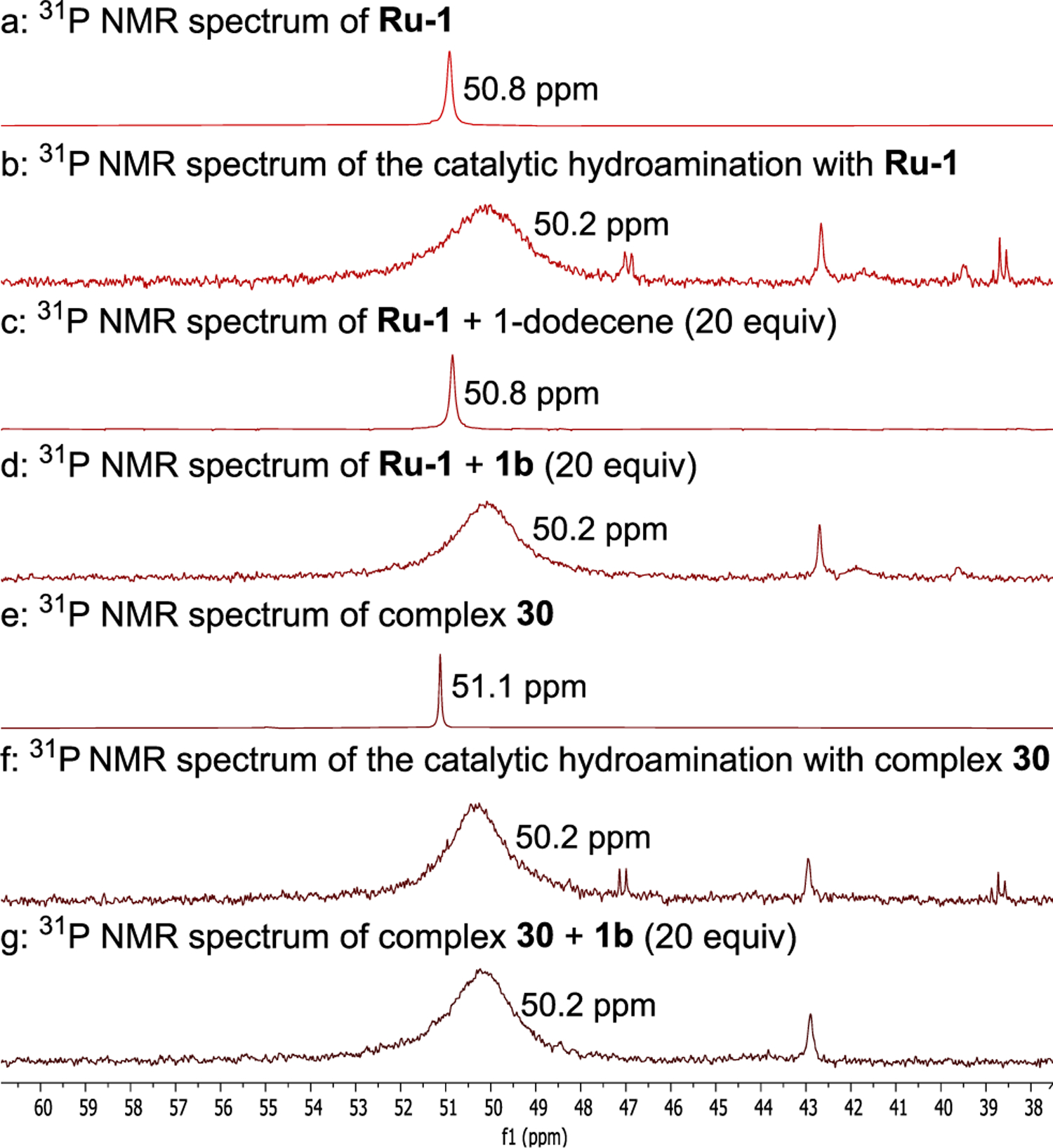
(a) 31P NMR spectrum Ru-1. (b) 31P NMR spectrum of the catalytic hydroamination of 1-dodecene with Ru-1 as the catalyst. (c) 31P NMR spectrum of the mixture of Ru-1 and 1-dodecene (20 equiv). (d) 31P NMR spectrum of the mixture of Ru-1 and 1b (20 equiv). (e) 31P NMR spectrum of complex 30. (f) 31P NMR spectrum of the catalytic hydroamination of 1-dodecene with complex 30 as the catalyst. (g) 31P NMR spectrum of the mixture of complexes 30 and 1b (20 equiv). All of the above spectra were acquired at 80 °C.
Further information on the identity of the resting state was obtained by allowing Ru-1 to react with 1-dodecene and 1b separately and monitoring the reaction mixtures by 31P NMR spectroscopy. Treatment of Ru-1 with excess 1-dodecene led to no new Ru complexes, as determined by 31P NMR spectroscopy at 80 °C (a and c of Figure 1). However, treatment of Ru-1 with excess aminopyridine 1b led to the observation of a broad resonance at 50 ppm in the 31P NMR spectrum of the reaction mixture at 80 °C, and this resonance was identical to that of the resting state of the Ru catalyst (d of Figure 1). Therefore, the major ruthenium complex in the reaction system forms from the combination of Ru-1 with one or more equiv of aminopyridine 1b.
An independent synthesis of the resting state was pursued (Figure 2) because we were unable to isolate it in pure form directly from the mixture of Ru-1 and excess 1b. Treatment of Ru-1 with 1 equiv of 1b afforded the aminopyridine-coordinated ruthenium complex 29 in 82% yield after recrystallization (Figure 2a). The 31P NMR spectrum of complex 29 contained a single 31P resonance at 38 ppm, and the structure of 29 was determined by single-crystal X-ray diffraction. The Ru center in 29 is ligated by three PEt3 ligands, one O-bound triflimide, and one κ2-bound aminopyridine in an octahedral geometry (Figure 2c). The 31P NMR chemical shift of 38 ppm for complex 29 is distinct from that of the catalyst resting state (50 ppm).
Figure 2.
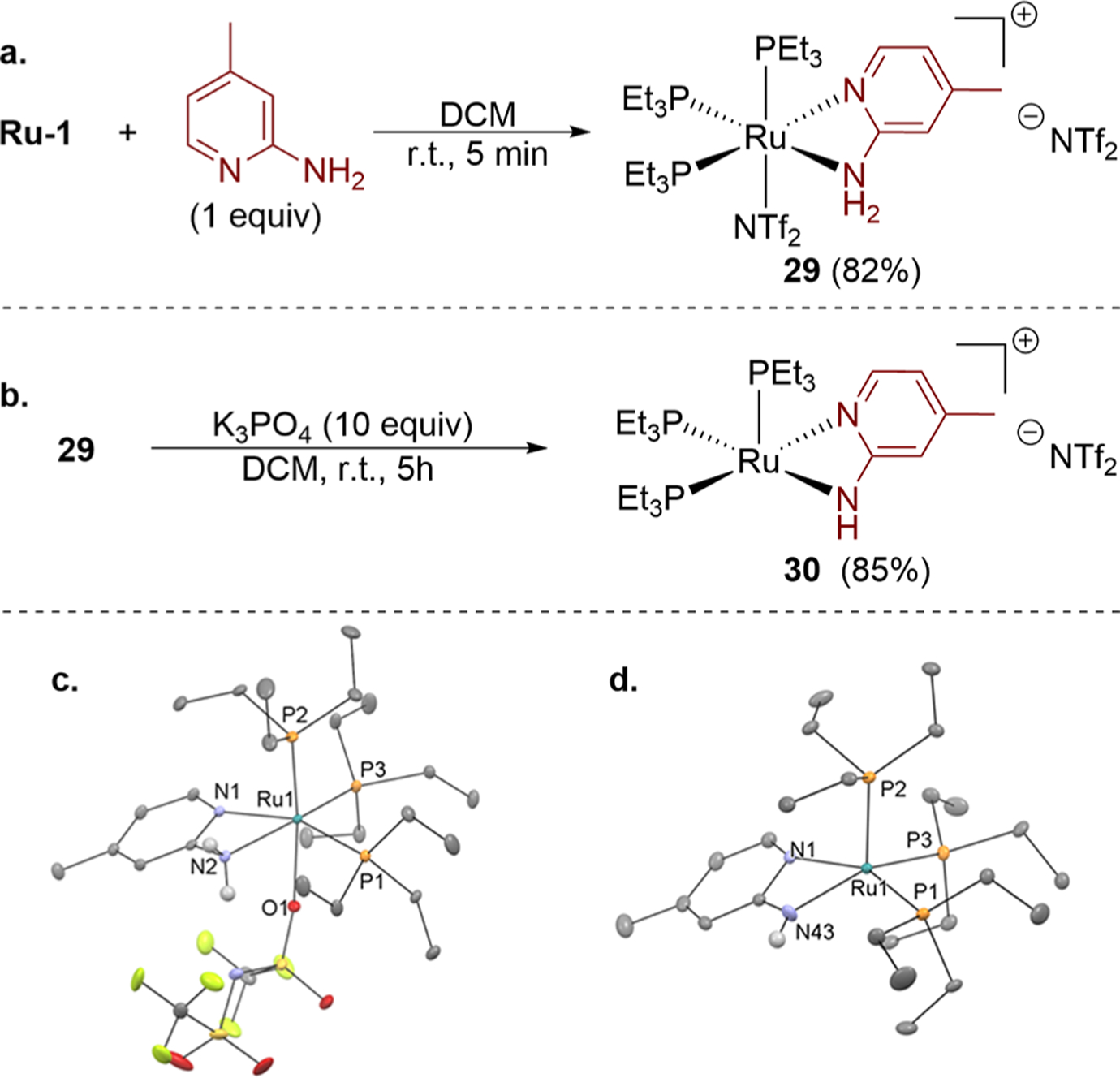
(a) Synthesis of complex 29. (b) Synthesis of complex 30. (c) Solid-state structure of complex 29 with ellipsoids set at 30% and selected hydrogen atoms and free triflimide anion omitted for clarity. (d) Solid-state structure of complex 30 with ellipsoids set at 30% and selected hydrogen atoms and free triflimide anion omitted for clarity.
