

Abstract
The regulation of pre-mRNA processing has important consequences for cell division and the control of cancer cell proliferation, but the underlying molecular mechanisms remain poorly understood. We report that three splicing factors, SPF45, SR140, and CHERP, form a tight physical and functionally coherent complex that regulates a variety of alternative splicing events, frequently by repressing short exons flanked by suboptimal 3′ splice sites. These comprise alternative exons embedded in genes with important functions in cell-cycle progression, including the G2/M key regulator FOXM1 and the spindle regulator SPDL1. Knockdown of either of the three factors leads to G2/M arrest and to enhanced apoptosis in HeLa cells. Promoting the changes in FOXM1 or SPDL1 splicing induced by SPF45/SR140/CHERP knockdown partially recapitulates the effects on cell growth, arguing that the complex orchestrates a program of alternative splicing necessary for efficient cell proliferation.
Keywords: CHERP, SPF45, SR140, alternative splicing, cell proliferation
INTRODUCTION
The generation of mature mRNAs requires the removal of introns and ligation of exons through pre-mRNA splicing by the spliceosome. The spliceosome is a complex macromolecular ribonucleoprotein composed of five snRNPs (U1, U2, U4, U5, and U6) and more than 150 proteins (Wahl et al. 2009), which assemble onto the pre-mRNA in a step-wise manner to catalyze the splicing reaction (Wan et al. 2019; Wilkinson et al. 2019). Recognition of the 5′ splice site (ss) by U1 snRNP, and of the branch site, polypyrimidine tract and 3′ ss AG by BBP/SF1 and the two subunits of U2AF, leads to E complex formation. BBP/SF1 is subsequently displaced from the branch point by U2 snRNP, whose SF3B1 subunit interacts with the branch point in prespliceosomal (A) complexes. Several key protein–protein interactions occurring at these early steps of 3′ss recognition are mediated by atypical RNA Recognition Motifs (RRMs) known as U2AF Homology Motifs (UHMs) (Kielkopf et al. 2001, 2004; Selenko et al. 2003; Spadaccini et al. 2006). Complex B is assembled upon binding of the U4/5/6 tri-snRNP and substantial compositional and conformational rearrangements lead to a series of transition states (Bact, B*, C, and C*) where the RNA-based catalysis of the two consecutive transesterification reactions of the splicing process takes place (Wan et al. 2019; Wilkinson et al. 2019).
Alternative splicing (AS), the differential use of splice sites in pre-mRNAs to produce different mRNAs, occurs in over 95% of human genes (Pan et al. 2008; Wang et al. 2008), greatly expanding transcriptome complexity. This, in turn, can increase proteome diversity, control gene expression through nonsense-mediated-decay (NMD)-mediated mRNA degradation or regulate mRNA translation (Lewis et al. 2003; Mockenhaupt and Makeyev 2015; Yang et al. 2016). AS includes the alternative use of 3′ or 5′ ss (alt 3′/5′ss), inclusion or skipping of cassette exons (CEx) and microexons or the retention of introns (RI). AS can be controlled by cis-acting sequences bound by trans-acting factors which modulate access of the core spliceosomal components to the splice sites (Chen and Manley 2009). AS can also be regulated by pre-mRNA secondary structures, coupling with transcriptional changes or by chromatin structure (Kornblihtt et al. 2013). Variations in the levels of core spliceosomal components can also contribute to AS regulation (Saltzman et al. 2011; Papasaikas et al. 2015).
SPF45 (RBM17) is a U2AF homology domain (UHM)-containing splicing factor associated with U2 snRNP and present in complex E (Neubauer et al. 1998; Will et al. 2002). SPF45 has been linked to different biological processes and pathways. Thus, SPF45 was found to activate a distal AG in Sex-lethal (Sxl) pre-mRNA and be relevant for Sxl AS autoregulation (Lallena et al. 2002). UHM-mediated interactions of SPF45 with U2AF65, SF1, and SF3B1 were found to be required for its modulation of FAS exon 6 splicing in mammalian cells (Corsini et al. 2007). This event is also regulated by Saf lncRNA and its interaction with SPF45, which contributes to apoptosis resistance (Villamizar et al. 2015). While constitutive deletion of SPF45 in mice is embryonic lethal, conditional depletion SPF45 in Purkinje cells causes neurodegeneration (Tan et al. 2016). Related to this, SPF45 plays a direct role in the neuropathology of spinocerebellar ataxia 1, which predominantly affects Purkinje neurons (Lim et al. 2008).
Phosphorylation of SPF45 by several kinases influences adhesion, migration and invasion as well as AS choices (Al-Ayoubi et al. 2012; Liu et al. 2013). Two recent studies have reported that SPF45 controls proliferation, cell-cycle progression and apoptosis of hypopharyngeal carcinoma and glioma cell lines and its down-regulation in a mouse xenograft model suppresses tumor growth (Han et al. 2018; Lu et al. 2018). Additionally, increased SPF45 levels found in several carcinomas are associated with the gain of multidrug resistance (Sampath et al. 2003; Perry et al. 2005).
An intriguing connection has also been established between Drosophila SPF45 and DNA damage repair. Thus, dSPF45 increases resistance to DNA damaging agents of bacteria lacking RecG, a helicase that resolves Holliday junctions generated during DNA damage repair, and genetically and physically interacts with the DNA damage repair enzyme RAD201 (Chaouki and Salz 2006). Furthermore, human SPF45 binds Holliday junctions and interacts with RAD51, RAD51B, and DMC1 (Horikoshi et al. 2010).
Here we build upon a functional link between SPF45 and splicing factors SR140 and CHERP (Papasaikas et al. 2015) to show that SPF45, SR140, and CHERP stabilize each other, coimmunoprecipitate and cosediment in glycerol gradients, indicating that they are part of the same functional splicing complex, consistent with a recent report by De Maio et al. (2018). Knockdown of each of these three factors decreases the clonogenic and proliferative capacities and impairs cell-cycle progression of HeLa and U2OS cells, correlating with changes in AS in cell-cycle-control genes, including the G2/M master control factor FOXM1 and the spindle regulator SPDL1.
RESULTS
SPF45/RBM17, SR140/U2SURP, and CHERP form a functional physical complex
Previous work from our group using a functional network reconstruction approach identified a module of three splicing factors, SPF45/RBM17, SR140/U2SURP, and CHERP, which displayed highly similar profiles of splicing perturbations upon their individual siRNA-mediated knockdown in HeLa cells in a panel of AS events important for cell proliferation and/or apoptosis (Papasaikas et al. 2015). The knockdowns displayed mostly consistent changes—in extent and direction—on the 39 CEx included in our assay (Fig. 1A). For example, each of the knockdowns enhanced inclusion of NUMB exon 9, MADD exon 16, MINK1 exon 18 and MAP4K3 exon 15, while induced skipping of MCL1 exon 2, both in HeLa and in U2OS cells (Supplemental Fig. S1B,C). A systematic screen to discover AS regulators of cell fate in mouse cells also identified functional links between SPF45, SR140, and CHERP, as well as with the helicase DHX15 (Han et al. 2017), involved in the disassembly and proofreading of spliceosomes (Arenas and Abelson 1997; Martin et al. 2002; Mayas et al. 2010). The functional link with DHX15 was however weaker in our system (Supplemental Fig. S1A–C).
FIGURE 1.
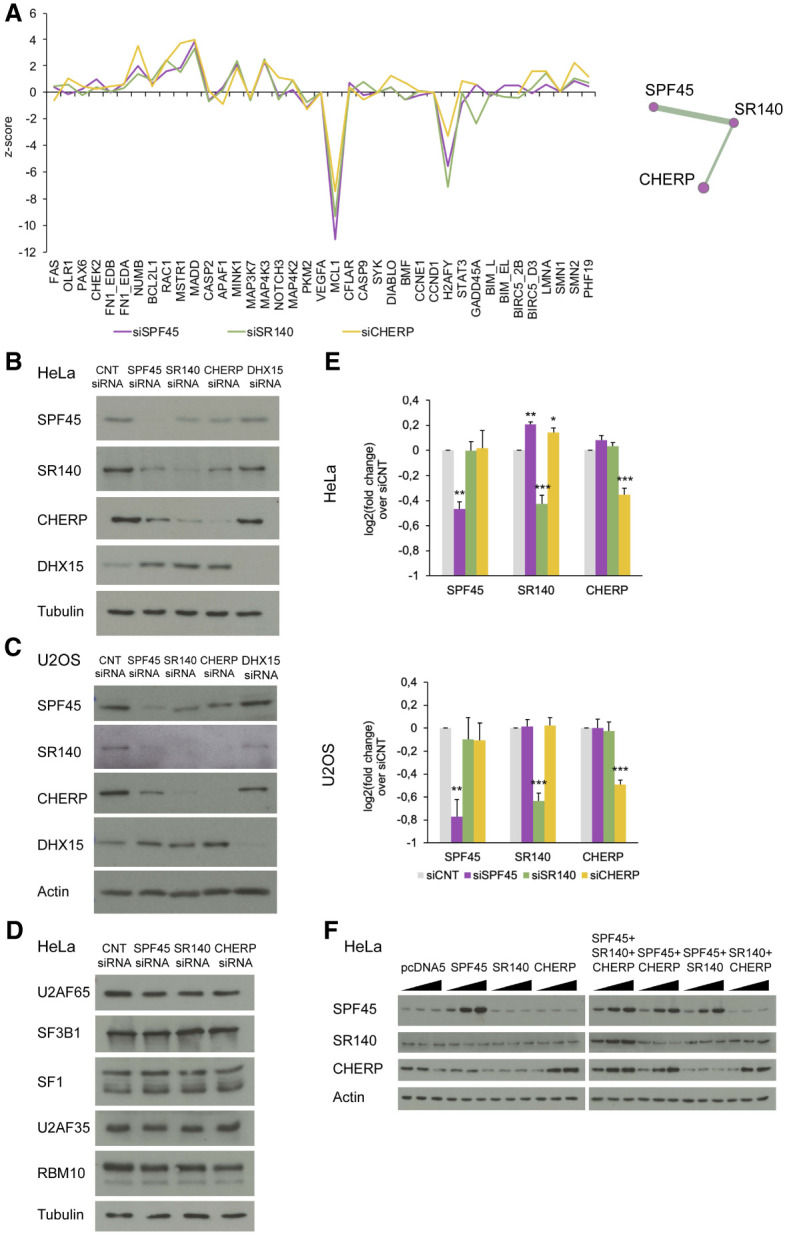
SPF45, SR140, and CHERP cross-regulate each other at the protein level. (A) Similar splicing perturbation profiles upon knockdown of SPF45, SR140, or CHERP. Robust z-score values of changes in alternative splicing (y-axis, >0: inclusion <0: skipping) for 39 exons in the indicated genes (x-axis) were extracted from Papasaikas et al. (2015). The high similarity between the profiles of splicing changes of the three factors identified a functional module (right panel) in a systematic comparison across splicing factors (Papasaikas et al. 2015). (B,C) Western blot analyses of SPF45, SR140, CHERP, and DHX15 (and Tubulin/actin as loading controls) upon their individual siRNA-mediated knockdowns in HeLa and U2OS cells. The results shown are representative of three independent experiments. (D) Western blot analysis of interactors of SPF45, SR140, or CHERP (and tubulin as loading control) of the same samples as in B. The result shown is representative of two independent experiments. (E) RT-qPCR-measurements of the fold change of mRNA levels of SPF45, SR140, and CHERP upon their individual knockdowns in HeLa and U2OS cells compared to siCNT. Data are represented as mean ± SD of three biological replicates, and P-values are calculated by one-way ANOVA followed by Dunnett's multiple comparison test on ΔΔCt values ([*] P < 0.01; [**] P < 0.001, [***] P < 0.0001). (F) Western blot analysis of SPF45, SR140, and CHERP (and actin as a loading control) upon individual or combined overexpression of these factors in HeLa cells. A total of 250 ng, 500 ng, or 1 µg of DNA was transfected (in individual overexpression assays these amounts correspond to single plasmids, while in combined overexpression assays they correspond to the sum of equal amounts of plasmids). See also Supplemental Figure S1.
