

ABSTRACT
Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever, is an enzootic, obligate, intracellular bacterial pathogen. Nitric oxide (NO) synthesized by the inducible NO synthase (iNOS) is a potent antimicrobial component of innate immunity and has been implicated in the control of virulent Rickettsia spp. in diverse cell types. In this study, we examined the antibacterial role of NO on R. rickettsii. Our results indicate that NO challenge dramatically reduces R. rickettsii adhesion through the disruption of bacterial energetics. Additionally, NO-treated R. rickettsii cells were unable to synthesize protein or replicate in permissive cells. Activated, NO-producing macrophages restricted R. rickettsii infections, but inhibition of iNOS ablated the inhibition of bacterial growth. These data indicate that NO is a potent antirickettsial effector of innate immunity that targets energy generation in these pathogenic bacteria to prevent growth and subversion of infected host cells.
KEYWORDS: Rickettsia, host defense, innate immunity, nitric oxide
INTRODUCTION
Members of the genus Rickettsia are small, Gram-negative, rod-shaped, obligate intracellular bacteria of the class Alphaproteobacteria. Rickettsia species include pathogens as well as endosymbionts that are not known to infect humans. Ticks and other hematophagous arthropods are the major reservoirs and vectors of pathogenic Rickettsia species. Their zoonotic life cycle includes transmission between arthropods and vertebrate hosts. Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever, is endemic to the American continents and is the most severe of the spotted fever group rickettsioses (1). Spotted fever rickettsioses are considered emerging infectious diseases in the United States by the CDC. In their close association with eukaryotic hosts, R. rickettsii must resist the defenses of the vertebrate innate immune system.
Many types of mammalian and tick cells can express nitric oxide synthases (NOSs), which participate in diverse physiological functions, including host defense (2–6). During inflammatory processes, the mammalian NOS2 gene can be expressed in phagocytes, fibroblasts, and endothelial cells, and the high enzymatic activity of this inducible NO synthase (iNOS) produces nitric oxide (NO) at antimicrobial levels (2, 4, 5). iNOS expression requires stimulation with microbial products such as lipopolysaccharides (LPS), mycolic acids, peptidoglycan, nucleic acids, or lipoproteins (5, 7–10). iNOS expression is typically enhanced in combination with proinflammatory cytokines (such as interferon gamma [IFN-γ], tumor necrosis factor-α, and interleukin-1β) (5, 6, 8, 11). NO and other reactive nitrogen species (RNS) have broad, direct antimicrobial activity, in both medium- and cell culture-based experimental systems, against phylogenetically diverse microbes, including viruses, fungi, parasites, and bacteria, as well as Rickettsia spp. (8, 12–14). Specifically, NO production is critical to restricting the growth of Rickettsia conorii and Rickettsia prowazekii in infected macrophages and endothelial cells, respectively (15, 16). However, the role of host-derived NO in the control of virulent R. rickettsii infections has not been determined. Furthermore, the molecular basis for the antimicrobial activity on Rickettsia spp. is poorly understood.
In this study, we determined that NO is a potent inhibitor of R. rickettsii in cell-free medium, endothelial cells, and macrophage-like cell lines. Activated macrophages require iNOS expression and NO production to reduce rickettsial burden. NO treatment dramatically reduces R. rickettsii adhesion and is mediated by depletion of bacterial ATP pools. ATP supplementation partially rescues attachment of NO-treated bacteria. In R. rickettsii-infected Vero cells, NO exposure is sufficient to inhibit bacterial protein synthesis, which subsequently represses downstream known and suspected virulence determinants, including actin polymerization (17) and dispersal of the trans-Golgi network (TGN) (18). These results indicate that NO inhibits several critical and fundamental aspects of R. rickettsii biology.
RESULTS
R. rickettsii is susceptible to NO.
J774 macrophage-like cells were stimulated with Escherichia coli LPS and/or the proinflammatory cytokine IFN-γ to examine iNOS expression. LPS and IFN-γ were separately insufficient to induce iNOS (Fig. 1A). However, the combination of the two stimulants synergistically induced iNOS expression and nitrite (NO detoxification product) accumulation in the medium (Fig. 1A). J774 cells were infected with R. rickettsii in the presence or absence of LPS and IFN-γ. R. rickettsii replicated robustly in unstimulated J774 cells or those stimulated with only LPS (Fig. 1B), conditions that did not elicit iNOS or nitrite production (Fig. 1C). IFN-γ-stimulated J774 cells produced some nitrite and restricted the growth of R. rickettsii, but macrophages stimulated with both LPS and IFN-γ dramatically reduced R. rickettsii burdens, which coincided with nitrite accumulation (Fig. 1B and C). These results indicate that R. rickettsii does not induce iNOS in unstimulated macrophage-like cells but IFN-γ stimulation causes a moderate increase in nitrite production, with a concomitant reduction in rickettsial replication.
FIG 1.