Thus, we postulated that coordination of 1b to the cationic ruthenium center sufficiently acidifies the N–H bonds to cause deprotonation of 29 by another equivalent of aminopyridine. To test this hypothesis, we treated complex 29 with an insoluble base (K3PO4). This reaction led to the isolation of the Ru–amido complex 30 in 85% yield after recrystallization (Figure 2b). The 31P NMR spectrum of complex 30 consisted of a single sharp resonance at 51 ppm (e of Figure 1). The solid-state structure of complex 30 was determined by X-ray diffraction (Figure 2d). This complex adopts a square pyramidal geometry with an empty coordination site trans to a PEt3 ligand.
The hydroamination of 1-dodecene by aminopyridine 1b catalyzed by 5 mol % of the isolated complex 30 afforded the product 13a in 65% yield in 48 h. This result indicates that complex 30 is kinetically competent to be an intermediate in this hydroamination reaction. Furthermore, a broad resonance at 50 ppm, which matched that observed for the catalytic reaction initiated with Ru-1, was observed in the 31P NMR spectrum of the hydroamination reaction initiated with complex 30 as catalyst (f of Figure 1).
These data suggest that complex 30 could be the resting state of the catalyst, but the resonance corresponding to isolated 30 is much sharper and slightly more downfield (0.9 ppm) than that of the major ruthenium complex in the catalytic system. We determined that this difference in line shape and chemical shift depends on the presence or absence of an excess of the aminopyridine; the 31P NMR signal of isolated 30 broadened and shifted slightly upfield in the presence of 20 equiv of aminopyridine 1b at 80 °C (g of Figure 1).
To investigate the origin of this difference in line shape and chemical shift further, we conducted variable-temperature NMR spectroscopy with a 20:1 mixture of aminopyridine 1b and complex 30 (Figure 3). At −40 °C, the 31P NMR spectrum of the mixture contained three triplet resonances. A potential structure for the complex corresponding to the three triplet resonances is 31a shown at the bottom of Figure 3. This structure contains an additional 2-aminopyridine occupying the sixth coordination site and a hydrogen bond to the amido ligand. The hydrogen-bonding interaction in 31a is evidenced by the observation of a broad downfield proton resonance (14 ppm) in the 1H NMR spectrum of this mixture. The integration of the three triplet resonances in the 31P NMR spectrum decreased at higher temperatures, and we were unable to detect the triple resonances when the temperature of the mixture reached 40 °C, indicating that complex 31a is a minor species in the catalytic reaction at 80 °C.
Figure 3.
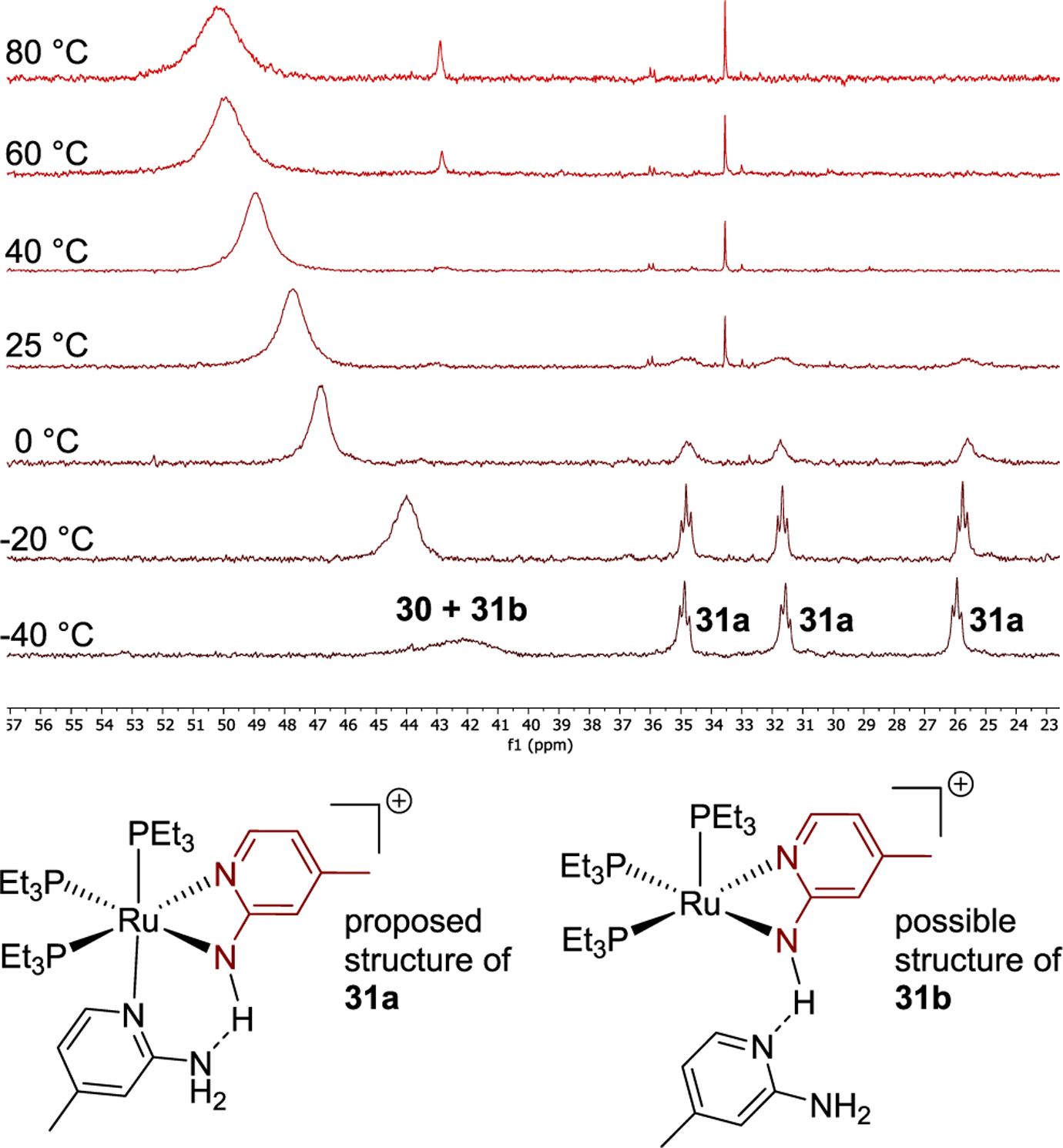
31P NMR spectra of the mixture of complexes 30 and 1b (20 equiv) at different temperatures and possible structures for complexes 31a and 31b.
The chemical shift of the broad resonance corresponding to the major ruthenium complex at 80 °C migrated significantly upfield at lower temperature. The origin of this line shape and change in chemical shift is not clear, but could result from hydrogen bonding between the amide ligand NH and the aminopyridine without coordination of the pyridine nitrogen to the metal as we suggest is present in complex 31b. The chemical shift of this resonance lies further upfield in solutions containing higher concentrations of aminopyridine, implying that the resonance results from an equilibrium between 30 and an adduct formed between 30 and the aminopyridine that is distinct from complex 31a. At the same time, the similarity in chemical shift between pure complex 30 and the resonance in the presence of aminopyridine at the concentration and 80 °C temperature of the reaction implies that the major component in the catalytic reaction is complex 30.
Kinetic Studies on Catalytic Hydroamination.
Kinetic experiments were conducted to gain further information on the mechanism of the hydroamination. Kinetic experiments with 1-dodecene as the substrate were complicated by the observation of alkene isomerization during the reaction. Therefore, we conducted kinetic studies with vinylcyclohexane, which did not undergo competing isomerization.
