

Abstract
The first FDA-approved engineered chimeric antigen receptor (CAR) T cell therapies are remarkably effective in a subset of hematological malignancies with few therapeutic options. While these clinical successes have been exciting, CAR T cells have hit roadblocks in solid tumors that include the lack of highly tumor-specific antigens to target, opening up the possibility of life-threatening “on-target/off-tumor” toxicities, and there are problems with T cell entry into solid tumor and persistent activity in suppressive tumor microenvironments. Here we improve the specificity and persistent anti-tumor activity of therapeutic T cells with synthetic Notch (synNotch) CAR circuits. We identify Alkaline Phosphatase Placental-like 2 (ALPPL2) as a tumor-specific antigen expressed in a spectrum of solid tumors, including mesothelioma and ovarian cancer. ALPPL2 can act as a sole target for CAR therapy or be combined with tumor-associated antigens such as melanoma cell adhesion molecule (MCAM), mesothelin, or human epidermal growth factor receptor 2 (HER2) in synNotch CAR combinatorial antigen circuits. SynNotch CAR T cells display superior control of tumor burden when compared to T cells constitutively expressing a CAR targeting the same antigens in mouse models of human mesothelioma and ovarian cancer. This was achieved by preventing CAR-mediated tonic signaling through synNotch controlled expression, allowing T cells to maintain a long-lived memory and non-exhausted phenotype. Collectively, we establish ALPPL2 as a clinically viable cell therapy target for multiple solid tumors and demonstrate the multi-faceted therapeutic benefits of synNotch CAR T cells.
ONE SENTENCE SUMMARY:
SynNotch CAR circuits targeting highly-specific solid tumor antigens enhance specificity and improve therapeutic efficacy by regulating T cell exhaustion.
INTRODUCTION
Genetically modified T cells armed with chimeric antigen receptors (CARs) have shown unparalleled clinical efficacy in certain B cell malignancies by targeting lineage-restricted surface molecules (1). However, CAR T cell therapies for solid tumors have been largely ineffective due to the lack of high-quality antigen targets and poor tumor infiltration and activity. Most solid tumor antigens that have been targeted by CAR T cells are tumor-associated, rather than tumor-specific, meaning that they are highly expressed on the tumor but also expressed at low abundance in normal tissues, which can lead to severe on-target/off-tumor toxicities. For example, human epidermal growth factor receptor 2 (HER2)-targeted CAR T cells caused massive toxicity in the lungs of a patient within a half hour of administration, leading to death within days (2). Thus, discovery of new tumor-specific antigens or combinations of antigens that could be paired with HER2 or other tumor-associated antigens for more specific recognition of tumors is of particular interest in cell therapy.
Beyond enhanced tumor recognition, the phenotypic character and state of CAR T cells can greatly affect their therapeutic potency. CAR T cells with a more long-lived memory phenotype exhibit superior anti-tumor efficacy when compared to terminal effector-like CAR T cells, emphasizing the importance of persistent activity for clinical success (3). CAR T cell phenotype and fitness is influenced by many factors, but chronic antigen-independent tonic signaling is now a well-appreciated problem that plagues many CARs, leading to premature CAR T cell differentiation and exhaustion (4). Tonic signaling is a function of the specific CAR architecture and high receptor cell surface densities that can facilitate receptor aggregation prior to antigen-binding (5). Temporary suspension of CAR signaling or receptor redesigns have been proposed means to enhance anti-tumor activity by reduction of tonic signaling (6–8). Alternatively, tonic signaling can be reduced through targeted genomic integration into the TRAC locus, which allows for more controlled and dynamically regulated expression of CARs similar to the natural T cell receptor (9).
Here we focus on the development of more specific and potent next generation engineered T cell therapies for mesothelioma and ovarian cancer. Mesothelioma is a highly aggressive chronic inflammation-driven cancer with an exceedingly poor prognosis that has three histological subtypes; epithelioid (69% of cases, median overall survival ~14 months), biphasic (12%, ~10 months), and sarcomatoid (19%, ~4 months) (7, 8). Mesothelioma is inherently hard to treat with traditional modes of cancer therapy, and recent trials exploring mono- or combination immune checkpoint inhibitors (ICI) have also had limited impact (10, 11). CAR T cells targeting tumor-associated antigens such as mesothelin or fibroblast activation protein have shown preclinical promise, and are now in clinical trials alone or in combination with ICI (ClinicalTrials.gov Identifiers: NCT02414269 and NCT01722149) (12, 13). Ovarian cancer is also an aggressive tumor type and the fifth leading cause of cancer-related deaths among females (14). Current clinical CAR T cell trials have focused on tumor-associated antigens such as HER2, mesothelin, and MUC16 (also known as CA125) (ClinicalTrials.gov Identifiers: NCT01935843, NCT01583686, and NCT02498912) but little success has been observed thus far (15).
We have taken a comprehensive approach to develop potentially more safe and efficacious engineered T cell therapies for mesothelioma and ovarian cancer. To accomplish this, we identified Alkaline Phosphatase Placental-like 2 (ALPPL2) as a tumor-specific antigen that is expressed in a spectrum of solid tumors including mesothelioma and ovarian cancer. We then engineered synthetic Notch (synNotch) CAR combinatorial antigen circuit T cells that recognize the combination of ALPPL2 and the tumor-associated antigens melanoma cell adhesion molecule (MCAM), mesothelin, or HER2 to more precisely target the T cells to the tumors. We also show that synNotch CAR T cells have surprising benefits beyond enhanced tumor specificity as they display more persistent and potent activity in vivo compared to conventional CAR T cells through dynamically regulated CAR expression that prevents tonic signaling and maintains a long-lived memory and non-exhausted T cell phenotype.
RESULTS
ALPPL2 is a highly specific and broadly applicable tumor antigen for combinatorial antigen recognition of solid tumors using synNotch CAR T cells.
We have previously described ALPPL2 as a highly specific and targetable cell surface antigen in all subtypes of mesothelioma (16, 17). We can now show that ALPPL2 is not only relevant to mesothelioma but is also expressed on other solid tumor types. Immunohistochemistry (IHC) on tumor tissue arrays revealed that ALPPL2 is expressed in 43% of mesotheliomas (39/91), 60% of ovarian cancers (36/60), 36% of pancreatic cancers (18/50), 18% of gastric cancers (13 of 72) and 100% of seminomas (11/11) (Fig. 1A and table S1). Analysis of IHC data from the public protein expression database, the Human Protein Atlas, yielded similar results (table S1). ALPPL2 expression appears to be homogeneous in seminoma. In mesothelioma, ovarian, and gastric cancer, limited intra-specimen variation is observed. However, in pancreatic cancer, ALPPL2 exhibits more intra-specimen variation.
Figure 1. ALPPL2 is a highly specific and broadly applicable tumor antigen for combinatorial antigen recognition of solid tumors with synNotch CAR T cells.
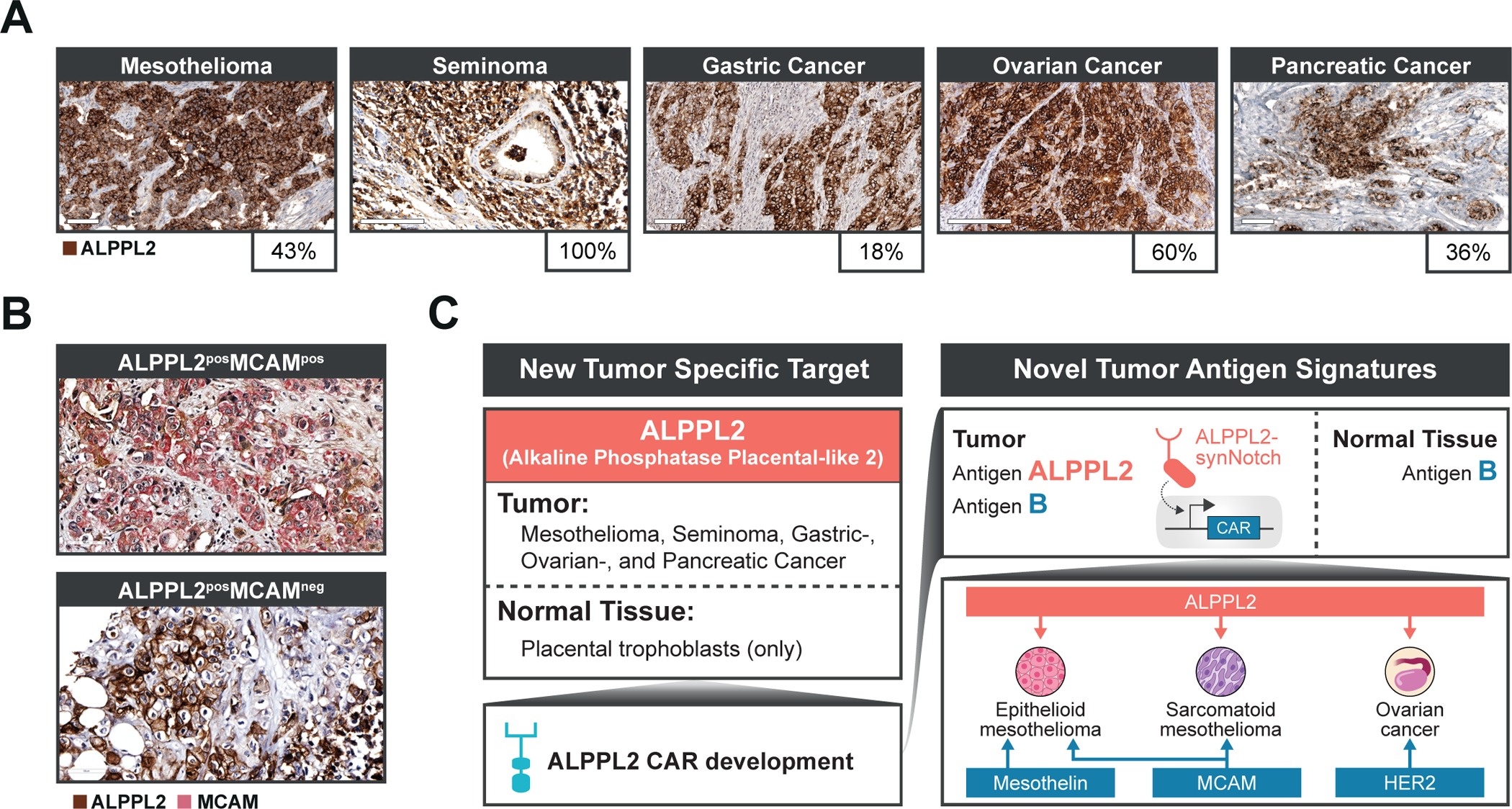
(A) ALPPL2 expression in FFPE sections of solid tumor tissues was measured by IHC. Positive staining (brown) was observed in mesothelioma, seminoma, gastric cancer, ovarian cancer, and pancreatic cancer. Scale bar: 100μm for mesothelioma, pancreatic cancer, and gastric cancer; 200μm for ovarian cancer and seminoma. (B) Representative images of ALPPL2 and MCAM co-expression in mesothelioma tissues are shown. Scale bar: 50μm. (C) ALPPL2 is a highly tumor-specific antigen found in a range of solid tumor types that display minimal expression in normal tissues. ALPPL2 can be targeted directly using an ALPPL2 CAR but can also function as a priming switch for CARs targeting other tumor-associated antigen through synNotch CAR circuits to minimize on-target/off-tumor toxicity. We identified ALPPL2/MCAM as an antigen signature with coverage across mesothelioma subtypes. Mesothelin is mainly restricted to epithelioid mesothelioma. In addition, ALPPL2/HER2 is a potential combinatorial antigen signature for ovarian cancer.
To design a CAR targeting ALPPL2, we utilized an ALPPL2 binding single-chain variable fragment M25 (16, 17). Second generation anti-ALPPL2 M25 scFv 4-1BB-based (BBζ) CARs or two affinity matured clones (M25ADLF and M25FYIA) (17) all effectively triggered activation of CD8+ T cells when stimulated with ALPPL2 positive tumor cells. With the exception of M25ADLF, CARs carrying M25 or M25FYIA responded with high specificity to ALPPL2 but not the closely related (89%) homolog, alkaline phosphatase, intestinal (ALPI), expressed in the healthy intestine (fig. S1A). To further verify the tissue specificity of M25FYIA, a human tissue array that covers 20 different organs in duplicates (40 tissue cores total) was stained with M25FYIA or a control non-binding scFv (YSC10). Positive staining by M25FYIA was only observed in placental trophoblasts, with no staining observed in any other normal tissues (fig. S1B and table S2), in line with what we have previously observed for the corresponding full IgG1 antibodies (17). Although the functional role of ALPPL2 in tumor cells is unknown, ALPPL2 expression was recently described to be associated with and functionally essential for the establishment and maintenance of naïve pluripotency in various types of human pluripotent stem cells (18). The broad tumor expression and highly restricted normal tissue expression of ALPPL2, combined with the proposed function of ALPPL2 in various types of stem cells indicates that it might serve as an oncofetal antigen and carry high potential as immunotherapeutic target.
