

Abstract
Getting from a GWAS “hit” to an actionable gene remains a challenge in complex disease genetics. In this issue of the JID, Sobczyk et al. use a wide variety of genomic data to generate a prioritization algorithm to tackle this problem in atopic dermatitis (AD), calling upon the “wisdom of the genome” to generate promising results.
The past decade has yielded great progress in the genetic analysis of “complex genetic disorders”, in which multiple genes and environmental factors interact to determine risk. Prominent among these are the immune-mediated inflammatory diseases (IMIDs), several of which involve the skin, including atopic dermatitis (AD), acne, alopecia areata, psoriasis, lupus, and vitiligo. While providing new insights into disease pathogenesis, genome-wide association studies (GWAS) of atopic dermatitis, psoriasis, and other IMIDs have uncovered several challenges. In addition to the modest odds ratios associated with most IMID susceptibility loci, most of these genetic signals occur in putative regulatory regions (Farh et al., 2015). While the discovery that many genetic variants exert their effects via gene regulation is very exciting, the most elegant feature of genetic analysis—the ability to work with genomic DNA from blood or other sources—is replaced by a more complex scenario which requires analysis of multiple molecular readouts (e.g. mRNA, protein, DNA methylation) in disease-relevant cell types, preferably studied in their normal physiologic contexts. Besides uncertainty as to which cell types are disease-relevant, these signals may emanate from only a small fraction of the cell types present in diseased tissue, and the pathogenic cell types are often hard to access experimentally in humans. Moreover, the existence of linkage disequilibrium (LD)—the nonrandom segregation of closely-spaced genetic markers due to our common evolutionary history—complicates the identification of the “best” genetic markers and candidate genes within a “genetic signal” generated by GWAS. Adding further complexity, each disease-associated region can contain multiple genetic signals independent of LD (Mahajan et al., 2018) and a given genetic signal may influence the expression of multiple coordinately-regulated genes by both cis- and trans- mechanisms (Võsa et al., 2018). Clearly, we cannot naively interpret localization of “interesting” genes to the vicinities of association signals as proof that these genes exert causal roles. Indeed, overcoming this critical barrier to progress is the key to unlocking the “treasure chest” of genetic signals revealed by GWAS, requiring strategic re-tooling of available resources.
In this issue of the JID, Sobczyk et al. have made an important step towards this goal. They developed a bioinformatics pipeline to systematically prioritize candidate causal genes at 25 AD loci that emerged from their earlier multi-ancestry GWAS of AD (Paternoster et al., 2015). Their pipeline (Figure 1) utilizes over 100 molecular resources relevant to AD, including RNA, protein and DNA methylation quantitative trait locus (QTL) datasets derived from skin or other immune-relevant tissues, as well as other, less skin-specific datasets for regulatory variant prediction, including promoter-enhancer interactions, expression studies, and variant fine mapping. The authors weighted the prioritization algorithm to emphasize the most robust datasets and de-emphasize those in which there was a high a priori likelihood of false-positive results (such as individual eQTL signals, which are abundant throughout the genome). While the choice of weighting schemes reflects the judgement of the authors, their rationale is based in well-founded assumptions that are not specific to AD, including the use of statistical methods such as TWAS and coloc that formally compare the association patterns in QTL studies and GWAS. Their results are reinforced by the striking differences in aggregate scores between individual genes and their near neighbors (for example, 1q21.3-IL6R, 10q21.2-ADO, 11p13-PRR5L, 5p13.2-IL7R, 11q24.3-ETS1, 2q37.1-INPP5D, 12q15-MDM1, and 14q32.32-TRAF3). Indirect validation studies using functional enrichment and interaction tools revealed strong enrichment for cytokine-mediated signaling pathways and JAK-STAT signaling, consistent with the clinical efficacy of biologicals targeting IL-4 receptor-α, blocking IL-4 and IL-13 intracellular signaling.
Figure 1.