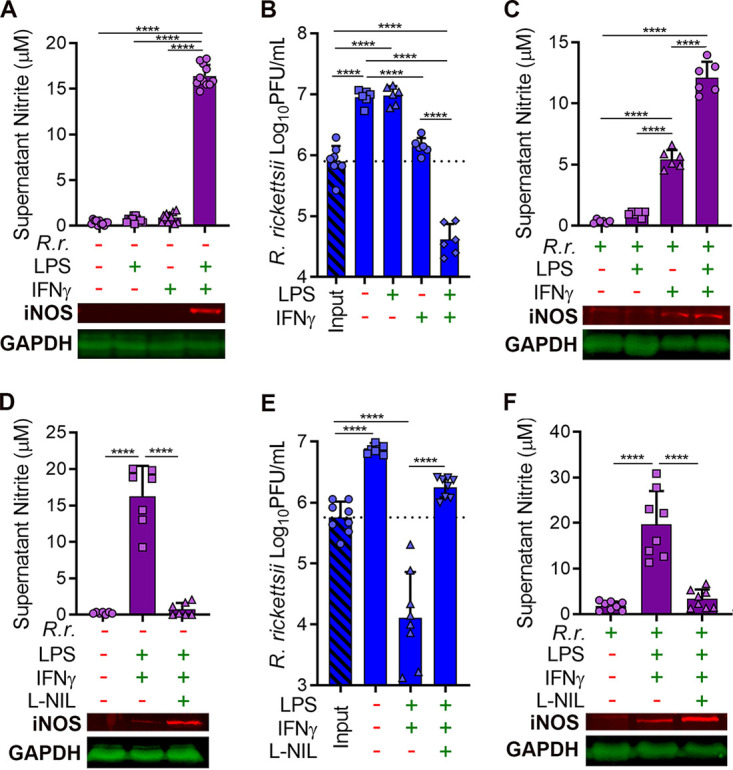
NO production is essential for clearance of R. rickettsii in activated J774 macrophages. (A) Culture supernatants were collected from J774 cells stimulated with LPS (1.5 ng/ml) or IFN-γ (15 ng/ml) for 24 h. The Griess reaction was used to determine nitrite concentrations (mean ± standard deviation [SD], n = 10). Additionally, monolayers were collected and analyzed by Western blotting for iNOS and GAPDH (representative images, n = 4). (B) Infectivity of R. rickettsii populations (PFU per milliliter) in infected J774 cells (MOI of 1 to 2) after 2 h (input) or 24 h with or without LPS or IFN-γ stimulation (mean ± SD, n = 6). (C) The Griess reaction was used to determine nitrite concentrations in culture supernatants from panel B, and Western blotting was used to examine iNOS and GAPDH expression (mean ± SD and representative images, n = 6). (D) J774 cells were stimulated as in panel A but with the addition of the iNOS inhibitor l-NIL (500 μM) and were analyzed similarly with the Griess reaction and Western blotting (mean ± SD and representative images, n = 7). (E) J774 cells were infected and stimulated as described for panel B but with l-NIL, and R. rickettsii populations were determined similarly (mean ± SD, n = 8). (F) Nitrite concentrations in culture supernatants and iNOS and GAPDH expression from samples described for panel E (mean ± SD and representative images, n = 8). Significance was determined with one-way ANOVA. ****, P < 0.0001.
Stimulation of J774 cells was repeated with the iNOS inhibitor N6-(1-iminoethyl)-l-lysine (l-NIL). l-NIL reduced nitrite accumulation in supernatants of J774 cells stimulated with both LPS and IFN-γ (Fig. 1D). J774 cells were infected with R. rickettsii and treated with LPS, IFN-γ, and l-NIL. l-NIL-mediated inhibition of NO production in LPS- and IFN-γ-stimulated J774 cells restored replication of R. rickettsii (Fig. 1E and F). These results demonstrate that NO synthesized by iNOS is an essential aspect of an antirickettsial response in J774 macrophage-like cells.
NO is directly inhibitory to R. rickettsii in vitro.
To explore whether NO could directly reduce R. rickettsii infectivity, bacteria in cell-free, brain heart infusion (BHI) medium were challenged with increasing concentrations of diethylamine NONOate (DEA-NO), which undergoes chemical decomposition reactions to release NO at a regular rate. While a host cell is essential for Rickettsia sp. replication, these bacteria can maintain their infectivity in rich, cell-free broth for short periods. DEA-NO was selected as an NO donor for these experiments because of its short half-life (2 min at 37°C). Treatment with 800 μM DEA-NO for only 10 min reduced R. rickettsii infectivity nearly 100-fold, while treatment with 800 μM levels of the control vehicle amine diethylamine (DEA) did not affect bacterial infectivity, as assessed by plaque assay (Fig. 2A). These results demonstrate that NO is directly antimicrobial to R. rickettsii.
FIG 2.

NO decreases R. rickettsii adhesion to host cells. (A) R. rickettsii was challenged with DEA (800 μM) or DEA-NO in BHI medium for 10 min at 34°C in 5% CO2, and infectivity was determined by plaque counts (mean ± SD, n = 4). (B) Immunofluorescence microscopy of external and internal R. rickettsii cells treated with DEA or DEA-NO (images are representative of three or four independent experiments). (C and D) Quantification of images in panel B. The total number of R. rickettsii bacteria per cell was determined (C) and the proportion of internalized R. rickettsii was calculated (D) (five images were quantified from three or four independent experiments). (E) Nucleotides were extracted from R. rickettsii cells that had been challenged with DEA or DEA-NO for 10 min in BHI medium. Extracts were neutralized, and ATP content was determined with firefly luciferase and luminescence measurements (mean ± SD, n = 3). (F). R. rickettsii cells were challenged with or without 800 μM DEA-NO for 10 min in BHI medium. Samples were diluted into BHI medium or BHI medium supplemented with 1 mM ATP and incubated at 34°C in 5% CO2 for 30 min, and plaque assays determined infectivity (mean ± SD, n = 4). (G) R. rickettsii cells were challenged with DEA or DEA-NO (1 mM in BHI medium) for 10 min at 34°C and were analyzed by Western blotting. Antibodies against spotted fever rickettsial LPS (LPS), rOmpA, rOmpB, and Sca1 were used to detect R. rickettsii outer membrane antigens (representative blots, n = 2). Statistical analyses were performed using one-way ANOVA. *, P < 0.05; ***, P < 0.001; ****, P <0.0001; ns, not significant.
