

Abstract
We describe two complementary approaches based on a convergent [4+2] logic toward the synthesis of amorfrutins, cannabinoids, and related plant metabolites. An anionic cascade cyclization employing β-methoxycrotonates and β-chloro-α,β-unsaturated esters yielded amorfrutins in four linear steps and demonstrated utility of β-alkoxycrotonate-derived nucleophiles as functional equivalents of β-ketoester-derived dianions. Analogously, tandem Diels–Alder/retro-Diels–Alder cycloaddition of dimedone-derived bis(trimethylsiloxy)-dienes and α,β-alkynyl ester dienophiles provided facile access to resorcinol precursors of amorfrutins and cannabinoids, avoiding late-stage installation of prenyl or geranyl moieties as in previous approaches.
Graphical Abstract

Amorfrutins are plant-derived natural products based on an isoprenoid-substituted β-resorcylic acid core, with a wide range of biological activities.1,2 Amorfrutin A (1) and B (2) (Figure 1a), isolated from Amorpha fruticosa and Glycyrrhiza foetida, increase insulin production in mice by targeting the peroxisome proliferator-activated receptor gamma (PPARγ), a key regulator of fat and glucose metabolism, and further decrease inflammation.3–5 A structural variant, amorfrutin 2 (3), also demonstrates high affinity to PPARγ despite bearing a pentyl group instead of the phenethyl moiety. Amorfrutin C (4), isolated from the plant Glycyrrhiza foetida, reduces colon cancer progression by disrupting mitochondrial function via an unknown mechanism.6,7 Corresponding resorcinol derivatives of amorfrutin A and B have been isolated from R. complanata and H. umbraculigerum,8–11 and similar resorcinol-based natural products have been described, such as grifolic acid (5) first identified from the fungus Albatrellus confluens.12 Amorfrutins are closely related to cannabinoids, such as CBGA (6), with identical substitution positions of geranyl and pentyl groups about the resorcinol.11
Figure 1.

(a) Examples of amorfrutin and cannabinoid derivatives. (b) Representative previous approaches to amorfrutins (left) and this work (right).
Given their potential pharmaceutical significance, numerous approaches to amorfrutins and related derivatives have been reported over the past decade.3,4,13–22 Earlier syntheses were based on extensive derivatization of phloroglucinol carboxylic acid (7) via Mitsunobu, Sonogashira coupling, and orthoalkylation steps (Figure 1b, approach A).3,4 In application to amorfrutin A (1), an elegant tandem Michael–Claisen condensation approach using ethyl acetoacetate (8) and Michael acceptor (9) provided a 1,3-cyclohexadione adduct, which was converted to the corresponding resorcinol by oxidative aromatization using mercury(II) acetate (approach B).13 Notably, the majority of previous amorfrutin syntheses include a problematic late-stage installation of prenyl or geranyl (R1) substituents, requiring potassium phenolates/hydrophobic solvents to favor C- vs O-alkylation.3,4,13,14 Subsequent iterations of this approach employed a Claisen rearrangement of dimethylallyl ether precursors15 or a decarboxylative prenyl migration–aromatization sequence.16
We envisioned developing shorter and more broadly applicable routes that would minimize the use of activation steps and avoid late-stage installation of the R1 moiety, while increasing the scope to include cannabinoid derivatives such as CBGA (6). Whereas most previous approaches employ aromatic precursors and proceed in a linear fashion, we here describe two routes to amorfrutins and related cannabinoids that both utilize a convergent [4+2] assembly strategy for the aromatic core. Approach C is based on a novel tandem anionic cascade addition sequence using enolates of β-methoxycrotonate methyl esters (10) and β-chloro-α,β-unsaturated Michael acceptors (11). Approach D is based on a Diels–Alder/retro-Diels–Alder step employing dimedone-derived 1,3-bis-(trimethylsiloxy)-dienes (12) and α,β-alkynyl esters (13).23–25
Approach C was inspired by a tandem Michael–Aldol sequence recently described for the synthesis of amorfrutin C (4), which furnished the resorcinol precursor (14) in 56% yield from treatment of the β-chloro-α,β-unsaturated malonate derivative (15) with approximately two equivalents of lithiated 1,3-diprenylacetone (16) (Scheme 1a).7 However, when applied to unsymmetrically substituted ketones, this reaction produced mixtures of difficult-to-separate regioisomers. Next, we considered applicability of a tandem Michael-addition–Dieckmann condensation/annulation sequence as previously employed for the synthesis of mycophenolic acid,26 suggesting a retrosynthetic cut in the ring formation step between C1–C2 (Scheme 1b) as opposed to C2–C3 for the Michael–Aldol approach (Scheme 1a). We envisioned that ring formation to provide amorfrutin esters and derivatives (17) could result from a Dieckmann cyclization of an intermediate (18) derived from Michael addition of deprotonated β-methoxy-α-alkylcrotonate derivatives (10) to (Z)-β-chloro-α,β-unsaturated ester Michael acceptors (11) (Scheme 1b), analogous to a previously described method for the preparation of p-hydroxybenzoates.27
Scheme 1.
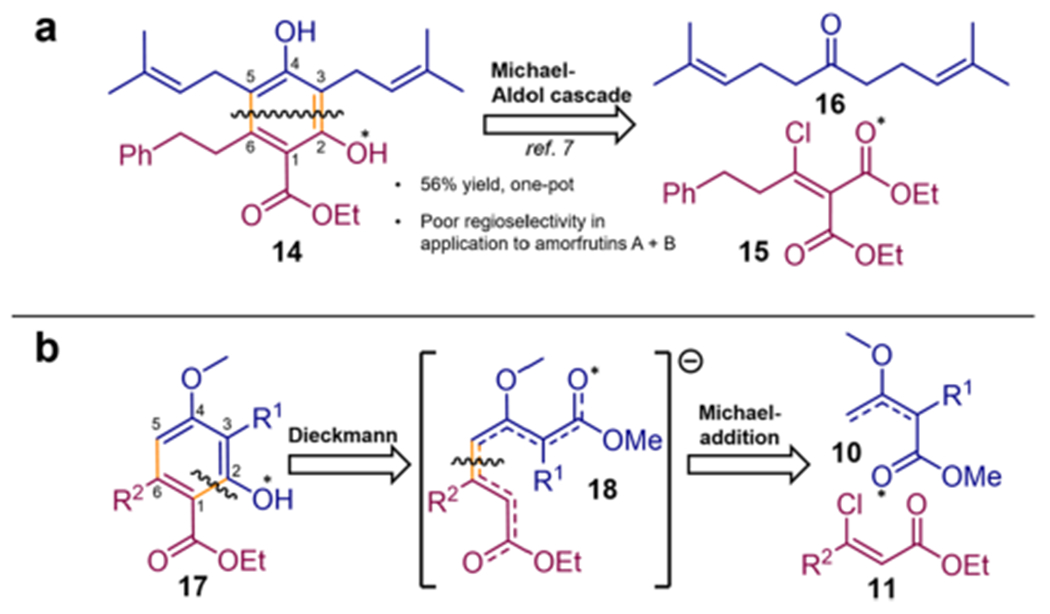
Retrosynthetic Analyses for Tandem Anionic Cascade Addition Approaches
α-Alkylation of methyl β-methoxycrotonate (20)28 derived from commercially available methyl acetoacetate (19) provided facile access to mixtures of α,β- and β,γ-unsaturated enol ethers 10a and 10b (Scheme 2a).29 (Z)-β-Chloro-α,β-unsaturated esters 11a and 11b were prepared in a high yield in one step from terminal alkynes, e.g., 21a and 21b, using a rhodium-catalyzed chloroesterification approach using ethyl chloroformate (22).30
Scheme 2.