Initial rates of the hydroamination reaction were measured at a series of concentrations of vinylcyclohexane, aminopyridine 1b, and catalyst Ru-1 (see the Supporting Information for details). Plots of initial rates against the concentration of the vinylcyclohexane, 1b, and Ru-1 are shown in Figure 4a–c. We found that the hydroamination reaction is first order in the concentration of vinylcyclohexane, zero order in the concentration of 1b, and first order in the concentration of Ru-1. The orders in alkene and catalyst imply that these species react in the turnover-limiting step. The zero-order dependence of the reaction on the concentration of aminopyridine 1b suggests that the hydroamination does not proceed by turnover-limiting nucleophilic attack of 1b on the alkene in a Ru complex like 32 (Scheme 3, pathway a). Instead, the kinetic data are consistent with a migratory insertion of the alkene into the Ru–N bond of the Ru–alkene complex 32 to generate a Ru–alkyl complex 33 (Scheme 3, pathway b).
Figure 4.
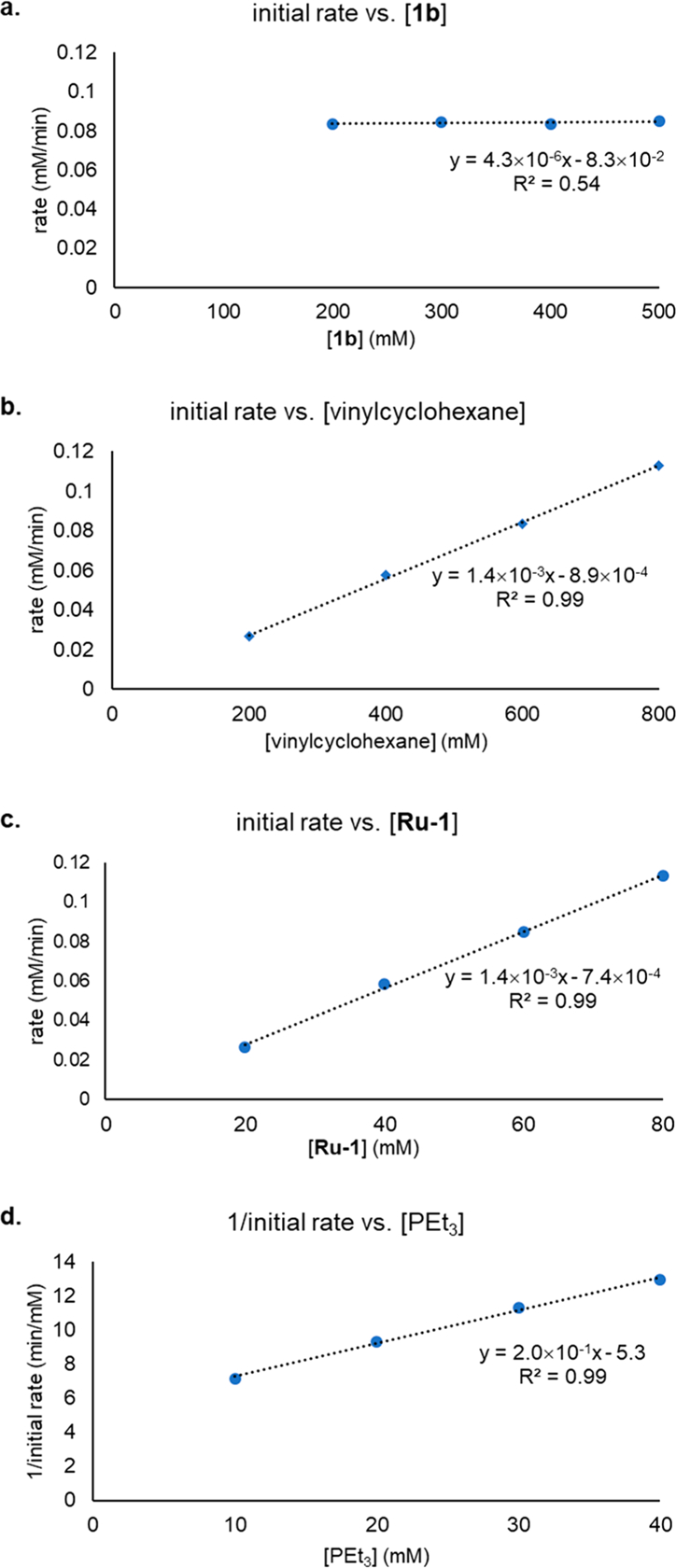
(a) Initial rates of product formation as a function of [1b]. (b) Initial rates of product formation as a function of [vinylcyclohexane]. (c) Initial rates of product formation as a function of [Ru-1]. (d) 1/initial rates of product formation as a function of [PEt3].
Scheme 3.
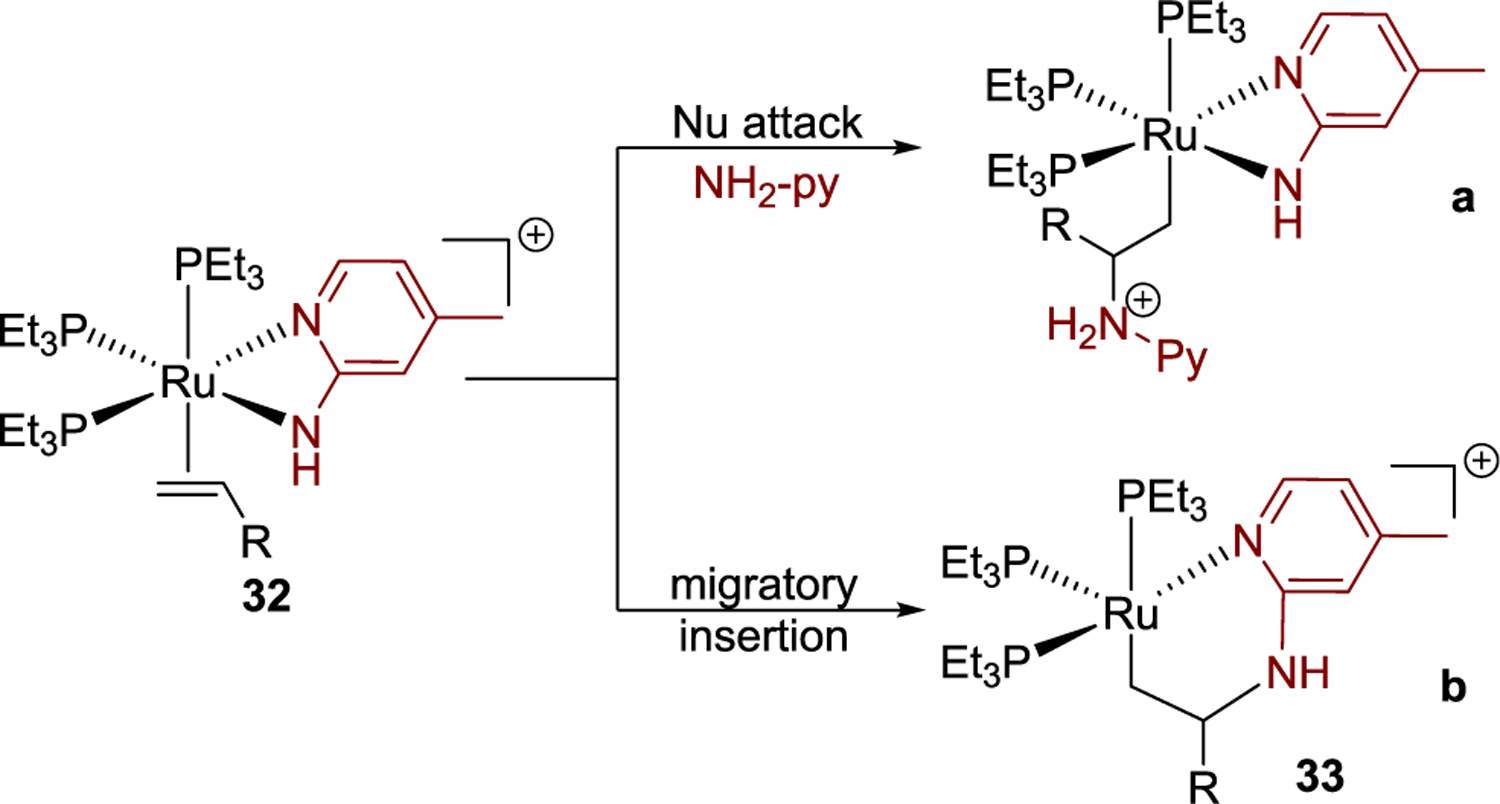
Possible Pathways for the Formation of the C–N Bond
The effect of the concentration of PEt3 on the rate of the catalytic hydroamination was also studied. Added PEt3 inhibited the reaction, and a plot of 1/initial rate against the concentration of PEt3 was linear (Figure 4d). To account for this observation, we considered two hypotheses. First, the added PEt3 could inhibit the reaction by reversibly coordinating to complex 30 to generate complex 34 (Scheme 4a). Second, reversible dissociation of PEt3 from complex 30 could generate the catalytically active complex 35 (Scheme 4b). To test these hypotheses, variable-temperature NMR spectroscopy analysis was conducted with a 5:1 mixture of PEt3 and complex 30. Full conversion of 30 to complex 34 was observed by 31P NMR spectroscopy of this mixture at −20 °C (see the Supporting Information for details). In addition, the association of PEt3 to complex 30 was computed by DFT to be exergonic by 0.3 kcal/mol at −20 °C, whereas the dissociation of PEt3 from complex 30 was calculated to be endergonic by an energy (31.5 kcal/mol at 80 °C) that exceeds the entire 30.5 kcal/mol barrier for the reaction determined from the experimental rates. Therefore, we conclude that the origin of the inhibition of the hydroamination by added PEt3 results from the reversible coordination of PEt3 to complex 30 to form the inactive tetraphosphine complex 34 (Scheme 4a).