Solid tumors can be targeted with ALPPL2 synNotch CAR combinatorial antigen recognition circuits.
Advances in synthetic biology and immune cell engineering have led to approaches to engineer combinatorial antigen sensing capabilities into therapeutic T cells (19). We have previously engineered a new class of synthetic receptors based on the Notch receptor we call synNotch that enables custom gene regulation in response to a tissue or disease-related antigen cue. SynNotch receptors can be engineered to sense a tumor antigen and induce the expression of a CAR to a second tumor-related antigen (20). These synNotch - CAR inducible circuits confine CAR expression and thus T cell activation to the site of disease. Here we sought to develop a clinically relevant synNotch - CAR circuit for mesothelioma that senses the combination of the recently identified tumor-specific antigen ALPPL2 in addition to MCAM (also known as CD146 or MUC18) a mesothelioma-associated antigen found expressed in both epithelioid and sarcomatoid mesothelioma, as well as tumor associated blood vessels (16, 21). MCAM is reported to be restricted to few normal tissues (22, 23). To determine the co-expression pattern of ALPPL2 and MCAM, we performed dual-antibody IHC on human mesothelioma arrays. Using two different MCAM antibodies (table S3), we observed ~52–81% MCAM co-staining in ALPPL2 positive tissue cores (Fig. 1B and table S4). ALPPL2/MCAM cell surface co-expression was also seen on the mesothelioma cell lines M28 (epithelioid) and VAMT-1 (sarcomatoid) using our anti-ALPPL2 scFv (M25FYIA) and anti-MCAM scFv (M1) (fig. S1C) (24). Indeed, ALPPL2 CAR expressing CD8+ T cells effectively recognized and killed both mesothelioma cell lines (fig. S1D). Together, these results suggest that the majority of ALPPL2 expressing tumors also express MCAM, providing a rationale for dual targeting of ALPPL2/MCAM using an ALPPL2 synNotch design (Fig. 1C). Additionally, for patients with epithelioid mesothelioma, an alternative opportunity would be to use the well-established mesothelioma biomarker mesothelin as a secondary target, thereby reducing the risk of on-target/off-tumor toxicity associated with mesothelin (25). This approach would be mainly restricted, but generally applicable, to epithelioid mesothelioma for which mesothelin is found expressed in 84–93% of patients (26, 27) (Fig. 1C).
ALPPL2 synNotch circuits can also be applied to cancers beyond mesothelioma as 60% of ovarian cancer is ALPPL2 positive. Epithelial ovarian cancer constitutes the majority of ovarian cancers and is the type of gynecological cancer with the highest mortality rate (14). Mesothelin is expressed in about 70% of epithelial ovarian cancer (28) and HER2 is found at immunologically relevant expression in primary ascites and solid tumor samples from epithelial ovarian cancer (29), but with some controversy (30). Thus, we have engineered ALPPL2 synNotch CAR circuits that target the combination of ALPPL2 and MCAM, mesothelin, or HER2 to show the power of ALPPL2 to enhance the recognition of multiple solid tumors with high unmet therapeutic need (Fig. 1C).
SynNotch CAR T cells exhibit multi-antigen specificity and paced elimination of mesothelioma and ovarian tumor cells.
We first constructed BBζ CARs targeting either MCAM, mesothelin, or HER2 as well as a CD28 based (28ζ) mesothelin CAR, which is currently in clinical trials (fig. S2A). To confine CAR expression locally to tumor tissues, we generated an ALPPL2 sensing synNotch using the M25FYIA scFv and linked it to inducible genetic circuits containing either the MCAM (BBζ) CAR, a mesothelin (BBζ or 28ζ) CAR, or HER2 (BBζ) CAR (Fig. 2A). Both CD4+ and CD8+ T cells equipped with these circuits were all able to selectively drive expression of the respective CARs when stimulated with target cells expressing ALPPL2 (Fig. 2A and fig. S2B–S2E). As with T cells constitutively expressing the CARs, both the ALPPL2 synNotch - MCAM, mesothelin, and HER2 CAR circuits induced T cell activation, proliferation, and Th1 cytokine production (fig. S3 and S4). We also compared synNotch CAR T cells to CAR T cells in their ability to simultaneously produce several cytokines (interleukin (IL)-2, interferon gamma (IFN-γ), and tumor necrosis factor alpha (TNFα)), a phenotype referred to as polyfunctionality. Polyfunctionality of the synNotch CAR T cells were comparable to the CAR T cells with the exception of the ALPPL2 synNotch - HER2 CAR T cells, which exhibited a lower fraction of polyfunctionality than the constitutive HER2 CAR T cells (fig. S5). Target K562 killing was dependent on expression of the correct antigen combination (fig. S6A). In general, the full set of synNotch CAR T cells exhibited dual antigen recognition and robust activity in response to target tumor cells.
Figure 2. SynNotch CAR circuit T cells exhibit multi-antigen specificity and paced elimination of mesothelioma.
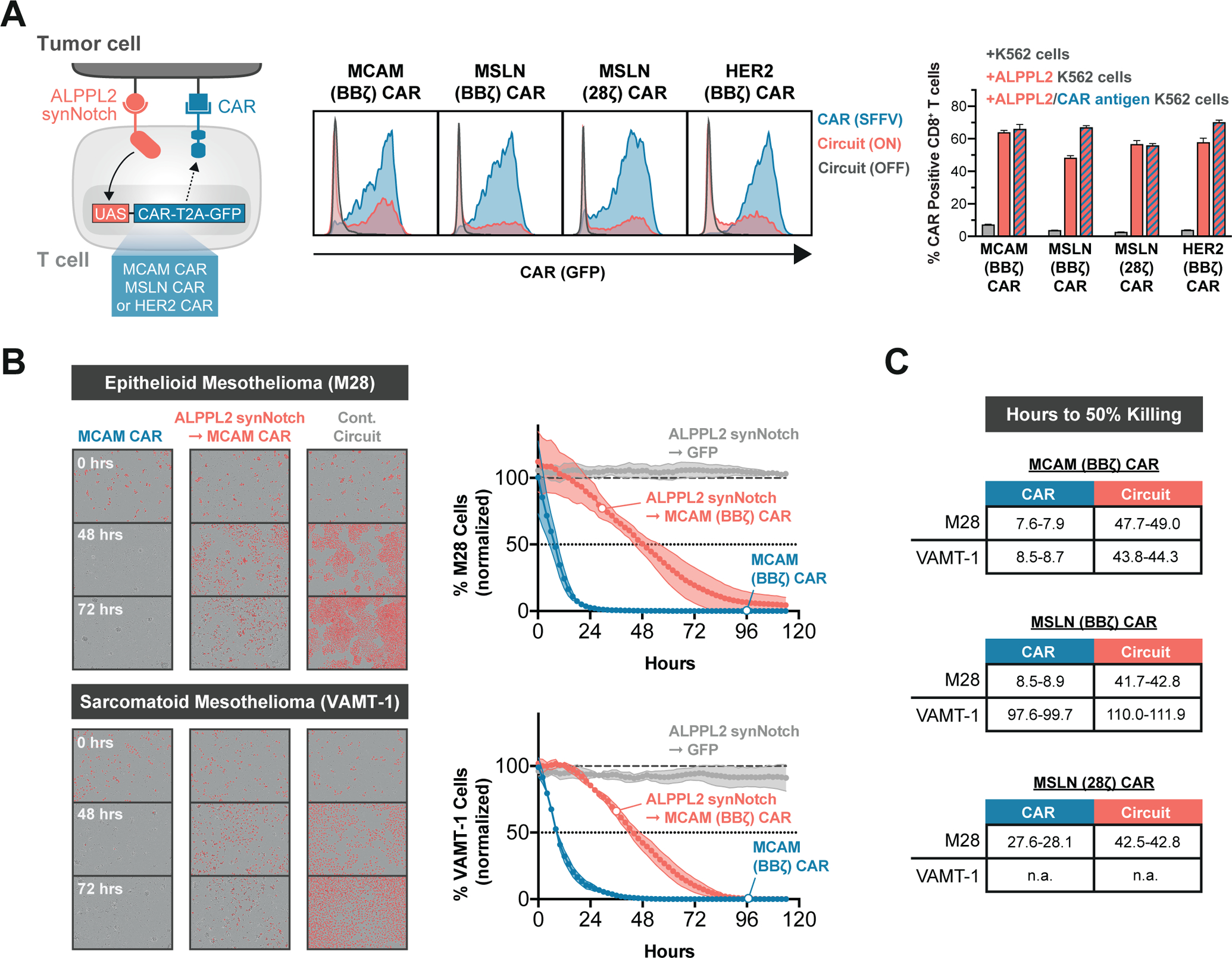
(A) Primary human T cells were engineered with ALPPL2 sensing synNotch with genetic circuits encoding for either an MCAM (BBζ), mesothelin (MSLN, BBζ or 28ζ), or HER2 (BBζ) CAR. Antigen specific CAR expression in CD8+ T cells was determined by GFP expression after 48 hours of stimulation with K562 cells expressing MCAM, mesothelin, and HER2 ± ALPPL2 (representative of at least two independent experiments). (B) Killing kinetics of epithelioid (M28) and sarcomatoid (VAMT-1) mesothelioma tumor cells by CD8+ T cells expressing a MCAM CAR constitutively or through an ALPPL2 synNotch circuit. ALPPL2 synNotch regulating GFP expression acted as a control circuit (representative of at least two independent experiments). (C) Hours to 50% killing of M28 and VAMT-1, as compared to untransduced CD8+ T cells, were recorded for constitutive or ALPPL2 synNotch regulated expression of MCAM (BBζ) or MSLN (BBζ or 28ζ) CARs. n.a.; not applicable. Data are shown as mean±SD.
Both constitutive- and ALPPL2-primed expression of MCAM CAR was able to elicit an effective cytotoxic response towards M28 and VAMT-1 tumor cell lines in vitro (Fig. 2B), whereas mesothelin CARs only effectively targeted the epithelioid M28 cell line (fig. S6B). There was a marked difference in the kinetics of cytotoxicity between constitutive- and circuit-controlled CAR expression, with the latter displaying slower tumor killing rates (Fig. 2C). Similarly, ALPPL2 synNotch - HER2 CAR circuit T cells showed more paced killing of epithelial ovarian SK-OV-3 tumor cells compared to ALPPL2 and HER2 CAR T cells (fig. S6C). This slower cytotoxic response of the synNotch - CAR circuit T cells can be attributed to the requirement for transcriptional regulation of CAR expression and as a population the circuit T cells do not upregulate the CAR in perfect unison.
ALPPL2 SynNotch circuits are sensitive to a range of antigen densities expressed by multiple tumor cell types.
Protein expression of ALPPL2 varies within and between tumors based on IHC of various tumor types. Therefore, we wanted to confirm that our ALPPL2 synNotch remained responsive to lower antigen densities. For this purpose, we designed target cells for which the amount of ALPPL2 expression could be controlled through titration of doxycycline (Fig. 3A and fig. S7A). Using this system, we show that low expression of ALPPL2 is sufficient to induce robust expression of the MCAM CAR through the anti-ALPPL2 M25FYIA-based synNotch, allowing for T cell activation through combinatorial antigen sensing (Fig. 3A). Beyond the priming antigen ALPPL2, varying densities may also be encountered by the secondary CAR antigen. To ensure that ALPPL2 synNotch CAR T cells showed comparable sensitivity to low antigen expression to constitutively expressed CARs, we further developed ALPPL2+ target cells able to display varying densities of MCAM, mesothelin, or HER2 (Fig. 3B and fig. S7B). Indeed, the ALPPL2 synNotch CAR T cells and conventional CAR T cells exhibited robust and comparable activity to the full spectrum of antigen expression we were able to achieve (Fig. 3C, 3D, 3E, and fig. S7C). We also engineered adherent A549 lung epithelial tumor cells to express ALPPL2 in combination with either low or high densities of MCAM or HER2 to assess the cytotoxicity potential of synNotch CAR T cells to low and high CAR antigen expression with time (Fig. 3E). ALPPL2 synNotch MCAM or HER2 CAR circuit T cells were able to effectively recognize and kill the tumor cells regardless of the amount of CAR antigen, albeit with varying kinetics (Fig. 3F). Collectively, our ALPPL2 synNotch CAR T cells are capable of driving CAR expression to a broad range of ALPPL2 densities, and the induced CAR responds to low antigen expression, similar to that of a constitutively expressed CAR.