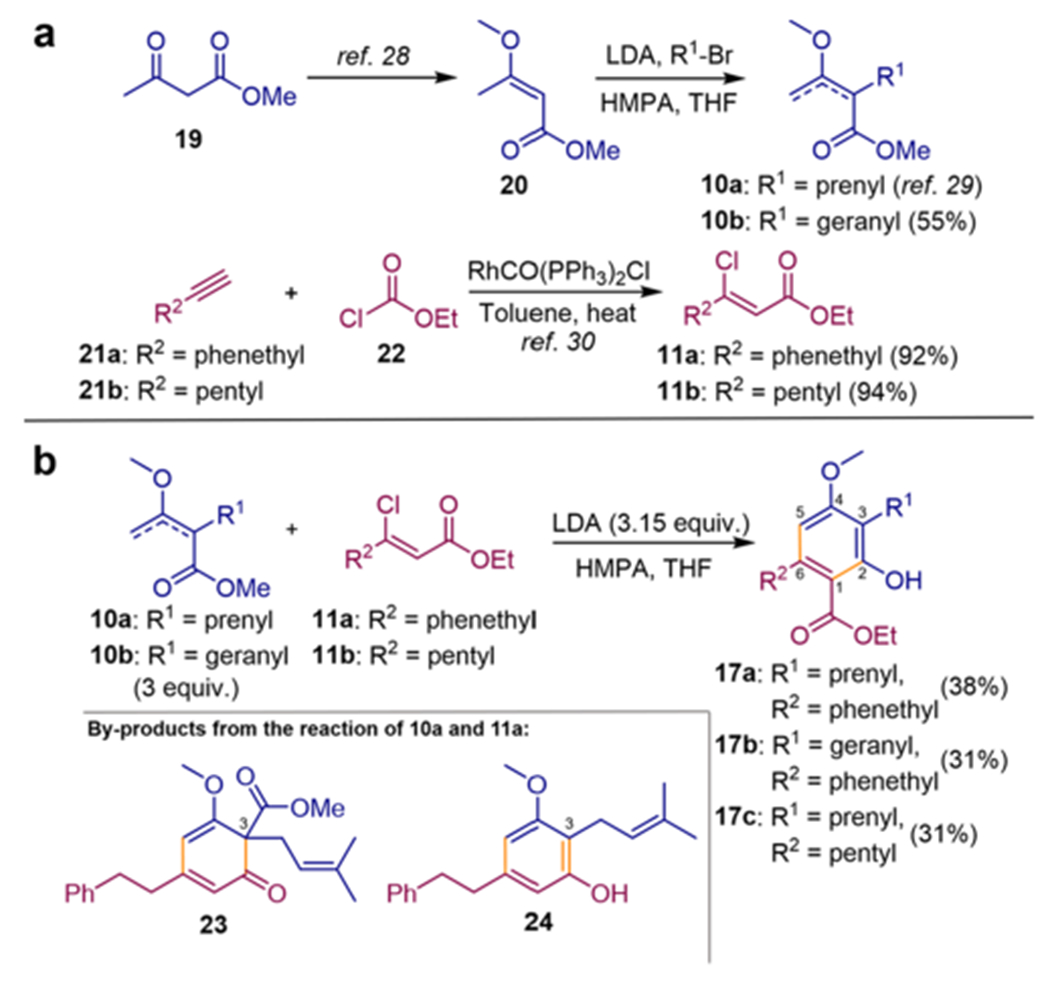
Synthesis of Amorfrutin Derivatives via the Anionic Cascade Approach
Provided that initial Michael addition to β-chloroester Michael acceptors consumes one equivalent of base, it appeared that at least two equivalents of base were required to allow further deprotonation of the Michael adduct for subsequent Dieckmann cyclization, such as for the Michael–Aldol annulation (Scheme 1a). Varying equivalents of LDA and enol ether 10a/b, we found that use of three equivalents of base and enol ether was necessary for complete consumption of β-chloroesters 11, affording the desired amorfrutin ethyl esters 17a–c as well as byproducts (i.e., 23 and 24) derived from alternative cyclization via C3 and the ethyl ester moiety (see Figure S1), which could be easily separated. Byproduct formation could be partially suppressed by addition of a stoichiometric amount of HMPA. The excess of enol ethers 10a/b was recoverable and recyclable. Although modest yielding, this annulation sequence provided two carbon–carbon bonds in one pot, furnishing esters of amorfrutins A, B, and 2 (17a–c) in three linear steps starting from methyl acetoacetate (19).
Next, we examined a related retrosynthetic approach based on the formation of the same carbon–carbon bonds as in the Michael–Dieckmann cascade approach, but instead using a Diels–Alder reaction. Tandem Diels–Alder/retro-Diels–Alder approaches using 1,3-bis(trimethylsiloxy)-dienes derived from dimedone (i.e., 12a) have been reported as highly regioselective for ene-ynes24 as well as α,β-alkynyl esters (13), e.g., in application to the syntheses of psymberin (25)25 and 1-oxo-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (26) (Scheme 3a).23 We reasoned that the application of this methodology to 2-alkyldimedone derivatives should provide access to diverse amorfrutin and cannabinoid derivatives through their resorcinol precursors (27) (Scheme 3b).
Scheme 3.
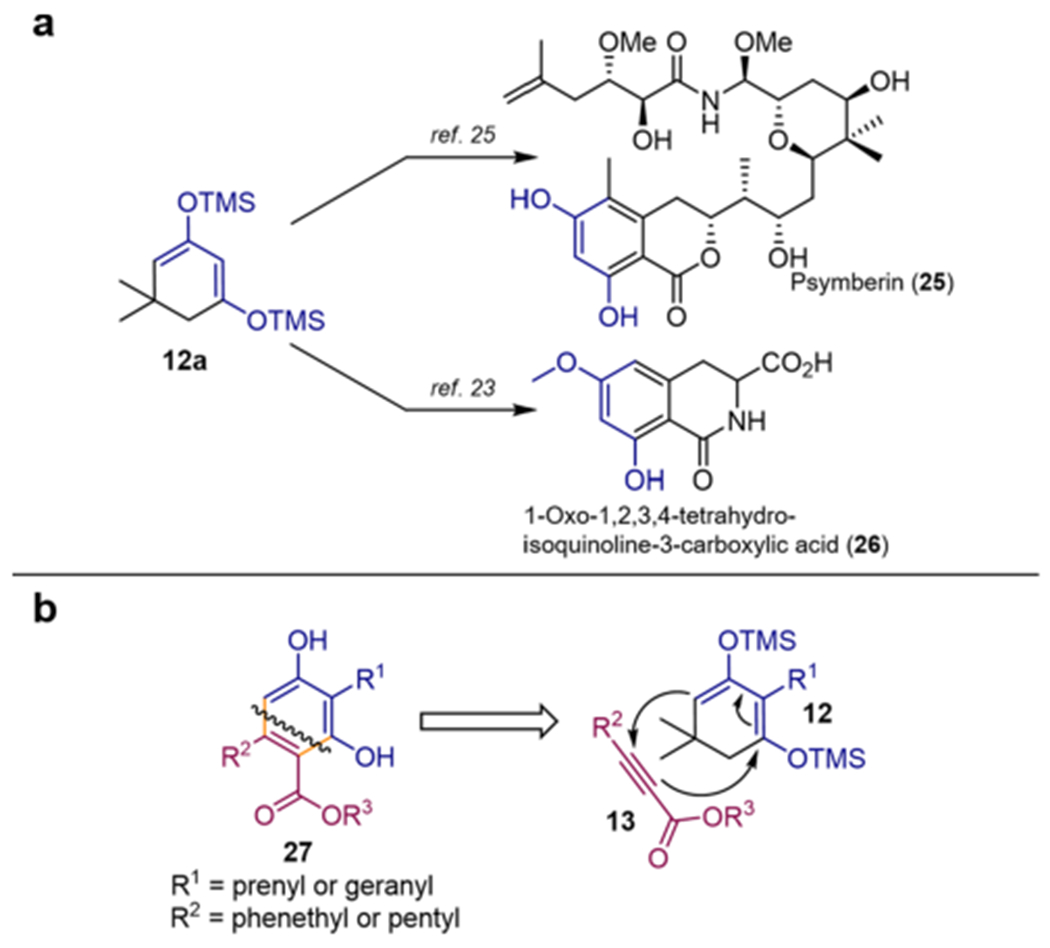
Utility of Tandem Diels–Alder/Retro-Diels–Alder Approaches to Resorcinol Derivatives
The requisite 1,3-bis(trimethylsiloxy)-dienes were prepared in two steps from dimedone (28) (Scheme 4a). Alkylation of dimedone (28) with prenyl and geranyl bromides furnished 2-substituted derivatives 29a and 29b, in addition to O-alkylation and bis-alkylation byproducts,31 which could be removed easily by crystallization of the product from the reaction mixture. The corresponding bis(trimethylsiloxy)-dienes 12b and 12c were prepared analogous to known procedures.23 Silylation proceeds quantitatively, and products require no purification for use in the subsequent Diels–Alder reaction. Dienophile (13a), targeting amorfrutin A and B, was prepared via acylation of 4-phenyl-1-butyne (21a) using nBuLi and ethyl chloroformate (22),32 whereas methyl-2-octynoate (13b), used for the preparation of amorfrutin 2 and cannabinoids, was commercially available.