To better understand which R. rickettsii processes are inhibited by NO, bacteria were treated with DEA-NO in BHI medium and then added to Vero cell monolayers for assessment of attachment and internalization. Samples were fixed and processed for immunofluorescence microscopy using separate anti-rickettsial antibodies in the absence and presence of permeabilization, to detect external and internalized R. rickettsii, respectively (Fig. 2B). Control R. rickettsii or bacteria treated with the DEA carrier were observed in association with Vero cells, while approximately 35-fold fewer NO-treated bacteria were found in association with Vero cells (Fig. 2C). Approximately 30% of untreated control or carrier-alone-treated R. rickettsii cells had been internalized. Fewer NO-treated R. rickettsii cells were associated with the Vero cells but, of those that were cell associated, only a slightly smaller proportion were internalized (∼18%) (Fig. 2D). Thus, NO treatment potently inhibits R. rickettsii adherence to Vero cells and may modestly inhibit bacterial invasion.
NO treatment reduces rickettsial energy charge.
The bo and bd cytochrome oxidase complexes within the electron transport chain of enteric bacteria are major targets of NO (19, 20). Despite their limited metabolic capacity and reduced genomes, rickettsiae can generate their own ATP (21–23) and have maintained the genes encoding the tricarboxylic acid (TCA) cycle and electron transport chain, including the cytochrome c oxidase and cytochrome bd complexes (21, 24, 25). ATP levels in R. rickettsii extracts were determined after in vitro treatment with DEA-NO. DEA did not dramatically affect ATP pools, while DEA-NO treatment caused greater than 10-fold reductions in R. rickettsii ATP pools (Fig. 2E). These data indicate that NO depletes the R. rickettsii energy charge.
To determine whether ATP supplementation could rescue R. rickettsii infectivity after NO challenge, R. rickettsii cells were challenged with DEA-NO and were then diluted into either BHI medium or BHI medium supplemented with 1 mM ATP; prior to plaque assays, the cultures were incubated at 34°C in 5% CO2 for 30 min. Cytosolic Rickettsia spp. can obtain ATP directly from their eukaryotic host cells with their ATP/ADP antiporters, which should enable acquisition of extracellular ATP (26, 27). ATP supplementation partially rescued the infectivity of NO-challenged R. rickettsii (Fig. 2F), which demonstrates that NO acts bacteriostatically against R. rickettsii. Thus, ATP depletion appears to be a major aspect of the antimicrobial activity of NO against R. rickettsii, and these data are consistent with previous studies that indicate that Rickettsia cells require active metabolism to adhere to host cells (28, 29).
Western blotting of major outer membrane R. rickettsii epitopes, including recombinant OmpA (rOmpA), rOmpB, Sca1, and Rickettsia LPS, from control and DEA-NO-treated R. rickettsii did not reveal any observable differences in antigenicity (Fig. 2G), which suggests that NO does not cause widespread destruction of the rickettsial outer membrane.
NO is directly inhibitory to R. rickettsii in vivo.
While NO can exert antibacterial activity against extracellular bacterial pathogens during mammalian infection (30), R. rickettsii is more likely to be exposed to NO in an intracellular environment. Vero cells were infected with R. rickettsii and challenged with increasing concentrations of spermine NONOate (SPR-NO), host cells were lysed after 3 h, and bacterial infectivity was assessed by plaque assay. NO is lipophilic and can freely diffuse across lipid bilayers. SPR-NO was chosen as the NO donor for these experiments because of its longer half-life (39 min at 37°C), which would lead to prolonged NO exposure. Similar to the observations in cell-free medium, challenge of intracellular rickettsiae with SPR-NO decreased the infectivity of R. rickettsii obtained directly from Vero cells (Fig. 3A). The assay was repeated but, after SPR-NO challenge, select cultures were given fresh medium and the bacteria were allowed 1 day to recover. The addition of this recovery step resulted in nearly complete restoration of R. rickettsii infectivity challenged with high micromolar amounts of SPR-NO (Fig. 3A), indicating that the NO-mediated inhibition of host cell adhesion is bacteriostatic and transient if these bacteria have time and the nutrient-rich environment of the host cytosol to recover.
FIG 3.

NO inhibits R. rickettsii intracellular growth. (A) Vero cells were infected for 24 h with R. rickettsii and then challenged with SPR or SPR-NO for 3 h. Select samples were analyzed for PFU or were given fresh medium for 21 h before being analyzed for PFU (mean ± SD, n = 3 or 6). (B) Vero cells were infected with R. rickettsii for 2 h, input samples were harvested, and the remaining samples were challenged with SPR or SPR-NO (150 mM) for 24 h prior to harvesting for PFU determinations (mean ± SD, n = 4). (C) Samples were treated as described in panel B but were analyzed by Western blotting with anti-rOmpA, anti-rOmpB, and anti-GAPDH antibodies. Relative levels of rOmpA and rOmpB were quantified and normalized to GAPDH loading controls (mean ± SD, n = 3; representative images are Fig. 4A). (D) Vero cells were infected with R. rickettsii for 2 h and then challenged with increasing amounts of SPR-NO. Samples were harvested at 0, 8, 16, and 24 hpt, analyzed by Western blotting, and quantified as described for panel C (mean ± SD, n = 5; representative images are Fig. 4B). Statistical analyses were performed using one- or two-way ANOVA. **, P < 0.01; ***, P < 0.001; ****, P <0.0001; ns, not significant.