Figure 3. ALPPL2 synNotch circuits are sensitive to a range of antigen densities across multiple tumor types.
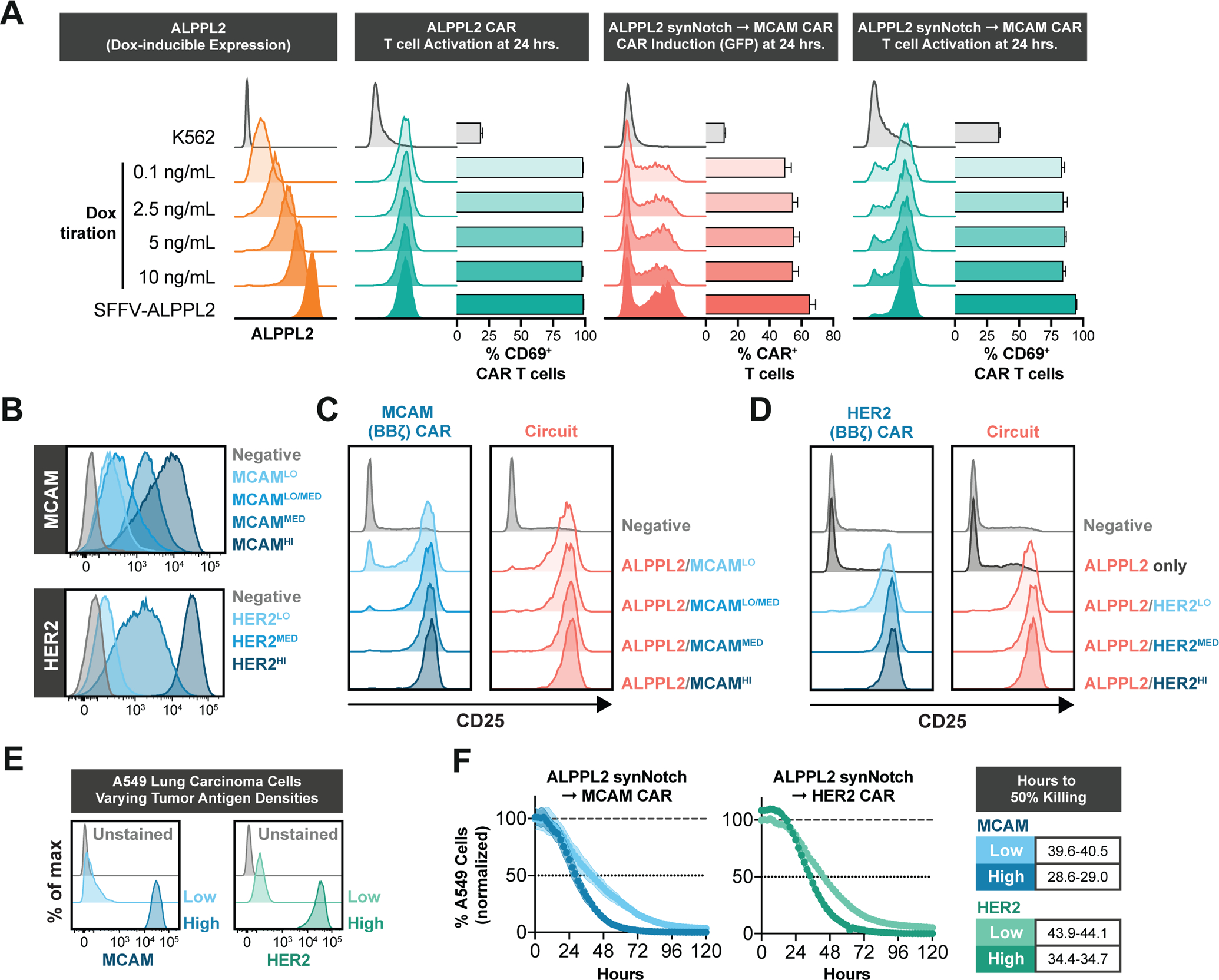
(A) K562 cells constructed with doxycycline-inducible ALPPL2 expression show dose-dependent induction of ALPPL2 after 72 hours of doxycycline (Dox) treatment. CD8+ T cells engineered with an ALPPL2 (BBζ) CAR or ALPPL2 synNotch MCAM (BBζ) CAR circuit were challenged with K562 cells displaying a range of ALPPL2 expression and analyzed for antigen specific CAR expression, as determined by GFP expression, and T cell activation, as determined by CD69 expression (representative of at least two independent experiments). (B) ALPPL2+ K562 cells were constructed to display dose-dependent expression of either MCAM or HER2 upon treatment with doxycycline. MCAMMED represents endogenous expression and HER2HI represents cells expressing HER2 driven by the SFFV promoter. (C,D) CD8+ T cells engineered with either an MCAM (BBζ) CAR (C) or HER2 (BBζ) CAR (D) either under a constitutive SFFV promoter or ALPPL2 synNotch circuit were challenged with doxycycline-treated K562 target cells and assessed for CD25 expression after 48 hours. (E) ALPPL2+ A549 tumor cells were engineered to express either low or high densities of MCAM or HER2. (F) Cytotoxic kinetics for CD8+ T cells engineered with ALPPL2 synNotch circuits driving either a MCAM (BBζ) CAR or HER2 (BBζ) CAR challenged with A549 tumor cells expressing low or high densities of MCAM or HER2. Data is shown as mean±SD.
SynNotch regulation of CAR expression promotes maintenance of long-lived T cells.
Another limiting factor in CAR T therapy of solid tumors is the intrinsic maintenance of multifunctional T cell states and the prevention of T cell exhaustion/dysfunction. In the absence of antigen, constitutive expression of CARs is known to elicit tonic signaling. This low intensity signaling is linked to detrimental effects on the phenotypic state of CAR T cells, such as differentiation of long-lived T cell memory phenotypes towards short-lived effector states and the upregulation of inhibitory receptors (4, 5, 9). Although most of the components of CAR domain architecture impact tonic signaling, including the scFv, hinge, and signaling domains, surface density remain as the unifying determinant (4). We thus reasoned that restricting the timing of CAR expression and location of expression to the tumor tissue with synNotch could prevent CAR-mediated tonic signaling and the detrimental effects thereof. Indicative of tonic signaling, we observed increased antigen-independent expansion for constitutive CAR T cells in both CD4+ (p<0.001) and CD8+ (p<0.001 donor 1 and p=0.03 for donor 2) as opposed to corresponding ALPPL2 synNotch CAR T cells (Fig. 4A, 4B, and fig. S8A)
Figure 4. SynNotch regulation of CAR expression maintains T cell stemness prior to therapeutic administration.
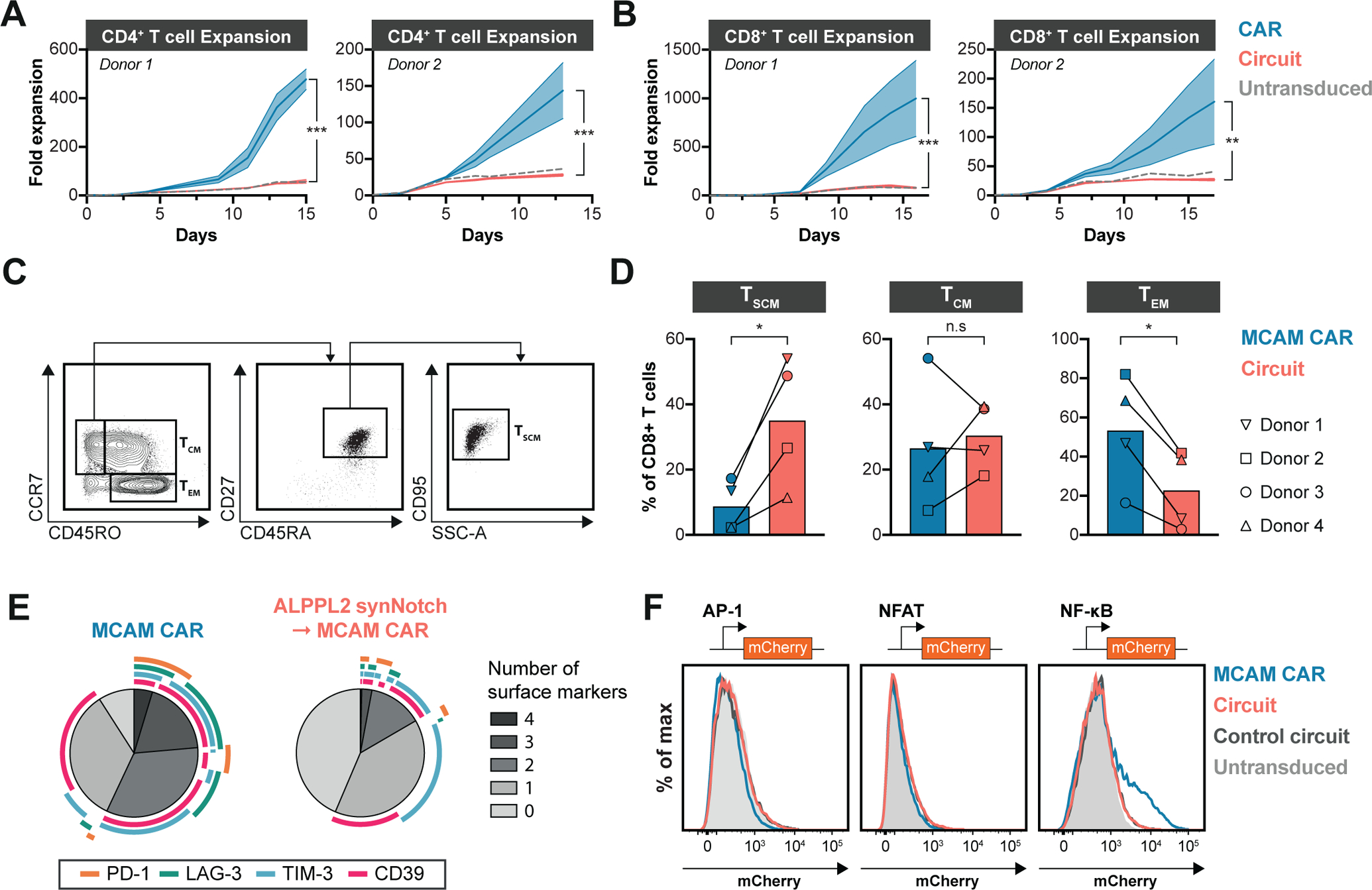
(A,B) Expansion of engineered CD4+ (A) or CD8+ (B) T cells expressing a MCAM (BBζ), mesothelin (MSLN, BBζ or 28ζ), or HER2 (BBζ) CAR either constitutively or through ALPPL2 synNotch circuit in two donors after removal of CD3/CD28 Dynabead stimulation. (C) Gating strategy for identifying T cells immunophenotypically resembling T stem cell memory-like (TSCM), T central memory (TCM), and T effector memory (TEM) cells. SSC-A, side scatter area. (D) Composition of TSCM, TCM, and TEM in non-antigen exposed CD8+ T cells from four different donors engineered to express a MCAM (BBζ) CAR either constitutively or through an ALPPL2 synNotch circuit 14 days post initial CD3/CD28 Dynabead stimulation. (E) Expression pattern analysis of CD39, LAG-3, PD-1, and TIM-3 in non-antigen exposed CD8+ T cells from three different donors engineered to express a MCAM (BBζ) CAR either constitutively or through ALPPL2 synNotch circuit 14 days post initial CD3/CD28 Dynabead stimulation. (F) Jurkat cells carrying AP-1, NFAT, or NF-κB response elements expressing an MCAM (BBζ) CAR constitutively or through an ALPPL2 synNotch circuit. The control circuit is ALPPL2 synNotch driving GFP expression (representative of two independent experiments). Statistics were calculated using two-way ANOVA with Šidák’s post-hoc test comparing CAR and Circuit (A,B) or unpaired (C) Student’s t-test. *P ≤0.05; **P ≤0.01; ***P ≤0.001; n.s.; not significant.