Scheme 4.

Synthesis of Amorfrutin Derivatives via the Diels–Alder/Retro-Diels–Alder Approach
The Diels–Alder/retro-cycloaddition step was performed neat using dienes 12b or 12c and alkynoates 13a or 13b (Scheme 4b). Complete consumption of dienophiles required high temperatures (~170 °C) and use of up to 4.3 equivalents of dienes 12b and 12c. Gratifyingly, 1H NMR spectra of the crude reaction mixtures revealed formation of a single aromatic product, indicating near 100% regioselectivity (Figure S2). Following desilylation of the reaction crudes using SiO2/MeOH,25 pure resorcinols were obtained in excellent yields, including amorfrutin A and B precursors, 27a (89%, based on dienophile) and 27b (84%), respectively, from dienophile 13a as well as amorfrutin 2 precursor 27c (85%), and cannabinoid precursor 27d (80%) from methyl-2-octynoate (13b). Excess diones 29a and 29b could be recovered almost quantitatively by crystallization and/or chromatography from the mixture following desilylation.
Methylation of resorcinols 27a and 27b afforded amorfrutin ethyl esters 17a (66%, 77% BRSM) and 17b (60%, 67% BRSM) (Scheme 4b). Although regioselective, we did observe formation of up to 20% of bis-methylated derivatives, even when methylation was terminated prior to complete consumption of starting material. Subsequent alkaline hydrolysis14,15 afforded amorfrutins A (1) and B (2) in four linear steps from methyl acetoacetate (19) and five linear steps from dimedone (28). Further, resorcinol 27d was hydrolyzed and decarboxylated to afford pure cannabigerol (CBG, 30) in 81% yield. Alternatively, careful treatment of 27d with concentrated aqueous sodium sulfide has been reported to allow hydrolysis to CBGA (5), providing access to CBG and CBGA in four steps from dimedone.33 While short synthetic approaches to CBG have been described from the condensation of geraniol and olivetol, additional undesired regioisomers and bisgeranylated products have been reported.34,35
In summary, we here present two complementary convergent approaches to amorfrutins and related resorcinol derivatives, starting from simple, acyclic precursors. The tandem anionic cascade cyclization approach provides access to amorfrutins in only four linear steps from inexpensive precursors. Although yields of the anionic cyclization step are modest, the approach is scalable and products can be isolated easily in high purity. Further, this cyclization reaction demonstrates novel reactivity for a carbonyl-conjugated enol ether, which following deprotonation serves as a functional equivalent to a β-keto ester-derived dianion. In the context of amorfrutin synthesis, the use of the enol ether in this step removed the need for late-stage monomethylation of a resorcinol intermediate; however, substitution of β-alkoxycrotonate-derived nucleophiles for β-keto ester-derived dianions may have more widespread utility. The tandem Diels–Alder/retro-Diels–Alder cycloaddition provided resorcinol derivatives with complete regiospecificity and higher yields than the anionic cascade cyclization approach; however, the preparation of the diene from dimedone is somewhat inefficient. Both routes presented here avoid the use of protection/deprotection steps, accommodate a wide range of substituents R1 and R2, and are shorter than previous approaches to amorfrutins A, B, and 2, demonstrating the advantages of convergent [4+2] strategies for this class of natural products. The methods are applicable to a wide range of amorfrutins and cannabinoids as well as non-natural derivatives and probes, which will facilitate exploration of their biological activities and modes of action.
EXPERIMENTAL SECTION
General Procedures, Materials, and Instrumentation.
All oxygen- and moisture-sensitive reactions were carried out under an argon (Ar) atmosphere in flame-dried glassware. Solutions and solvents sensitive to moisture and oxygen were transferred via standard syringe and cannula techniques. Reaction mixtures were cooled using dry ice–acetone or ice–water baths and heated using a mineral oil bath. Unless stated otherwise, all chemicals and reagents used for synthetic compound preparation were purchased from Sigma-Aldrich and used without further purification. Tetrahydrofuran (THF), toluene, dichloromethane (DCM), and hexamethylphosphoramide (HMPA) were dried and stored over 3 or 4 Å molecular sieves prior to use. Diisopropylamine (DIPA) was passed through a plug of silica and dried over 4 Å molecular sieves. 4-Phenyl-1-butyne and bis(triphenylphosphine)rhodium(I) carbonyl chloride were purchased from BeanTown Chemical. Ethyl chloroformate was purchased from Alfa Aesar. Chloroform (CHCl3), DCM, diethyl ether (Et2O), ethyl acetate (EtOAc), ethanol (EtOH), hexanes, and toluene were purchased from Fisher Scientific. Thin-layer chromatography (TLC) was performed using J. T. Baker Silica Gel IB2F plates. Flash chromatography was performed using Teledyne Isco CombiFlash systems with Teledyne Isco RediSep Rf silica or manually using aluminum oxide, activated, neutral, Brockmann grade 1, 58 Å (Alfa Aesar). All deuterated solvents were purchased from Cambridge Isotopes. Nuclear magnetic resonance (NMR) spectra were recorded on Varian INOVA 600 (600 MHz), Varian INOVA 400 (400 MHz), and Bruker AV500 spectrometers at Cornell University’s NMR facility. 1H NMR chemical shifts are reported in ppm (δ) relative to residual solvent peaks (7.26 ppm for chloroform-d, 7.16 ppm for benzene-d6, 2.05 for acetone-d6). NMR spectroscopic data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants (Hz), and integrals. 13C NMR chemical shifts are reported in ppm (δ) relative to residual solvent peaks (77.16 ppm for chloroform-d, 128.06 ppm for benzene-d6, and 29.84 for acetone-d6). All NMR data processing was done using MNOVA 12.0.1 (https://mestrelab.com/). All high-resolution mass spectrometry data were acquired using a Thermo Scientific Q Exactive HF hybrid quadrupole-orbitrap mass spectrometer and reported to four decimal places.