To further examine whether lower concentrations of SPR-NO could impede intracellular R. rickettsii growth, Vero cells were infected with R. rickettsii for 2 h or treated with SPR-NO and incubated for 24 h. Control and spermine (SPR)-treated samples replicated over 10-fold (Fig. 3B), but those treated with 150 μM SPR-NO showed inhibition of rickettsial replication. Western blotting for rOmpA and rOmpB was performed on similarly treated samples to examine rickettsial mass (31). Both control and SPR-treated samples increased their rOmpA and rOmpB abundance 10- and 15-fold, respectively, over the input controls 24 h later (Fig. 3C and Fig. 4A). These changes are similar to the increase in R. rickettsii PFU seen under these conditions (Fig. 3B). SPR-NO at 100 and 150 μM inhibited accumulation of rOmpA and rOmpB, compared to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) controls, over a 24-h period (Fig. 3C and D and Fig. 4A and B). SPR-NO at 25 and 50 mM was sufficient to reduce rOmpA and rOmpB accumulation for 8 and 16 h, respectively, before increases in these outer membrane proteins could be observed (Fig. 3C and Fig. 4B). These results further suggest that NO acts bacteriostatically on R. rickettsii residing in Vero cells.
FIG 4.
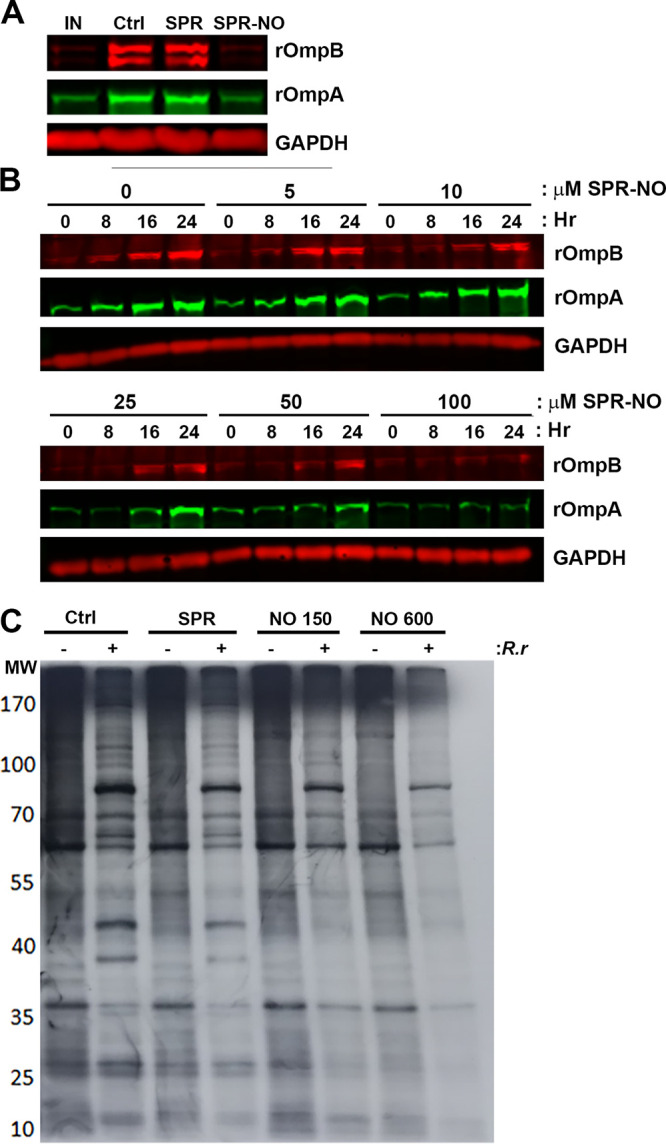
NO inhibits protein synthesis of intracellular R. rickettsii. (A) Vero cells were infected with R. rickettsii for 2 h, and select samples were collected immediately (IN) or were treated with 150 μM SPR or SPR-NO and then collected 24 h later. Samples were processed for Western blotting with anti-rOmpA, anti-rOmpB, and anti-GAPDH antibodies (representative blot, n = 3). (B) Vero cells were infected with R. rickettsii for 2 h and treated with increasing concentrations of SPR-NO. After 0, 8, 16, and 24 h of treatment, samples were collected for Western blotting. Anti-rOmpA, anti-rOmpB, and anti-GAPDH antibodies were used (representative images, n = 5). (C) Vero cells were infected with R. rickettsii for 24 h, treated with SPR or SPR-NO for 3 h, and then labeled with [35S]Met for 3 h. Samples were scraped into Laemmli sample buffer, boiled, and separated by SDS-PAGE, gels were dried and exposed to film, and autoradiograms were visualized (representative autoradiogram, n = 3).
Inhibition of rickettsial protein synthesis by NO.
To determine whether this reduction in rOmpA and rOmpB accumulation was due to a general inhibition of R. rickettsii translation within Vero cells, samples were challenged with SPR-NO and then pulsed with emetine and 35S-labeled methionine for 3 h to examine total bacterial protein synthesis. SDS-PAGE autoradiograms indicated that NO-treated R. rickettsii exhibited greatly reduced incorporation of the isotope into proteomes (Fig. 4C). Specifically, at least 13 R. rickettsii-specific bands were observed in control and SPR-treated samples, of which 12 were not observed in SPR-NO-treated samples. Translation is one of the most energy-demanding processes in the cell; thus, the depletion of R. rickettsii ATP could be the mechanism for downregulation of translation in intracellular NO-treated bacteria.