To explore the molecular basis for the similar splicing perturbation profiles of SPF45, SR140, and CHERP, their protein levels were examined upon each individual knockdown. Depletion of each factor decreased the protein levels of the three factors in HeLa and U2OS cells (Fig. 1B,C; Supplemental Fig. S1D), but not of other spliceosome components known to interact with them (Fig. 1D; Corsini et al. 2007; Hegele et al. 2012; Crisci et al. 2015; De Maio et al. 2018) and increased the levels of DHX15 (Fig. 1B,C). As mRNA levels were only affected upon each specific knockdown (Fig. 1E), the results suggest that SPF45, SR140, and CHERP are part of a protein complex whose components become destabilized upon reduction of the levels of any of its subunits, with CHERP and SR140 displaying the most apparent cross-regulatory effects. This is consistent with cross-regulation of SR140 and CHERP in HEK293 cells (Lin-Moshier et al. 2013) and RKO cells (Wang et al. 2019), and of the three proteins in mice cerebellar extracts and HEK293T cells (De Maio et al. 2018). Also consistent with this concept were results of overexpression experiments showing that, while transfection of an expression plasmid of SR140 did not result in increased levels of the protein, coexpression with both SPF45 and CHERP did (Fig. 1F), suggesting that accumulation of SR140 protein depends upon the availability of its protein partners.
To further document the functional links between these factors, rescue experiments were carried out in which knockdown of SPF45, SR140, or CHERP was complemented by stable overexpression of siRNA-resistant versions of the depleted factor in Flp-In T-Rex HeLa cells. As controls, we used cells integrating an empty vector (pcDNA5) or a version of SPF45 cDNA containing a hairpin (SPF45hp) which prevents translation of its mRNA. The results showed that overexpression of SPF45 or CHERP cDNAs increased the levels of the three factors and restored the patterns of splicing of MADD exon 16, while overexpression of SR140 cDNA did not increase the levels of any of the three proteins and failed to restore splicing of MADD exon 16 (Fig. 2A,B), once again suggesting that accumulation of SR140 and function of the complex requires interactions between the three proteins.
FIGURE 2.

SPF45, SR140, and CHERP form a functional physical complex. (A, upper panel) Western blot analysis of SPF45, SR140, and CHERP (and tubulin as loading control) upon 3′UTR-targeted siRNA knockdowns in Flp-In T-Rex HeLa cells stably overexpressing either an empty vector (pcDNA5), a SPF45 cDNA containing a hairpin to prevent translation (SPF45hp), or cDNAs of SPF45, SR140, or CHERP with 3′UTRs not targeted by the siRNAs utilized. Twenty-four hours after siRNA transfection, protein expression was induced using tetracycline, and total cell extracts were prepared 48 h later. The results shown are representative of three independent experiments. (Lower panel) Quantification of western blot results, represented as mean ± SD of three biological replicates. P-values calculated comparing to pcDNA5 by one-way ANOVA followed by Dunnett's multiple comparison test ([*] P < 0.01; [**] P < 0.001). (B) Quantification of MADD exon 16 AS by RT-PCR followed by capillary electrophoresis, represented as fold change compared to siCNT in the same experimental conditions of A. Data are represented as mean ± SD of three biological replicates, and P-values are calculated comparing to pcDNA5 by Student's t-test ([**] P < 0.01). (C) Immunoprecipitation of endogenous SPF45 or CHERP in total protein extracts from HeLa cells and detection of coimmunoprecipitating proteins by western blot analyses. (D) Fractionation of HeLa nuclear extract (NE) through a 5%–40% glycerol gradient followed by western blot analysis of the indicated proteins. The approximate size of the fractions was estimated by profiling a molecular weight marker kit through a parallel gradient and staining it with Coomassie. The result shown is representative of three independent experiments.
To further document physical interactions between these factors, coimmunoprecipitation and cosedimentation assays were carried out. Both SR140 and CHERP were detected in SPF45 immunoprecipitates from HeLa extracts, and both SPF45 and SR140 were detected in CHERP immunoprecipitates (Fig. 2C). These results are consistent with previous data documenting interactions between SPF45 and SR140 in two-hybrid assays (Hegele et al. 2012), between CHERP and SR140 in HEK293 and RKO cells (Lin-Moshier et al. 2013; Wang et al. 2019) and between the three factors in mouse cerebellar extracts (De Maio et al. 2018). Fractionation of HeLa nuclear extracts by centrifugation through a glycerol gradient and western blot analyses detected the presence of SPF45, SR140 and CHERP in both low and high molecular fractions (Fig. 2D), the latter corresponding to a molecular mass of about 660 kDa. This mass would be compatible with a complex containing two molecules of each of the three factors or with a complex of each of the three proteins with other factors. SF3B1 and DHX15, two known interactors of SPF45 (Corsini et al. 2007; De Maio et al. 2018) sediment in fractions partially overlapping with the high molecular weight complex and may therefore be part of such a complex in a yet to be defined stoichiometry. Similar results were reported in mouse brain lysates by size exclusion chromatography (De Maio et al. 2018).
The SPF45/SR140/CHERP complex is required for cell proliferation, cell-cycle progression, and inhibition of apoptosis
To study cellular phenotypes associated with SPF45, SR140, and CHERP depletion, we set up a lentivirus system to transduce shRNAs targeting these factors (five different shRNAs were tested for each, two of each were selected for further analyses) in HeLa and U2OS cells. The effects of lentiviral-mediated knockdown recapitulated the cross-regulation of protein levels between the three proteins observed using siRNAs (Fig. 3A; Supplemental Fig. S2A). Very significant inhibition of the kinetics of cell proliferation and particularly of the clonogenic capacity (10–20-fold) of HeLa and U2OS cells was observed upon knockdown of each of the three factors (Fig. 3B,C; Supplemental Fig. S2B,C). These results are consistent with studies on different cell lines (Laplante et al. 2000; O'Rourke et al. 2003; Zhang et al. 2017a; Han et al. 2018; Lu et al. 2018; Wang et al. 2019).
FIGURE 3.

The SPF45/SR140/CHERP complex is required for cell proliferation, cell-cycle progression, and inhibition of apoptosis. (A, left panels) Western blot analyses of SPF45, SR140, or CHERP (and tubulin as a loading control) upon their individual knockdowns using lentiviral transduction of shRNAs in HeLa and U2OS cells. The results shown are representative of three independent experiments, which are quantified in the right panels and represented as mean ± SD with P-values calculated comparing to shCNT by one-way ANOVA followed by Dunnett's multiple comparison test ([*] P < 0.05; [**] P < 0.001; [***] P < 0.0001). (B) Cell proliferation measured by direct cell number count of HeLa or U2OS cells upon lentivirus-mediated knockdown of SPF45, SR140, or CHERP measured 24, 48, 72, and 96 h after plating. Data are represented as mean ± SD of three biological replicates. (C, left) Representative clonogenic assays (left) of HeLa or U2OS cells upon lentivirus-mediated knockdown of SPF45, SR140, or CHERP. (C, right) Quantification of the number of colonies formed after seeding 1000 HeLa or 2000 U2OS cells, represented as mean ± SD of three biological replicates with P-values calculated comparing to shCNT by one-way ANOVA followed by Dunnett's multiple comparison test ([*] P < 0.01; [**] P < 0.001; [***] P < 0.0001). (D) Distribution of HeLa or U2OS cells in different phases of cell cycle upon lentivirus-mediated knockdown of SPF45, SR140, or CHERP, analyzed by propidium iodide (PI) staining and flow cytometry. Data are represented as mean ± SD of three biological replicates with P-values calculated comparing to shCNT by χ2 test ([*] P < 0.01; [**] P < 0.001; [***] P < 0.0001). (E, upper panels) Western blot analysis of the indicated cell-cycle markers (and GAPDH as loading control) upon knockdown of SPF45, SR140, or CHERP or upon treatment with UV light (UV-C at 10 J/m2 for 6 h) as a control of Chk1 phosphorylation. (Lower panels) Quantification of the results, represented as mean ± SD of three biological replicates.
Such a decline in proliferative and clonogenic capacities could be due to defective progression through the cell cycle or/and induction of apoptosis. Flow cytometry analyses revealed that, for HeLa cells, knockdown of each of the three factors resulted in a slight reduction of cells in G1 phase and a significant accumulation of cells in G2/M (Fig. 3D; Supplemental Fig. S2D). While some differences were observed between shRNA constructs, these are likely due to different quantitative effects on target knockdown. Interestingly, knockdown of each of the three factors in U2OS cells resulted in a significant increase of cells in G1 and a decrease in S phase (Fig. 3D; Supplemental Fig. S2D). These results indicate that the activity of the SPF45/SR140/CHERP complex is relevant for proper cell-cycle progression, impinging on different stages in different cell lines. This is consistent with depletion of SPF45 inducing G2/M arrest in FaDu cells (Han et al. 2018) but G1 arrest in glioblastoma cell lines (Lu et al. 2018) and CHERP depletion promoting G1 accumulation in neuroblastoma cells (Zhang et al. 2017a).
Next, we used propidium iodide and Annexin V to study apoptosis upon SPF45, SR140 or CHERP knockdowns. A significant reduction in the fraction of viable HeLa cells was accompanied by an increase in early apoptotic as well as late apoptotic/necrotic cells for the three knockdowns (Supplemental Fig. S3A), in agreement with previous results reporting increased apoptosis upon knockdown of these factors in HeLa and other cell lines (Villamizar et al. 2015; Zhang et al. 2017a; Han et al. 2018; Lu et al. 2018; Wang et al. 2019). However, SPF45, SR140, or CHERP knockdown did not induce apoptosis in U2OS cells (Supplemental Fig. S3A), once again arguing for different activities of the SPF45/SR140/CHERP complex in different cellular contexts, depending on the impact of the targets of the complex on cell type-specific rate-limiting steps for cell growth/viability.
To further characterize cell-cycle alterations, we determined the levels and/or phosphorylation status of some key cell-cycle regulatory factors. Interestingly, while phosphorylation of Chk1 kinase was not affected upon knockdown of SPF45/SR140/CHERP, increased phosphorylation of Histone H3 Serine 10 and increased accumulation of Cyclin B1 (CCNB1) were observed under these conditions, at least in HeLa cells (Fig. 3E; Supplemental Fig. S2E) (see Discussion). Flow cytometry results were also consistent with increased accumulation of cells in G2 and H3S10 phosphorylation (mitosis) upon knockdown of SPF45/SR140/CHERP (Supplemental Fig. S3B).