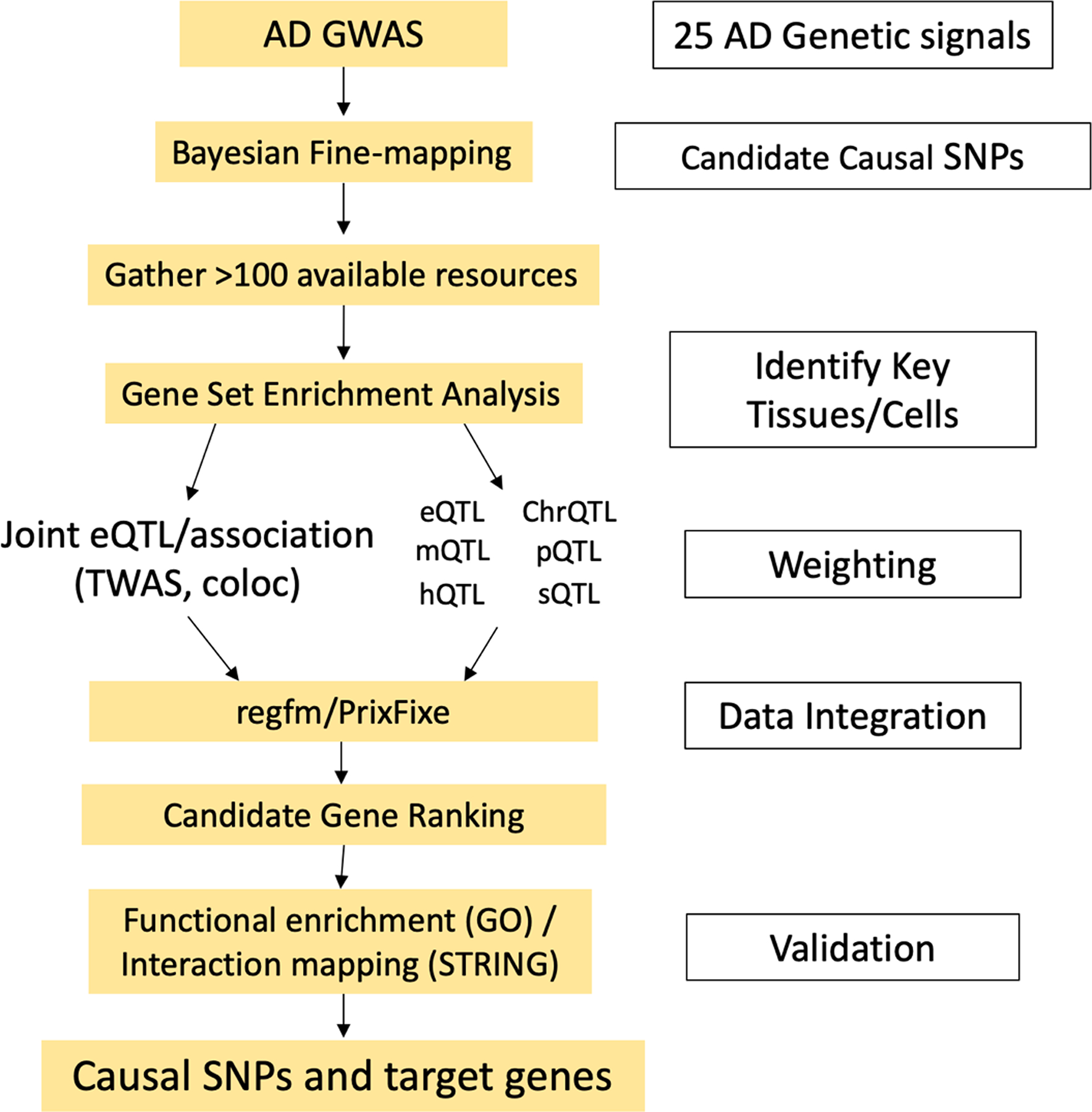
Gene prioritization workflow. Abbreviations: AD, atopic dermatitis; GWAS, genome-wide association study; SNP, single nucleotide polymorphism; chrQTL, chromatin accessibility quantitative trait locus; eQTL, expression quantitative trait locus; hQTL, histone quantitative trait locus; mQTL, DNA methylation quantitative trait locus; pQTL, protein quantitative trait locus; sQTL, splicing quantitative trait locus
Despite these successes, some ambiguities remain. The pipeline of Sobczyk and colleagues produced examples of similarly high-scoring genes that are closely-spaced and functionally-related, including IL18R1, IL18RAP, and IL1R1 in the interleukin-1 like gene cluster on chr 2q12.1, and IL2RA vs. IL15RA on chr 10p15.1. Recently, IL2RA has been independently implicated by CRISPR-Cas mutagenesis experiments demonstrating an effect of the T-allele at rs61839660 on IL2RA gene expression (Simeonov et al., 2017). In other cases, the structural and functional relatedness of closely-spaced, high-scoring gene candidates was less clear, including LRRC32 and EMSY on Chr 11q13.5; KIF3A, PDLIM4, SLC22A4, and IRF1 in the 5q31.1 cytokine gene cluster; and STMN3, LIME1, and ARFRP1 on 20q13.33. Supporting the candidacy of KIF3A, the derived (non-ancestral) alleles at two implicated SNPs near KIF3A are CpG dinucleotides that can become methylated, reducing KIF3A expression when present, decreasing barrier function, and increasing risk for allergic skin responses (Stevens et al., 2020).