Further, we observed that constitutive expression of the MCAM CAR in CD8+ T cells impacted T cell differentiation and displayed a smaller fraction of T cells immunophenotypically resembling long-lived T stem cell memory-like (TSCM) cells, defined as CC-chemokine receptor 7 (CCR7)+CD45RO−CD27+CD45RA+CD95+ and a higher proportion of T effector memory cells defined as CCR7−CD45RO+ (TEM) in contrast to the ALPPL2 synNotch - MCAM CAR circuit (p=0.03 for TSCM and p=0.02 for TEM, Fig. 4C, 4D). In line with this, constitutive CAR expression led to higher expression and co-expression of surface markers linked to T cell exhaustion that was avoided by synNotch transcriptional regulation (p<0.001 for T cells expressing no markers, Fig. 4E and fig. S8B, S8C, and S8D). Further, Jurkat T cell reporter systems for activator protein 1 (AP-1), nuclear factor of activated T-cells (NFAT), and nuclear factor kappa B (NF-κB) transcriptional activity showed that constitutive expression of the MCAM CAR was sufficient to induce NF-κB-mediated transcription (Fig. 4F and fig. S9A). NF-κB transcriptional activity was not observed when the MCAM CAR was under control of the ALPPL2 synNotch suggesting that synNotch regulated CAR expression maintains the engineered T cell population in a superior functional state by circumventing unfavorable tonic signaling. NF-κB signaling has previously been associated with tonic signaling in BBζ CARs and has been linked to high CAR expression (31).
For lentiviral vector expression of CARs, the constitutive promoter dictates the strength of transgene expression (32). Since high CAR expression can contribute to tonic signaling, we tested whether a weaker constitutive promoter could mitigate tonic signaling similar to a synNotch CAR circuit. Therefore, we expressed the MCAM (BBζ), mesothelin (BBζ or 28ζ), and HER2 (BBζ) CARs under the strong spleen focus-forming virus (SFFV) and elongation factor 1-alpha (EF1α) promoters or the weaker human phosphoglycerate kinase (hPGK) promoter (fig. S9B). Generally, T cells with weaker CAR expression through the hPGK promoter more closely resembled those carrying the corresponding CAR in synNotch circuits in memory phenotype and CD39 expression (fig. S9C, S9D), similar to what have been shown before (9, 33). In line with this, hPGK promoter MCAM (BBζ) and mesothelin (BBζ) CAR expression caused lower tonic NF-κB-mediated transcriptional activity as opposed to SFFV and EF1α in the jurkat reporter system (fig. S9E). It has been suggested that weak CAR expression through a truncated PGK promoter display better anti-tumor activity and increased persistence to that of an EF1α driven CAR (33). However, the CAR expression profile from the hPGK promoter is highly variable, leading to a less uniform T cell therapeutic (fig. S9B).
SynNotch CAR circuits regulate T cell differentiation pre and post antigen stimulation in vitro.
We next sought to determine whether dynamically regulated CAR expression by synNotch is a general approach to prevent premature T cell differentiation beyond the MCAM CAR. Thus, we performed a long-term assay in which CD4+ and CD8+ T cells expressed the MCAM (BBζ), mesothelin (BBζ or 28ζ), or HER2 (BBζ) CARs constitutively or under the control ALPPL2 synNotch. The T cells were left unstimulated or underwent two rounds of stimulation over the course of two weeks and the differentiation state of the T cells was assessed periodically to longitudinally map the evolution of the memory and effector T cell populations (Fig. 5A). At baseline, T cells with constitutive expression of CARs showed a higher presence of effector memory phenotype than their ALPPL2 synNotch regulated counterpart. This was true in both CD4+ and CD8+ T cells, except for the mesothelin (28ζ) CAR in CD4+ T cells (Fig. 5B, 5C). When unstimulated, this phenotypic difference was conserved and with an enhanced accumulation of TEM (defined as CD45RA−CD62L−) in CD4+ T cells (p<0.001 at day 14) and T effector memory cells re-expressing CD45RA (TEMRA, defined as CD45RA+CD62L−) in CD8+ T cells (p<0.001 at day 14) with constitutively expressed CARs (Fig. 5D, 5E and fig. S10A, S10B). With two rounds of stimulation with dual antigen positive target cells, the synNotch circuit CAR T cells followed a similar differential pattern to that of regular CAR T cells, but with a higher fraction of retained TCM (defined as CD45RA−CD62L+) (p<0.001 at day 14, Fig. 5D, 5E and fig. S10A, S10B). Constitutively expressed CARs also displayed higher fractions of T cells co-expressing CD39, LAG-3, PD-1, and TIM-3 compared to synNotch circuit CAR T cells after the initial stimulation (p<0.001 for T cells co-expressing all 4 markers, fig. S10C). Thus, synNotch-mediated CAR regulation is a general means to control tonic signaling and prevent detrimental T cell differentiation prior to antigen exposure, and this effect is sustained during long-term ex vivo T cell culture and repeated antigen exposure.
Figure 5. SynNotch CAR circuits regulate T cell differentiation pre and post antigen stimulation in vitro.
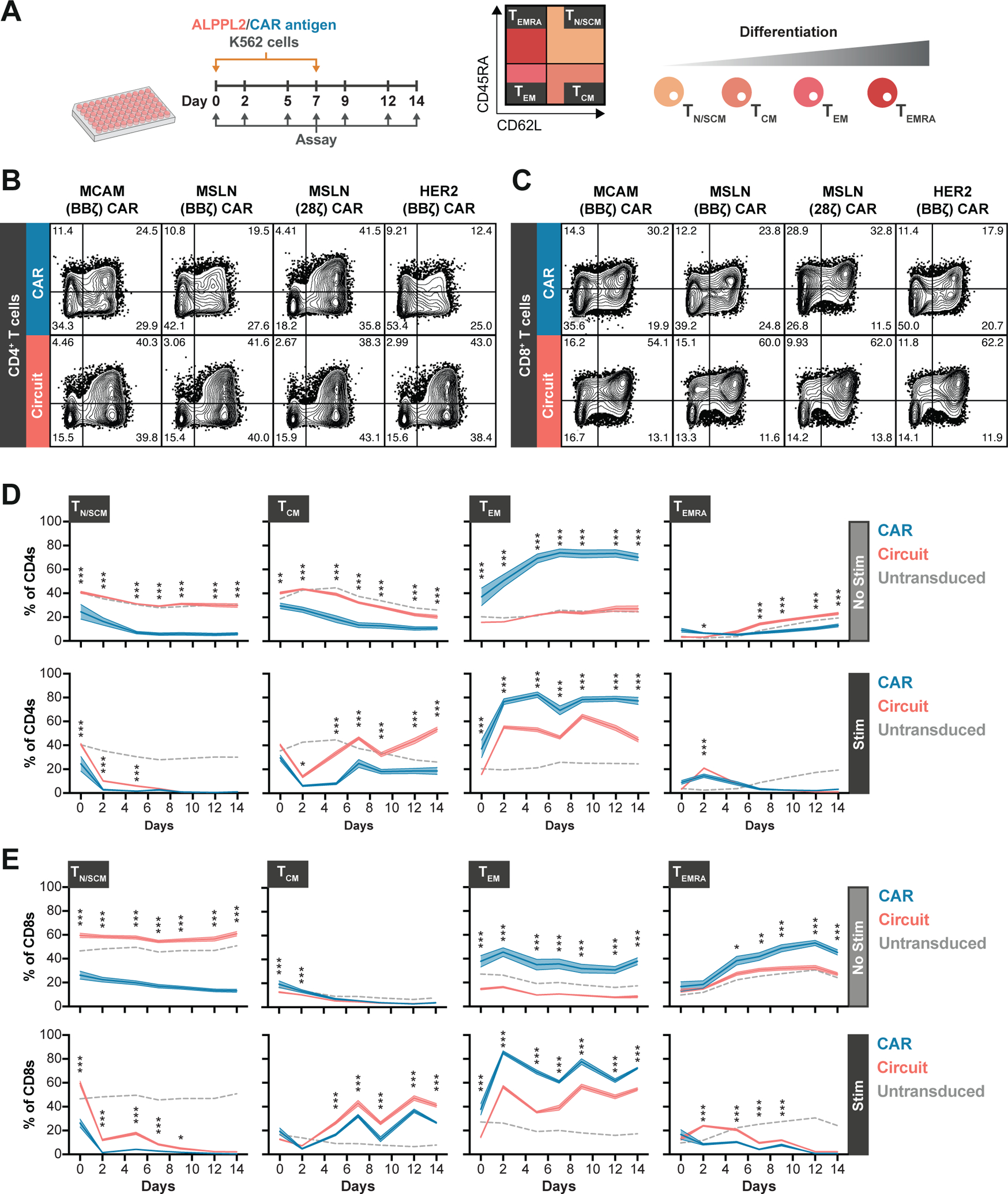
(A) Experimental timeline of longitudinal T cell memory subset analysis for CD4+ or CD8+ T cells engineered to express either an MCAM (BBζ), mesothelin (MSLN, BBζ or 28ζ), or HER2 (BBζ) CAR constitutively or through ALPPL2 synNotch circuits without or after two stimulations with ALPPL2+ K562 cells expressing MCAM, mesothelin, and HER2. TN/SCM, naïve/stem cell memory-like T cells (CD45RA+CD62L+); TCM, central memory-like T cells (CD45RA−CD62L+); TEM, effector memory-like T cells (CD45RA−CD62L−); TEMRA, effector memory-like T cells re-expressing CD45RA (CD45RA+CD62L−). (B,C) Baseline memory phenotype of CD4+ (B) and CD8+ (C) T cells expressing an MCAM (BBζ), mesothelin (BBζ or 28ζ), or HER2 (BBζ) CAR constitutively or through ALPPL2 synNotch circuits 14 days post initial CD3/CD28 Dynabead stimulation. (D,E) Phenotypic evolvement for engineered CD4+ (D) and CD8+ (E) T cells expressing an MCAM (BBζ), mesothelin (BBζ or 28ζ), or HER2 (BBζ) CAR constitutively or through ALPPL2 synNotch circuits upon culture without (No Stim) or after two stimulations (Stim) of ALPPL2+ K562 cells expressing the cognate CAR antigens. Data are presented as mean±SEM and statistics were calculated using two-way ANOVA with Šidák’s post-hoc test. *P ≤0.05; **P ≤0.01; ***P ≤0.001 comparing T cells expressing CAR or Circuit.
SynNotch CAR Circuit T cells exhibit superior efficacy and persistence relative to conventional CAR T cells in vivo.
To verify that ALPPL2 synNotch - MCAM CAR circuit T cells selectively targeted tumors expressing the combination of ALPPL2 and MCAM, we subcutaneously implanted NSG mice contralaterally with either ALPPL2 wild-type (WT) M28 tumor cells or ALPPL2 knock-out (KO) epithelioid M28 tumor cells (Fig. 6A). Seven days later mice were infused T cells engineered with the ALPPL2 synNotch - MCAM CAR or untransduced T cells as control. Mice treated with circuit CAR T cells showed selective reduction of the tumor expressing both ALPPL2 and MCAM (p<0.001), which was not seen for the tumor only expressing MCAM or for mice treated with untransduced T cells. This dual antigen dependent tumor reduction indicates exclusive AND-gated targeting of tumors expressing the correct antigen combination.
Figure 6. SynNotch CAR circuit T cells exhibit superior efficacy and persistence in vivo.