Methyl 2-Prenyl-3-methoxy-2- and 3-butenoate (10a).
This reagent was prepared according to a previously published procedure using enol ether 2028 and prenyl bromide.29
Methyl 2-Geranyl-3-methoxy-2- and 3-butenoate (10b).
This reagent was prepared according to a previously published procedure.29 To a freshly prepared solution of LDA from nBuLi (4.30 mL, 10.75 mmol, 2.5 M in hexanes, 1.16 equiv) and DIPA (1.40 mL, 10.0 mmol, 1.08 equiv) in 10 mL of THF was added enol ether 20 (1.20 g, 9.23 mmol, 1.00 equiv)28 in 2 mL of THF at −78 °C dropwise under Ar. The resulting solution was stirred up to 0 °C (formation of a white precipitate was observed). HMPA (1.70 mL, 9.78 mmol, 1.06 equiv) was added; then, the solution was cooled down to −78 °C to which geranyl bromide (3.00 g, 13.8 mmol, 1.49 equiv) was added dropwise in 3 mL THF. The reaction mixture was stirred up to room temperature over a 1 h period and then stirred at that temperature for 42 h. The solution was poured into cold sat. aqueous NaHCO3 and extracted with EtOAc (3×). Combined organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on alumina using 3% EtOAc in hexanes afforded 10b (1.40 g, 55%, primarily β,γ isomer) as a colorless oil, with up to ~9% (mol/mol) bis-alkyl impurity. 1H NMR (500 MHz, benzene-d6): δ 5.27 (t, J = 7.1 Hz, 1H), 5.19 (t, J = 7.0 Hz, 1H), 4.14 (d, J = 2.4 Hz, 1H), 3.92 (d, J = 2.4 Hz, 1H), 3.37 (s, 3H), 3.24 (t, J = 7.7 Hz, 1H), 3.14 (s, 3H), 2.82 (ddd, J = 14.6, 7.4 Hz, 1H), 2.66 (ddd, J = 14.6, 7.4 Hz, 1H), 2.12 (q, J = 7.6 Hz, 2H), 2.03 (t, J = 7.3 Hz, 2H), 1.67 (br s, 3H), 1.58 (s, 3H), 1.54 (s, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.3, 161.4, 137.3, 131.1, 124.8, 121.8, 83.0, 54.8, 51.9, 51.5, 40.2, 29.2, 27.1, 25.9, 17.8, 16.1. HRMS (ESI) m/z: [M + H]+ calcd for C16H27O3, 267.1955; found, 267.1951.
(Z)-Ethyl-3-chloro-5-phenylpent-2-enoate (11a).
Chloroester 11a was prepared according to a previously reported procedure.30 Ethyl chloroformate (22, 5.74 mL, 60.3 mmol, 3.0 equiv), 4-phenyl-1-butyne (21a, 2.80 mL, 20.1 mmol, 1.0 equiv), and bis(triphenylphosphine)rhodium(I) carbonyl chloride (139 mg, 0.20 mmol, 0.01 equiv) were dissolved in 16 mL of dry toluene in a sealed container and heated to 110 °C for 18 h. The reaction mixture was cooled to room temperature, quenched with the addition of sat. aqueous NaHCO3, and extracted 3× with EtOAc. Organics were dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–20% EtOAc in hexanes was performed, affording 11a (4.40 g, 92%) as a yellow oil. 1H NMR (500 MHz, benzene-d6): δ 7.13–7.05 (m, 2H), 7.06–6.99 (m, 1H), 6.90–6.84 (m, 2H), 5.71 (t, J = 0.9 Hz, 1H), 3.95 (q, J = 7.1 Hz, 2H), 2.59–2.51 (m, 2H 2.28–2.20 (m, 2H), 0.94 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 163.3, 148.7, 140.0, 128.7, 128.6, 126.6, 117.5, 60.2, 43.0, 33.6, 14.2. HRMS (ESI) m/z: [M + H]+ calcd for C13H16ClO2, 239.0833; found, 239.0830.
(Z)-Ethyl-3-chlorooct-2-enoate (11b).
Chloroester 11b was prepared according to a previously reported procedure.30 Ethyl chloroformate (22, 2.86 mL, 30.0 mmol, 3.00 equiv), 1-heptyne (21b, 1.31 mL, 10.0 mmol, 1.00 equiv, Sigma-Aldrich), and bis(triphenylphosphine)rhodium(I) carbonyl chloride (69.0 mg, 0.10 mmol, 0.01 equiv) were dissolved in 6 mL of dry toluene in a sealed container and heated to 110 °C for 24 h. The reaction mixture was cooled to room temperature, quenched by the addition of sat. aqueous NaHCO3, and extracted 3× with EtOAc. Organics were dried with Na2SO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–20% EtOAc in hexanes afforded 11b (1.93 g, 94%) as a light yellow oil. 1H NMR (500 MHz, benzene-d6): δ 5.85 (t, J = 1.0 Hz, 1H), 4.01 (q, J = 7.1 Hz, 2H), 1.97 (t, J = 7.6 Hz, 2H), 1.29 (p, J = 7.6 Hz, 2H), 1.13–1.02 (m, 2H 1.00–0.92 (m, 2H), 0.98 (t, J = 7.1 Hz, 3H), 0.76 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 163.5, 150.2, 116.9, 60.2, 41.2, 30.8, 27.0, 22.6, 14.3, 14.0. HRMS (ESI) m/z: [M + H]+ calcd for C10H18ClO2, 205.0990; found, 205.0987.
General Procedure for the Anionic Cascade Approach.
To a freshly prepared solution of LDA from nBuLi (0.53 mL, 1.32 mmol, 2.5 M in hexanes, 3.15 equiv) and DIPA (0.18 mL, 1.32 mmol, 3.15 equiv) in 20 mL of THF was added enol ether 10a or 10b (1.26 mmol, 3.00 equiv) at −30 °C dropwise under Ar. The resulting solution was stirred up to approximately 0–5 °C (total time roughly 1 h) and then cooled to −20 °C after which HMPA (0.22 mL, 1.26 mmol, 3.0 equiv) was added, followed by dropwise addition of chloroester 11a or 11b (0.42 mmol, 1.00 equiv) in 2 mL of THF up to approximately −10 to 0 °C. The resulting solution was stirred up to room temperature over a 4 h period. The reaction was quenched at 0 °C with the addition of cold sat. aqueous NaHCO3 solution, and the organics were extracted three times with EtOAc (3×). Combined organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo.
Amorfrutin A Ethyl Ester (17a).
According to the general procedure, enol ether 10a (250 mg, 1.26 mmol, 3.00 equiv), chloride 11a (100 mg, 0.42 mmol, 1.00 equiv), and 22 mL of THF were used. Purification by flash column chromatography on silica using a gradient of 0–10% EtOAc in hexanes (w/0.3% TEA) was performed (to recover clean enol ether 10a), followed by additional purification with 0–50% DCM in hexanes, affording 17a (59 mg, 38%) as a colorless oil, and recovered 10a (150 mg). 1H NMR (500 MHz, benzene-d6): δ 12.55 (s, 1H), 7.20–7.17 (m, 2H), 7.12–7.06 (m, 3H), 6.00 (s, 1H), 5.70 (t, J = 7.3 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.80 (d, J = 7.1 Hz, 2H), 3.26 (s, 3H), 3.22–3.17 (m, 2H), 2.86–2.81 (m, 2H), 1.90 (br s, 3H), 1.69 (br s, 3H), 0.89 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.1, 163.1, 161.6, 144.3, 142.3, 131.2, 128.7, 126.3, 123.5, 116.0, 106.2, 105.9, 61.3, 55.0, 39.0, 38.5, 26.0, 22.8, 18.1, 14.0. HRMS (ESI) m/z: [M + H]+ calcd for C23H29O4, 369.2060; found, 369.2051. See Tables S1 and S2 for NMR spectroscopic data of byproducts 23 and 24.