Taking a longer view, all of these findings seem to relate to the “wisdom of the genome”, which favors spatial co-localization of functionally related genes. Prime examples of this wisdom include the Major Histocompatibility Complex on chr 6p21.3, the Epidermal Differentiation Complex on chr 1q21.3, the α- and β-globin gene clusters, and the 2q12.1 (IL-1-like) and 5q31.1 (Th2-related) cytokine gene clusters. It is attractive to speculate that the evolutionary reasons for such clustering extend beyond simple gene duplication to encompass important features of 3-dimensional chromatin organization. These include the formation of topologically associating domains (TADs) (Delaneau et al., 2019) and even higher orders of chromatin structure, including “A and B compartments” of active vs. inactive chromatin and “chromosome territories” within the nucleus (Szabo et al., 2019). These higher-order levels of genomic organization do not appear to be broadly conserved across phyla, and likely depend upon physical properties of chromatin that are not yet fully understood (Szabo et al., 2019). In mammals, these features of higher-order chromatin organization function to bring together promoters and enhancers in different combinations, which in turn depend on the cellular context via the coordinated expression of transcription factors, chromatin remodeling proteins, and chromatin loop “anchors”, notably the CTCF/cohesion complex. Indeed, DNA methylation has been shown to influence the anchoring function of CTCF across various cell lineages, via two specific positions in the CTCF binding site (Wang et al., 2012), and “grand canyons” with markedly reduced DNA methylation are found in stem cells and early progenitor cells (Zhang et al., 2020). Thus, inclusion of DNA methylation datasets appears to have been a wise choice for prioritizing AD loci.
The spatially co-located gene sets nominated by the prioritization algorithm of Sobczyk et al. clearly occupy a smaller fraction of genomic space than do the large gene clusters exemplified above. Nevertheless, based on the “wisdom of the genome” principle, we can expect to find more and more examples of disease-associated genetic signals in which genes not created by simple duplication may prove to work together in particular functional contexts. If, as we now appreciate, most disease-associated variants are regulatory, it will not be surprising to learn that more than one structurally-unrelated gene may be responsible for mediating the effect of certain genetic signals, not only in AD but in many other IMIDs as well. With the rapid recent advances in gene expression, DNA methylation, and epigenetic profiling, including the use of single cells and spatially-defined sequencing from tissue sections, we can expect that the months and years to come will see extensive use of all of these tools in taking the “lemons” of genetic complexity identified by GWAS, to create a tasty “lemonade” of functional genomic insights for AD and other IMIDs.
Bullet Points.
Most genetic signals identified by GWAS are regulatory in nature and often reside in regions of high linkage disequilibrium, making the identification of causal genes a challenge.
To meet this challenge, Paternoster and colleagues compiled over 100 molecular resources relevant to AD and developed a scoring system for prioritization of candidate genes across 25 AD-associated genetic regions, yielding clear top candidates for multiple AD loci, but also several other regions in which genes with similarly high scores were closely-spaced and functionally-related.
Indirect validation using functional enrichment and interaction tools revealed strong enrichment for cytokine-mediated signaling pathways and JAK-STAT signaling.
Clustering of functionally-related genes likely reflects the higher-order structure of the genome in addition to gene duplication events.
Footnotes
Conflicts of Interest: none
References:
- Delaneau O, Zazhytska M, Borel C, Giannuzzi G, Rey G, Howald C, et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science 2019;364(6439). [DOI] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518(7539):337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50(11):1505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015;47(12):1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 2017;549(7670):111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens ML, Zhang Z, Johansson E, Ray S, Jagpal A, Ruff BP, et al. Disease-associated KIF3A variants alter gene methylation and expression impacting skin barrier and atopic dermatitis risk. Nat Commun 2020;11(1):4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo Q, Bantignies F, Cavalli G. Principles of genome folding into topologically associating domains. Sci Adv 2019;5(4):eaaw1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra HJ, Bonder M, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018:447367. [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 2012;22(9):1680–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jeong M, Huang X, Wang XQ, Wang X, Zhou W, et al. Large DNA Methylation Nadirs Anchor Chromatin Loops Maintaining Hematopoietic Stem Cell Identity. Mol Cell 2020;78(3):506–21 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]