SPF45, SR140, and CHERP co-regulate a program of AS events in genes relevant for cell-cycle progression
To characterize the regulatory program of SPF45, SR140, and CHERP, total RNA was isolated from HeLa cells knocked down for each individual factor via lentivirus-mediated shRNA transduction and analyzed by RNA-seq. First, differential gene expression analyses identified 1325, 1288, and 1549 differentially expressed genes (log2 fold change ≥0.6 or ≤−0.6; ΔFPKM >1) between SPF45, SR140, or CHERP knockdowns and control shRNA, respectively, 54%–59% corresponding to down-regulation (Supplemental Fig. S4A). Two hundred and four differentially expressed genes were common to the three knockdowns, and 40% (CHERP), 59% (SR140), and 61% (SPF45) of changes were common between at least two knockdowns (Supplemental Fig. S4B). Gene ontology (GO) analysis of the deregulated genes revealed many overlapping categories, with knockdown of SPF45 and SR140 sharing extracellular space/matrix among the most enriched categories, and knockdown of CHERP being enriched in plasma membrane-related components (Supplemental Fig. S4C).
AS analysis of the RNA-seq data sets using VAST-TOOLS (Irimia et al. 2014; Tapial et al. 2017) revealed 2920 (SPF45), 1763 (SR140), and 2574 (CHERP) regulated events (|ΔPSI| ≥ 15%, min range 5%) (Fig. 4A). Independent validation by RT-PCR in biological replicates for two different shRNAs showed an excellent correlation between PSI values estimates by the two techniques (Fig. 4B; Supplemental Fig. S5A) and similar results were also obtained in U2OS cells (Supplemental Fig. S5B). The vast majority of these events correspond to either RI or regulation of CEx. Among CEx, 60% (SPF45) and 72% (SR140) corresponded to increased exon inclusion, while knockdown of CHERP resulted in more (63%) skipped exons (Fig. 4A). All three knockdowns resulted in a higher proportion of distal 3′ ss utilization, while for alternative 5′ ss there was a more similar distribution of proximal/distal site usage (Fig. 4A). Only 13%–19% of genes displaying AS changes showed also changes in gene expression upon knockdown of these factors (Supplemental Fig. S4D), some of which could be due to CDS disruption and mRNA degradation via nonsense-mediated decay (see below).
FIGURE 4.

SPF45, SR140, and CHERP co-regulate a program of AS events in genes relevant for cell-cycle progression. (A) Summary of AS changes upon knockdown of SPF45, SR140, or CHERP in HeLa cells. Total RNA from duplicates of each condition was used for RNA-seq analysis and AS changes were quantified using VAST-TOOLS. The number of AS events (y-axis) displaying increased inclusion (ΔPSI ≥ 15%) or skipping (ΔPSI ≤ −15%) are indicated for each type of AS event (x-axis). (B) Validation of events detected by VAST-TOOLS in RNA-seq data sets using independent RNA samples and RT-PCR followed by capillary electrophoresis upon knockdown of SPF45, SR140, or CHERP in HeLa cells. RT-PCR PSI values (bars) and VAST-TOOLS PSI values from RNA-seq data sets (orange line) display an overall correlation (R2) of 0.98 (shCNT), 0.73 (shSPF45), 0.76 (shSR140), and 0.57 (shCHERP). Data are represented as mean ± SD for three biological replicates. (C) Pairwise correlations of PSI values from AS events (|ΔPSI| ≥ 15%) upon knockdown of SPF45, SR140, or CHERP. Pearson correlation coefficients are shown in the upper left corner. (D) Venn diagram of the overlap between differentially spliced cassette exons (CEx) upon knockdown of SPF45, SR140, and CHERP. (E) Matt analyses of splicing regulatory sequence features enrichment, GC content, and length of CEx more included (UP) or more skipped (DOWN) upon knockdown of SPF45, SR140, CHERP, or all three (Triple overlap) compared to a stratified control set of nonchanging alternative exons. Sequence features include 5′ss and 3′ss strengths (maximum entropy scores), best predicted branch point (BP) sequence score, and polypyrimidine tract (Py) sequence score. Normalized differences in the median of the scores of each feature between each condition and nonchanging alternative exons were tested by Mann–Whitney U-tests ([*] P < 0.05; [**] P < 0.01; [***] P < 0.001). See also Supplemental Figure S4E,F. (F) GO analysis of alternatively spliced exons upon SPF45 knockdown in HeLa cells performed using GOrilla. In purple, “G2/M transition” categories. (G) Word cloud depicting the genes from the “G2/M transition” categories from F. The size of genes is proportional to the ΔPSI of the contained events in SPF45 knockdown samples and pink genes correspond to events regulated by the three knockdowns (triple overlap). See also Supplemental Figures S4, S5.
All validated changes showed consistent effects of the three knockdowns (Fig. 4B; Supplemental Fig. S5A,B) and pairwise comparisons of significant PSI changes showed correlations of 0.9 or higher in all three knockdown combinations (Fig. 4C). Although differences in profiles between individual components were observed—likely reflecting independent functions or in coordination with other regulatory complexes—in general, at least 50% of the AS changes for any individual knockdown were shared with at least another knockdown, the percentage of common events being higher for SR140 (68% for all AS events, 63% for RI, 72% for cassette exons—including microexons) and lower for CHERP (49% for all AS events or alternative exons, 50% for RI) (Fig. 4D; Supplemental Fig. S5C,D). Taken together, the results reveal a strong correlation of AS changes between SPF45, SR140 and CHERP knockdowns, suggesting coordinated regulation of AS events.
To explore the mechanistic basis of AS regulation by SPF45, SR140, or CHERP, we carried out an analysis of sequence features enriched in AS regions using Matt (Gohr and Irimia 2018). We first focused on alternative exons more included or more skipped upon knockdown of these factors. The most conspicuous and consistent features include relatively weak 3′ ss signals (maximum entropy model, Yeo and Burge 2004) in both of the introns flanking more included alternative exons, as well as a trend toward a stronger branch site and polypyrimidine tract in the preceding intron (Fig. 4E, left panel), with a partially reverse arrangement of features for more skipped exons. These observations suggest that SPF45/SR140/CHERP tend to repress exons flanked by weak 3′ ss. Repressed exons tend to be shorter (median length of 90 nt for those in the triple overlap) and reside in genomic regions of overall lower GC content, while the opposite is true for exons down-regulated upon depletion of any of the components of the complex (median length of 125 nt for those in the triple overlap) (Fig. 4E, right and middle panels and Supplemental Fig. S5E). The introns flanking regulated exons tend to be shorter (Supplemental Fig. S5F). Introns retained upon knockdown of SPF45, SR140, or CHERP also tend to be shorter, they feature weaker polypyrimidine tracts/3′ ss and reside in genomic regions of higher GC content; the opposite trends are observed for introns more spliced upon knockdown of these factors (Supplemental Fig. S6A,B).
GO analysis of genes harboring alternative exons affected upon SPF45, SR140, or CHERP knockdowns revealed an enrichment in terms related to cilium organization and assembly (Fig. 4F; Supplemental Fig. S6C,D). In the case of SPF45 we also find, interestingly, G2/M cell-cycle transitions categories (Fig. 4F), with genes with important functions in cell-cycle control (e.g., PLK3, CDK7, CLASP1, and FOXM1) contributing prominently to this membership (Fig. 4G and see below).
SPF45/SR140/CHERP regulate FOXM1 AS
Out of cell-cycle regulatory genes whose AS is regulated by SPF45, SR140, and CHERP, we first focused on FOXM1 because it encodes a transcription factor with key regulatory roles in cell-cycle progression and particularly at the G2/M transition (Laoukili et al. 2005). Knockdown of these factors results in increased inclusion of FOXM1 exon 9, or of a shorter version of exon 9 that uses an alternative 5′ ss (Fig. 5A), both in HeLa and U2OS cells, using two independent shRNAs (Fig. 5B; Supplemental Fig. S7A). In contrast, no major changes were observed under the same conditions in FOXM1 exon 6, which is also an alternatively spliced exon (Supplemental Fig. S7B).
FIGURE 5.

SPF45/SR140/CHERP regulate FOXM1 AS. (A) Scheme of AS events involving FOXM1 exon 9 (upper panel) and their changes upon knockdown of SPF45, SR140, or CHERP measured by RT-PCR followed by capillary electrophoresis (lower panel). The isoform identity of the amplification products is indicated. (B) Validation of FOXM1 exon 9 AS events (solid bars: full exon inclusion; striped bars: shorter exon inclusion) changes measured as in A in HeLa and U2OS cells. Data are represented as mean ± SD of three biological replicates, and P-values are calculated comparing to shCNT by Student's t-test ([*] P < 0.05; [**] P < 0.01; [***] P < 0.001). (C) Structural domain organization of FOXM1 isoforms. The different domains are indicated as well as the location and likely functional impact of inclusion of sequences corresponding to exon 6 and exon 9 inclusion. PTC: premature termination codon. (D) Fold change over shCNT of mRNA levels of FOXM1 isoforms and total FOXM1 quantified by RT-qPCR upon knockdown of SPF45, SR140, or CHERP in HeLa and U2OS cells. Data are represented as mean ± SD of three biological replicates, and P-values are calculated by comparing to shCNT by one-way ANOVA followed by Dunnett's multiple comparison test ([*] P < 0.05; [**] P < 0.01; [***] P < 0.001). (E) Relative abundance of the indicated common or isoform-specific peptides in control versus SPF45-depleted cells by targeted proteomics, normalized by the levels of ACLY, and corresponding fold changes (in log2) between the two conditions. (F) Relative expression levels of known targets of transcriptional activation by FOXM1, quantified by Cuffdiff in RNA-seq data sets of knockdown of SPF45, SR140, or CHERP in HeLa cells. Gene expression is displayed as FPKM values (y-axis) and significant values—according to Cuffdiff differential gene expression test—are indicated by an asterisk. See also Supplemental Figure S7.
AS of FOXM1 exons 6 and 9 generate a variety of isoforms (Korver et al. 1997; Ye et al. 1997; Laoukili et al. 2007; Zhang et al. 2017b): FOXM1a contains only exon 9, FOXM1b neither exon 6 nor 9, FOXM1c only exon 6 and FOXM1d contains both (not analyzed in this study due to its low expression) (Fig. 5C). Inclusion of exon 9 has been reported to disrupt the transcriptional transactivation domain, producing transcriptionally inactive FOXM1 isoforms (Ye et al. 1997), while FOXM1b and FOXM1c remain functional. Inclusion of the 34 bp shorter version of exon 9 causes a frameshift which leads to the emergence of a premature termination codon (PTC) that could generate a truncated protein lacking most of the transcription activation domain or might target the mRNA for NMD (Fig. 5C).
Profiling of the four detectable isoforms in HeLa and U2OS cells by RT-qPCR using isoform-specific primer pairs confirmed that functional isoforms (FOXM1b and 1c) were decreased, while nonfunctional isoforms (FOXM1a and 1a short) were increased upon knockdown of SPF45, SR140, or CHERP (Fig. 5A,D). The total levels of FOXM1 transcripts or FOXM1 protein were either not affected or slightly increased (Fig. 5D; Supplemental Fig. S7C). However, results of targeted proteomic analyses designed to detect peptides common or specific to different isoforms were consistent with a nearly fourfold decrease in the ratio between functional and nonfunctional (c /1a) isoforms (Fig. 5E).