Inhibition of protein synthesis restricts R. rickettsii host cell subversion.
It is likely that NO-induced bacteriostasis and inhibition of protein synthesis could prevent R. rickettsii from subverting processes of the host cells that are normally disrupted during infection. We examined two known or suspected virulence properties of R. rickettsii. Spotted fever group rickettsia use actin-based motility to propel themselves throughout the cytoplasm and to invade neighboring cells, without exposing themselves to the extracellular environment (32, 33). Genetic loss of the ability of R. rickettsii to polymerize actin results in avirulent bacteria (17). Actin filaments in close association with R. rickettsii were observed at 2 h postinfection (hpi) and after subsequent 12 h of treatment with SPR but were less common in SPR-NO-treated R. rickettsii (Fig. 5A and B). Highly virulent strains of R. rickettsii (e.g., Sheila Smith) disperse the TGN through delivery of the type IV secretion system effector rickettsial ankyrin repeat protein 2 (RARP2) into host cells (18). Dispersal of the TGN reduces the ability of infected cells to transport proteins to their plasma membranes, a suspected mechanism of immune evasion (18). The TGN was dispersed in control and SPR-treated R. rickettsii-infected cells (Fig. 5C and D). However, R. rickettsii-infected cells that were treated with SPR-NO were observed with intact TGN structures (Fig. 5C and D). These data confirm that NO-mediated inhibition of rickettsial protein synthesis counteracts subversion of the host cell by R. rickettsii.
FIG 5.

NO disrupts host cell subversion by intracellular R. rickettsii. (A) Vero cells on coverslips were infected for 2 h with R. rickettsii and were harvested (Input) or were untreated (Untr) or treated with 150 μM SPR or SPR-NO for 12 h. Samples were visualized by immunofluorescence microscopy. rOmpB antibody 13-2, phalloidin-647, and DAPI, were used to detect R. rickettsii (green), F-actin (red), and DNA (blue), respectively (representative images, n = 3). (B) The proportions of R. rickettsii cells with evident actin tails were calculated from data in panel A (mean ± SD, n = 6). (C) Vero cells were treated as described for panel B, but samples were inactivated at 17 hpt, and rOmpB antibody 13-2, anti-TGN46, and DAPI were used to detect R. rickettsii (green), the TGN (red/orange), and DNA (blue), respectively (representative images, n = 3). (D) Cells were assessed for intact or dispersed TGN in images from C, and the proportions of cells with intact TGN were calculated (mean ± SD, n = 3). Statistical analyses were performed using one-way ANOVA. ****, P <0.0001; ns, not significant.
DISCUSSION
Eukaryotic hosts have evolved multiple mechanisms to combat invading microbial pathogens. Identifying and understanding these mechanisms are of great importance and may guide the development of novel therapeutics and treatments for infections. As is the case with most bacteria, Rickettsia spp. are sensitive to the antimicrobial activity of host-derived NO (15). Here, we demonstrate that NO inhibits rickettsial adherence, intracellular growth, and host cell subversion. Fundamentally, NO challenge depletes ATP in R. rickettsii and prevents bacterial translation. Synthesis of NO is essential for J774 macrophages to control the growth of R. rickettsii. Understanding the interplay of the host innate immune system with R. rickettsii provides unique insights into rickettsial pathogenesis.
Concentrations of NO donors required to reduce R. rickettsii infectivity when treated in cell-free broth culture (800 μM DEA-NO) are similar to concentrations that are inhibitory to other microorganisms. Specifically, 750 μM SPR-NO (34) or 500 mM levels of several other NO donors (35) induced bacteriostasis of Salmonella enterica serovar Typhimurium, 2.5 mM DEA-NO reduced extracellular Borrelia burgdorferi viability (36), and Staphylococcus aureus, in contrast to commensal Staphylococcus epidermidis or Staphylococcus saprophyticus, can replicate in the presence of NO at concentrations of up to 1 mM (37). Candida albicans is inhibited by 1 mM nitrite (14). NO is typically bacteriostatic (14, 34, 35, 37), although some species, such as Burkholderia mallei (38) and Burkholderia pseudomallei (39), are killed by concentrations of SPR-NONOate as low as 25 μM. Somewhat lower SPR-NO concentrations (300 to 600 μM) were sufficient to induce R. rickettsii bacteriostasis in Vero cells. The concentrations of the NO donor that were effective against R. rickettsii are similar to the effective concentrations of the NO donor S-nitroso-acetyl-penicillamine (SNAP) against several viral pathogens, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (200 to 400 μM) (13), herpes simplex virus 1 (250 to 500 μM) (40), hantavirus (100 μM) (41), and coxsackievirus (200 μM) (42). Although the precise NO targets for bacterial, fungal, and viral pathogens likely differ, the effective concentration ranges of the various NO donors are comparable.