Scheme 4.
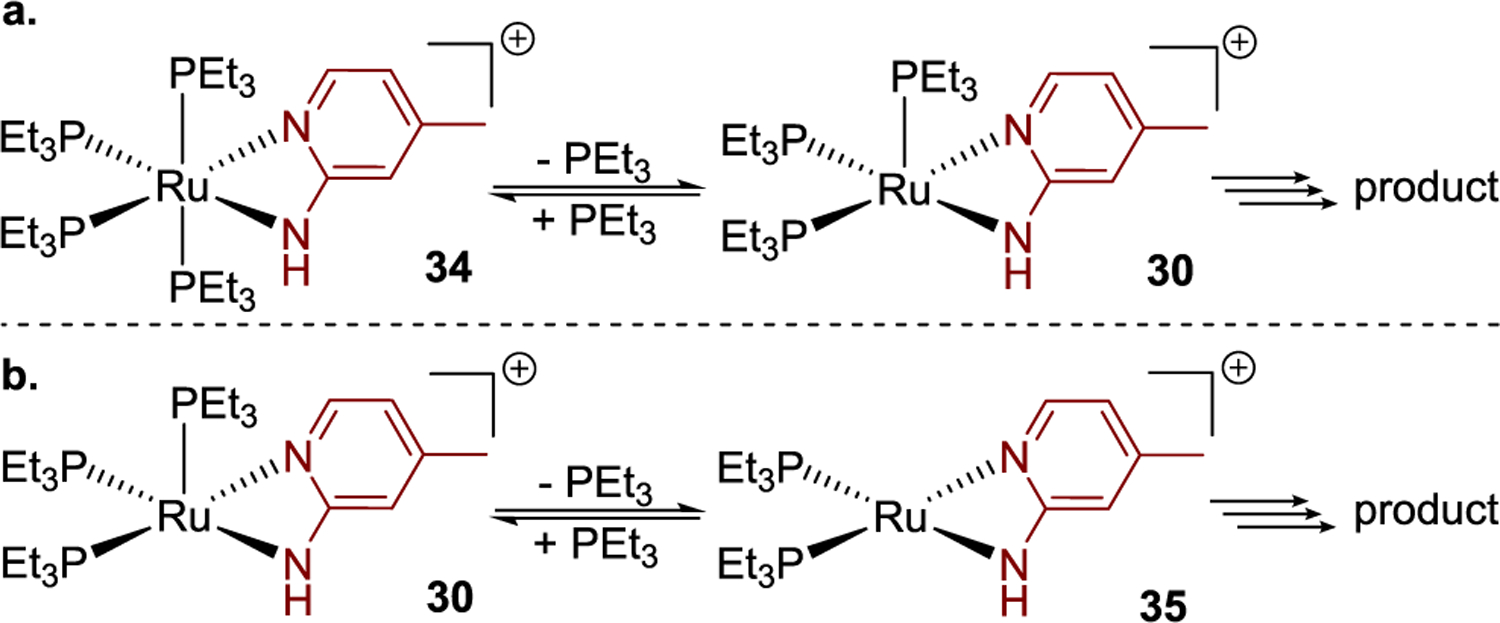
Two Hypotheses to Explain the Rate Inhibition by Additional PEt3
Studies on the Pathway for the Formation of the Product from Ru–Alkyl Complex 33.
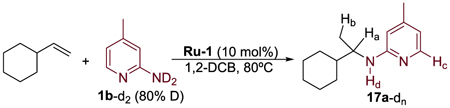
To elucidate the pathway by which the product of this hydroamination reaction was formed from the Ru–alkyl complex 33, the hydroamination of vinylcyclohexane was performed with 1b-d2 under the standard catalytic conditions (Table 4). Deuterium incorporation was observed at four different positions in the product 17a-dn. The ratios of the percentage of deuterium incorporation at the Ha and Hb positions were 1:1 at various time points during the reaction (Table 4), and the accumulation of deuterium into the starting vinylcyclohexane was not observed.
Table 4.
Hydroamination of Vinylcyclohexane with 1b-d2a
%D incorporation = moles of D atoms/moles of product.
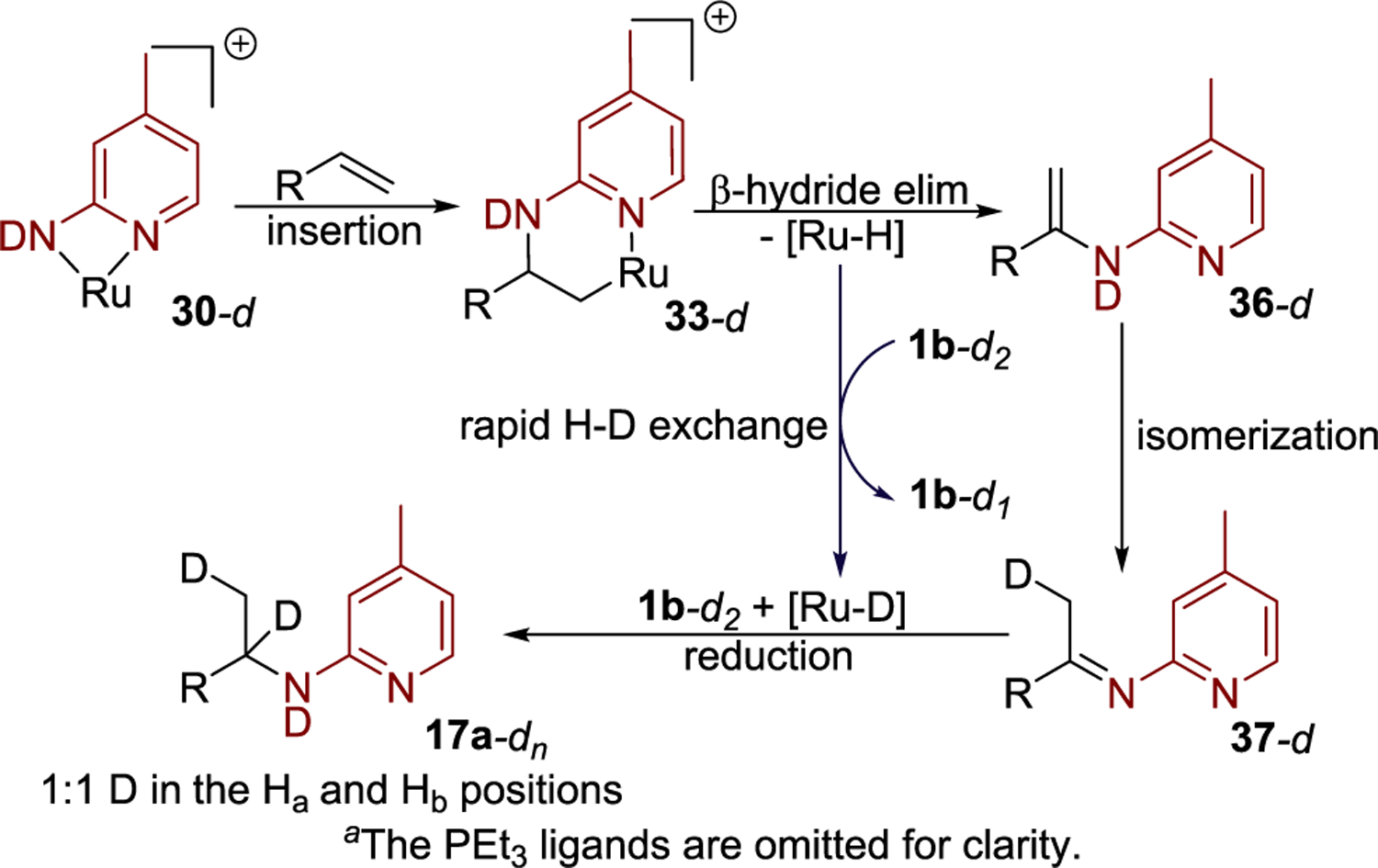
The results of this labeling study are consistent with the pathway in Scheme 5. By this pathway, coordination and migratory insertion of the alkene occur to form Ru–alkyl complex 33-d from the catalyst resting state 30-d, after which complex 33-d undergoes a subsequent β-hydride elimination to generate the ruthenium–hydride species 44 ([Ru–H]) (Figure 5a) and enamine 36-d. The enamine 36-d would then undergo tautomerization catalyzed by protonated aminopyridine, which was formed during the generation of the catalyst resting state 30-d. This tautomerization forms the β-deuterated imine 37-d. Intramolecular H–D exchange between the Ru–H position and bound aminopyridine in complex 44 would generate a ruthenium–deuteride. Subsequent reduction of imine 37-d by complex 44 containing a Ru–D bond would generate the final hydroamination product 17a-dn, containing an enhanced level of deuterium at the Hα and Hβ positions. Because the tautomerization step and H/D exchange of the hydride position of complex 44 occur by reversible proton transfers involving the amino group in 1b, the amounts of deuterium in the Ha and Hb positions of the product are equal to each other. Furthermore, the observation of the incorporation of deuterium at the α position of the product 17a-dn provides evidence against a nucleophilic attack of 1b on the metal-bound alkene complex, like 32 (Scheme 3, pathway a), because the coordinative saturation of the complex formed after a nucleophilic attack of the aminopyridine would disfavor β-hydrogen elimination.