Given that FOXM1 functional isoforms are reduced upon SPF45, SR140, or CHERP knockdowns, we analyzed the expression of some FOXM1 transcriptional targets (Sadasivam et al. 2012; Grant et al. 2013). A general trend toward decreased expression of FOXM1 targets was observed both examining RNA-seq data (Fig. 5F) or by RT-qPCR analyses of AURKB, CENPA, and CCNB2 in HeLa and U2OS (Supplemental Fig. S7E), the effects being typically less significant for SR140 knockdown, consistent with the more limited effects of this knockdown on FOXM1 AS (Fig. 5A,B). The milder effects of the knockdowns on FOXM1 AS in U2OS cells (Fig. 5B) may explain that depletion of the complex does not lead to G2/M arrest in this cell line.
Taken together, the results indicate that SPF45/SR140/CHERP repress the expression of nonfunctional FOXM1 isoforms by inhibiting inclusion of FOXM1 exon 9.
AS of the SPF45/SR140/CHERP targets FOXM1 and SPDL1 influence cell proliferation
Deregulation of FOXM1 AS upon knockdown of SPF45, SR140 or CHERP might contribute to the defects in cell proliferation observed upon the knockdown of these factors. To test this hypothesis, we aimed to reverse the effects of the knockdowns on FOXM1 AS by using 2′-O-methyl phosphorothioated antisense oligonucleotides (AONs) targeting either the alternative 5′ ss of exon 9 or both of the 5′ ss associated with this exon (Fig. 6A). Transfection of these AONs into cells knocked down for SPF45 reduced the levels of exon 9 inclusion in both HeLa and U2OS cells (Fig. 6B). Clonogenic assays under the same conditions showed that the AONs also induced a reproducible, albeit partial, rescue of the decrease in colony formation caused by SPF45 depletion in both cell lines (Fig. 6C), as well as a trend toward restoring the levels of FOXM1 target genes CENPA and CCNB2 (Supplemental Fig. S7E). These results suggest that FOXM1 AS contributes, but is not the only—nor the main—alteration causing cell proliferation defects induced by the knockdown of SPF45/SR140/CHERP. Interestingly, however, the AONs also caused a reduction in FOXM1 inactive isoforms in control cells, which also showed a trend toward increased clonogenic capacity (Fig. 6B,C), again arguing that FOXM1 AS does have an impact on cell proliferation under normal conditions.
FIGURE 6.

AS of the SPF45/SR140/CHERP target FOXM1 exon 9 influences cell proliferation. (A) Schematic diagram of antisense oligonucleotides (AONs) targeting FOXM1 exon 9 5′ss and alternative 5′ss to promote exon skipping. Upper/lower case indicates exonic/intronic nucleotides, respectively. (B, upper panels) FOXM1 exon 9 isoforms inclusion (PSI) upon FOXM1 AONs (50 nM) transfection after SPF45 knockdown, assessed by RT-PCR and capillary electrophoresis. AONs predicted not to target human sequences (control), targeting the alternative 5′ss of FOXM1 and an equimolar combination of AONs targeting the 5′ss and alternative 5′ss (total 50 nM) were used in HeLa and U2OS cells. (Lower panel) quantification of these results, represented as mean ± SD of four biological replicates; P-values were calculated by Student's t-test ([*] P < 0.05; [**] P < 0.01) comparing the PSI values to those for CNT AON within each condition. (C) Percentage of plated cells forming colonies (clonogenic capacity, y-axis) upon FOXM1 AONs (50 nM) transfections in HeLa and U2OS cells knocked down for SPF45. Data are represented as mean ± SD of four biological replicates, and P-values are calculated by one-way ANOVA followed by Dunnett's multiple comparison test (ns = not significant; [**] P < 0.001; [***] P < 0.0001). (D) Schematic representation of predicted base-pairing interactions for canonical U1 snRNA and for engineered U1 snRNAs (alt 5′ss U1, 5′ss U1, and shifted U1) with FOXM1 exon 9 alternative 5′ss, 5′ss and downstream intronic region. Bases in bold indicate sequences different from the canonical U1 sequence and black dots depict wobble base-pairings. (E) Quantification of FOXM1 exons 9 inclusion (PSI) upon transfection of wild type or variant U1 snRNAs, measured by RT-PCR and capillary electrophoresis in HeLa cells. Cells were cotransfected with a GFP plasmid to allow sorting of GPF-positive cells, which coexpress U1 snRNAs, 24 h after transfection. Data are represented as mean ± SD of three biological replicates, and P-values are calculated comparing to GFP by Student's t-test ([**] P < 0.01; [***] P < 0.001). (F) Clonogenic assay of GFP-positive HeLa cells sorted 24 h after cotransfection of GFP- and U1 snRNA-expressing plasmids. The number of colonies is normalized to that of control samples transfected only with the GFP plasmid (y-axis). Data are represented as mean ± SD of three biological replicates, and P-values are calculated by one-way ANOVA followed by Dunnett's multiple comparison test ([***] P < 0.0001). See also Supplemental Figure S7.
To further test this idea, variants of U1 snRNA were engineered to promote the inclusion of FOXM1 exon 9 as previously described (Alanis et al. 2012; Rogalska et al. 2016). The U1 snRNAs harbored sequences at their 5′ end complementary to the two 5′ ss of exon 9 (5′ ss U1 and alt 5′ ss U1) or complementary to an intronic sequence downstream from exon 9 (shifted U1). These sequences replace those located at the 5′ end of U1 snRNA that recognize 5′ splice sites in general (canonical U1), which present several mismatches with the 5′ ss of exon 9 (Fig. 6D). Expression of 5′ ss U1—or shifted U1—plasmids in HeLa cells promoted a significant increase in exon inclusion (of both the short and long isoforms), while transfection of alt 5′ss U1 preferentially enhanced inclusion of the shorter exon 9 isoform when compared to expression of GFP or canonical U1 (Fig. 6E). Induced inclusion of the longer exon 9 isoform correlated with decreased clonogenic capacity (Fig. 6F), particularly for the shifted U1 construct which, by targeting a unique intronic sequence rather than 5′ ss, can exert its effects in a more selective and specific manner (Rogalska et al. 2016). These results further argue that modulation of FOXM1 AS, which is regulated by SPF45/SR140/CHERP, plays a role in the control of cell proliferation and the capacity of cells to form colonies.
Given the variety of possible targets impacting cell division, an interesting question is whether the SPF45/SR140/CHERP complex itself is regulated through the cell cycle and if this has an impact on the profiles of AS regulation during cell-cycle progression. As a first test of this possibility we used RNA-seq data from synchronized HeLa cells through two cell division cycles (Dominguez et al. 2016) and analyzed the gene expression pattern of SPF45, SR140, and CHERP. The three factors appear to fluctuate across the different stages but only SR140, the likely rate-limiting factor for complex formation (Fig. 1F), was categorized as cell-cycle-regulated by Dominguez et al. (Fig. 7A). The peak of SR140 expression at G2/M might induce the highest levels of the complex to control stage-specific AS events. To analyze the cycling nature of AS events regulated by the complex, we overlapped the events changing upon the three knockdowns with the cycling events defined by Dominguez et al. and retrieved 28 AS events. These can be grouped in two distinct clusters of profiles, the larger one including AS events in cell-cycle regulators like CCDC99 and DUSP16 and in FOXM1 exon 9 (Fig. 7B). This exon is more skipped right before G2/M and becomes gradually more included after G2/M, consistent with functional FOXM1 isoforms peaking at the early stages of G2/M. Another interesting AS event in this category affects exon 3 of the gene SPDL1, a regulator of mitotic spindle assembly and orientation (Fig. 7C). Knockdown of SPF45/SR140/CHERP increases exon 3 inclusion (Fig. 7D), which contains stop codons that interrupt SPDL1 ORF and therefore can induce decreased expression of the full length SPDL1 protein and/or mRNA degradation by NMD. Indeed, knockdown of SPF45 or CHERP leads to decreased levels of SPDL1 protein (Fig. 7E). To assess the functional impact of this AS event, we engineered U1 snRNA variants designed to enhance inclusion of SPDL1 exon 3, similar to the experiments aimed to increase FOXM1 exon 9 described above (Fig. 6F). Expression of two different shifted U1-plasmids (Fig. 7F) in HeLa cells promoted a very significant increase in exon 3 inclusion (Fig. 7G) and a significant decrease in clonogenic capacity, proportional to the level of exon inclusion achieved by the different U1 variants (Fig. 7H). These results argue that modulation of different cell-cycle-regulated AS events by SPF45/SR140/CHERP plays a role in the control of cell proliferation.
FIGURE 7.

AS of the SPF45/SR140/CHERP target SPDL1 exon 3 influences cell proliferation. (A) Expression levels of SPF45, SR140, and CHERP in a data set monitoring gene expression levels through two continuous cell division cycles of synchronized HeLa cells (Dominguez et al. 2016). Gene expression values are displayed as normalized FPKM (y-axis) throughout two cell cycles after double thymidine block (x-axis). (B) Splicing pattern of the 28 AS events regulated by SPF45, SR140, and CHERP that overlap with cycling events as defined by Dominguez et al. Splicing changes are quantified as scaled PSIs (y-axis) through two cell division cycles after double thymidine block (x-axis). Events are grouped in two clusters (dark and light gray lines) according to their cycling pattern; cluster 1 includes FOXM1 exon 9 (red line). (C) Schematic representation of the alternatively spliced exon 3 of the SPDL1 gene, which includes an in-frame premature termination codon (PTC) that down-regulates SPDL1 protein expression. (D) Increased inclusion of SPDL1 exon 3 upon knockdown of the indicated components of the SPF45/SR140/CHERP complex, measured by RT-PCR and gel electrophoresis (left panel), quantified in the right panel as mean ± SD of three technical replicates, and P-values are calculated by one-way ANOVA test ([***] P < 0.001). (E, left panels) Representative western blot analysis of the levels of SPDL1 protein upon knockdown of the indicated factors. (Right panels) Quantification of western blot analysis for three biological replicates. (F) Schematic representation of predicted base-pairing interactions for canonical U1 snRNA and for shifted U1 snRNAs with SPDL1 exon 3 5′ss and downstream intronic region. Bases in bold indicate sequences different from the canonical U1 sequence. (G) Quantification of SPDL1 exon 3 inclusion (PSI) upon transfection of wild type or variant U1 snRNAs, measured by RT-PCR and capillary electrophoresis in HeLa cells. Cells were cotransfected with a GFP plasmid to allow sorting of GPF-positive cells, which coexpress U1 snRNAs shown in F, 24 h after transfection. Data are represented as mean ± SD of three biological replicates and P-values were calculated by Student's t-test ([***] P < 0.001). (G) Clonogenic assay of GFP-positive HeLa cells sorted 24 h after cotransfection of GFP- and U1 snRNA-expressing plasmids shown in F. The number of colonies is normalized to that of control samples transfected only with the GFP plasmid (y-axis). Data are represented as mean ± SD of three biological replicates and P-values calculated by one-way ANOVA test ([***] P < 0.001).