Amorfrutin B Ethyl Ester (17b).
According to the general procedure, enol ether 10b (335 mg, 1.26 mmol, 3.00 equiv), chloride 11a (100 mg, 0.42 mmol, 1.00 equiv), and 22 mL of THF were used. Purification by flash column chromatography on silica using a gradient of 0–10% Et2O in hexanes afforded 17b (57.0 mg, 31%). 1H NMR (400 MHz, benzene-d6): δ 12.58 (s, 1H), 7.26–7.17 (m, 2H), 7.15–7.00 (m, 3H), 6.00 (s, 1H), 5.75 (t, J = 7.0 Hz, 1H), 5.20 (t, J = 7.1 Hz, 1H), 3.96 (q, J = 7.4 Hz, 2H), 3.84 (d, J = 7.3 Hz, 2H), 3.26 (s, 3H), 3.24–3.15 (m, 2H), 2.87–2.79 (m, 2H), 2.23–2.08 (m, 4H), 1.96 (br s, 3H), 1.61 (br s, 3H), 1.49 (br s, 3H), 0.87 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.0, 163.1, 161.6, 144.3, 142.3, 135.0, 130.9, 128.7, 126.3, 125.1, 123.3, 116.0, 106.2, 105.9, 61.3, 55.0, 40.4, 39.0, 38.5, 27.3, 25.8, 22.7, 17.7, 16.5, 14.0. HRMS (ESI) m/z: [M + H]+ calcd for C28H37O4, 437.2686; found, 437.2679.
Amorfrutin 2 Ethyl Ester (17c).
According to the general procedure, enol ether 10a (250 mg, 1.26 mmol, 3.00 equiv), chloride 11b (86.0 mg, 0.42 mmol, 1.00 equiv), and 22 mL of THF were used. Purification by flash column chromatography on silica using a gradient of 0–50% DCM in hexanes afforded 17c (44.0 mg, 31%). 1H NMR (500 MHz, benzene-d6): δ 12.57 (s, 1H), 6.16 (s, 1H), 5.71 (t, J = 7.0 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.82 (d, J = 7.2 Hz, 2H), 3.32 (s, 3H), 2.91–2.87 (m, 2H), 1.91 (s, 3H), 1.69 (s, 3H) 1.62–1.52 (m, 2H), 1.38–1.30 (m, 4H), 0.93 (m, 6H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.2, 163.1, 161.6, 145.6, 131.1, 123.5, 115.7, 105.9, 105.8, 61.1, 55.0, 38.0, 32.6, 32.5, 26.0, 23.1, 22.8, 18.1, 14.4, 14.0. HRMS (ESI) m/z: [M + H+ calcd for C20H31O4, 335.2217; found, 335.2209.
2-Prenyldimedone (29a).
To a mixture of dimedone (28, 6.0 g, 42.8 mmol, 1.00 equiv, Sigma-Aldrich) in 170 mL of CHCl3 was added K2CO3 (11.00 g, 79.6 mmol, 1.86 equiv), followed by prenyl bromide (5.60 mL, 48.5 mmol, 1.13 equiv). The reaction mixture was stirred at room temperature for 60 h and was then diluted with 50 mL of CHCl3 and 50 mL of H2O followed by acidification to pH ~ 5 using aqueous 1 M HCl. The organic layer was separated, and the aqueous layer was extracted with three 100 mL portions of CHCl3. Combined organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo. The residue was stirred with hexanes to remove O-alkyl and bis-alkyl products along with excess 3,3-dimethylallyl bromide resulting in the formation of a precipitate, which was collected via vacuum filtration, affording 29a (3.77 g, 42%), as a white solid containing ~1% of unreacted dimedone as an impurity. 1H NMR (400 MHz, chloroform-d, enol): δ 6.78 (br s, 1H), 5.20 (t, J = 7.3 Hz, 1H), 3.08 (d, J = 7.4 Hz, 2H), 2.27 (s, 4H), 1.79–1.71 (m, 6H), 1.06 (s, 6H). 1H NMR (400 MHz, chloroform-d, keto): δ 5.06 (t, J = 7.2 Hz, 1H), 3.28 (t, J = 6.1 Hz, 1H), 2.63 (d, J = 13.7 Hz, 2H), 2.54–2.44 (m, 2H), 2.48 (d, J = 13.7 Hz, 2H), 1.66 (s, 3H), 1.64 (s, 3H), 1.14 (s, 3H), 0.87 (s, 3H). 13C{1H} NMR (151 MHz, chloroform-d, keto–enol): δ 204.7, 136.3, 133.9, 122.0, 121.1, 112.6, 67.0, 54.3, 31.9, 31.1, 30.1, 28.5, 28.4, 26.8, 25.9, 22.6, 21.3, 18.0, 17.9. HRMS (ESI) m/z: [M + H]+ calcd for C13H21O2, 209.1536; found, 209.1538.
2-Geranyldimedone (29b).
To a mixture of dimedone (28, 2.80 g, 20.0 mmol, 1.00 equiv) in 75 mL of CHCl3 was added K2CO3 (5.50 g, 39.8 mmol, 1.99 equiv), followed by geranyl bromide (4.40 mL, 22.1 mmol, 1.10 equiv). The reaction mixture was stirred at room temperature for 70 h and was then diluted with 50 mL of CHCl3 and 10 mL of H2O, followed by acidification to pH ~ 6 using aqueous 1 M HCl. The organic layer was separated, and the aqueous layer was extracted with two 50 mL portions of CHCl3. Combined organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo. The crude oil was stored at −20 °C for 24 h, and the resulting solid was separated from the remaining yellow oil (O-alkyl and bis-alkyl products along with any excess geranyl bromide) by washing with hexanes via vacuum filtration, affording 29b (2.52 g, 45%) as a white solid. NMR spectroscopic data were identical to those reported previously.36
Diene (12b).23
To a suspension of 29a (2.00 g, 9.60 mmol, 1.0 equiv) in 14 mL of DCM was added hexamethyldisilazane (HMDS) (2.60 mL, 12.4 mmol, 1.29 equiv). The resulting solution was concentrated in vacuo after 12 h and added in 5 mL of THF to a freshly prepared solution of LDA from nBuLi (4.21 mL, 10.5 mmol, 2.5 M in hexanes, 1.09 equiv) and DIPA (1.47 mL, 10.5 mmol, 1.09 equiv) in 25 mL of THF at −78 °C over a 10 min period under Ar. After stirring up to −40 °C over a 1 h period, the solution was cooled to −78 °C, TMSCl (1.40 mL, 11.0 mmol, 1.14 equiv) was added dropwise, and the resulting solution was stirred up to room temperature over a 2 h period. The reaction mixture was concentrated in vacuo, diluted in hexanes, filtered, and concentrated in vacuo, affording bis-TMS diene 12b (3.37 g, 100%) as a yellow oil. 1H NMR (400 MHz, benzene-d6): δ 5.49 (t, J = 6.9 Hz, 1H), 4.60 (s, 1H), 3.22 (d, J = 6.7 Hz, 2H), 2.20 (s, 2H), 1.77 (br s, 3H), 1.69 (br s, 3H), 1.08 (s, 6H), 0.24 (s, 9H), 0.16 (s, 9H). 13C{1H} NMR (151 MHz, benzene-d6): δ 149.6, 147.7, 129.9, 125.0, 115.0, 107.9, 45.0, 32.2, 29.2, 25.9, 23.7, 18.2, 1.06, 0.37.