Scheme 5. Proposed Pathway for the Formation of Hydroamination Product from 33a.
aThe PEt3 ligands are omitted for clarity.
Figure 5.
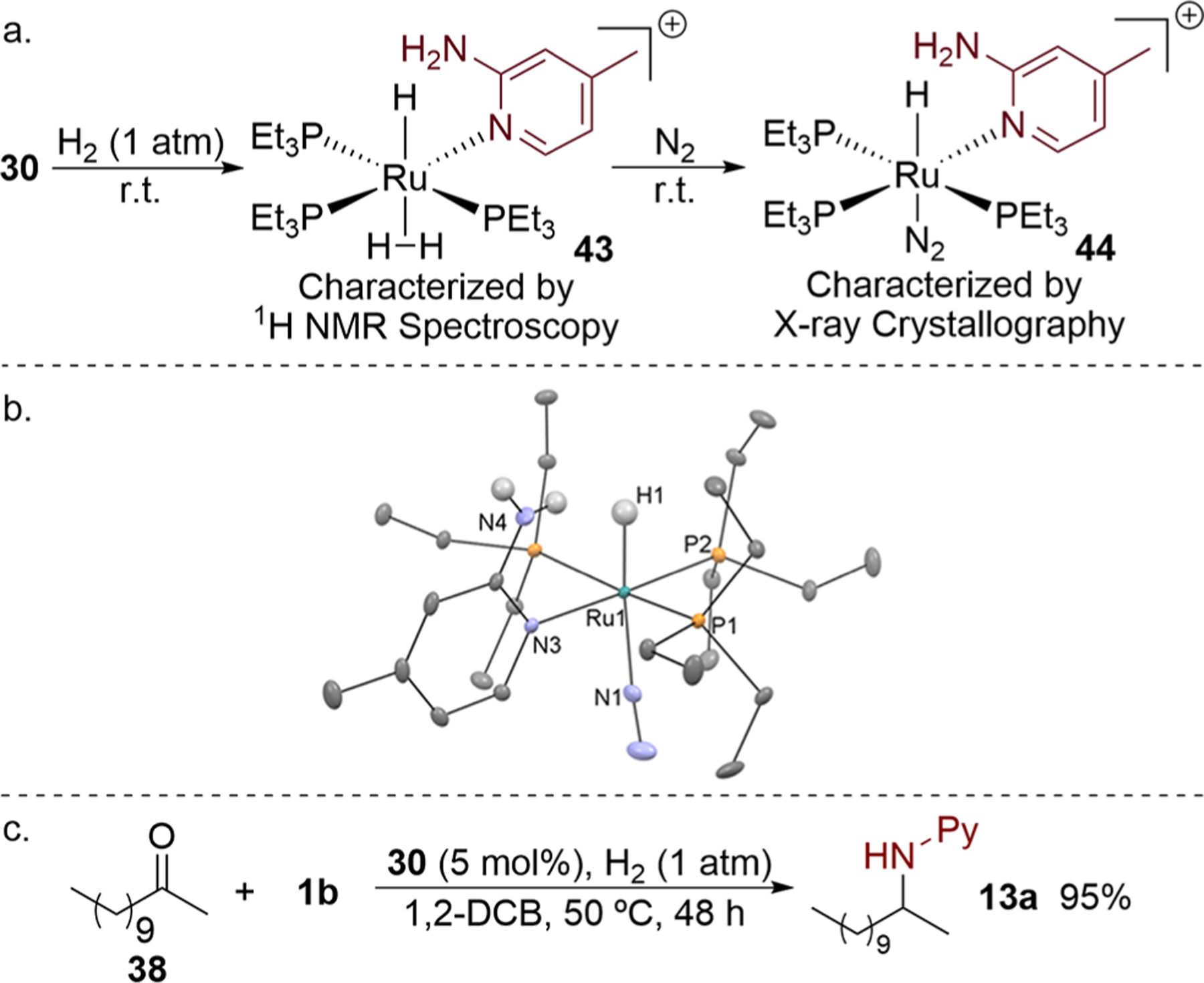
(a) Synthesis of the [Ru–H] intermediate 44. (b) Solid-state structure of complex 44 with ellipsoids set at 30% and selected hydrogen atoms and free triflimide anion omitted for clarity. (c) Catalytic imine hydrogenation with complex 30 in the presence of H2.
To investigate the pathway for the formation of the hydroamination product further, we conducted the hydroamination of 1-dodecene with 1b in the presence of 1 equiv of acetone. 2-Dodecanone 38 and the product from reductive amination of acetone 39 were observed, along with the hydroamination product 13a (Scheme 6). The formation of 38 and 39 implies that the reaction occurs through an imine intermediate. Dodecyl enamine 40 is generated by migratory insertion of the alkene into the amidopyridine complex 30 and β-hydride elimination from metallacycle 33. A subsequent tautomerization of the enamine 40 forms dodecyl imine 42. Isopropyl imine 41 and water are generated by reversible condensation of acetone with aminopyridine 1b. The imines 41 and 42 undergo competitive reduction to yield amines 13a and 39, respectively. The portion of imine 42 that is not reduced is hydrolyzed to give ketone 38, which is detected by 1H NMR spectroscopy.
Scheme 6.