DISCUSSION
Our results and those of De Maio et al. establish that splicing factors SPF45, SR140, and CHERP form a physical complex, as they stabilize each other in a variety of cell lines (Fig. 1B,C) and, in cerebellum (De Maio et al. 2018), coimmunoprecipitate (Fig. 2C; De Maio et al. 2018) and form a high order complex in sedimentation (Fig. 2D) and size exclusion chromatography analyses (De Maio et al. 2018). This can explain the close functional effects of their individual knockdowns, revealed by a splicing network analysis (Papasaikas et al. 2015) and further confirmed by extensive transcriptome profiling (Fig. 4; Supplemental Figs. S3–S5; De Maio et al. 2018). The three proteins are considered 17S U2 snRNP-associated factors and are found in complexes E, A and B (Wahl et al. 2009). Moreover, some of them interact directly with U2 snRNP components such as SF3B1–4, with the branch point/polypyrimidine tract-recognizing factors SF1/BBP and U2AF65 and with other spliceosome components and regulators such as DHX15, SNRNP200, PRP38, RBM5, and RBM10 (Corsini et al. 2007; Hegele et al. 2012; Lin-Moshier et al. 2013; Crisci et al. 2015; De Maio et al. 2018). Nevertheless, SPF45, SR140, and CHERP form a physically distinct complex, because these other interactors are not significantly affected by SPF45, SR140, or CHERP knockdown (Fig. 1D; De Maio et al. 2018) nor display equivalent sedimentation profiles (Fig. 2D). However, the native molecular mass of the complex (∼660 kDa) is compatible with the presence of several copies of one or more of the three factors and/or with the presence of some of the other interacting proteins. For example, the DEAD-box helicase DHX15 shows a sedimentation profile alike to that of the SPF45/SR140/CHERP complex (Fig. 2D) and clustered together with these factors in a high-throughput screen for regulators of AS events linked to cell fate in mouse cells (Han et al. 2017), but its knockdown has a very weak effect on the AS events of our splicing network (Supplemental Fig. S1B,C) and its protein levels seem to increase, rather than decrease, upon SPF45, SR140, or CHERP knockdowns (Supplemental Fig. S1D,E). It is possible that DHX15, or other interactors of the SPF45/SR140/CHERP complex, form physical and functional complexes only in particular cell types or cellular contexts. Other peaks in the sedimentation profile of SPF45, SR140, or CHERP (∼160 kDa) (Fig. 2D) would be compatible with monomers of SR140 and CHERP and with oligomers of SPF45 (Frenal et al. 2006). Based upon the requirement of coexpression with SPF45 and CHERP for its overexpression (Fig. 1F), and the extensive overlap of SR140-regulated AS events with SPF45 and CHERP targets (Supplemental Fig. S4C), we propose that SR140 principally accumulates as part of the SPF45/SR140/CHERP complex, as well as in an approx. 160 KDa complex revealed by sedimentation assays (Fig. 2D) in which SR140 is likely stabilized by other partners and possibly involved in other functions.
Mechanism of splicing regulation
Consistent with the association of the SPF45/SR140/CHERP complex with 17S U2 snRNP (Wahl et al. 2009), alternative 3′ss and CEx feature prominently among the AS classes commonly affected upon SPF45, SR140, and CHERP knockdowns, while alternative 5′ss were the least affected (Fig. 4D; Supplemental Fig. S4C). Interestingly, the alternative CEx included upon each of the knockdowns correspond to short exons (including many microexons), harboring weak 3′ss and often leading to CDS disruption upon exon inclusion (Fig. 4E; Supplemental Fig. S6E), in a number of cases involving cell-cycle genes. We propose that the SPF45/SR140/CHERP complex is a repressor of short, CDS-disrupting exons, which acts as a built-in proofreading activity within the spliceosome. This activity has resemblances with the proposed role of SPF45 in cryptic splice site repression in Purkinje cells (Tan et al. 2016) and of the three factors in HEK293T cells (De Maio et al. 2018). Motif analysis of the cryptic splice sites repressed by SPF45 in Purkinje neurons revealed A-rich motifs and three consensus AG motifs near the 3′ss (Tan et al. 2016). The interplay between AG dinucleotides is likely to be important for SPF45 function, as SPF45 has been shown to bind and activate an AG upstream of the polypyrimidine tract in Drosophila SXL exon 3, in this case resulting in exon inclusion (Lallena et al. 2002). Recently, Keiper et al. reported that splicing factors Smu1 and RED facilitate the activation of spliceosomal B complexes assembled on short introns (Keiper et al. 2019). Interestingly, recent results by the Mayeda laboratory also revealed a role for SPF45 in splicing of short introns (Fukumura et al. 2019). It seems likely that architectural built-in features of the spliceosome, including the need for multiple UHM-ULM interactions of SPF45 with SF1/BBP, U2AF65, and SF3B1 for SPF45-mediated control (Corsini et al. 2007) are behind the regulatory properties of SPF45/SR140/CHERP and the substrate length features (activation of short introns, repression of short exons) of its targets.
Biological effects on cell proliferation
In what physiological context are the splicing regulatory activities of the complex displayed? Disruption of the SPF45/SR140/CHERP complex strongly inhibits cell proliferation, clonogenicity and cell-cycle progression and stimulates apoptosis (Fig. 3; Supplemental Fig. S2). Several genes changing in expression or in AS upon the knockdown of these factors play important roles in cell proliferation and mitotic progression and could mediate these effects. Because of the G2/M arrest observed in HeLa cells, we inititally focused on FOXM1, a master regulator of this transition whose exon 9 (in two length versions) becomes included upon SPF45, SR140, or CHERP knockdown (Fig. 5B–D), concomitant with a reduction in the expression of FOXM1 transcriptional targets, including key regulators of cell-cycle progression such as AURKB or CCNB2 (Fig. 5F). Reverting FOXM1 exon 9 repression in HeLa and U2OS SPF45-depleted cells using AONs results only in partial—but reproducible—rescue of the clonogenic capacity of cells (Fig. 6C). However, the converse experiment of inducing FOXM1 exon 9 inclusion through targeting of modified U1 snRNP downstream from the exon (Alanis et al. 2012; Rogalska et al. 2016) mimics the effects of SPF45/SR140/CHERP depletion, causing significant decreases in the clonogenic capacity of HeLa cells (Fig. 6F). Thus, FOXM1 is a relevant target of SPF45/SR140/CHERP-mediated regulation of cell cycle, but other targets must contribute to the phenotype.
Attractive additional targets include members of the mitotic apparatus such as CLASP1, PLK3, LATS1, and SDCCAG8, which are modulated by the three factors (Fig. 4G) and feature prominently as members of the G2/M GO categories of genes alternatively spliced upon SPF45 knockdown. Given their essential functions in mitosis (Neumann et al. 2010), AS modulation of AURKB and INCENP could have important implications as well. The knockdown of SPF45, SR140, or CHERP also promotes MDM4 exon 6 skipping, which inhibits cancer growth when induced using AONs (Dewaele et al. 2016). Several genes involved in the maintenance of sister chromatid cohesion are also alternatively spliced upon the three knockdowns, including NIPBL, POGZ, SMC4, and CHTF8. UPF3A exon 4, which is activated by SR140 and CHERP in colon carcinoma cell lines by binding to a motif within the exon (Wang et al. 2019), could also contribute to the phenotype, as overexpression of the long UPF3A isoform, a NMD repressor, but not of the short nonfunctional isoform, greatly rescued the clonogenic capacity upon siSR140 or siCHERP treatment (Wang et al. 2019). Our analysis of the consequences of inducing inclusion of SPDL1 exon 2, which reduces the levels of SPDL1 protein, show that this regulator of mitotic spindle assembly and orientation is another functionally relevant target of the SPF45 complex in the control of cell growth (Fig. 7).
Alternative mechanisms for regulating the activity of the SPF45/SR140/CHERP complex during cell cycle could also operate, for example through the control of the interactions between its components by phosphorylation (Al-Ayoubi et al. 2012; Liu et al. 2013).
Another attractive physiological context for modulation of SPF45/SR140/CHERP activity is the DNA Damage Response (DDR). A recent study has shown an increase in double-strand breaks upon SR140 or CHERP knockdowns in RKO cells and mouse xenografts (Wang et al. 2019). On the other hand, SPF45 is recruited to DNA damage sites, binds DNA and interacts with DNA damage repair proteins (Horikoshi et al. 2010) and Drosophila SPF45 can rescue the sensitivity to a mutagen and repair the DNA breaks induced during transposition (Chaouki and Salz 2006). One possible scenario would be that upon DNA damage SPF45 is mobilized to DNA damage sites and leaves the SPF45/SR140/CHERP complex, which in turn would lead to the production of a nonfunctional FOXM1 isoform and G2/M arrest, which is a feature of DDR. Further work will be needed to explore the interplay between DDR and RNA processing factors and the roles of SPF45/SR140/CHERP complex components in these processes.
MATERIALS AND METHODS
Cell culture
HeLa cells were cultured in Glutamax Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS), 500 U/mL penicillin and 500 µg/mL streptomycin. U2OS cells were cultured in McCoy's 5A (modified) medium (Gibco) supplemented with 10% FBS, 500 U/mL penicillin and 500 µg/mL streptomycin. Flp-In T-Rex HeLa host cell line was cultured in Glutamax DMEM supplemented with 10% FBS, 500 U/mL penicillin, 500 µg/mL streptomycin, 50 µg/mL zeocin, and 5 µg/mL blasticidin. Flp-In T-Rex HeLa stable cell lines were cultured in Glutamax DMEM supplemented with 10% Tet System Approved FBS (Takara), 500 U/mL penicillin, 500 µg/mL streptomycin, 150 µg/mL hygromycin, and 5 µg/mL blasticidin. All cells were maintained at 37°C in a 5% CO2 humidified incubator.
siRNA and plasmid transfection
For siRNA transfection, 300,000 HeLa or 250,000 U2OS cells per well were plated on six-well plates and cultured overnight. Cells were incubated with 60 nM of siRNA complexed with 5 µL of Lipofectamine RNAiMAX (Invitrogen) in OPTI-MEM (Gibco) for 6 to 8 h and then media was replaced. Cells were collected 72 h after transfection.
For plasmid transient transfection, 350,000 HeLa cells per well were plated on six-well plates and cultured overnight. Cells were incubated with 3 µL of Lipofectamine 2000 (Invitrogen) complexed with a maximum of 1 µg of plasmid DNA in 1 mL of OPTI-MEM for 6 h and media was replaced after that. Cells were collected 48 h after transfection.
RNA extraction and reverse transcription
Routinely, total RNA was extracted using Maxwell RNAeasy kit (Promega) following the manufacturer's instructions. For RNA-seq experiments, total RNA extraction was carried out using RNeasy Mini Kit (Qiagen) following the manufacturer's instructions.
Reverse transcription (RT) was performed with 200 ng–1 µg of total RNA in the presence of 125 ng of random primers (Invitrogen), 50 pmol of oligo dT (Sigma) and 0.5 µL of Superscript III reverse transcriptase (Invitrogen) in a total volumen of 20 µL.