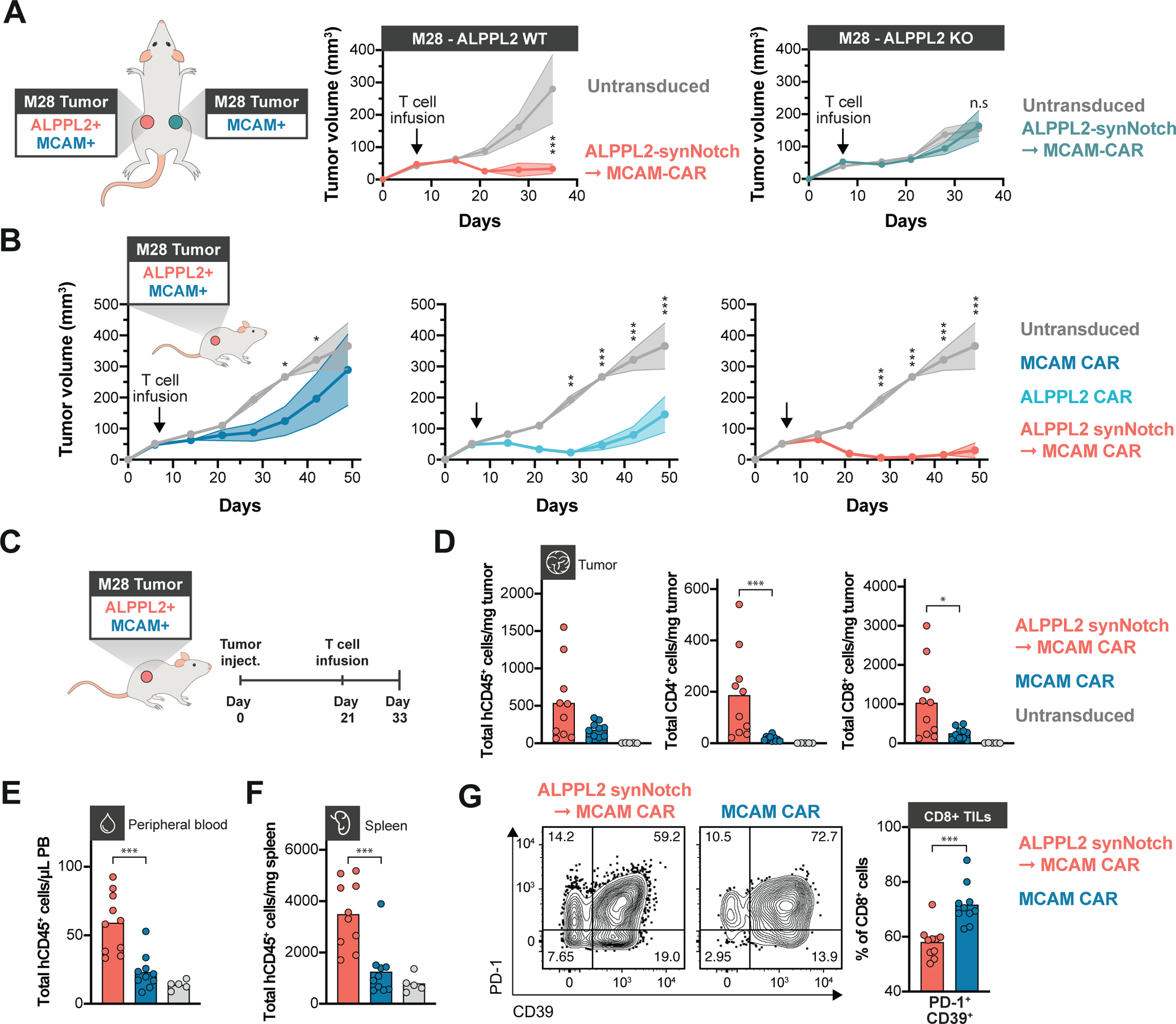
(A) NSG mice bearing contralateral subcutaneous tumors with ALPPL2 wild-type (WT) M28 tumors on the left side and with ALPPL2 knock-out (KO) M28 tumors on the right side received an adoptive transfer of a mix of CD4+ and CD8+ T cells engineered with an ALPPL2 synNotch→MCAM CAR circuit (n=5) or untransduced T cells (n=5). Tumor size was monitored over 35 days (representative of two independent experiments). (B) NSG mice bearing subcutaneous M28 tumors were injected i.v. with 1:1 ratio of CD4+:CD8+ T cells engineered with an ALPPL2 CAR (n=5), MCAM CAR (n=5), ALPPL2 synNotch regulating MCAM-CAR expression, or untransduced T cells (n=5). Tumor size was monitored over 49 days. (C) NSG mice bearing subcutaneous M28 tumors received an i.v. infusion of a 1:1 ratio of CD4+:CD8+ T cells engineered with either the MCAM CAR (n=10), the ALPPL2 synNotch→MCAM-CAR circuit (n=10), or untransduced T cells 21 days after tumor inoculation (n=5). Tumors, peripheral blood, and spleens were collected for phenotypic and numeric analysis of infused T cells 12 days post T cell infusion. (D) Number of tumor-infiltrating T cells (TILs) per mg tumor was determined by expression of human CD45 (hCD45) and CD4 or CD8. (E,F) Number of T cells per μL peripheral blood (E) and mg spleen tissue (F) was determined by expression of hCD45 (representative of two independent experiments). (G) Expression of PD-1 and CD39 in CD8+ TILs expressing a MCAM CAR either constitutively or through an ALPPL2 synNotch (representative of two independent experiments). Data are presented as mean±SEM. Statistics were calculated using either two-way ANOVA with Šidák’s (A) or Tukey’s post-hoc test (B), or unpaired Mann-Whitney’s t-test (D-G). *P ≤0.05; **P ≤0.01; ***P ≤0.001; n.s.; not significant.
To further evaluate the therapeutic efficacy of our new CARs and our clinically viable synNotch circuits, NSG mice were implanted subcutaneously with epithelioid M28 tumors seven days prior to receiving an infusion of ALPPL2 CAR T cells, MCAM CAR T cells, or ALPPL2 synNotch - MCAM CAR circuit T cells. Constitutive MCAM CAR T cells exhibited inconsistent ability to control M28 tumor growth, whereas ALPPL2 CAR T cells showed a distinct reduction in tumor growth (p=0.001 at the end of the experiment, Fig. 6B). However, the greatest effect was observed for the ALPPL2 synNotch - MCAM CAR circuit T cells, for which complete response was observed in a majority of the mice (p<0.001 at the end of the experiment, Fig. 6B). There was a clear correlation between both the number of CD4+ (p=0.010) and CD8+ (p=0.002) engineered T cells in the spleen and the ability to control tumor growth (fig. S11A), suggesting that T cell persistence plays a pivotal role in therapeutic efficacy. In the spleens of tumor bearing mice 9 days post T cell infusion, synNotch regulated MCAM CAR expression was highly associated with increased presence of T cells with a long-lived memory phenotype in both CD8+ and CD4+ T cells (p<0.001, fig. S11B). To enable isolation of tumor-infiltrating T cells (TILs), M28 tumors were implanted subcutaneously in NSG mice and allowed to establish and grow for three weeks prior to infusion of the engineered T cells (Fig. 6C). In line with their enhanced anti-tumor activity, ALPPL2 synNotch - MCAM CAR circuit T cells displayed higher numbers of total CD4+ (p<0.001) and CD8+ (p=0.03) TILs in contrast to MCAM CAR T cells (Fig. 6D). Further, circuit CAR T cells displayed higher numbers of circulating T cells (p<0.001) as well as T cells residing in the spleen (p<0.001, Fig. 6E and 6F), consistent with a higher persistence for these cells. CD8+ circuit CAR TILs displayed a smaller fraction of PD-1+CD39+ T cells as opposed to T cells constitutively expressing the MCAM CAR (p<0.001, Fig. 6G and fig. S11C), a population linked to exhausted T cells (34, 35).
ALPPL2 synNotch CAR circuits are adaptable to other tumor-associated antigens.
Combinatorial antigen targeting will likely be of benefit for precise recognition of a range of solid tumors (36). We therefore wanted to validate the therapeutic feasibility of ALPPL2 synNotch CAR circuits that target other tumor-associated antigens such as mesothelin. Thus, we treated NSG mice implanted with epithelioid M28 mesothelioma with T cells engineered with ALPPL2 synNotch → mesothelin CAR circuits where the CAR either had a 41BB or CD28 costimulatory domain. Both circuit options allowed T cells to reliably target and effectively clear M28 mesothelioma tumors in all mice (Fig. 7A).
Figure 7. ALPPL2 synNotch CAR circuit T cells effectively target mesothelin and HER2 positive tumors and exhibit persistent activity upon rechallenge.
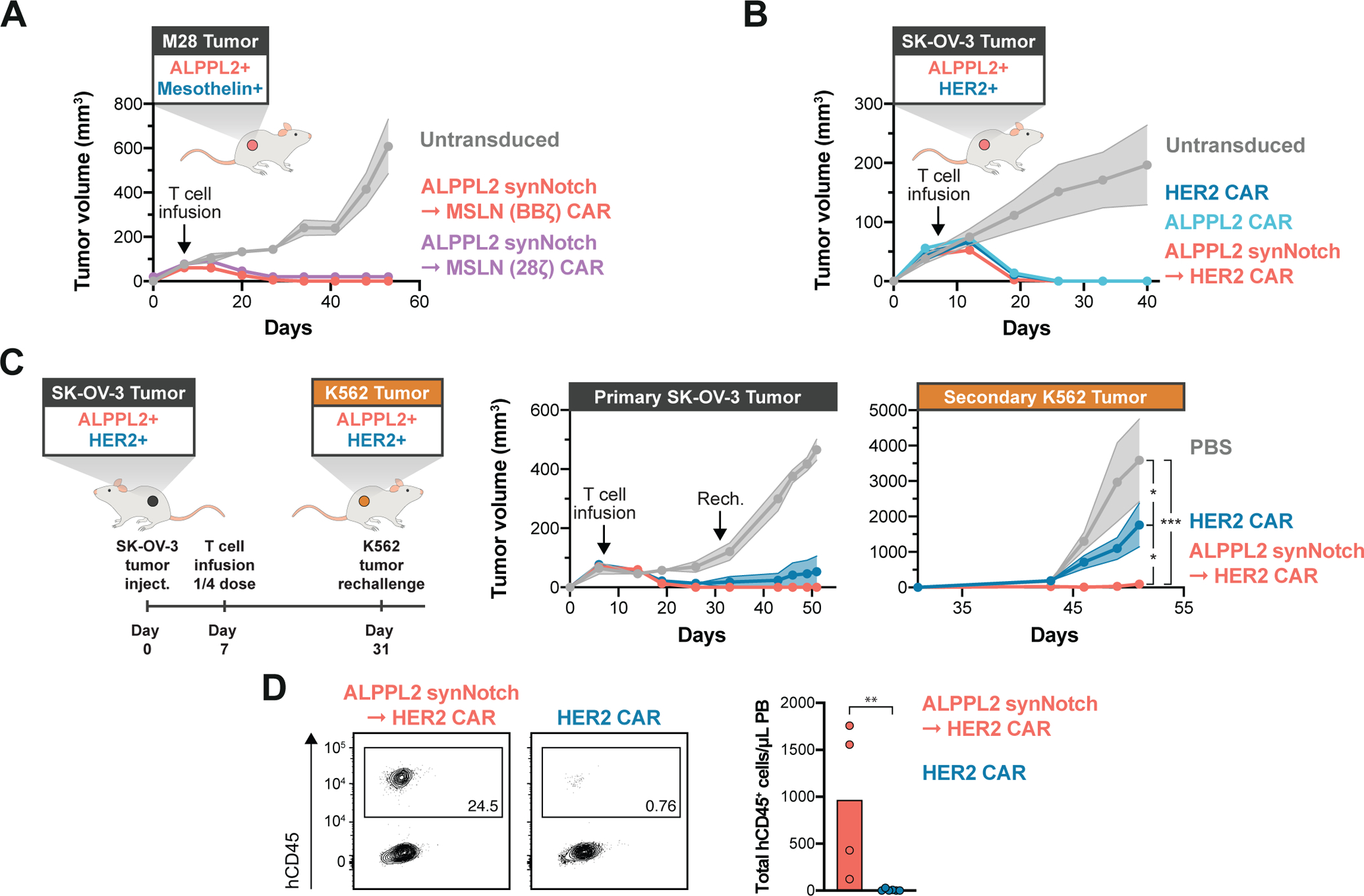
(A) NSG bearing subcutaneous M28 tumors were injected i.v. with 1:1 ratio of CD4+:CD8+ T cells engineered with an ALPPL2 synNotch regulating mesothelin (MSLN, BBζ or 28ζ) CAR expression (n=5 and n=3, respectively), or untransduced T cells (n=5) at 7 days post tumor implantation. Tumor size was monitored over 53 days. ALPPL2 synNotch - MSLN (28ζ) CAR is offset by +20 on the y-axis. (B) NSG mice bearing subcutaneous ALPPL2+ SK-OV-3 tumors were injected i.v. with 1:1 ratio of CD4+:CD8+ T cells engineered with an ALPPL2 CAR (n=5), HER2 CAR (n=5), an ALPPL2 synNotch regulating HER2 CAR expression (n=5), or untransduced T cells (n=5) at 7 days post tumor implantation. Tumor size was monitored over 40 days. (C) NSG mice bearing subcutaneous ALPPL2+ SK-OV-3 tumors on the left flank were injected i.v. with 1:1 ratio of CD4+:CD8+ T cells engineered with a HER2 CAR (n=6) or an ALPPL2 synNotch regulating HER2 CAR expression (n=4) at 7 days post tumor implantation. Control mice were injected with PBS (n=4). After 24 days, mice were contralaterally rechallenged with a subcutaneous injection of ALPPL2+HER2+ K562 tumor cells on the right flank. Tumor sizes were monitored over 51 days. (D) At 44 days post T cell infusion, presence of persistent human CD45+ (hCD45+) engineered T cells in the peripheral blood (PB) was assessed in tumor rechallenged mice by flow cytometry. Data are presented as mean±SEM. Statistics were calculated using two-way ANOVA with Tukey’s multiple comparison (C) or unpaired Mann-Whitney’s t-test (D). *P ≤0.05; **P ≤0.01; ***P ≤0.001.