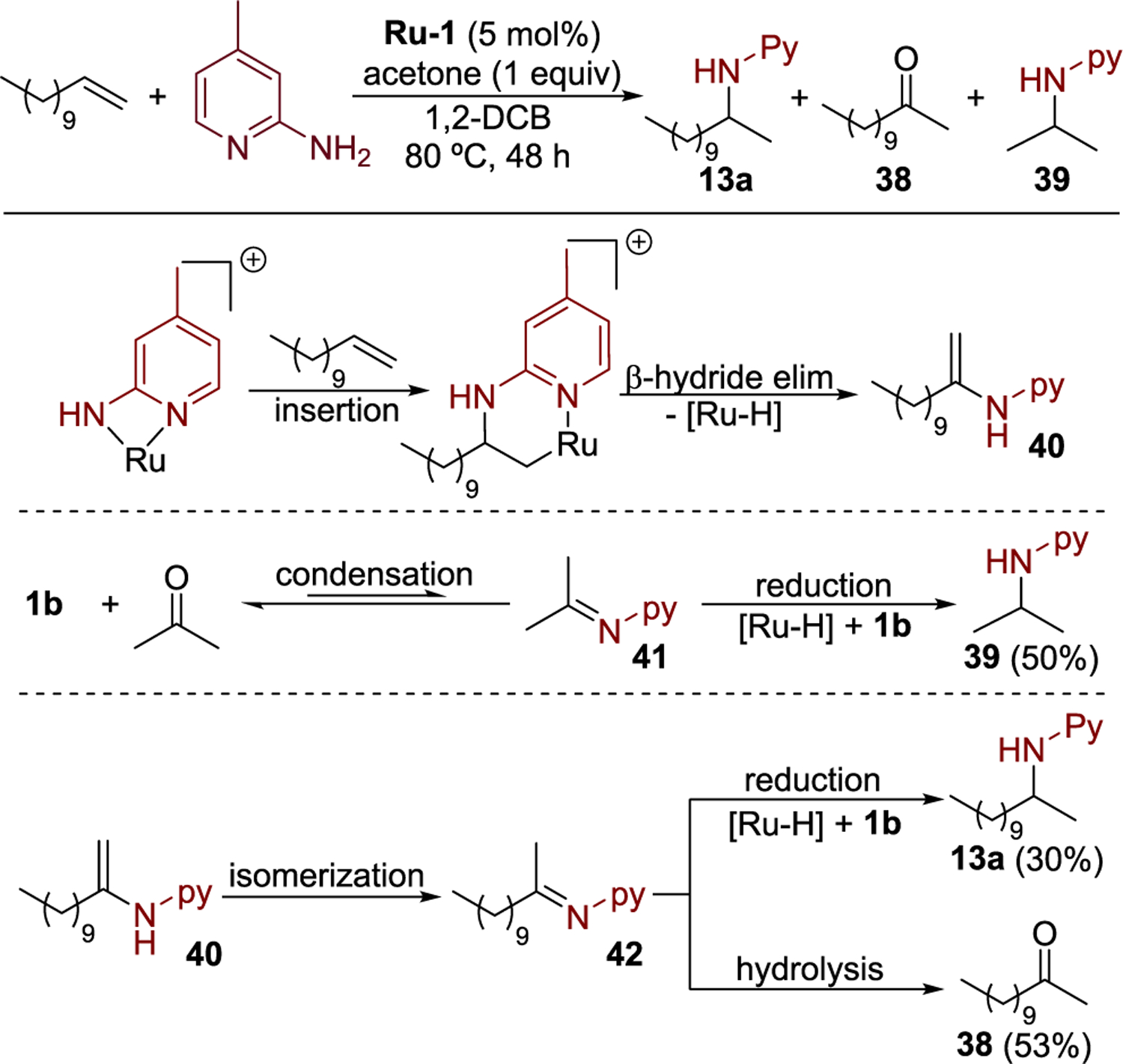
Hydroamination of 1-Dodecene in the Presence of Acetone
To elucidate the identity of the proposed [Ru–H] intermediate that would result from β-H elimination of the aminoalkyl intermediate, we exposed complex 30 to 1 atm of H2 (Figure 5a). Complex 43, which resulted from the overall addition of H2 across the Ru–N bond of complex 30 and coordination of H2, was observed. The structure of this complex was deduced by the observation of two distinct ruthenium hydride signals in the 1H NMR spectrum. The H2 ligand in 43 was slowly replaced by N2 in a nitrogen atmosphere to afford complex 44, which was characterized by single-crystal X-ray diffraction (Figure 5a,b).
Under an atmosphere of H2, the imine generated in situ from ketone 38 and aminopyridine 1b underwent hydrogenation to form amine 13a catalyzed by the amido complex 30 in 95% yield (Figure 5c). This result indicates that the hydridoruthenium complex 43 that would form in situ during the catalytic process from complex 30 and H2 catalyzes the reduction of imines, such as 41 that would be formed in the catalytic process, to generate the final amine 13a.
DFT Computational Studies of the Proposed Catalytic Cycle for the Ruthenium-Catalyzed Hydroamination of Terminal Alkenes.
Our mechanistic experiments imply that this Ru-catalyzed hydroamination of terminal alkenes with 2-aminopyridine occurs by coordination of alkene to the catalyst resting state 30, followed by migratory insertion of the alkene into the Ru–N bond, β-hydride elimination to generate an enamine, tautomerization of the enamine to an imine, and reduction of the imine to afford to amine product. Our kinetic studies indicate that the alkene and the ruthenium catalyst are involved in the turnover-limiting step. Hydrogenation of the imine cannot be turnover limiting because this step occurs faster than the overall hydroamination of the alkene (Figure 5c). To determine whether migratory insertion or β-hydride elimination following a reversible insertion process is turnover-limiting, DFT computations were conducted on the oxidative amination portion of the mechanism.
This mechanism was calculated for the hydroamination of propene, as a model alkene, with aminopyridine 1b. An energy diagram for this process is shown in Figure 6. The coordination of propene to the catalyst resting state 30 was calculated to be endergonic at 16.2 kcal/mol. The free-energy barrier to the elementary migratory insertion of propene into the Ru–N bond was computed to be 12.5 kcal/mol (32 to TS1). These energies lead to an overall barrier for coordination and insertion of 28.7 kcal/mol. The metallacycle 33 formed from insertion was computed to lie 8.1 kcal/mol uphill of the starting complex and free propene, and the barrier for β-hydride elimination from complex 33 was computed to be 11.2 kcal/mol (33 to TS2). Thus, the transition state for the β-hydride elimination lies 19.3 kcal/mol uphill of the starting species and nearly 10 kcal/mol below the transition state for migratory insertion. The oxidative amination portion of the hydroamination reaction was calculated to be uphill by 5.9 kcal/mol, and the overall hydroamination was computed to be exergonic by 3.8 kcal/mol. These calculations strongly suggest that the combination of coordination of alkene and migratory insertion of the alkene into the Ru–N bond is irreversible and the turnover-limiting portion of this hydroamination reaction.
Figure 6.
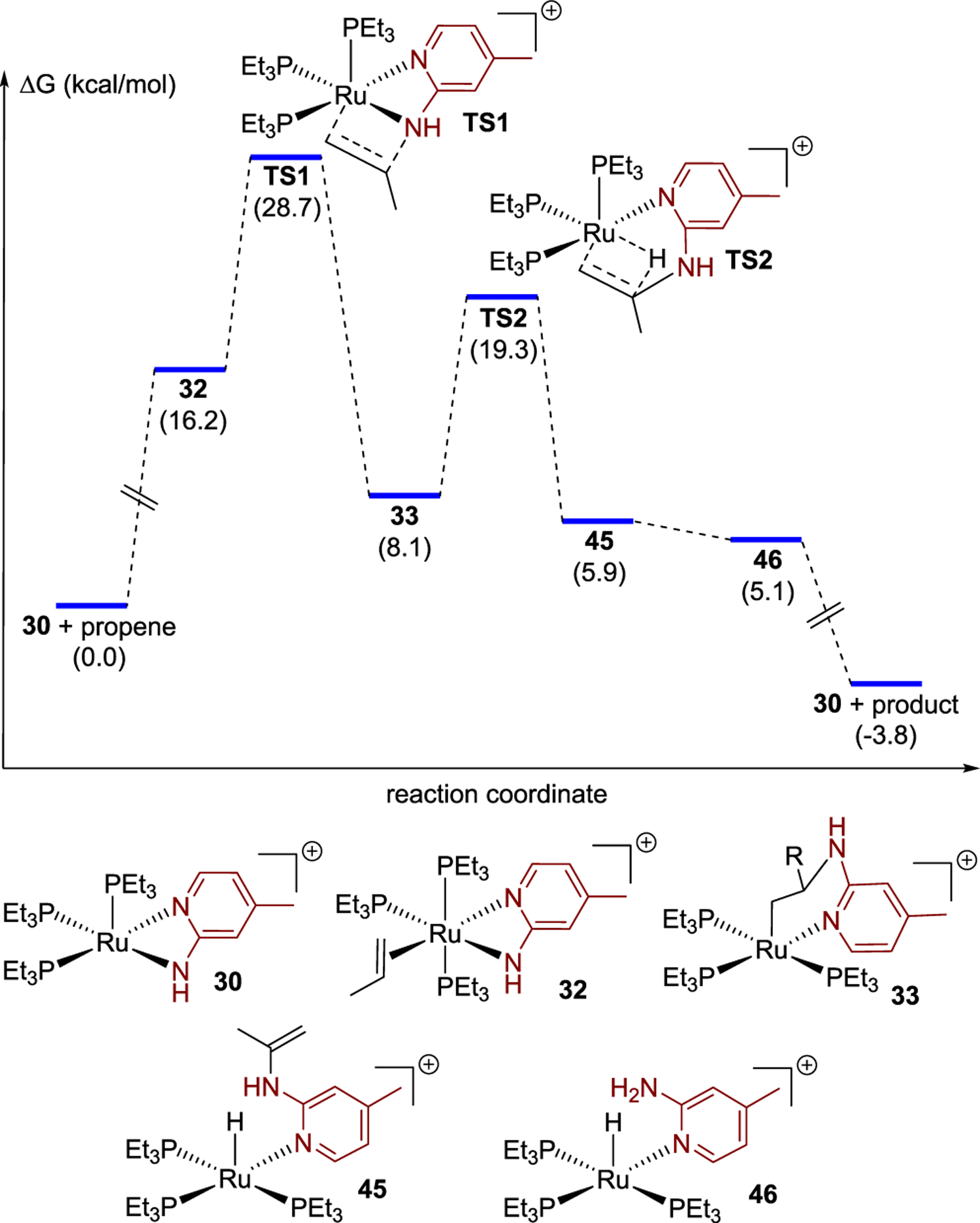
DFT computational studies on the ruthenium-catalyzed hydroamination of terminal alkenes. Free energies in kcal/mol at 80 °C are provided in parentheses.