NO and its related radical congeners are cytotoxic to bacteria, killing some species and leaving others in stasis (9, 39). While NO is a general bacterial inhibitor, it does so by inactivating specific prosthetic groups in specific proteins. Solvent-exposed iron-sulfur clusters are particularly sensitive to NO, such as the [4Fe-4S] cluster in dihydroxyacid dehydratase, an essential enzyme in the biosynthesis of branched-chain amino acids (34, 43–46). Thiols are also key targets of NO, particularly when they coordinate metal cofactors, such as zinc in the cases of DksA, a transcriptional regulator, and the glycolytic enzyme fructose bisphosphate aldolase (36, 47–49). Heme cofactors can be used by proteins to bind O2, and NO can occupy these O2 binding sites, as is the case with cytochrome oxidase complexes, which are among the most sensitive targets of NO in enteric bacteria (50). Reversible saturation of these targets by NO can transiently cause decreases in oxygen consumption, the proton motive force (PMF), and ATP pools, thereby depleting the energetics of the bacterial cell (19, 49, 51). Here, we documented that NO can also deplete ATP in R. rickettsii, suggesting that these evolutionarily conserved complexes may indeed be prime targets across phylogenetically diverse bacterial pathogens. Previous studies in Gram-negative and Gram-positive pathogens indicated that the glycolytic pathway is important for resisting NO-mediated stress, and they suggested that microbes lacking these enzymes are more sensitive to the inhibitory properties of NO (49, 52). Consistent with this hypothesis, R. rickettsii lacks most of the genes encoding glycolysis and is indeed sensitive to NO. The antirickettsial activity of NO is unlikely to be strictly limited to the inhibition of bacterial energetics. However, ATP synthesis is a critical target of NO that likely underlies many of the observations in this study.
Several redox-active transcription factors can directly or indirectly sense NO or NO-derived oxidants, including (but not limited to) NsrR, SoxR, and OxyR (44, 53, 54). Additionally, the alternative transcription factors RpoS, RpoE, and RpoN have been suggested to partially mediate bacterial responses to nitrosative stress (55–57). The regulons of these transcription factors include NO-detoxifying enzymes, chaperones, and repair proteins (58). Of these transcriptional regulators, R. rickettsii has only a putative nsrR homologue (accession number A1G_04620). This emphasizes the genomic reduction of this obligate intracellular parasite but also suggests that retaining at least one redox-active transcriptional regulator may be advantageous.
Diverse microbes from bacteria to yeast to eukaryotic parasites use flavohemoglobins (Hmp) to detoxify NO to nitrate (59). The putative R. rickettsii hmp gene (accession number A1G_04610) is severely truncated (predicted 11-kDa protein, compared to the 44-kDa Hmp of E. coli). However, we observed that R. rickettsii can recover from NO challenges; therefore, other mechanisms of detoxifying or resisting nitrosative stress must be in play.
Previous studies with R. prowazekii indicated that bacterial adhesion and invasion are active processes that require both bacteria and host cells to have functioning metabolisms. Specifically, rickettsial metabolic activity is required for adhesion of the bacteria to the host cell, and active host cell metabolism is required for bacterial invasion (28, 29). Our data with R. rickettsii are consistent with those previous observations, in that NO depletes bacterial ATP and decreases adhesion to host cells but the few rickettsiae that are cell associated invade at similar rates, compared to untreated controls. Although the requirement for active rickettsial metabolism for attachment has been recognized for over 45 years, the mechanism requiring rickettsial ATP to promote bacterium-host cell attachment is unknown. Rickettsiae are now known to possess a type IV secretion system, which would be energy requiring (60). Whether unknown effectors are involved in the invasion process is an intriguing possibility.
Here, we examined the antimicrobial effects of NO on diverse aspects of Rickettsia biology. A major target of NO appears to be ATP generation, whose inhibition is likely fundamental to the various impediments NO places on the biology of Rickettsia.
MATERIALS AND METHODS
Cell lines and rickettsiae.
Vero 76 cells (ATCC CCL-81) were grown at 37°C in RPMI 1640 medium with 5% fetal bovine serum (FBS). Rickettsia rickettsii Sheila Smith (GenBank accession number CP000848.1) was grown at 34°C in Vero 76 cells in M199 medium with 2% FBS. For purification of rickettsiae, Vero cells were lysed by Dounce homogenization, followed by centrifugation through a 30% Renografin pad. Rickettsiae were washed twice in 250 mM sucrose and either used directly for infections or stored in BHI medium at −80°C. J774A.1 murine macrophage-like cells (ATCC TIB-67) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS (DMEM-10% FBS), and R. rickettsii infections were performed in the same medium. Both cell lines were grown at 37°C in 5% CO2, and R. rickettsii infections or stimulations were performed at 34°C in 5% CO2.
Plaque assays.
Briefly, Vero cells were grown to monolayers in 6-well plates in RPMI 1640 medium with 5% FBS, as described previously (17, 61). The medium was removed, and 100 μl of R. rickettsii-containing BHI medium dilutions was added directly to each well. Plates were incubated for 30 min at 34°C in 5% CO2. Vero cells and R. rickettsii dilutions were overlaid with M199 medium with 5% FBS and molten 0.5% low-melt agarose and were grown for 5 days at 34°C in 5% CO2. Wells with visible plaques were stained overnight with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (0.33 mg/ml), PFU were counted the next day, and PFU per milliliter values were calculated.
Antibodies.
Antibodies against Rickettsia included a rabbit polyclonal antibody raised against formalin-fixed whole Rickettsia montanensis (αRick); a rabbit polyclonal antibody raised against gel-purified R. rickettsii rOmpB; a custom-synthesized rabbit polyclonal antibody against Sca1 (Thermo Fisher Scientific); and monoclonal antibodies to rOmpA (13-3 and 14-14), rOmpB (13-2), and rickettsial LPS (14-4) (62). Primary commercial antibodies were directed against iNOS (ab3523), GAPDH (ab161802), and GAPDH (ab83956) (from Abcam); iNOS (13120; Cell Signaling Technology), and TGN46 (PA5-23068; Invitrogen). Secondary antibodies included anti-mouse Ig-Alexa Fluor 488 (150117) and anti-rabbit Ig-Alexa Fluor 568 (ab175696) (from Abcam). Alexa Fluor 647-phalloidin stain (A22287; Invitrogen) was used to detect actin filaments. For immunoblotting, secondary antibodies were from LI-COR, including anti-rabbit Ig-IRDye 680RD (925-68071), anti-mouse Ig-IRDye 800CW (926-32210), and anti-chicken Ig-IRDye 800CW (926-32218).