To address the applicability of ALPPL2 as cell therapy target beyond mesothelioma, NSG mice were implanted with subcutaneous ALPPL2+HER2+ SK-OV-3 epithelial ovarian tumors seven days prior to receiving an infusion of ALPPL2 CAR T cells, HER2 CAR T cells, or ALPPL2 synNotch - HER2 CAR circuit T cells. At a dose of 3×106 CD4+ + 3×106 CD8+ (total of 6e6 T cells) all experimental groups effectively cleared the SK-OV-3 tumors (Fig. 7B). To more stringently assess whether synNotch regulated CAR expression enhanced T cell persistence for HER2 CAR T cells, ALPPL2+HER2+ SK-OV-3 tumor-bearing mice with a lower dose of engineered T cells (total of 1.5×106 T cells, 1:1 ratio of CD4+:CD8+) seven days after tumor implantation, for which a majority of mice still controlled the primary tumor growth (Fig. 7C). Therefore, to determine the functionality and persistence of the infused T cells, we rechallenged the mice with a fast-growing ALPPL2+HER2+ K562 tumor on the contralateral flank 31 days post initial tumor inoculation. ALPPL2 synNotch - HER2 CAR circuit T cells displayed superior tumor control compared to HER2 CAR T cells upon rechallenge (p=0.021, Fig. 7C), suggesting that synNotch CAR T cells persist and retain activity longer than the CAR T cells in vivo. Indeed, mice receiving ALPPL2 synNotch → HER2 CAR circuit T cells retained more circulating T cells than mice with HER2 CAR T cells, where the CAR T cells were barely detected at endpoint (p=0.010, Fig. 7D). Collectively, we show that ALPPL2 synNotch CAR circuits are a viable and potentially superior T cell therapy to conventional CAR T cells for solid tumors, as they enhance specificity and persistent activity of the T cells through controlled CAR expression, which was associated with maintenance of a T cell memory phenotype.
DISCUSSION
The field of engineered T cell therapy suffers from the lack of tumor-specific antigens, but it has recently been reported that there are many potential opportunities to increase specificity when combinations of antigens are considered (36, 37). In particular, T cells that can reliably perform AND or AND-NOT logic could be of great benefit to improve tumor targeting (37). Here we identified ALPPL2 as a highly tumor-specific antigen that can act as a singular target for CAR T cell therapy or can be sensed in combination with a second highly expressed tumor-associated antigens such as MCAM, mesothelin, or HER2 with synNotch CAR circuits (fig. S12). Of note, the exquisite specificity of ALPPL2 was defined by IHC using our anti-ALPPL2 antibodies as well as commercial antibodies (17). When we consider public databases with RNA-seq information (https://www.proteinatlas.org), there is a residual amount of ALPPL2 transcripts in some normal tissues such as lung and colon. In these tissues, transcripts per million (pTPM) on average are 1 for lung and 0.2 for colon. However, our IHC studies using multiple methods and antibody forms did not detect ALPPL2 expression in normal lung and colon (ref. 17 and this study). There is a possibility that the lack of detection is due to inherent limitations of IHC such as epitope alteration during tissue processing even though we used frozen tissue samples or the residual pTPMs seen in lung and colon could also be due to rare cells not detectable by IHC. Nonetheless, ALPPL2 shows a much higher degree of tumor specificity when compared to many of the antigen currently targeted by CAR T cells in trials, including mesothelin and HER2. Thus, ALPPL2 is an antigen that warrants further exploration for therapeutic targeting by cell therapies.
Although synNotch CAR circuits enhance specificity through multi-antigen targeting, there is a chance that this feature could increase the risk of tumor escape due to antigen loss. An approach to mitigate this potential problem is presented in our companion study by Choe et. al. where synNotch CAR circuits were designed to achieve OR-gated tumor killing through induction of a bi-specific CAR (38). The study also demonstrated that a small subset of synNotch-priming tumor cells is sufficient to eliminate surrounding single antigen tumor cells, showing that synNotch CAR circuits can eliminate tumors with heterogenous antigen expression patterns.
We also explored the multidimensional advantages of synNotch CAR circuits, which circumvent some of the critical problems thought to prevent efficacy of cell therapies for solid tumors. SynNotch CAR circuits not only provide improved specificity through multi-antigen sensing, but are a general means to enhance therapeutic efficacy through cell-autonomous and context-dependent regulation of CAR expression and prevention of tonic signaling, leading to the maintenance of T cell memory subsets important for persistence and sustained activity of the cell therapy (fig. S12B).
Although there are several strategies to develop T cells with AND-logic for improved tumor recognition, most rely on continuous expression of CAR components prone to tonic signaling (19). An alternative is drug-controlled CAR signaling activity, either through small molecule-dependent receptor assembly or degradation or dampening of CAR signaling activity through the Src family kinase-inhibitor Dasatinib (7, 39–41). Induced rest of CAR T cells by pulsed inhibitor administration can improve persistence and memory formation (7, 42), further highlighting the benefit and need for dynamic CAR regulation for enhanced function. However, dasatinib is known to promote immunosuppression and to impair CD8+ T cell function (43, 44), warranting caution for its use in patients with solid tumors.
It is also unclear whether solely maintaining a beneficial T cell phenotypic state, either through genetic circuits or drug-control, will be sufficient to combat the plethora of functional challenges presented to CAR T cells in solid tumors (45). Beside contextual and localized CAR expression, synNotch circuits offer the ability to drive tumor-localized production of genetically encoded therapeutic payloads such as shRNAs, cytokines, chemokines, or antibodies along with the CAR (19, 46). These payloads can be used to enhance intrinsic T cell function or modulate immunosuppressive and refractory tumor microenvironments. However, relevant and immunologically intact experimental tumor models are needed to fully assess the true impact and potential risk of such additions. These models will also be crucial to study actual memory formation of CAR T cells, a field with limited insight. Additionally, more systematic and in-depth characterization of the tumor microenvironment in patient-derived tumor samples is needed to help prioritize payloads for specific tumor types.
Above we have discussed the limitations of our study including the inability to rule out low levels of ALPPL2 on normal tissue, lack of data addressing antigen escape, and incomplete modeling of the tumor microenvironment in our xenograft studies. Despite these limitations, we show marked benefits of synNotch CAR circuit T cells when compared to the current state-of-the-art CAR T cells. Dynamic antigen-dependent control of CAR expression with synNotch can lead to a T cell therapy that exhibits persistent anti-tumor activity at lower T cell doses concomitant with enhanced specificity. Because of these features, synNotch circuit T cells may prove to have therapeutic benefit in patients with solid tumors where conventional CAR T cells have been ineffective to date.
MATERIALS AND METHODS
Study design
The aim of this study was to determine the therapeutic potential of ALPPL2 as target in cell-based immunotherapy of solid tumors. We started by generating a second-generation ALPPL2 BBζ CAR based on affinity matured scFv targeting ALPPL2 that we have previously described (17). We then determined clinically relevant antigen partners to ALPPL2 in mesothelioma and ovarian cancer that would allow for combinatorial targeting. We engineered synNotch CAR circuits for the resultant antigen signatures and assessed their ability to perform a range of T cell functionalities in vitro. We then investigated whether synNotch CAR circuits could circumvent the detrimental tonic signaling commonly seen for constitutively expressed CARs. Circuit CAR T cells were assessed for their specificity, efficacy, and persistence in a range of xenograft mice models. Sample sizes were selected on the basis of previous experience and published experiments and experiments included a minimum of three mice per group. For TIL immunophenotyping, only subsets with at least 100 recorded events were included. Number of biological replicates and experiment replicates is described in the figure legends. For in vivo mouse experiments, mice were randomly assigned to each experimental group. Investigators were not blinded to the experiments.
Recombinant scFv production
ScFv expression and purification was done as previously described (16, 47). Briefly, the scFv gene was cloned into the secretion vector pUC119mycHis to impart a c-myc epitope and a hexahistidine tag at the C-terminus. Soluble scFv was harvested from the bacterial periplasmic space and purified by immobilized metal affinity (HiTrap His, GE HealthCare) and ion-exchange (DEAE, GE HealthCare) chromatography.
Immunohistochemistry
IHC studies on frozen tissues and formalin-fixed paraffin-embedded (FFPE) tissue arrays were performed as previously described (17). Briefly, frozen normal human tissue arrays (US Biomax) were blocked with 3% H2O2 and avidin/biotin (Vector Laboratories), washed three times with phosphate buffered saline (PBS) containing 0.1% Tween-20 (PBST), and incubated with 10μg/ml biotinylated scFvs (M25FYIA and a non-binding C10) at 4°C overnight, followed by detection with streptavidin-horseradish peroxidase (HRP, Jackson ImmunoResearch Laboratories) using DAKO liquid 3,3-Diaminobenzidine (DAB)+ (Agilent). For MCAM/ALPPL2 co-staining on FFPE tissue arrays (US Biomax), the ImmPRESS Duet Double Staining HRP/ AP Polymer Kit was used (HRP anti-Mouse IgG and alkaline phosphatase [AP] anti-Rabbit IgG, Vector Laboratories). Following deparaffinization and antigen retrieval with Tris Buffer (10mM Tris Base, 0.05% Tween 20, pH 10.0), endogenous peroxidase activity was blocked by BLOXALL blocking solution for 15 minutes followed by PBST wash. The slides were further blocked with 2.5% normal horse serum at RT for 1 hour, washed three times with PBST and incubated with the following primary antibody pairs diluted into 2.5% normal horse serum and incubated with the tissue slides at 4°C overnight: pair 1: anti-MCAM rabbit mAb and anti-ALPPL2 mouse mAb (both at 1:200 dilution); and pair 2: anti-MCAM rabbit pAb and anti-ALPPL2 mouse mAb (both at 1:100 dilution). Slides were washed three times with PBST, incubated with ImmPRESS Duet reagent at RT for 30 minutes, followed by sequential detection with ImmPACT DAB EqV and ImmPACT Vector Red substrates. Slides were counterstained with hematoxylin followed by bluing reagent (Scytek). After dehydration, the slides were mounted using Permount mounting media (Thermo Fischer Scientific) and scanned by Aperio XT and AT2 digital scanners (Leica).
Vector construction designs
All CARs contained the CD8α signaling peptide (MALPVTALLLPLALLLHAARP) followed by a FLAG-tag (DYKDDDDK) and either an anti-CD19 (FMC63) (48), anti-ALPPL2 (M25, M25ADLF, or M25FYIA) (16, 17), anti-MCAM (M1 or M40) (16), anti-mesothelin (m912) (49), or anti-HER-2 (4D5–8) (50) scFv. CARs were then fused to either the CD8α hinge/transmembrane domain, 4-1BB intracellular domain, and CD3ζ intracellular domain or CD28 hinge/transmembrane domain and CD3ζ intracellular domain. In order to determine expression, a T2A self-cleaving peptide preceding enhanced green fluorescent protein (eGFP) was attached to the CAR coding sequence. All constitutive CARs were cloned into a modified pHR’SIN:CSW vector containing a SFFV promoter. For some experiments, CARs were cloned into a modified pHR’SIN:CSW vector containing a EF1α or hPGK promoter. The ALPPL2 synNotch was constructed using the anti-ALPPL2 (M25FYIA) scFv as previously described (20). The ALPPL2-synNotch was cloned into a modified pHR’SIN:CSW vector containing a mPGK promoter. Inducible CAR response elements were constructed by subcloning the CAR-T2A-eGFP sequences into a modified pHR’SIN:CSW vector carrying five copies of the Gal4 DNA binding domain target sequence (GGAGCACTGTCCTCCGAACG) 5′ to a minimal cytomegalovirus (CMV) promoter. These vectors also included a mPGK promoter constitutively driving the expression of either of the fluorescent reporters mCherry or blue fluorescent protein. For ectopic expression of ALPPL2, ALPI, mesothelin, and truncated HER2, coding sequences corresponding to NP_112603.2 (ALPPL2), NP_001622.2 (ALPI), NP_001170826.1 (mesothelin), and NP_004439.2 (HER-2; amino acid 1–730) were cloned into a modified pHR’SIN:CSW vector containing a SFFV promoter. For ALPPL2, either a FLAG-tag or HA-tag (YPYDVPDYA) was inserted 3´ of its predicted signaling peptides (MQGPWVLLLLGLRLQLSLG). For ALPI, a FLAG-tag was inserted 3´ of its predicted signaling peptide (MQGPWVLLLLGLRLQLSLG). To establish non-perturbing nuclear fluorescent labeling, Sv40 NLS (PKKKRKV) was fused 5´ to mKate2 via a linker sequence (DPPVAT) and cloned into a modified pHR’SIN:CSW vector containing a EF1α promoter. To establish doxycycline controlled expression of ALPPL2 (FLAG-tag), MCAM, mesothelin, and HER2 were cloned into a modified pHR’SIN:CSW vector containing a TRE3GS inducible promoter with constitutive downstream cassette of a SFFV promoter driving the expression of rtTA3. To enable genetic knockout (KO) of ALPPL2 and MCAM, the gRNA sequence TGGGAGTGGTAACCACCACA and AGGCGCAGCTCCCGGGCTGG were cloned into the pL-CRISPR.EFS.GFP vector, which was a gift from Benjamin Ebert (Addgene plasmid #57818). AP-1, NFAT, and NFκB response elements and the minimal promoters used for T cell activity reporters were a gift from Peter Steinberger (Addgene plasmid #118031, 118094, and 118095) and were cloned together with mCherry into a modified pHR’SIN:CSW vector. All constructs were cloned using In-Fusion cloning (Takara Bio).