The results of our mechanistic investigation are summarized in Figure 7. By this mechanism, the major component of the catalyst resting state, complex 30, is formed by coordination of the aminopyridine 1b to the ruthenium precatalyst Ru-1, followed by deprotonation of the aminopyridine ligand by another molecule of 1b. The first step of the catalytic cycle, then, involves reversible coordination of the alkene to 30 to form the alkene complex 32. Turnover-limiting migratory insertion of the alkene into the Ru–N bond of 32 then affords the Ru–alkyl intermediate 33, which undergoes β-hydride elimination to generate the hydridoruthenium enamine intermediate 45. Subsequent exchange of the enamine for aminopyridine 1b in intermediate 45 forms the hydridoruthenium aminopyridine intermediate 46 and free enamine 36. Eliminated enamine 36 undergoes tautomerization to generate imine 37, which is computed to be more stable thermodynamically than enamine 36 by 3.3 kcal/mol and would be expected to react with amine-ligated ruthenium hydride complexes by a metal–ligand bifunctional mechanism.40 Thus, reduction of imine 37 by intermediate 46 affords the final hydroamination product.
Figure 7.
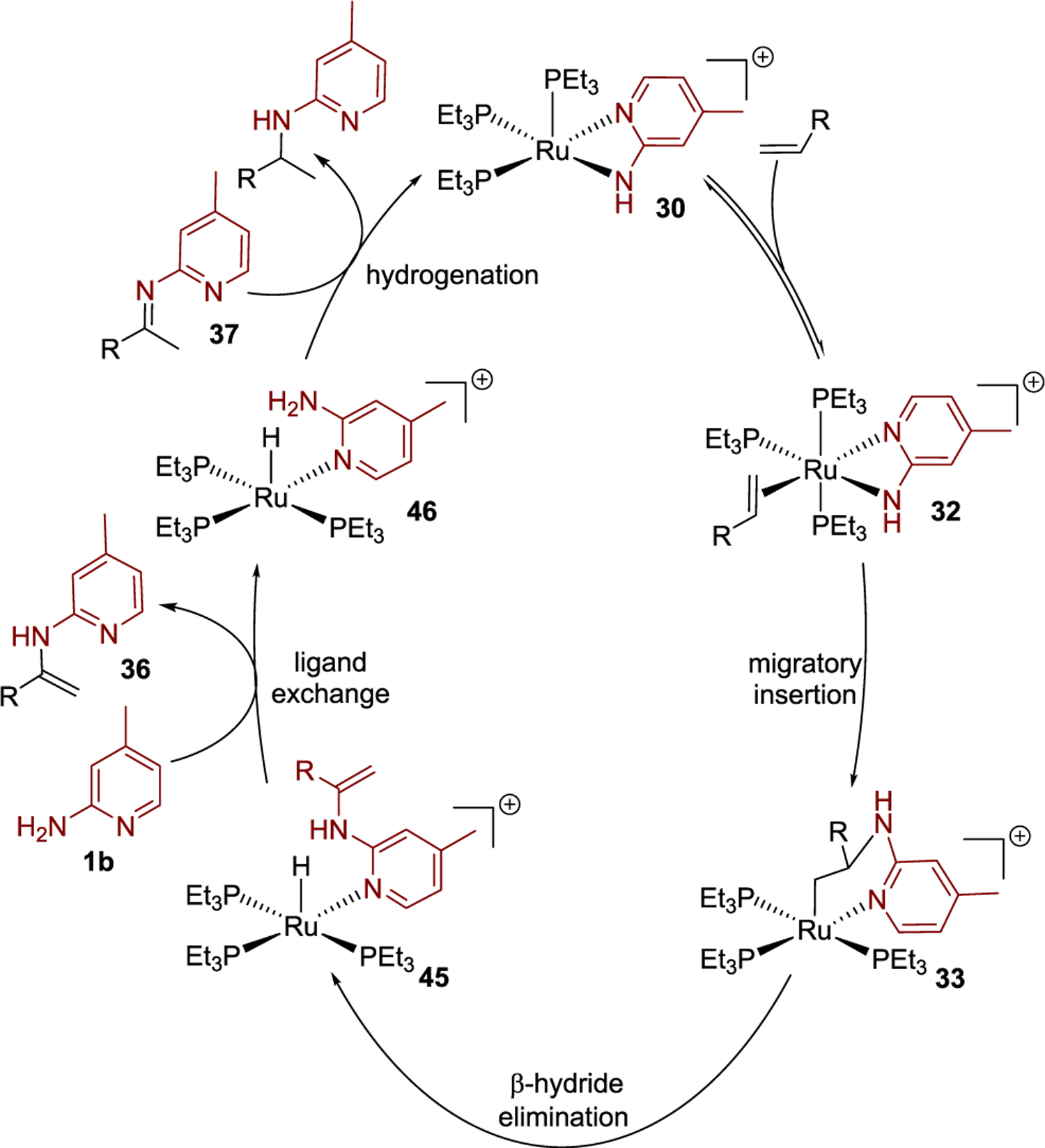
Proposed catalytic cycle for the ruthenium-catalyzed hydroamination of terminal alkenes.
The proposed catalytic cycle is supported by a large body of experimental data. The catalyst resting state 30 was identified by 31P NMR spectroscopy and an independent synthesis. The kinetic measurements support a mechanism by which migratory insertion of the alkene into the Ru–N bond occurs, rather than a potential nucleophilic attack on a coordinated alkene by the amine. Deuterium labeling experiments indicate that the Ru–alkyl complex 33 undergoes β-hydride elimination, rather than a potential direct protonation by 1b, and hydroamination reactions conducted with added acetone imply the formation of an imine intermediate. Independent studies of the reactivity of the ruthenium–hydride complex arising from β-hydride elimination with the imine resulting from tautomerization of the initially formed enamine show that the reduction of the imine is much faster than the overall catalytic cycle. Finally, DFT calculations indicate that the turnover-limiting step of the reaction is migratory insertion of the alkene into a ruthenium amido complex, rather than β-hydride elimination from a reversibly formed insertion product.
CONCLUSION
Ruthenium-catalyzed Markovnikov hydroamination of both unactivated and activated terminal alkenes occurs with 2- aminopyridine as a surrogate for ammonia with a stoichiometric amount of alkene by an unusual pathway for hydroamination. This process constitutes a rare example of hydroamination of alkenes with ruthenium, and it is enabled by a combination of a cationic metal center and a carefully designed aminopyridine as an ammonia surrogate. This combination facilitates the deprotonation of the aminopyridine coordinated to an electron-deficient ruthenium center, the migratory insertion of the alkene into the strained four-member ruthenacycle, and the cooperative reduction of the imine intermediate generated from β-hydrogen elimination to lead to an overall redox-neutral addition process. This reaction proceeds with a variety of terminal alkenes to afford the amine products under conditions with the alkene in stoichiometric quantities. A combination of experimental and computational mechanistic studies reveals that this hydroamination reaction occurs by turnover-limiting migratory insertion of the alkene into the Ru–N bond, followed by β-hydride elimination to generate an enamine, tautomerization of the enamine to an imine, and reduction of the imine by the hydridoruthenium aminopyridine complex to generate the amine product. This pathway implies that an enantioselective process could be developed if the step involving reduction of the imine intermediate can be rendered enantioselective. Studies to achieve such a process by this mechanism are ongoing.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH (R35 GM130387), the Molecular Graphics and Computation Facility at UC Berkeley (NSF CHE-0840505) for DFT calculations, and the College of Chemistry’s NMR facility at UC Berkeley (NIH S10OD024998) for NMR experiments. We gratefully acknowledge Dr. Hasan Celik for assistance with NMR experiments, Dr. Nicholas Settineri for X-ray crystallography (NIH S10-RR027172), Dr. David Small and Yehao Qiu for assistance with DFT calculations, and Dr. Yumeng Xi for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c11043.
Experimental procedures, characterization of new compounds, information about computational studies, and crystallographic information (PDF)
X-ray crystallographic data for Ru(PEt3)3(NTf2)2 (CIF)
X-ray crystallographic data for Ru(PEt3)3(amine)-(NTf2)2 (29) (CIF)
X-ray crystallographic data for Ru(PEt3)3(amido)(NTf2) (30) (CIF)
X-ray crystallographic data for Ru(H)(PEt3)3(amine)-(NTf2)2 (44) (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c11043
The authors declare no competing financial interest.