NO-releasing polyamines.
DEA-NO and SPR-NO were purchased from Cayman Chemical (Ann Arbor, MI). These compounds were resuspended to 150 mM in 10 mM NaOH, stored at −80°C in single-use aliquots, and directly added to R. rickettsii-containing cultures. The control amine and polyamine compounds DEA and SPR were resuspended and used similarly to the NONOates.
Rickettsia DEA-NO challenge.
Frozen aliquots of previously purified and enumerated R. rickettsii samples were thawed and diluted to approximately 1 × 106 to 2 × 106 PFU/ml in BHI medium and were challenged with the indicated concentrations of DEA or DEA-NO for 10 min at 34°C in 5% CO2. After challenge, Rickettsia bacteria were serially diluted 10-fold into BHI broth, and 100 μl of Rickettsia dilutions was assessed by plaque assay. Where indicated, DEA-NO-challenged R. rickettsii bacteria were serially diluted into BHI medium or BHI medium supplemented with 1 mM ATP; the dilutions were incubated for 30 min at 34°C in 5% CO2 and were then used in plaque assays.
Firefly luciferase intracellular ATP assay.
R. rickettsii cells were thawed, diluted into BHI medium (∼2 × 107 PFU/ml), and challenged with DEA or DEA-NO as described above. After 10 min, nucleotides were extracted by mixing aliquots (50 μl) with ice-cold, freshly prepared 760 mM formic acid-17 mM EDTA (60 μl). Extracts were incubated for 30 min on ice in the dark, centrifuged at 15,300 × g for 5 min, neutralized with 5 M KOH (4.5 ml), and then diluted 10-fold into 100 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) buffer (pH 7.6). Ten-microliter samples were mixed with 90 μl of firefly luciferase master mix (Molecular Probes) in 96-well white-well plates, and luminescence was recorded over a 0.1-s interval with a SpectraMax iD3 plate reader (Molecular Devices, San Jose, CA). Solutions of known ATP standards and linear regression were used to calculate ATP concentrations in the original samples. Samples were quantified as moles of ATP per R. rickettsii PFU.
Rickettsia SPR-NO challenge in Vero cells. (i) Infectivity assay.
Confluent Vero cell monolayers in 6-well plates were infected with R. rickettsii at a multiplicity of infection (MOI) of ∼1 in M199-2% FBS. Bacteria were attached by centrifugation (1,136 × g) for 5 min at room temperature in a Thermo Fisher Scientific Sorvall Legend X1R centrifuge with an M-20 microplate rotor. Plates were incubated at 34°C in 5% CO2 for 30 min to allow invasion, after which the infection medium was replaced with fresh medium, and R. rickettsii was allowed to grow overnight at 34°C in 5% CO2. At 24 hpi, select samples were treated with SPR or SPR-NO, and the plate was returned to 34°C in 5% CO2 for 3 h. Afterwards, the medium was replaced with 1 ml BHI medium, into which the cells were scraped. Alternatively, where indicated, the medium was replaced with fresh M199-2% FBS, and the cells were incubated at 34°C in 5% CO2 for an additional 21 h and then scraped into 1 ml BHI medium. The Vero cell-BHI medium solutions were lysed by bead beating (1-mm beads, with one 10-s burst), and lysates were stored at −80°C. Thawed lysates were serially diluted 10-fold into BHI medium and plated for PFU determinations as described above.
(ii) Growth assays.
Vero cells were infected with R. rickettsii (MOI of 1) as described above except that the bacteria were allowed to invade for 2 h at 34°C in 5% CO2, after which the medium was replaced with M199-2% FBS with or without the indicated concentrations of SPR or SPR-NO. Samples were incubated at 34°C in 5% CO2 for 0 to 24 h posttreatment (hpt), as indicated for each experiment.
Rickettsial growth in J774 cells.
J774 cells were plated in 12-well plates (∼4 × 105 cells/well) and the following day were infected with R. rickettsii (MOI of 1) in DMEM-10% FBS. R. rickettsii cells were attached by centrifugation as described above, followed by incubation at 34°C in 5% CO2 for 30 min for invasion. The medium was replaced with fresh DMEM-10% FBS supplemented with either LPS (1.5 ng/ml Escherichia coli O111:B4 antigen; Sigma-Aldrich), murine IFN-γ (15 ng/ml; Abcam), or l-NIL (500 μM; Cayman Chemical). At 2 and 24 hpi, the medium was removed and saved for the Griess reaction, macrophages were scraped into 1 ml of BHI medium and subjected to bead beating, and lysates were stored at −80°C until plaque assays.
Nitrite determination assays.
Nitrite, a detoxification product of NO in cell culture, was measured with the Griess reaction. Briefly, in 96-well plates, 100 μl of culture supernatants or NaNO2 standards was mixed with 100 μl of freshly prepared Griess reagent, i.e., a 1:1 (vol/vol) mixture of 1% (wt/vol) sulfanilamide in 5% (vol/vol) phosphoric acid and 0.1% (wt/vol) N-(1-naphthyl)ethylenediamine. The A550 was recorded with a Molecular Devices SpectraMax iD3 plate reader, and nitrite concentrations were determined through linear regression.