Diene (12c).23
To a suspension of 29b (1.05 g, 3.80 mmol, 1.0 equiv) in 6 mL of DCM was added HMDS (1.00 mL, 4.78 mmol, 1.26 equiv). The resulting solution was concentrated in vacuo after 24 h and added in 3 mL of THF to a freshly prepared solution of LDA from nBuLi (1.74 mL, 4.35 mmol, 2.5 M in hexanes, 1.14 equiv) and DIPA (0.61 mL, 4.35 mmol, 1.14 equiv) in 10 mL of THF at −78 °C over a 10 min period under Ar. After stirring up to −10 °C over a 1 h period, the solution was cooled to −78 °C, TMSCl (0.70 mL, 5.51 mmol, 1.45 equiv) was added dropwise, and the resulting solution was stirred up to room temperature over a 1 h period. The reaction mixture was concentrated in vacuo, diluted in hexanes, filtered, and concentrated in vacuo, affording bis-TMS diene 12c (1.60 g, 100%) as a yellow oil. 1H NMR (400 MHz, benzene-d6): δ 5.53 (t, J = 6.9 Hz, 1H), 5.24 (t, J = 7.0 Hz, 1H), 4.60 (s, 1H), 3.24 (d, J = 6.7 Hz, 2H), 2.24–2.09 (m, 4H 2.21 (s, 2H), 1.81 (br s, 3H), 1.67 (br s, 3H), 1.55 (br s, 3H), 1.08 (s, 6H), 0.25 (s, 9H), 0.17 (s, 9H). 13C{1H} NMR (151 MHz, benzene-d6): 149.6, 147.7, 133.8, 130.9, 125.1, 124.8, 115.0, 107.8, 45.0, 40.3, 32.3, 29.2, 27.3, 25.9, 23.6, 17.8, 16.6, 1.07, 0.40.
Ethyl 5-Phenylpent-2-ynoate (13a).
This reagent was prepared according to a previously published procedure starting from 4-phenyl-1-butyne (21a) and ethyl chloroformate (22) using nBuLi.32
General Procedure for Diels–Alder Approach.
Excess diene (12b or 12c) and alkyne (13a or 13b) were combined and heated in a sealed tube at 170 °C for the indicated time period, allowing for complete consumption of alkyne. The reaction was then diluted with a mixture of MeOH/DCM, SiO2 was added, and the resulting mixture was refluxed for several hours, until all TMS–ethers had been deprotected, as determined by TLC. The reaction mixture was then filtered and concentrated in vacuo.
Resorcinol (27a).
According to the general procedure, diene (12b, 3.30 g, 9.35 mmol, 4.08 equiv) and alkyne (13a, 465 mg, 2.29 mmol, 1.00 equiv) were heated for 42 h. TMS deprotection was performed for 5 h with 60 mL of MeOH/6 mL of DCM and 5.6 g of SiO2. The reaction mixture was filtered and concentrated in vacuo. To precipitate excess 2-prenyldimedone (29a), a 1:1 mixture of hexanes/DCM (100 mL) was added to the crude, and the mixture was then vacuum filtered. The collected solid was washed with hexanes, affording pure recovered 2-prenyldimedone (29a, 1.34 g, 6.44 mmol, 91% of expected recovery). The filtrate was concentrated in vacuo, and flash column chromatography on silica using a gradient of 0–20% EtOAc in hexanes afforded 27a (721 mg, 89%) as an off-white solid. 1H NMR (500 MHz, benzene-d6): δ 12.81 (s, 1H), 7.22–7.17 (m, 2H), 7.14–7.07 (m, 3H), 5.93 (s, 1H), 5.45 (t of septets, J = 7.1, 1.4 Hz, 1H), 5.21 (s, 1H), 3.95 (q, J = 7.1 Hz, 2H), 3.64 (d, J = 7.3 Hz, 2H), 3.14–3.09 (m, 2H), 2.82–2.77 (m, 2H), 1.69 (br s, 3H), 1.57 (d, J = 1.0 Hz, 3H, 0.87 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.2, 163.9, 159.5, 144.5, 142.4, 133.7, 128.7, 128.6, 126.3, 122.5, 113.1, 111.2, 105.2, 61.3, 38.6, 38.5, 25.8, 22.7, 17.8, 14.0. HRMS (ESI) m/z: [M – H]− calcd for C22H25O4, 353.1758; found, 353.1753.
Resorcinol (27b).
According to the general procedure, diene (12c, 1.03 g, 2.45 mmol, 4.30 equiv) and alkyne (13a, 115 mg, 0.57 mmol, 1.00 equiv) were heated for 23 h. TMS deprotection was performed for 4.5 h with 15 mL of MeOH/2 mL of DCM and 1.7 g of SiO2. The reaction mixture was filtered and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–30% EtOAc in hexanes afforded 27b (203 mg, 84%) as a yellow solid, along with recovered 2-geranyldimedone (29b, 380 mg, 1.38 mmol, 73% of expected recovery). 1H NMR (500 MHz, benzene-d6): δ 12.84 (s, 1H), 7.22–7.18 (m, 2H), 7.13–7.08 (m, 3H), 5.99 (s, 1H), 5.49 (t of septets, J = 7.3, 1.4 Hz, 1H), 5.32 (s, 1H), 5.13 (t of septets, J = 7.1, 1.4 Hz, 1H), 3.95 (q, J = 7.1 Hz, 2H), 3.66 (d, J = 7.2 Hz, 2H), 3.16–3.08 (m, 2H), 2.83–2.76 (m, 2H), 2.09 (q, J = 7.5 Hz, 2H), 2.00 (t, J = 7.5 Hz, 2H), 1.72 (br s, 3H), 1.66 (br s, 3H), 1.50 (d, J = 1.3 Hz, 3H), 0.86 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.2, 163.9, 159.8, 144.6, 142.4, 137.8, 131.5, 128.7, 128.6, 126.3, 124.6, 122.4, 113.0, 111.3, 105.2, 61.3, 40.1, 38.6, 38.5, 26.9, 25.9, 22.7, 17.7, 16.2, 14.0. HRMS (ESI) m/z: [M – H]− calcd for C27H33O4, 421.2384; found, 421.2376.
Resorcinol (27c).
According to the general procedure, diene (12b, 1.70 g, 4.81 mmol, 4.15 equiv) and alkyne (13b, 176 mg, 1.16 mmol, 1.00 equiv) were heated for 36 h. TMS deprotection was performed for 4 h with 30 mL of MeOH/3 mL of DCM and 2.7 g of SiO2. The reaction mixture was filtered and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–20% EtOAc in hexanes afforded 27c (279 mg, 85%) as an off-white solid. 1H NMR (500 MHz, benzene-d6): δ 12.67 (s, 1H), 6.04 (s, 1H), 5.42 (t of septets, J = 7.3, 1.4 Hz, 1H), 5.27 (s, 1H), 3.62 (d, J = 7.3 Hz, 2H), 3.33 (s, 3H), 2.80–2.74 (m, 2H), 1.67 (br s, 3H), 1.55 (d, J = 1.0 Hz, 3H), 1.54–1.46 (m, 2H), 1.33–1.46 (m, 2H), 0.92–0.86 (m, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.7, 163.8, 159.7, 145.7, 133.7, 122.6, 112.7, 111.0, 105.0, 51.3, 37.1, 32.5, 32.0, 25.8, 22.9, 22.7, 17.8, 14.3. HRMS (ESI) m/z: [M – H]− calcd for C18H25O4, 305.1758; found, 305.1750.
Resorcinol (27d).