Contributor Information
Senjie Ma, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
Christopher K. Hill, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
Casey L. Olen, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
REFERENCES
- (1).Bagley MC; Dale JW; Merritt EA; Xiong X Thiopeptide Antibiotics. Chem. Rev 2005, 105, 685–714. [DOI] [PubMed] [Google Scholar]
- (2).Nugent TC Chiral Amine Synthesis: Methods, Developments and Applications; Wiley-VCH: Weinheim, 2010. [Google Scholar]
- (3).Wang C; Bai X; Wang R; Zheng X; Ma X; Chen H; Ai Y; Bai Y; Liu Y Synthesis of Imatinib by C-N Coupling Reaction of Primary Amide and Bromo-Substituted Pyrimidine Amine. Org. Process Res. Dev 2019, 23, 1918–1925. [Google Scholar]
- (4).Shen Q; Hartwig JF [(CyPF-tBu)PdCl2]: An Air-Stable, One-Component, Highly Efficient Catalyst for Amination of Heteroaryl and Aryl Halides. Org. Lett 2008, 10, 4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Afanasyev OI; Kuchuk E; Usanov DL; Chusov D Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev 2019, 119, 11857–11911. [DOI] [PubMed] [Google Scholar]
- (6).Tripathi RP; Verma SS; Pandey J; Tiwari VK Recent Development on Catalytic Reductive Amination and Applications. Curr. Org. Chem 2008, 12, 1093–1115. [Google Scholar]
- (7).Müller TE; Beller M Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev 1998, 98, 675–704. [DOI] [PubMed] [Google Scholar]
- (8).Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- (9).Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- (10).Pawlas J; Nakao Y; Kawatsura M; Hartwig JF A General Nickel-Catalyzed Hydroamination of 1,3-Dienes by Alkylamines: Catalyst Selection, Scope, and Mechanism. J. Am. Chem. Soc 2002, 124, 3669–3679. [DOI] [PubMed] [Google Scholar]
- (11).Yang X-H; Lu A; Dong VM Intermolecular Hydroamination of 1,3-Dienes to Generate Homoallylic Amines. J. Am. Chem. Soc 2017, 139, 14049–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Utsunomiya M; Hartwig JF Intermolecular, Markovnikov Hydroamination of Vinylarenes with Alkylamines. J. Am. Chem. Soc 2003, 125, 14286–14287. [DOI] [PubMed] [Google Scholar]
- (13).Utsunomiya M; Hartwig JF Ruthenium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc 2004, 126, 2702–2703. [DOI] [PubMed] [Google Scholar]
- (14).Zhou J; Hartwig JF Intermolecular, Catalytic Asymmetric Hydroamination of Bicyclic Alkenes and Dienes in High Yield and Enantioselectivity. J. Am. Chem. Soc 2008, 130, 12220–12221. [DOI] [PubMed] [Google Scholar]
- (15).Dorta R; Egli P; Zürcher F; Togni A The [IrCl-(Diphosphine)]2/Fluoride System. Developing Catalytic Asymmetric Olefin Hydroamination. J. Am. Chem. Soc 1997, 119, 10857–10858. [Google Scholar]
- (16).Teng H-L; Luo Y; Nishiura M; Hou Z Diastereodivergent Asymmetric Carboamination/Annulation of Cyclopropenes with Aminoalkenes by Chiral Lanthanum Catalysts. J. Am. Chem. Soc 2017, 139, 16506–16509. [DOI] [PubMed] [Google Scholar]
- (17).Teng H-L; Luo Y; Wang B; Zhang L; Nishiura M; Hou Z Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem., Int. Ed 2016, 55, 15406–15410. [DOI] [PubMed] [Google Scholar]
- (18).Sevov CS; Zhou J; Hartwig JF Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. J. Am. Chem. Soc 2012, 134, 11960–11963. [DOI] [PubMed] [Google Scholar]
- (19).Sevov CS; Zhou J; Hartwig JF Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc 2014, 136, 3200–3207. [DOI] [PubMed] [Google Scholar]
- (20).Nguyen HN; Lee H; Audörsch S; Reznichenko AL; Nawara-Hultzsch AJ; Schmidt B; Hultzsch KC Asymmetric Intra- and Intermolecular Hydroamination Catalyzed by 3,3′-Bis-(trisarylsilyl)- and 3,3′-Bis(arylalkylsilyl)-Substituted Binaphtholate Rare-Earth-Metal Complexes. Organometallics 2018, 37, 4358–4379. [Google Scholar]
- (21).Miller DC; Ganley JM; Musacchio AJ; Sherwood TC; Ewing WR; Knowles RR Anti-Markovnikov Hydroamination of Unactivated Alkenes with Primary Alkyl Amines. J. Am. Chem. Soc 2019, 141, 16590–16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang Z; Lee SD; Widenhoefer RA Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes. J. Am. Chem. Soc 2009, 131, 5372–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science 2017, 355, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xi Y; Ma S; Hartwig JF Catalytic asymmetric addition of an amine N-H bond across internal alkenes. Nature 2020, 588, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang J; Yang C; He C Gold(I)-Catalyzed Intra- and Intermolecular Hydroamination of Unactivated Olefins. J. Am. Chem. Soc 2006, 128, 1798–1799. [DOI] [PubMed] [Google Scholar]
- (26).Reznichenko AL; Nguyen HN; Hultzsch KC Asymmetric Intermolecular Hydroamination of Unactivated Alkenes with Simple Amines. Angew. Chem., Int. Ed 2010, 49, 8984–8987. [DOI] [PubMed] [Google Scholar]
- (27).Gurak JA; Yang KS; Liu Z; Engle KM Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc 2016, 138, 5805–5808. [DOI] [PubMed] [Google Scholar]
- (28).Vanable EP; Kennemur JL; Joyce LA; Ruck RT; Schultz DM; Hull KL Rhodium-Catalyzed Asymmetric Hydroamination of Allyl Amines. J. Am. Chem. Soc 2019, 141, 739–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhu S; Buchwald SL Enantioselective CuH-Catalyzed Anti-Markovnikov Hydroamination of 1,1-Disubstituted Alkenes. J. Am. Chem. Soc 2014, 136, 15913–15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Guo S; Yang JC; Buchwald SL A Practical Electrophilic Nitrogen Source for the Synthesis of Chiral Primary Amines by Copper-Catalyzed Hydroamination. J. Am. Chem. Soc 2018, 140, 15976–15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Liu Z; Yamamichi H; Madrahimov ST; Hartwig JF Rhodium Phosphine-π-Arene Intermediates in the Hydroamination of Alkenes. J. Am. Chem. Soc 2011, 133, 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cheung HW; So CM; Pun KH; Zhou Z; Lau CP Hydro(trispyrazolyl)borato-Ruthenium(II) Diphosphinoamino Complex-Catalyzed Addition of β-Diketones to 1-Alkynes and Anti-Markovnikov Addition of Secondary Amines to Aromatic 1-Alkynes. Adv. Synth. Catal 2011, 353, 411–425. [Google Scholar]
- (33).Johns AM; Sakai N; Ridder A; Hartwig JF Direct Measurement of the Thermodynamics of Vinylarene Hydroamination. J. Am. Chem. Soc 2006, 128, 9306–9307. [DOI] [PubMed] [Google Scholar]
- (34).Hill CK; Hartwig JF Site-selective oxidation, amination and epimerization reactions of complex polyols enabled by transfer hydrogenation. Nat. Chem 2017, 9, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McBee JL; Bell AT; Tilley TD Mechanistic Studies of the Hydroamination of Norbornene with Electrophilic Platinum Complexes: The Role of Proton Transfer. J. Am. Chem. Soc 2008, 130, 16562–16571. [DOI] [PubMed] [Google Scholar]
- (36).Smout V; Peschiulli A; Verbeeck S; Mitchell EA; Herrebout W; Bultinck P; Vande Velde CML; Berthelot D; Meerpoel L; Maes BUW Removal of the Pyridine Directing Group from α-Substituted N-(Pyridin-2-yl)piperidines Obtained via Directed Ru-Catalyzed sp3 C-H Functionalization. J. Org. Chem 2013, 78, 9803–9814. [DOI] [PubMed] [Google Scholar]
- (37).Ronson TO; Renders E; Van Steijvoort BF; Wang X; Wybon CCD; Prokopcová H; Meerpoel L; Maes BUW Ruthenium-Catalyzed Reductive Arylation of N-(2-Pyridinyl)amides with Isopropanol and Arylboronate Esters. Angew. Chem., Int. Ed 2019, 58, 482–487. [DOI] [PubMed] [Google Scholar]
- (38).Pan S; Endo K; Shibata T Ir(I)-Catalyzed Enantioselective Secondary sp3 C-H Bond Activation of 2-(Alkylamino)pyridines with Alkenes. Org. Lett 2011, 13, 4692–4695. [DOI] [PubMed] [Google Scholar]
- (39).Bruneau C; Dixneuf PH Metal Vinylidenes and Allenylidenes in Catalysis: Applications in Anti-Markovnikov Additions to Terminal Alkynes and Alkene Metathesis. Angew. Chem., Int. Ed 2006, 45, 2176–2203. [DOI] [PubMed] [Google Scholar]
- (40).Cobley CJ; Henschke JP Enantioselective Hydrogenation of Imines Using a Diverse Library of Ruthenium Dichloride-(diphosphine)(diamine) Precatalysts. Adv. Synth. Catal 2003, 345, 195–201. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.