PCR and quantitative PCR
Semiquantitative PCR reactions were carried out with 1–2 µL of cDNA and 0.2 µL of GoTaq DNA Polymerase (Promega) in a total reaction volume of 30 µL according to the manufacturer's instructions. Typical parameters for the reactions were: 3 min at 94°C, 30 cycles of 30 sec at 94°C, 30 sec at 60°C, 30 sec at 72°C and a final incubation of 1 min at 72°C.
Quantitative PCR (qPCR) reactions were carried out with 4 µL of diluted cDNA, 0.4 µM of each primer, and 5 µL of Power SYBR Green PCR master mix (Applied Biosystems) in a total reaction volume of 10 µL. Technical replicates for each experimental sample were analyzed in a ViiA 7 Real-Time PCR system (Applied Biosystems) using the standard curve method. Relative expressions were calculated using the quantity mean normalized by GAPDH.
Protein extraction and western blot analysis
Protein extraction was performed by resuspending cells in RIPA buffer (50 mM Tris-HCl pH 7,4; 100 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 2 mM EDTA; 0.1% SDS; 1 mM DTT; 1× protease inhibitors), incubating them on ice for 30 min, vortexing every 5–10 min and centrifuging the lysates at 15,700g at 4°C for 15 min. Supernatants were collected and quantified using the Bradford Protein Assay Kit (Bio-Rad). To study phospho-proteins, protein extracts were prepared by resuspending freshly collected cells in RIPA buffer (as before) supplemented with β-glycerolphosphate and sodium orthovanadate and snapped frozen before keeping them at −80°C.
For western blot analysis, 20 µg of total protein were resolved in 8% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes (Amersham Biosciences). Membranes were blocked for 1h at room temperature with 10% (w/v) skimmed milk or BSA (Sigma) diluted in PBST (PBS, 0.1% Tween-20) and probed overnight at 4°C with primary antibodies diluted in 5% skimmed milk or BSA in PBST. The following day membranes were washed three times for 10 min with PBST and incubated with horseradish peroxidase-conjugated secondary antibodies diluted in 5% skimmed milk in PBST for 1 h at room temperature. Protein signal was detected by chemiluminiscence using Amersham Hyperfilm ECL (GE Healthcare). Bands were quantified using ImageJ. For Figure 3E and Supplemental Figure S2E, protein extracts from the same number of cells (counted before protein extraction) were loaded per lane. Western blots of phospho-proteins were performed as described before but using TBST (TBS, 0.1% Tween-20) and BSA instead of PBST or milk. Li-COR secondary antibodies were used and analyzed on an Odyssey CLX.
Targeted proteomics
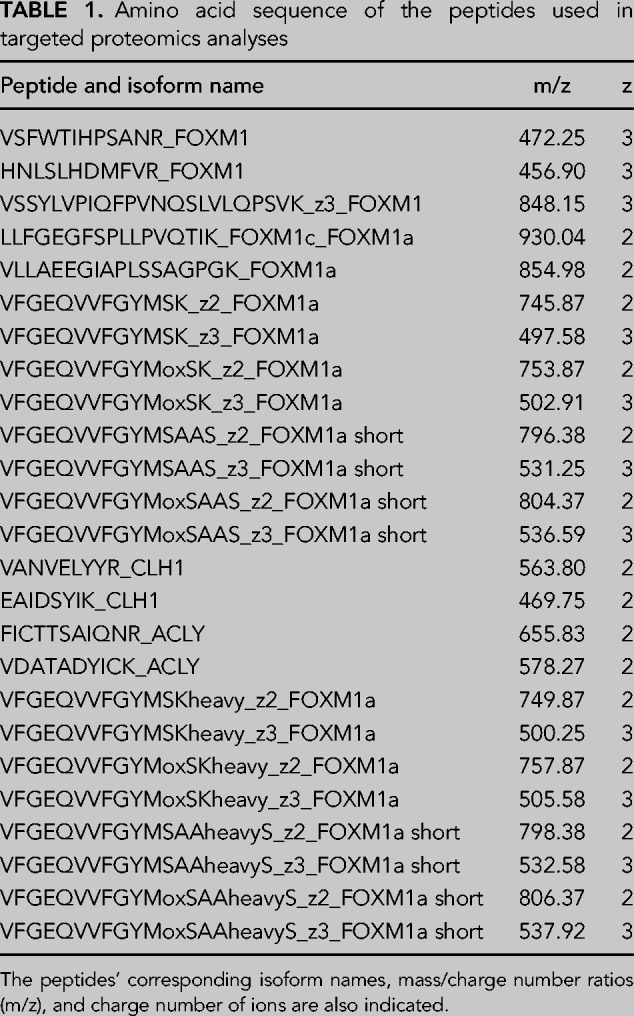
After electrophoresis in SDS polyacrylamide gels, gel slices corresponding to the region of migration of FOXM1 proteins were destained with 40%ACN/100 mM ABC, reduced with dithiothreitol (2 mmols, 30 min, 56°C) and alkylated in the dark with iodoacetamide (11 mmols, 30 min, 25°C). Gel slices were then dehydrated with ACN and digested with 0.8 μg of trypsin (Promega, cat # V5113) overnight at 37°C. After digestion, peptides were extracted, acidified with formic acid, and desalted with a MicroSpin C18 column (The Nest Group, Inc.). Unique peptides for isoform FOXM1a short and FOXM1a (VFGEQVVFGYMSAAS and VFGEQVVFGYMSK) peptides were chemically synthesized using carboxy-terminal 13C6, 15N2-Lysine or 13C3, 15N1-Arginine fourteen (Thermo Fisher Scientific, Germany) and spiked into the sample.
Samples were analyzed using an Orbitrap Eclipse (Thermo Fisher Scientific) coupled to an EASY-nanoLC 1200 UPLC system (Thermo Fisher Scientific). Peptides were loaded directly onto the analytical column and were separated by reversed-phase chromatography using a 50-cm column with an inner diameter of 75 µm, packed with 2 µm C18 particles spectrometer (Thermo Scientific). Chromatographic gradients started at 95% buffer A and 5% buffer B with a flow rate of 300 nL/min for 5 min and gradually increased to 25% buffer B and 75% A in 79 min and then to 40% buffer B and 60% A in 11 min. After each analysis, the column was washed for 10 min with 10% buffer A and 90% buffer B. Buffer A: 0.1% formic acid in water. Buffer B: 0.1% formic acid in 80% acetonitrile. The mass spectrometer was operated in positive ionization mode with an EASY-Spray nanosource at 2.4 kV and at a source temperature of 305°C. A full MS scan with 1 micro scan at a resolution of 30,000 was used over a mass range of m/z 350–1400 with detection in the Orbitrap mass analyzer. A PRM (parallel reaction monitoring method) was used for data acquisition with a quadrupole isolation window set to 1.4 m/z and MSMS scans over a mass range of m/z 300–2000, with detection in the Orbitrap at a resolution of 60,000. MSMS fragmentation was performed using HCD at 30 NCE, and the auto gain control (AGC) was set to 1 × 105 and maximum injection time of 118 msec. Peptide masses (m/z) were defined in the PRM method for further fragmentation (Table 1) as follows:
TABLE 1.
Amino acid sequence of the peptides used in targeted proteomics analyses
Skyline software (MacLean et al. 2010) (v20.2.0.343) was used to generate the libraries (observed as the output of the MaxQuant [1.6.10] search and predicted with Prosit [Gessulat et al. 2019]) and to extract the fragment areas of each peptide.
Generation of stable Flp-In T-Rex HeLa cell lines and rescue experiment
For stable cell line generation, 700,000 Flp-In T-Rex HeLa cells (Invitrogen) were plated on 60 mm plates and transfected the following day with 6 µL of Lipofectamine 2000 (Invitrogen), 0.3 µg of pcDNA5/FRT/TO expression vector and 2.7 µg of pOG44 plasmid following the standard procedure. Cells were incubated for 8 h, then media was replaced and 24 h after transfection cells were trypsinized and selected with 150 µg/mL hygromycin and 5 µg/mL blasticidin. After 12 d of selection, colonies were picked using a micropipette with 5 µL of trypsin and expanded in 12-well plates in the presence of hygromycin and blasticidin. Clones were screened for overexpression levels upon tetracycline addition (1 µg/mL), and those with highest expression levels were expanded and stored.
For the rescue experiment, we used the established Flp-In T-Rex HeLa stable cell lines. 200,000 cells per well were plated on six-well plates and transfected the following day with 3′UTR-targeting siRNAs following the standard procedure. Twenty-four hours after siRNA transfection, expression of the flipped-in recombinant proteins was induced using tetracycline (1 µg/mL) and cells were collected after 48 h of tetracycline treatment.
Coimmunoprecipitation experiments
A total of 850 µg of total protein extract were diluted in RSB100 buffer (10 mM Tris-HCl pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% Triton X-100, 1 mM DTT, 1× protease inhibitors) and precleared with 20 µL of Dynabeads Protein A (Thermo Fisher Scientific) for 45min at 4°C with rotation. Antibody-coated beads were prepared by incubating 40 µL of beads in 1 mL of RSB100 with 2 µg of rabbit IgG, 2 µg of anti-CHERP or 20 µL of anti-SPF45 for 45 min at 4°C with rotation. The beads bound to the antibodies were washed with RSB100 three times and resuspended with the precleared extracts. After overnight incubation, beads were washed with RSB100 four times and resuspended in 50 µL of 2× SDS loading buffer. Immunoprecipitates were resolved by SDS-PAGE and coimmunoprecipitating proteins were identified by western blot analyses.
Glycerol gradient fractionation
A total of 1 mL of commercial HeLa nuclear extract was first concentrated using an Amicon 5k Filter Unit (Merck Millipore) and then applied to a 10 mL 5%–40% glycerol gradient prepared in a dilution buffer (20 mM HEPES pH 7.9, 150 mM KCl, 0.2 mM EDTA, 0.1% NP-40, 1× protease inhibitors and 0.5 mM DTT). After 22 h of centrifugation at 33,000 rpm in a SW41Ti rotor (Beckman) at 4°C, 24 fractions of 500 µL were manually collected from the top to the bottom of the gradient. A total of 20 µL of each fraction were analyzed by western blotting. To estimate the molecular weights of the fractions, a parallel gradient was loaded with molecular mass standards (Gel Filtration High Molecular Weight Calibration Kit), separated by SDS-PAGE and stained with Coomassie.
Lentiviral production and infection
For lentiviral particle production, 7.5 million low passage 293T/17 cells were seeded in 15 cm plates, incubated overnight and 2 h before transfection media was replaced. For each plate, 44 µg of pLKO.1-shRNA (Sigma), 13 µg of VSV-G envelope and 33 µg of delta 8.9 packaging vectors were mixed with 1125 µL of endotoxin-free water (Sigma, W-3500) and 125 µL of 2.5 M CaCl2 (Sigma). After incubating the mix for 5 min in a rotor, 1250 µL of 2× HBS pH 7.2 (281 mM NaCl, 100 mM HEPES, 1.5 mM Na2HPO4) were added in a drop-wise manner. After 10min of incubation at room termperature, the precipitate was added dropwise to the cells and incubated for 30 h. Virus-containing supernatants were collected, filtered (0.22 µm) and centrifuged at 20,000rpm at 4°C for 2 h in a SW32Ti rotor (Beckman). Lentiviral pellets were resuspended in 80 µL of PBS and kept at −80°C.