According to the general procedure, diene (12c, 0.93 g, 2.21 mmol, 4.25 equiv) and alkyne (13b, 79.0 mg, 0.52 mmol, 1.00 equiv) were heated for 36 h. TMS deprotection was performed for 4 h with 15 mL of MeOH/2 mL of DCM and 1.6 g of SiO2. The reaction mixture was filtered and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–10% EtOAc in hexanes afforded 27d (155 mg, 80%) as a yellow solid. 1H NMR (500 MHz, benzene-d6): δ 12.70 (s, 1H), 6.10 (s, 1H), 5.46 (t of septets, J = 7.3, 1.4 Hz, 1H), 5.39 (s, 1H), 5.12 (t of septets, J = 7.0, 1.4 Hz, 1H), 3.65 (d, J = 7.2 Hz, 2H), 3.33 (s, 3H), 2.80–2.74 (m, 2H). 2.08 (q, J = 7.4 Hz, 2H), 1.98 (t, J = 7.5 Hz, 2H), 1.70 (br s, 3H), 1.66 (br s, 3H). 1.54–1.47 (m, 2H), 1.49 (br s, 3H), 1.32–1.25 (m, 4H), 0.91–0.87 (m, 3H). 13C{1H} NMR (126 MHz, benzene-d6): δ 172.7, 163.7, 159.9, 145.8, 137.9, 131.6, 124.6, 122.4, 112.6, 111.1, 105.0, 51.3, 40.1, 37.1, 32.5, 32.0, 26.8, 25.9, 22.9, 22.6, 17.7, 16.2, 14.3. HRMS (ESI) m/z: [M – H]− calcd for C23H33O4, 373.2384; found, 373.2381.
Amorfrutin A Ethyl Ester (17a).
To a solution of resorcinol 27a (100 mg, 0.28 mmol, 1.00 equiv) in acetone (4.8 mL) was added K2CO3 (0.73 g, 5.29 mmol, 18.9 equiv) followed by MeI (26.0 μL, 0.42 mmol, 1.50 equiv). The resulting mixture was vigorously stirred at room temperature for 3 h, neutralized with 1 M aqueous HCl, and extracted with EtOAc (3 × 15 mL). The combined organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–10% EtOAc in hexanes afforded 17a (68.0 mg, 66%) as a colorless oil, along with recovered 27a (11.0 mg, 11%). NMR spectroscopic data were identical to those reported previously.
Amorfrutin B Ethyl Ester (17b).
To a solution of resorcinol 27b (100 mg, 0.23 mmol, 1.00 equiv) in acetone (4 mL) was added K2CO3 (0.62 g, 4.49 mmol, 19.5 equiv) followed by MeI (20.0 μL, 0.35 mmol, 1.52 equiv). The resulting mixture was vigorously stirred at room temp for 3.5 h, acidified with 1 M aqueous HCl, and extracted with EtOAc (3×). The organics were washed with brine, dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–10% EtOAc in hexanes was performed, affording 17b (62.0 mg, 60%) as a colorless oil, along with recovered 27b (7.0 mg, 7%). NMR spectroscopic data were identical to those reported previously.
Cannabigerol (CBG, 30).
Resorcinol 27d (57.0 mg, 0.15 mmol, 1.00 equiv) was combined with KOH (408 mg, 7.28 mmol, 48.5 equiv) in a 1:1 mixture of EtOH/H2O (5 mL) and was refluxed for 8 h. The resulting solution was concentrated in vacuo, acidified with 1 M aqueous HCl to pH ~ 5, and then extracted with EtOAc (3×). The combined organic layers were dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–30% EtOAc in hexanes afforded 30 (39.0 mg, 81%) as an off-white solid. NMR spectroscopic data were identical to those reported previously.34
Amorfrutin A (1).
A mixture of 17a (71 mg, 0.19 mmol, 1.0 equiv) and KOH (101 mg, 1.80 mmol, 9.47 equiv) in 3 mL of EtOH and 2 mL of H2O was refluxed for 2.5 h. The resulting solution was concentrated in vacuo, acidified with 1 M aqueous HCl to pH ~ 2, and then extracted with EtOAc (4×). The combined organic extracts were dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–60% EtOAc in hexanes afforded 1 (62 mg, 95%) as a white solid. NMR spectroscopic data were identical to those reported previously.3
Amorfrutin B (2).
A mixture of 17b (70 mg, 0.16 mmol, 1.0 equiv) and KOH (100 mg, 1.78 mmol, 11.12 equiv) in 3 mL of EtOH and 2 mL of H2O was refluxed for 1.75 h. The resulting solution was concentrated in vacuo, acidified with 1 M aqueous HCl to pH ~ 1, and then extracted with EtOAc (4×). The combined organic extracts were dried with MgSO4, filtered, and concentrated in vacuo. Flash column chromatography on silica using a gradient of 0–60% EtOAc in hexanes afforded 2 (61 mg, 94%) as a white solid. NMR spectroscopic data were identical to those reported previously.4
Supplementary Material
ACKNOWLEDGMENTS
We thank Tae Hyung Won for assistance with mass spectrometry and Ryan A. Woltornist and David B. Collum for helpful discussions. This work was partly supported by the NIH (P41GM79571 and R35GM131877 to F.C.S. and T32GM008500 to B.J.C.) and the Howard Hughes Medical Institute.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c03043.
Suppression of by-product formation in the anionic cascade reaction, regioselectivity of Diels–Alder/retro-Diels–Alder reaction, and NMR spectroscopic data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c03043
The authors declare the following competing financial interest(s): F.C.S. is a founder of Ascribe Bioscience, a company that develops plant treatments based on natural small molecules.
Contributor Information
Brian J. Curtis, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Robert J. Micikas, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Russell N. Burkhardt, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Rubin A. Smith, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Judy Y. Pan, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Katrina Jander, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Frank C. Schroeder, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
REFERENCES
- (1).Lefebvre P; Staels B Naturally improving insulin resistance with amorfrutins. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (19), 7136–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sauer S Amorfrutins: A Promising Class of Natural Products That Are Beneficial to Health. ChemBioChem 2014, 15 (9), 1231–1238. [DOI] [PubMed] [Google Scholar]
- (3).Weidner C; de Groot JC; Prasad A; Freiwald A; Quedenau C; Kliem M; Witzke A; Kodelja V; Han C-T; Giegold S; Baumann M; Klebl B; Siems K; Muller-Kuhrt L; Schurmann A; Schuler R; Pfeiffer AFH; Schroeder FC; Bussow K; Sauer S Amorfrutins are potent antidiabetic dietary natural products. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (19), 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).De Groot JC; Weidner C; Krausze J; Kawamoto K; Schroeder FC; Sauer S; Büssow K Structural Characterization of Amorfrutins Bound to the Peroxisome Proliferator-Activated Receptor γ. J. Med. Chem 2013, 56 (4), 1535–1543. [DOI] [PubMed] [Google Scholar]
- (5).Fuhr L; Rousseau M; Plauth A; Schroeder FC; Sauer S Amorfrutins Are Natural PPARγ Agonists with Potent Anti-inflammatory Properties. J. Nat. Prod 2015, 78 (5), 1160–1164. [DOI] [PubMed] [Google Scholar]
- (6).Weidner C; Rousseau M; Micikas RJ; Fischer C; Plauth A; Wowro SJ; Siems K; Hetterling G; Kliem M; Schroeder FC; Sauer S Amorfrutin C Induces Apoptosis and Inhibits Proliferation in Colon Cancer Cells through Targeting Mitochondria. J. Nat. Prod 2016, 79 (1), 2–12. [DOI] [PubMed] [Google Scholar]
- (7).Sauer S; Weidner C; Kliem M; Schroeder FC, Micikas RJ Amorfrutin analogs as ppargamma-modulators. U.S. Patent WO2014177593, 2014.