Lentiviral production and cell lines
To produce pantropic vesicular stomatitis virus-G pseudotyped lentivirus, Lenti-X 293T cells (Takara Bio) were transfected with a transgene expression vector and the viral packaging plasmids pCMVdR8.91 and pMD2.G using TransIT-Lenti Transfection Reagent (Mirus Bio LLC). M28 and VAMT-1 tumor cells were originally obtained from Dr. Brenda Gerwin’s lab at the National Cancer Institute and A549 (CCLZR013) and SK-OV-3 (CCLZR377) cells were obtained from the UCSF Cell and Genome Engineering Core. Expression of ALPPL2, ALPI, mesothelin, HER2, and/or CD19 on K562 cells (CCL-243, ATCC), ALPPL2 and overexpression of MCAM or HER2 on A549, mesothelin on M28 cell, and ALPPL2 on SK-OV-3 was performed by lentiviral transduction. Generation of ALPPL2 KO M28 tumor cells and MCAM KO K562 tumor cells was performed through lentiviral transduction with a vector carrying sgRNA-Cas9-P2A-eGFP. To enable real-time counting of M28, VAMT-1, and SK-OV-3 target cells, nuclei were tagged with a non-perturbing fluorescent mKate2 reporter through lentiviral transduction. Inducible ALPPL2, mesothelin, and HER2 K562 clonal target cells were generated through lentiviral transduction. Inducible MCAM was generated similarly but using MCAM KO K562 cells. AP-1, NFAT, and NFκB reporters were generated through lentiviral transduction of Jurkat (clone E6-1) cells (TIB-152, ATCC). Lenti-X 293T and A549 cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco) with 10% fetal bovine serum (FBS, MilliporeSigma), 50units/mL Penicillin and 50 μg/ml Streptomycin (MP biochemicals), and 1X Sodium Pyruvate (MilliporeSigma). K562 cells were cultured in Iscove’s Modified Dulbecco’s Medium (Gibco) with 10% FBS, 50units/mL Penicillin and 50μg/ml Streptomycin. Jurkat, M28, and VAMT-1 cells were cultured in RPMI-1640 (Gibco) with 10% FBS, 100units/mL Penicillin and 100μg/ml Streptomycin, and 1X Glutamax (Gibco). SK-OV-3 cells were cultured in McCoy’s 5a Medium Modified (Gibco) 10% FBS (MilliporeSigma), 50units/mL Penicillin and 50μg/ml Streptomycin (MP biochemicals), and 1X Glutamax (Gibco).
Doxycycline inducible surface ligand
Clonal lines of K562 cells carrying a doxycycline inducible ALPPL2, HER2, MCAM, or mesothelin cassette was treated with doxycycline (Abcam) at doses ranging between 0.1–100ng/mL for 2–3 days. Surface expression densities were assessed by flow cytometry right before being used in T cell stimulation assays.
Engineering of human T cells and functional assays
Primary CD4+ and CD8+ T cells were isolated, cultured, and lentivirally transduced as previously described (20). Transduced T cells were sorted to carry to correct transgene composition and expanded and rested until day 10 for in vivo experiments and day 14 to 16 for in vitro assays. For in vitro stimulation assays of engineered T cells, effector cells were combined with targets cells at a 1:1 ratio in 96-well U-bottom plates and centrifuged for 1 minute at 400×g to force interaction between effector and target cells. To assess T cell activation, cells were analyzed for surface expression of CD69 and CD25 16 to 48 hours post target challenge. For proliferation assays, engineered T cells were stained with CellTrace Violet (CTV) Cell Proliferation Kit (Thermo Fisher Scientific) prior to being combined with target cells. CTV dilution was assessed 3 days post target challenge. Prior to intracellular cytokine (IC) assays T cells were treated with Pierce Anti-c-Myc Magnetic Beads (Thermo Fisher Scientific) for 24 hours in order to prime CAR expression for inducible CAR circuits before blocking protein transport. After inducible CAR priming, K562 target cells were added in the presence of 1X Brefeldin A solution (Thermo Fisher Scientific) and 1X Monensin solution (Biolegend) for 6 hours. All experiments were performed in X-VIVO 15 (Lonza), 5% Human AB serum, and 10mM neutralized N-acetyl L-Cysteine (Millipore Sigma) supplemented with 30units/mL recombinant human IL-2 (R&D Systems).
Incucyte killing assay
For in vitro engineered T cell killing assays, mesothelin+ M28, VAMT-1, ALPPL2+ SK-OV-3, or engineered A549 tumor cells expressing nuclear mKate2 were seeded in 96-well flat-bottom plates. After 24 hours, engineered T cells were added at an expected effector:target ratio of 2:1. Plates were imaged every 2 hours using the IncuCyte S3 Live-Cell Analysis System (Essen Bioscience) for a duration of 5 days. Three images per well at 10X magnification were collected and analyzed using the IncuCyte S3 Software (Essen Bioscience) to detect and count the number of mKate2+ nuclei per image. Experiments were performed in RPMI-1640 with 10% FBS, 100units/mL Penicillin and 100μg/ml Streptomycin, 1X Glutamax supplemented with 30units/mL IL-2.
Flow cytometry antibodies, analysis, and sorting
For scFv binding analysis, M28 and VAMT-1 mesothelioma cells in exponential growth phase were incubated with M1 (MCAM) or M25FYIA (ALPPL2) scFv for 1 hour at RT, washed three times with PBS, further incubated with a secondary anti-6xHis (4E3D10H2/E3, MA1-135-A647, Thermo Fischer Scientific) at 1:1000 dilution for 1 hour at RT, and washed three times with PBS before analysis. For immunophenotyping, samples were stained in 50–100μL of PBS supplemented with 2% FBS for either 20 to 30 minutes at 4°C or 30 minutes at RT. Stains including CC-chemokine receptor 7 (CCR7) were performed for 15 minutes at 37°C followed by 15 minutes at RT. For extracellular stains, cell washes and final resuspension was performed with PBS supplemented with 2% FBS. IC stains were performed using an Intracellular Fixation & Permeabilization Buffer Set (Thermo Fisher Scientific) per manufacturer’s instructions. Dead cells were excluded with Draq7 (Abcam) or Zombie NIR Fixable Viability Kit (Biolegend).
Antibodies used for immunophenotyping can be found in table S5. Cells were analyzed using either Accuri C6, LSR II SORP, FACSymphony X50 SORP or, for cell sorting, FACSAria II SORP, FACSAria IIIu SORP, or FACSAria Fusion SORP (all BD Biosciences). Cell counts were performed using flow cytometry with CountBright Absolute Counting Beads (Thermo Fisher Scientific) per manufacturer’s instructions. Data was analyzed using FlowJo software (BD Biosciences).
Xenograft tumor models
All mouse experiments were conducted according to an Institutional Animal Care and Use Committee (IACUC)–approved protocol (AN177022) or through the UCSF Helen Diller Family Comprehensive Cancer Center Preclinical Core. Sex and age-matched NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) (RRID:IMSR_JAX:005557) mice were used for all experiments, starting at an age of 8–12 weeks. For unilateral tumors, NSG mice were implanted with either 4×106 M28, Mesothelin+ M28, or ALPPL2+ SK-OV-3 tumor cells subcutaneously. Seven days after tumor implantation, a total of 3–6×106 primary T cells at a 1:1 ratio of CD4+:CD8+ T cells were infused intravenously (i.v.) through tail vein injection. Tumor size was monitored using a caliper. To enable isolation of tumor infiltrating T cells from M28 tumors, mice were implanted with 4×106 M28 21 days prior to T cell infusion. Tumor discrimination experiments were performed by contralateral tumors, for which NSG mice were implanted with 4×106 ALPPL2 wild-type (WT) M28 tumor cells on the left side and with 4×106 ALPPL2 knock-out (KO) M28 tumor cells on the right side. Seven days after tumor implantations, a total of 3×106 primary T cells were infused i.v. through tail vein injection. Tumor sizes were monitored weekly using a caliper. For tumor rechallenge, NSG mice were initially challenged with 4×106 ALPPL2+ SK-OV-3 on the left flank 7 days prior to a T cell infusion of 1.5×106 primary T cells at a 1:1 ratio of CD4+:CD8+. After an additional 24 days, 1×106 ALPPL2+HER2+ K562 tumor cells were implanted on the right flank. For immunophenotypic analysis, peripheral blood was collected with EDTA coated Capillary Blood Collection tubes (RAM scientific) and subjected to red blood cell lysis (ACK; KD medical); spleens were manually dissociated and subjected to red blood cell lysis (ACK; KD medical); and tumors were finely minced and digested for 30 minutes at 37°C in RPMI-1640 (Gibco) supplemented with 0.1mg/mL DNase I (Sigma) and 4mg/mL Collagenase IV (Worthington Biochemical Corporation).
Statistical analysis
Data are presented as mean means ± standard error mean (SEM), means ± standard deviations (SD), or mean with individual data points as indicated in the figure legends. Data shows technical replicates unless otherwise stated. Statistics were calculated using paired or unpaired t-test and two-way ANOVA with Šidák’s (when comparing two groups) or Tukey’s (when comparing multiple groups) post-hoc test as stated in the figure legends. For Student’s t-test, normality was first determined using Shapiro-Wilk normality test. Correlation was determined using Spearman’s rank correlation coefficient. All statistical analyses were performed with Prism software version 9.0.1 (GraphPad software). Raw data are provided in data file S1.
Supplementary Material
Fig. S1. Characterization of ALPPL2 tissue expression and ALPPL2 CAR specificity.
Fig. S2. Characterization of ALPPL2 synNotch-regulated CAR expression.
Fig. S3. Activation and proliferation of antigen-stimulated ALPPL2 synNotch CAR circuits.
Fig. S4. Cytokine production by CAR and synNotch CAR circuit T cells.
Fig. S5. Polyfunctionality of CAR and synNotch CAR circuit T cells.
Fig. S6. SynNotch CAR circuit T cells exhibit multi-antigen specificity and paced tumor elimination.
Fig. S7. Development of tumor cell lines expressing titratable antigen densities and synNotch CAR circuit sensitivity toward mesothelin.
Fig. S8. Phenotypic analysis of CAR and synNotch CAR T cells prior to antigen exposure.
Fig. S9. Impact of CAR expression on tonic signaling.
Fig. S10. SynNotch CAR circuits regulate the evolution of T cell differentiation pre and post antigen stimulation.
Fig. S11. Persistence of CAR T cells and synNotch CAR circuit T cells in treated tumor-bearing mice.
Fig. S12. SynNotch enhancements for CAR T therapy.
Table S1. ALPPL2 expression in solid tumors.
Table S2. Tissue specificity of ALPPL2 expression.
Table S3. IHC of MCAM expression in FFPE mesothelioma tissue arrays.
Table S4. MCAM co-expression in ALPPL2 positive mesothelioma.
Table S5. Antibodies used for immunophenotypic analysis.
Data file S1. Primary data.
Acknowledgements
We thank the members of the Roybal lab for valuable input. We acknowledge the UCSF Parnassus Flow Core (PFCC, RRID:SCR_018206) for providing instruments and service related to flow cytometry.
Funding
The study was supported in part by a grant from the Parker Institute for Cancer Immunotherapy (K.T.R. & B.L.). A.H-W. has received funding from the Swedish Society for Medical Research and the Swedish Research Council. K.T.R. is funded by the Parker Institute for Cancer Immunotherapy, the UCSF Helen Diller Family Comprehensive Cancer Center, the Chan Zuckerberg Biohub, an NIH Director’s New Innovator Award (DP2 CA239143 to K.T.R), Cancer Research UK, and the Kleberg Foundation. The PFCC is supported in part by Grant NIH P30 DK063720 and NIH S10 Instrumentation Grant S10 1S10OD021822-01.