For lentiviral infections, cells were incubated with concentrated lentivirus overnight and subcultured with fresh medium the following day. Forty-eight hours after infection, cells were selected with puromycin (1 µg/mL for U2OS, 2 µg/mL for HeLa) for 2 d.
Phenotypic assays
After lentiviral infection and puromycin selection, HeLa or U2OS cells were cultured in media without puromycin for 1 d (cell proliferation and clonogenic assays) or 3 d (cell cycle and apoptosis assays), and phenotypic assays were performed.
Cell proliferation assays
In six-well plates, or 12-well plates, 150,000 or 65,000 cells were plated per well respectively, incubated overnight and counted at 24 h intervals using a Countess Automated Cell Counter (Invitrogen) for 4 d.
Clonogenic assays
In six-well plates, 1000 HeLa or 2000 U2OS cells per well were maintained in culture, replacing the medium every 3 d, until colonies were visible. Cells were fixed with 1 mL of methanol for 10 min at room temperature and stained with 5% Giemsa. After extensive washing, plates were dried and scanned. Colonies were counted using ImageJ.
Cell-cycle assays
After trypsinization, 500,000 cells were washed twice in PBS and resuspended in 150 µL of PBS. A total of 350 µL of absolute ethanol previously incubated at −20°C, was added slowly while vortexing. After overnight incubation at 4°C, cells were pelleted at 2320 g and washed twice with PBS. Cells were resuspended in 500 µL of PBS with 7.5 µg of propidium iodide (Molecular Probes) and 150 µg of RNase A (Sigma) and incubated overnight at 4°C. Cells were analyzed with a BD FACScalibur flow cytometer (BD Biosciences) and results were analyzed using ModFit.
Mitotic (pH3S10) cells staining
Cells were fixed following the same procedure as for the cell-cycle assays. After the second PBS wash, 106 cells were resuspended in 1 mL of PBS containing 0.25% Triton X-100 and incubated on ice for 15 min. Cells were spun at 5000 rpm for 5 min, washed once with 1 mL of PBS and resuspended in 100 µL of 1%BSA (in PBS) containing the antibody against phospho-H3 serine 10 (1/1000). Cells were incubated at 4°C ON followed by the addition of 1 mL of 1%BSA (in PBS) and spinning at 5000 rpm for 5 min. Cells were resuspended in 100 µL of anti-rabbit Alexa 488 (1/250) diluted in 1%BSA (in PBS) and incubated at RT for 30 min in the dark. After adding 1 mL of PBS and centrifuging the cells at 5000 rpm for 5 min, cells were stained with propidium iodide following the same procedure as in the cell-cycle assays but incubating for 30 min at 37°C in the dark. Cells were analyzed with a BD LSR II flow cytometer (BD Biosciences) and results were analyzed using FlowJo.
Apoptosis assays
After trypsinization, 100,000 cells were washed in PBS, resuspended in 195 µL of 1× binding buffer and incubated with 5 µL of annexin V-FITC (eBioscience) for 10 min at room temperature in the dark. After centrifugation, the cells were again washed with 1× binding buffer and resuspended in 190 µL of binding buffer and 0.2 µg of propidium iodide (eBioscience). Cells were analyzed with a BD FACScalibur flow cytometer (BD Biosciences) and results were analyzed using BD FACSDiva.
Antisense oligonucleotide transfection and rescue experiment
After lentiviral infection and 48 h of puromycin selection, ∼500,000 cells were plated in six-well plates for AON transfection. The following day, cells were incubated with 50 nM of AONs complexed with 3.75 µL of Lipofectamine RNAiMAX (Invitrogen) in a 1:1 mix of OPTI-MEM and media without antibiotics for 6 h and then media was replaced. Twenty-four hours after ASO transfection, cells (shSPF45: 15,000 for HeLa and 7500 for U2OS, shCNT: 3000 and 7500 for both) were plated for clonogenic assays performed as described above.
U1 snRNAs transfection
500,000 HeLa cells were cotransfected with 100 ng of pmaxGF and U1 2500 ng of modified U1snRNA plasmids using an Amaxa Nucleofector (Lonza). GFP-positive cells were sorted 24 h after nucleofection and used for RT-PCR and clonogenic experiments.
RNA-seq analysis
Data sets
Total RNA from three replicates per condition was analyzed using a Bioanalyzer (Agilent), and the best two replicates for each condition according to RNA quality were used for RNA sequencing. Strand-specific libraries were built and sequenced at the Center for Genomic Regulation (CRG) Genomics Unit using 125 bp paired-end reads. The RNA-seq data generated during the current study have been deposited in NCBI's Gene Expression Omnibus database and are accessible through GEO Series accession number GSE180892.
Differential gene expression
Reads alignment was performed using STAR (Dobin et al. 2013) (genome annotation hg19), and differential gene expression analyses were performed using CuffDiff (Trapnell et al. 2013) and setting the following parameters: log2 fold change ≥ 0.6 or ≤−0.6, ΔFPKM > 1, status OK, significant YES.
Gene ontology
Functional enrichment analyses were performed using GOrilla (http://cbl-gorilla.cs.technion.ac.il/). For differentially expressed genes the background used was set as: status OK. For alternatively spliced genes a background set provided by VAST-TOOLS was used as a control.
AS analysis
Splicing analyses of RNA-seq data were performed using VAST-TOOLS v2.2.2 using the hg19 library (Irimia et al. 2014; Tapial et al. 2017). For all events, a minimum read coverage of 10 actual reads per sample was required, as described in Irimia et al. (2014). PSI values for single replicates were quantified for all types of alternative events, including single and complex cassette exons (S, C1, C2, C3, ANN), microexons (MIC) alternative 5′ss and 3′ss (Alt5, Alt3) and retained introns (IR-S, IR-C). A minimum absolute ΔPSI of 15% was required to define differentially spliced events upon each knockdown, as well as a minimum range of 5% between the PSI values of the two samples. See https://github.com/vastgroup/vast-tools for details. For pairwise PSI comparisons, a Pearson correlation coefficient was calculated between the PSI values of the overlapping events [only AS events differentially spliced (|ΔPSI| ≥ 15%) in both conditions were considered].
Sequence features analysis
Splicing regulatory features, GC content and length analyses were performed using Matt v1.3.0 (Gohr and Irimia 2018), comparing differentially spliced events to a set of control alternative nonchanging events stratified to match the GC content of each set of events changing upon knockdown of SPF45/SR140/CHERP or the three of them (maximum deviation 0.01). Sequence features analyzed include 5′ss and 3′ss strength scores calculated according to the maximum entropy model (Yeo and Burge 2004) and best predicted branch point (BP) sequence and polypyrimidine tract (Py) sequence scores determined using the best predicted BP for each exon (Corvelo et al. 2010). Median score differences of each sequence feature (with the exclusion of GC content and lengths) were calculated between each condition and the set of control alternative nonchanging events and normalized on the maximum difference observed for the feature across the data set. Statistical significance was tested by Mann–Whitney U-tests.
ORF impact prediction
Potential ORF impact of alternative exons was predicted as described in Irimia et al. (2014). First, exons were mapped on the coding sequence (CDS) or 5′/3′ untranslated regions (UTR) of genes. Events mapping on the CDS were divided into CDS-preserving (exon length is multiple of 3, exon contains a PTC not predicted to undergo NMD or contains a start or stop codon) or CDS-disrupting. Exons can disrupt the CDS upon inclusion (exon causes frameshift, contains a PTC predicted to undergo NMD or a PTC that truncates the protein by >300 aa) or upon exclusion (exon causes a frameshift). Noncoding exons can affect noncoding RNAs, 5′UTRs, or 3′UTRs.
Cycling gene expression and AS events
To check for periodic changes in gene expression of the SPF45/SR140/CHERP complex associated with cell-cycle progression, we used normalized gene expression values from (Dominguez et al. 2016). To investigate the overlap between periodic AS changes and targets of SPF45/SR140/CHERP complex, we reanalyzed this RNA-seq data set using VAST-TOOLS v2.2.2 (Irimia et al. 2014) and overlapped previously identified cell-cycle-specific alternative exons with exons regulated by SPF45/SR140/CHERP.
Quantification and statistical analysis
Statistical analyses
P-values were calculated by two tailed Student's t-test (Excel), χ2-square test (Excel) or one-way ANOVA followed by Dunnett's multiple comparison test (GraphPad Prism). Statistical details of experiments can be found in figure legends.
PCR quantification
After RT-PCR, diluted PCR products were quantified by high-throughput capillary electrophoresis using a Labchip GX Caliper workstation (Caliper, PerkinElmer) and a HT DNA 5K LabChip chip (PerkinElmer). Isoform concentrations were calculated based on the nanomolar content of the corresponding band with LabChip GX software.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the CRG Genomics, Proteomics and Cell Sorting Core Facilities for their invaluable technical help and members of our group and Erwan Watrin for advice and critical reading of the manuscript. E.M. and J.H. are recipients of FPI Fellowships from the Spanish Ministry of Economy and Competitiveness. Work in our laboratory has been supported by the European Research Council (ERC AdvG 670146), AGAUR, Spanish Ministry of Economy and Competitiveness (BFU 2017 89308-P), and the Centre of Excellence Severo Ochoa. We acknowledge support from the Spanish Ministry of Science and Innovation to the EMBL partnership and the CERCA Programme/Generalitat de Catalunya.
Author contributions: J.V. conceived the study and E.M. and J.V. designed the experiments. E.M. performed the majority of experimental work, C.V. analyzed RNA-seq data and carried out downstream analyses, and M.R. and J.H.-V. performed experiments using modified U1 snRNAs and integrated analysis of RNA-seq data during cell cycle. E.M. and J.V. wrote the manuscript with input from all authors.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078935.121.
Freely available online through the RNA Open Access option.