- (8).Asakawa Y; Kusube E; Takemoto T; Suire C New bibenzyls from Radula complanata. Phytochemistry 1978, 17 (12), 2115–2117. [Google Scholar]
- (9).Asakawa Y; Toyota M; Takemoto T Seven new bibenzyls and a dihydrochalcone from Radula variabilis. Phytochemistry 1978, 17 (11), 2005–2010. [Google Scholar]
- (10).Asakawa Y; Takikawa K; Toyota M; Takemoto T Novel bibenzyl derivatives and ent-cuparene-type sesquiterpenoids from Radula species. Phytochemistry 1982, 21 (10), 2481–2490. [Google Scholar]
- (11).Bohlmann F; Hoffmann E Cannabigerol-ähnliche verbindungen aus Helichrysum umbraculigerum. Phytochemistry 1979, 18 (8), 1371–1374. [Google Scholar]
- (12).Zechlin L; Wolf M; Steglich W; Anke T Cristatsäure, ein modifiziertes Farnesylphenol aus Fruchtkörpern von Albatrellus cristatus. Liebigs Ann. Chem 1981, 1981, 2099–2105. [Google Scholar]
- (13).Song YY; He HG; Li Y; Deng Y A facile total synthesis of amorfrutin A. Tetrahedron Lett. 2013, 54 (21), 2658–2660. [Google Scholar]
- (14).Weber B; Brandes B; Powroznik D; Kluge R; Csuk R An efficient and robust synthesis of amorfrutin A. Tetrahedron Lett. 2019, 60 (20), 1379–1381. [Google Scholar]
- (15).Fujita T; Kuwahara S; Ogura Y Unified total synthesis of amorfrutins A and C via the Claisen rearrangement. Biosci., Biotechnol., Biochem 2019, 83 (9), 1635–1641. [DOI] [PubMed] [Google Scholar]
- (16).Laclef S; Anderson K; White AJP; Barrett AGM Total synthesis of amorfrutin A via a palladium-catalyzed migratory prenylation-aromatization sequence. Tetrahedron Lett. 2012, 53 (2), 225–227. [Google Scholar]
- (17).Fujita T; Kuwahara S; Ogura Y Synthesis of amorfrutins B and D from amorfrutin A ethyl ester. Tetrahedron Lett. 2020, 61 (6), 151477. [Google Scholar]
- (18).Aidhen IS; Mukkamala R; Weidner C; Sauer S A Common Building Block for the Syntheses of Amorfrutin and Cajaninstilbene Acid Libraries toward Efficient Binding with Peroxisome Proliferator-Activated Receptors. Org. Lett 2015, 17 (2), 194–197. [DOI] [PubMed] [Google Scholar]
- (19).Grandhi GS; Selvakumar J; Dana S; Baidya M Directed C-H Bond Functionalization: A Unified Approach to Formal Syntheses of Amorfrutin A, Cajaninstilbene Acid, Hydrangenol, and Macrophyllol. J. Org. Chem 2018, 83 (19), 12327–12333. [DOI] [PubMed] [Google Scholar]
- (20).Xu XJ; Zeng T; Huang ZX; Xu XF; Lin J; Chen WM Synthesis and Biological Evaluation of Cajaninstilbene Acid and Amorfrutins A and B as Inhibitors of the Pseudomonas aeruginosa Quorum Sensing System. J. Nat. Prod 2018, 81 (12), 2621–2629. [DOI] [PubMed] [Google Scholar]
- (21).Miao Q; Li Y; Xu J; Lin A; Tanabe G; Muraoka O; Wu X; Xie W First Total Syntheses of Amorfrutin C and pseudo-Amorfrutin A. Eur. J. Org. Chem 2018, 2018 (12), 1443–1448. [Google Scholar]
- (22).Patel C; Mies T; White AJP; Parsons PJ; Barrett A Biomimetic Syntheses of Amorfrutin C and C-5 Substituted Amorfrutin Analogues. Eur. J. Org. Chem 2021, 2021, 1258. [Google Scholar]
- (23).Quintiliano SAP; Silva LF Practical synthesis of a functionalized 1-oxo-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid. Tetrahedron Lett. 2012, 53 (29), 3808–3810. [Google Scholar]
- (24).Dai M; Sarlah D; Yu M; Danishefsky SJ; Jones GO; Houk KN Highly Selective Diels-Alder Reactions of Directly Connected Enyne Dienophiles. J. Am. Chem. Soc 2007, 129 (3), 645–657. [DOI] [PubMed] [Google Scholar]
- (25).Yu J; Yang M; Guo Y; Ye T Total Synthesis of Psymberin (Irciniastatin A). Org. Lett 2019, 21 (10), 3670–3673. [DOI] [PubMed] [Google Scholar]
- (26).Covarrubias-Zúñiga A; González-Lucas A A total synthesis of mycophenolic acid. Tetrahedron Lett. 1998, 39, 2881–2882. [Google Scholar]
- (27).Kileńyi SN; Smith AB III. Avermectin-milbemycin studies. 4. An expedient two-step preparation of p-hydroxybenzoate. Tetrahedron Lett. 1985, 26 (37), 4419–4422. [Google Scholar]
- (28).Essig S; Bretzke S; Müller R; Menche D Full Stereochemical Determination of Ajudazols A and B by Bioinformatics Gene Cluster Analysis and Total Synthesis of Ajudazol B by an Asymmetric Ortholithiation Strategy. J. Am. Chem. Soc 2012, 134 (47), 19362–19365. [DOI] [PubMed] [Google Scholar]
- (29).Caron B; Brassard P An integrated approach to the synthesis of contiguously substituted xanthopurpurins, pachybasins and purpurins. Tetrahedron 1993, 49 (4), 771–784. [Google Scholar]
- (30).Hua R; Shimada S; Tanaka M The First Example of Rhodium(I)-Catalyzed Regio- and Stereoselective Chloroesterification of Alkynes with Chloroformate Esters. J. Am. Chem. Soc 1998, 120 (47), 12365–12366. [Google Scholar]
- (31).Grabovyi GA; Mohr JT Total Synthesis of Grifolin, Grifolic Acid, LL-Z1272α, LL-Z1272β, and Ilicicolinic Acid A. Org. Lett 2016, 18 (19), 5010–5013. [DOI] [PubMed] [Google Scholar]
- (32).Zhang J; Cheng C; Wang D; Miao Z Regio- and Diastereoselective Construction of Spirocyclopenteneoxindoles through Phosphine-Catalyzed [3 + 2] Annulation of Methyleneindolinone with Alkynoate Derivatives. J. Org. Chem 2017, 82 (19), 10121–10128. [DOI] [PubMed] [Google Scholar]
- (33).Reekie T; Scott M; Kassiou M Synthesis of phytocannabinoids including a demethylation step. AU Patent WO2019033164, 2019.
- (34).Jentsch NG; Zhang X; Magolan J Efficient Synthesis of Cannabigerol, Grifolin, and Piperogalin via Alumina-Promoted Allylation. J. Nat. Prod 2020, 83, 2587–2591. [DOI] [PubMed] [Google Scholar]
- (35).Smeltzer T; David R; Colvin S; Black JC Synthesis of Cannabigerol. US Patent WO2020115306, 2020.
- (36).Takenaka K; Mohanta SC; Patil ML; Rao CVL; Takizawa S; Suzuki T; Sasai H Enantioselective Wacker-Type Cyclization of 2-Alkenyl-1,3-diketones Promoted by Pd-SPRIX Catalyst. Org. Lett 2010, 12 (15), 3480–3483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.