Footnotes
Competing Interests
K.T.R. is a cofounder, consultant, and scientific advisory board member of Arsenal Biosciences. K.T.R. is an inventor on patents for synthetic Notch receptors (WO2016138034A1, PRV/2016/62/333,106) and receives licensing fees and royalties. The patents were licensed by Cell Design Labs and are now part of Gilead. He was a founding scientist/consultant and stockholder in Cell Design Labs, now a Gilead Company. K.T.R. holds stock in Gilead. K.T.R is on the scientific advisory board of Ziopharm Oncology. K.T.R. was a scientific advisory board member and stockholder in Xyphos now an Astellas Company. K.T.R. is an advisor for Venrock. B.L. and Y.S. are inventors on patents (WO2017095823A1, US20180369409A1) held by University of California that cover ALPPL2-targeted anti-cancer therapy and ALPPL2-targeting antibodies. Unrelated to this work, B.L. is a founder and stockholder of Fortis Therapeutics and Vivace Therapeutics, and a consultant for Merck Sharpe & Dohme. The remaining authors declare no competing financial interests.
Data and Materials Availability
All data associated with this study are in the paper or the Supplementary Materials. Materials created in this study will be available for the scientific community by contacting the corresponding author and completion of a material transfer agreement.
References
- 1.Jacoby E, Shahani SA, Shah NN, Updates on CAR T-cell therapy in B-cell malignancies, Immunol. Rev 290, 39–59 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2, Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLellan AD, Ali Hosseini Rad SM, Chimeric antigen receptor T cell persistence and memory cell formation, Immunol. Cell Biol 97, 664–674 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Ajina A, Maher J, Strategies to Address Chimeric Antigen Receptor Tonic Signaling, Mol. Cancer Ther 17, 1795–1815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors, Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, Ng KY, Ghoddusi M, Purdon TJ, Wang X, Do T, Pham MT, Brown JM, De Larrea CF, Olson E, Peguero E, Wang P, Liu H, Xu Y, Garrett-Thomson SC, Almo SC, Wendel H-G, Rivière I, Liu C, Sather B, Brentjens RJ, GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells, Sci Transl Med 11, eaau7746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber EW, Lynn RC, Parker KR, Anbunathan H, Lattin J, Sotillo E, Good Z, Malipatlolla M, Xu P, Vandris P, Majzner RG, Qi Y, Chen L-C, Gentles AJ, Wandless TJ, Satpathy AT, Chang HY, Mackall CL, Transient “rest” induces functional reinvigoration and epigenetic remodeling in exhausted CAR-T cells, bioRxiv, 2020.01.26.920496 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Khericha M, Lakhani A, Meng X, Salvestrini E, Chen LC, Shafer A, Alag A, Ding Y, Nicolaou D, Park JO, Chen YY, Rational Tuning of CAR Tonic Signaling Yields Superior T-Cell Therapy for Cancer, bioRxiv, 2020.10.01.322990 (2020). [Google Scholar]
- 9.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M, Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection, Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolini F, Bocchini M, Bronte G, Delmonte A, Guidoboni M, Crinò L, Mazza M, Malignant Pleural Mesothelioma: State-of-the-Art on Current Therapies and Promises for the Future, Front Oncol 9, 1519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Gooijer CJ, Borm FJ, Scherpereel A, Baas P, Immunotherapy in Malignant Pleural Mesothelioma, Front Oncol 10, 187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M, Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity, Sci Transl Med 6, 261ra151–261ra151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, Soltermann A, Jüngel A, Marroquin Belaunzaran O, Stahel R, Renner C, Petrausch U, Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells, J Transl Med 11, 187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, CA Cancer J Clin 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Hartnett EG, Knight J, Radolec M, Buckanovich RJ, Edwards RP, Vlad AM, Immunotherapy Advances for Epithelial Ovarian Cancer, Cancers (Basel) 12, 3733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An F, Drummond DC, Wilson S, Kirpotin DB, Nishimura SL, Broaddus VC, Liu B, Targeted drug delivery to mesothelioma cells using functionally selected internalizing human single-chain antibodies, Mol. Cancer Ther 7, 569–578 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Y, Zhang X, Bidlingmaier S, Behrens CR, Lee N-K, Liu B, ALPPL2 Is a Highly Specific and Targetable Tumor Cell Surface Antigen, Cancer Res 80, 4552–4564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi Y, Tu Z, Zhang Y, Yang P, Guo M, Zhu X, Zhao C, Zhou J, Wang H, Wang Y, Gao S, Identification of ALPPL2 as a Naive Pluripotent State-Specific Surface Protein Essential for Human Naive Pluripotency Regulation, Cell Rep 30, 3917–3931.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Hyrenius-Wittsten A, Roybal KT, Paving New Roads for CARs, Trends Cancer 5, 583–592 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA, Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits, Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidlingmaier S, He J, Wang Y, An F, Feng J, Barbone D, Gao D, Franc B, Broaddus VC, Liu B, Identification of MCAM/CD146 as the target antigen of a human monoclonal antibody that recognizes both epithelioid and sarcomatoid types of mesothelioma, Cancer Res 69, 1570–1577 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shih IM, Nesbit M, Herlyn M, Kurman RJ, A new Mel-CAM (CD146)-specific monoclonal antibody, MN-4, on paraffin-embedded tissue, Mod. Pathol 11, 1098–1106 (1998). [PubMed] [Google Scholar]
- 23.Yan X, Lin Y, Yang D, Shen Y, Yuan M, Zhang Z, Li P, Xia H, Li L, Luo D, Liu Q, Mann K, Bader BL, A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth, Blood 102, 184–191 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Metcalf RA, Welsh JA, Bennett WP, Seddon MB, Lehman TA, Pelin K, Linnainmaa K, Tammilehto L, Mattson K, Gerwin BI, p53 and Kirsten-ras mutations in human mesothelioma cell lines, Cancer Res 52, 2610–2615 (1992). [PubMed] [Google Scholar]
- 25.Lv J, Li P, Mesothelin as a biomarker for targeted therapy, Biomark Res 7, 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eguchi T, Kadota K, Mayor M, Zauderer MG, Rimner A, Rusch VW, Travis WD, Sadelain M, Adusumilli PS, Cancer antigen profiling for malignant pleural mesothelioma immunotherapy: expression and coexpression of mesothelin, cancer antigen 125, and Wilms tumor 1, Oncotarget 8, 77872–77882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inaguma S, Wang Z, Lasota J, Onda M, Czapiewski P, Langfort R, Rys J, Szpor J, Waloszczyk P, Okoń K, Biernat W, Ikeda H, Schrump DS, Hassan R, Pastan I, Miettinen M, Comprehensive immunohistochemical study of mesothelin (MSLN) using different monoclonal antibodies 5B2 and MN-1 in 1562 tumors with evaluation of its prognostic value in malignant pleural mesothelioma, Oncotarget 8, 26744–26754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan R, Kreitman RJ, Pastan I, Willingham MC, Localization of mesothelin in epithelial ovarian cancer, Appl Immunohistochem Mol Morphol 13, 243–247 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Lanitis E, Dangaj D, Hagemann IS, Song D-G, Best A, Sandaltzopoulos R, Coukos G, Powell DJ, Hawkins SM, Ed. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels, PLoS ONE 7, e49829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo H, Xu X, Ye M, Sheng B, Zhu X, Sun L-Z, Ed. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies, PLoS ONE 13, e0191972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, Cabral JMS, Rooney CM, Orange JS, Brenner MK, Mamonkin M, Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent, Cell Rep 21, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH, Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo, Mol. Ther 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, Ang S, Cooper LJN, Platt JM, Johnson FB, Paulos CM, Zhao Y, Kalos M, Milone MC, June CH, Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells, Cancer Immunol Res 3, 356–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, Cosgrove C, Ledderose C, Junger WG, Robson SC, Wherry EJ, Alter G, Goulder PJR, Klenerman P, Sharpe AH, Lauer GM, Haining WN, Douek DC, Ed. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells, PLoS Pathog 11, e1005177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canale FP, Ramello MC, Núñez N, Araujo Furlan CL, Bossio SN, Gorosito Serrán M, Tosello Boari J, Del Castillo A, Ledesma M, Sedlik C, Piaggio E, Gruppi A, Acosta Rodríguez EA, Montes CL, CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8+ T Cells, Cancer Res 78, 115–128 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Dannenfelser R, Allen GM, VanderSluis B, Koegel AK, Levinson S, Stark SR, Yao V, Tadych A, Troyanskaya OG, Lim WA, Discriminatory Power of Combinatorial Antigen Recognition in Cancer T Cell Therapies, Cell Syst 11, 215–228.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, Owens B, Liu L, Roboz GJ, Guzman ML, Melnick AM, Wu S, Mason CE, The therapeutic landscape for cells engineered with chimeric antigen receptors, Nat. Biotechnol 38, 233–244 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, Cho J, Briones JD, Duecker JM, Goretsky YE, Dannenfelser R, Cardarelli L, Troyanskaya O, Sidhu SS, Roybal KT, Okada H, Lim WA, SynNotch-CAR T cells overcome challenges of specificity, heterogeneity and persistence in treating xenograft model of glioblastoma, Sci Transl Med, 10.1126/scitranslmed.abe3738 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL, Pharmacologic control of CAR-T cell function using dasatinib, Blood Adv 3, 711–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H, Hudecek M, The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells, Sci Transl Med 11, eaau5907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, Poirot L, Depil S, Duchateau P, Modulation of chimeric antigen receptor surface expression by a small molecule switch, BMC Biotechnol 19, 44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L, Nunez V, Ackerman CJ, Khialeeva E, Weglarz M, Lee SC, Woods AK, Young TS, Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory, Proc. Natl. Acad. Sci. U.S.A 115, E10898–E10906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sillaber C, Herrmann H, Bennett K, Rix U, Baumgartner C, Böhm A, Herndlhofer S, Tschachler E, Superti-Furga G, Jäger U, Valent P, Immunosuppression and atypical infections in CML patients treated with dasatinib at 140 mg daily, Eur J Clin Invest 39, 1098–1109 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Fei F, Yu Y, Schmitt A, Rojewski MT, Chen B, Greiner J, Götz M, Guillaume P, Döhner H, Bunjes D, Schmitt M, Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens, Exp. Hematol 36, 1297–1308 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Hegde PS, Chen DS, Top 10 Challenges in Cancer Immunotherapy, Immunity 52, 17–35 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, Lim WA, Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors, Cell 167, 419–432.e16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruan W, Sassoon A, An F, Simko JP, Liu B, Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection, Mol. Cell Proteomics 5, 2364–2373 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, Zola H, Hohmann AW, Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma, Mol. Immunol 34, 1157–1165 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Feng Y, Xiao X, Zhu Z, Streaker E, Ho M, Pastan I, Dimitrov DS, A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity, Mol. Cancer Ther 8, 1113–1118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM, Humanization of an anti-p185HER2 antibody for human cancer therapy, Proc. Natl. Acad. Sci. U.S.A 89, 4285–4289 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of ALPPL2 tissue expression and ALPPL2 CAR specificity.
Fig. S2. Characterization of ALPPL2 synNotch-regulated CAR expression.
Fig. S3. Activation and proliferation of antigen-stimulated ALPPL2 synNotch CAR circuits.
Fig. S4. Cytokine production by CAR and synNotch CAR circuit T cells.
Fig. S5. Polyfunctionality of CAR and synNotch CAR circuit T cells.
Fig. S6. SynNotch CAR circuit T cells exhibit multi-antigen specificity and paced tumor elimination.
Fig. S7. Development of tumor cell lines expressing titratable antigen densities and synNotch CAR circuit sensitivity toward mesothelin.
Fig. S8. Phenotypic analysis of CAR and synNotch CAR T cells prior to antigen exposure.
Fig. S9. Impact of CAR expression on tonic signaling.
Fig. S10. SynNotch CAR circuits regulate the evolution of T cell differentiation pre and post antigen stimulation.
Fig. S11. Persistence of CAR T cells and synNotch CAR circuit T cells in treated tumor-bearing mice.
Fig. S12. SynNotch enhancements for CAR T therapy.
Table S1. ALPPL2 expression in solid tumors.
Table S2. Tissue specificity of ALPPL2 expression.
Table S3. IHC of MCAM expression in FFPE mesothelioma tissue arrays.
Table S4. MCAM co-expression in ALPPL2 positive mesothelioma.
Table S5. Antibodies used for immunophenotypic analysis.
Data file S1. Primary data.
Data Availability Statement
All data associated with this study are in the paper or the Supplementary Materials. Materials created in this study will be available for the scientific community by contacting the corresponding author and completion of a material transfer agreement.