REFERENCES
- Alanis EF, Pinotti M, Dal Mas A, Balestra D, Cavallari N, Rogalska ME, Bernardi F, Pagani F. 2012. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet 21: 2389–2398. 10.1093/hmg/dds045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ayoubi AM, Zheng H, Liu Y, Bai T, Eblen ST. 2012. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol Cell Biol 32: 2880–2893. 10.1128/MCB.06327-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas JE, Abelson JN. 1997. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Biochemistry 94: 11798–11802. 10.1073/pnas.94.22.11798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouki AS, Salz HK. 2006. Drosophila SPF45: a bifunctional protein with roles in both splicing and DNA repair. PLoS Genet 2: 1974–1983. 10.1371/journal.pgen.0020178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL. 2009. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10: 741–754. 10.1038/nrm2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini L, Bonnal S, Basquin J, Hothorn M, Scheffzek K, Valcárcel J, Sattler M. 2007. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat Struct Mol Biol 14: 620–629. 10.1038/nsmb1260 [DOI] [PubMed] [Google Scholar]
- Corvelo A, Hallegger M, Smith CW, Eyras E. 2010. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol 6: 12–15. 10.1371/journal.pcbi.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisci A, Raleff F, Bagdiul I, Raabe M, Urlaub H, Rain JC, Krämer A. 2015. Mammalian splicing factor SF1 interacts with SURP domains of U2 snRNP-associated proteins. Nucleic Acids Res 43: 10456–10473. 10.1093/nar/gkv952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio AD, Yalamanchili HK, Adamski CJ, Gennarino VA, Liu Z, Qin J, Jung SY, Richman R, Orr H, Zoghbi HY. 2018. RBM17 interacts with U2SURP and CHERP to regulate expression and splicing of RNA-processing proteins. Cell Rep 25: 726–736.e7. 10.1016/j.celrep.2018.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewaele M, Tabaglio T, Willekens K, Bezzi M, Teo SX, Low DH, Koh CM, Rambow F, Fiers M, Rogiers A, et al. 2016. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest 126: 68–84. 10.1172/JCI82534.MDM4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. 2016. An extensive program of periodic alternative splicing linked to cell cycle progression. Elife 5: e10288. 10.7554/eLife.10288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frénal K, Callebaut I, Wecker K, Prochnicka-Chalufour A, Dendouga N, Zinn-Justin S, Delepierre M, Tomavo S, Wolff N. 2006. Structural and functional characterization of the TgDRE multidomain protein, a DNA repair enzyme from Toxoplasma gondii. Biochemistry 45: 4867–4874. 10.1021/bi051948e [DOI] [PubMed] [Google Scholar]
- Fukumura K, Yoshimoto R, Hirose T. 2019. A distinct subset of human short introns: U2AF heterodimer is replaced by SPF45 (RBM17) as general splicing factor. bioRxiv 10.1101/784868 [DOI] [Google Scholar]
- Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A, et al. 2019. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat Methods 16: 509–518. 10.1038/s41592-019-0426-7 [DOI] [PubMed] [Google Scholar]
- Gohr A, Irimia M. 2018. Matt: Unix tools for alternative splicing analysis. Bioinformatics 35: 130–132. 10.1093/bioinformatics/bty606 [DOI] [PubMed] [Google Scholar]
- Grant GD, Brooks L III, Zhang X, Mahoney JM, Martyanov V, Wood TA, Sherlock G, Cheng C, Whitfield ML. 2013. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol Biol Cell 24: 3634–3650. 10.1091/mbc.E13-05-0264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Braunschweig U, Gonatopoulos-Pournatzis T, Weatheritt RJ, Hirsch CL, Ha KCH, Radovani E, Nabeel-Shah S, Sterne-Weiler T, Wang J, et al. 2017. Multilayered control of alternative splicing regulatory networks by transcription factors. Mol Cell 65: 539–553.e7. 10.1016/j.molcel.2017.01.011 [DOI] [PubMed] [Google Scholar]
- Han Y, Zhang M, Chen D, Li H, Wang X, Ma S. 2018. Downregulation of RNA binding motif protein 17 expression inhibits proliferation of hypopharyngeal carcinoma faDu cells. Oncol Lett 15: 5680–5684. 10.3892/ol.2018.8012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. 2012. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. 10.1016/j.molcel.2011.12.034 [DOI] [PubMed] [Google Scholar]
- Horikoshi N, Morozumi Y, Takaku M, Takizawa Y, Kurumizaka H. 2010. Holliday junction-binding activity of human SPF45. Genes Cells 15: 373–383. 10.1111/j.1365-2443.2010.01383.x [DOI] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallières M, Tapial J, Raj B, O'Hanlon D, et al. 2014. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159: 1511–1523. 10.1016/j.cell.2014.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiper S, Papasaikas P, Will CL, Valcárcel J, Girard C, Lührmann R. 2019. Smu1 and RED are required for activation of spliceosomal B complexes assembled on short introns. Nat Commun 10: 3639. 10.1038/s41467-019-11293-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL, Rodionova NA, Green MR, Burley SK. 2001. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell 106: 595–605. 10.1016/S0092-8674(01)00480-9 [DOI] [PubMed] [Google Scholar]
- Kielkopf CL, Lücke S, Green MR. 2004. U2AF homology motifs: protein recognition in the RRM world. Genes Dev 18: 1513–1526. 10.1101/gad.1206204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. 2013. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14: 153–165. 10.1038/nrm3525 [DOI] [PubMed] [Google Scholar]
- Korver W, Roose J, Heinen K, Weghuis DO, de Bruijn D, van Kessel AG, Clevers H. 1997. The human TRIDENT/HFH-11/FKHL16 gene: structure, localization, and promoter characterization. Genomics 46: 435–442. 10.1006/geno.1997.5065 [DOI] [PubMed] [Google Scholar]
- Lallena MJ, Chalmers KJ, Llamazares S, Lamond AI, Valcárcel J. 2002. Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell 109: 285–296. 10.1016/S0092-8674(02)00730-4 [DOI] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. 2005. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136. 10.1038/ncb1217 [DOI] [PubMed] [Google Scholar]
- Laoukili J, Stahl M, Medema RH. 2007. FoxM1: at the crossroads of ageing and cancer. Biochim Biophys Acta 1775: 92–102. 10.1016/j.bbcan.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Laplante JM, O'Rourke F, Lu X, Fein A, Olsen A, Feinstein MB. 2000. Cloning of human Ca2+ homoeostasis endoplasmic reticulum protein (CHERP): regulated expression of antisense cDNA depletes CHERP, inhibits intracellular Ca2+ mobilization and decreases cell proliferation. Biochem J 348: 189–199. 10.1042/0264-6021:3480189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Green RE, Brenner SE. 2003. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci 100: 189–192. 10.1073/pnas.0136770100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. 2008. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452: 713–718. 10.1038/nature06731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Moshier Y, Sebastian PJ, Higgins L, Sampson ND, Hewitt JE, Marchant JS. 2013. Re-evaluation of the role of calcium homeostasis endoplasmic reticulum protein (CHERP) in cellular calcium signaling. J Biol Chem 288: 355–367. 10.1074/jbc.M112.405761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Conaway L, Rutherford Bethard J, Al-Ayoubi AM, Thompson Bradley A, Zheng H, Weed SA, Eblen ST. 2013. Phosphorylation of the alternative mRNA splicing factor 45 (SPF45) by Clk1 regulates its splice site utilization, cell migration and invasion. Nucleic Acids Res 41: 4949–4962. 10.1093/nar/gkt170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Li Q, Cai L, Zhu Z, Guan J, Wang C, Xia J, Xia L, Wen M, Zheng W, et al. 2018. RBM17 controls apoptosis and proliferation to promote Glioma progression. Biochem Biophys Res Commun 505: 20–28. 10.1016/j.bbrc.2018.09.056 [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968. 10.1093/bioinformatics/btq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Schneider S, Schwer B. 2002. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J Biol Chem 277: 17743–17750. 10.1074/jbc.M200762200 [DOI] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Semlow DR, Staley JP. 2010. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc Natl Acad Sci 107: 10020–10025. 10.1073/pnas.0906022107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockenhaupt S, Makeyev EV. 2015. Non-coding functions of alternative pre-mRNA splicing in development. Semin Cell Dev Biol 47–48: 32–39. 10.1016/j.semcdb.2015.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. 1998. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat Genet 20: 46–50. 10.1038/1700 [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464: 721–727. 10.1038/nature08869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke FA, LaPlante JM, Feinstein MB. 2003. Antisense-mediated loss of calcium homoeostasis endoplasmic reticulum protein (CHERP; ERPROT213-21) impairs Ca2+ mobilization, nuclear factor of activated T-cells (NFAT) activation and cell proliferation in Jurkat T-lymphocytes. Biochem J 373: 133–143. 10.1042/BJ20030013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcárcel J. 2015. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell 57: 7–22. 10.1016/j.molcel.2014.10.030 [DOI] [PubMed] [Google Scholar]
- Perry WL, Shepard RL, Sampath J, Yaden B, Chin WW, Iversen PW, Jin S, Lesoon A, O'Brien KA, Peek VL, et al. 2005. Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed: a phenotype partially reversed by selective estrogen receptor modulators. Cancer Res 65: 6593–6600. 10.1158/0008-5472.CAN-03-3675 [DOI] [PubMed] [Google Scholar]
- Rogalska ME, Tajnik M, Licastro D, Bussani E, Camparini L, Mattioli C, Pagani F. 2016. Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun 7: 11168. 10.1038/ncomms11168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, Duan S, DeCaprio JA. 2012. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 26: 474–489. 10.1101/gad.181933.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. 2011. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev 25: 373–384. 10.1101/gad.2004811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath J, Long PR, Shepard RL, Xia X, Devanarayan V, Sandusky GE, Perry WL, Dantzig AH, Williamson M, Rolfe M, et al. 2003. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am J Pathol 163: 1781–1790. 10.1016/S0002-9440(10)63538-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Krämer A, Sattler M. 2003. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol Cell 11: 965–976. [DOI] [PubMed] [Google Scholar]
- Spadaccini R, Reidt U, Dybkov O, Will C, Frank R, Stier G, Corsini L, Wahl MC, Lührmann R, Sattler M. 2006. Biochemical and NMR analyses of an SF3b155–p14–U2AF-RNA interaction network involved in branch point definition during pre-mRNA splicing. RNA 12: 410–425. 10.1261/rna.2271406.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, et al. 2016. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum Mol Genet 25: 5083–5093. 10.1093/hmg/ddw337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapial J, Ha KCH, Sterne-Weiler T, Gohr A, Braunschweig U, Hermoso-Pulido A, Quesnel-Vallières M, Permanyer J, Sodaei R, Marquez Y, et al. 2017. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res 27: 1759–1768. 10.1101/gr.220962.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villamizar O, Chambers CB, Riberdy JM, Persons DA, Wilber A. 2015. Long noncoding RNA Saf and splicing factor 45 increase soluble Fas and resistance to apoptosis. Oncotarget 7: 13810–13826. 10.18632/oncotarget.7329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- Wan R, Bai R, Zhan X, Shi Y. 2019. How is precursor messenger RNA spliced by the spliceosome? Annu Rev Biochem 89: 333–358. 10.1146/annurev-biochem-013118-111024 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wang Y, Liu Y, Zhang C, Luo Y, Guo R, Zhan Z, Wei N, Xie Z, Shen L, et al. 2019. U2-related proteins CHERP and SR140 contribute to colorectal tumorigenesis via alternative splicing regulation. Int J Cancer 145: 2728–2739. 10.1002/ijc.32331 [DOI] [PubMed] [Google Scholar]
- Wilkinson ME, Charenton C, Nagai K. 2019. RNA splicing by the spliceosome. Annu Rev Biochem 89: 359–388. 10.1146/annurev-biochem-091719-064225 [DOI] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Lührmann R. 2002. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21: 4978–4988. 10.1093/emboj/cdf480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, et al. 2016. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164: 805–817. 10.1016/j.cell.2016.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, Costa RH. 1997. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol 17: 1626–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Zhang D, Wang F, Pang Y, Ke XX, Zhu S, Zhao E, Zhang K, Chen L, Cui H. 2017a. Down-regulation of CHERP inhibits neuroblastoma cell proliferation and induces apoptosis through ER stress induction. Oncotarget 8: 80956–80970. 10.18632/oncotarget.20898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang L, Du Y, Zheng H, Zhang P, Sun Y, Wang Y, Chen J, Ding P, Wang N, et al. 2017b. A novel FOXM1 isoform, FOXM1D, promotes epithelial-mesenchymal transition and metastasis through ROCKs activation in colorectal cancer. Oncogene 36: 807–819. 10.1038/onc.2016.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.