Attachment and internalization (in/out) assay.
R. rickettsii samples (1 ml BHI medium with 106 PFU/ml) were challenged with either nothing (control), DEA, or DEA-NO (800 μM) for 10 min at 34°C in 5% CO2. Cultures were added directly to Vero cells on size 1.5 coverslips in a 24-well plate. Samples were centrifuged for 5 min at 1,136 × g and were incubated at 34°C in 5% CO2 for 2 h. Samples were washed with Hanks’ balanced salt solution (HBSS), inactivated with 4% paraformaldehyde for 20 min, and washed with phosphate-buffered saline (PBS). Prior to permeabilization, samples were probed with an antibody (αRick) to label extracellular R. rickettsii. Samples were washed with PBS, permeabilized with 0.1% Triton X-100 for 5 min, and probed with an antibody against rOmpB (62), which labeled both external and internal R. rickettsii cells. Samples were stained with 4′,6-diamidino-2-phenylindole (DAPI), washed, and mounted onto glass slides with ProLong Diamond antifade mountant (Invitrogen, Carlsbad, CA). Slides were visualized with a Nikon Ti2 motorized inverted microscope with an Apochromat Lambda 60× oil immersion objective and a Nikon DS-Qi2 16.25-megapixel scientific complementary metal oxide semiconductor (sCMOS) camera and were used to generate images with NIS Elements AR v5.11.02 software, which were further analyzed with Fiji (63). Five images per coverslip were captured from three or four independent experiments. Images were quantified by counting the number of Vero cells per image and the number of internal (single-positive) and external (double-positive) R. rickettsii cells. Adhesion was calculated as the total number of R. rickettsii cells per Vero cell per image, and invasion was calculated by normalizing the percentage of internal R. rickettsii to the total bacterial population per image.
SDS-PAGE and Western blotting.
After infection or stimulation of mammalian cells, medium supernatants were removed and samples were scraped into Laemmli sample buffer (64) with dithiothreitol (DTT) or β-mercaptoethanol. Samples were sealed and inactivated for 10 min at 100°C. For NO-treated R. rickettsii samples in BHI medium, 1-ml cultures of 107 PFU were centrifuged at 15,300 × g for 5 min, resuspended in sample buffer with DTT, and inactivated for 10 min at 100°C. Samples were electrophoresed at 80 V for 2 h in SDS-polyacrylamide (10 to 15%) mini gels. Proteins were transferred onto methanol-rinsed Immobilon-FL polyvinylidene difluoride (PVDF) transfer membranes (0.45-μm pore size) in a Bio-Rad semidry transfer apparatus with 25 mM Tris base, 190 mM glycine, 20% methanol, for 30 min at 25 V. Membranes were dried, rinsed with methanol and Tris-buffered saline (TBS), and blocked with LI-COR Odyssey or Intercept (TBS-based) blocking buffer for 1 h at room temperature or overnight at 4°C. Membranes were probed with primary commercial or custom antibodies diluted in blocking buffer supplemented with 0.2% Tween 20. Membranes were washed with TBS-Tween (TBST), and secondary LI-COR antibodies diluted in blocking buffer were added to the membranes. Membranes were washed with TBST and TBS, and fluorescence was imaged with a LI-COR Odyssey CLx imager with Image Studio v5.2 software. For quantification, boxes with fixed areas were drawn around bands of interest, and relative fluorescence units for rOmpA or rOmpB bands were normalized to GAPDH loading controls.
[35S]Met labeling of intracellular R. rickettsii.
Vero cells were infected with R. rickettsii (MOI of 1). At 24 hpi, select samples were treated with either 600 μM SPR or 150 or 600 μM SPR-NO for 3 h at 34°C in 5% CO2. Samples were then labeled for 3 h in RPMI 1640 medium lacking methionine, supplemented with 100 μCi/ml [35S]methionine and 2.5 μg/ml emetine. Select samples were treated with 600 μM SPR or 150 or 600 μM SPR-NO. After pulse labeling, samples were washed with HBSS, scraped into sample buffer, and heat inactivated at 100°C for 10 min. Samples (10 μl) were separated by SDS-PAGE on 12% polyacrylamide gels at 12 W/gel for approximately 2.5 h. Gels were washed with water and 1% (vol/vol) glycerol and dried onto filter paper at 70°C for 2 h in a model 583 Bio-Rad gel dryer. Dried gels were exposed to CL-XPosure film (Thermo Fisher Scientific) with a Kodak BioMax TranScreen LE intensifying screen. Autoradiograms were developed with a Kodak M35 X-Omat processor and were visualized with a Bio-Techne ProteinSimple AlphaImager HP imager.
Immunofluorescence assays.
Vero cells were plated onto 12-mm circular coverslips (size 1.5, Karl Hecht Assistant) and infected with R. rickettsii. Samples were given fresh medium, and select samples were treated with 150 μM SPR or SPR-NO for various times as described. After incubations, samples were inactivated with 4% paraformaldehyde in PBS at room temperature for 20 min and washed with PBS. Samples were permeabilized with 0.1% Triton X-100 for 5 min. Samples were blocked with 1% bovine serum albumin (BSA) in PBS-0.1% Tween 20 for 1 h at room temperature. Samples were probed with primary antibodies for 1 to 2 h, followed by appropriate secondary antibodies. Samples were counterstained with DAPI, washed with PBS, and mounted. Samples with imaged with a Nikon Ti2 microscope as described above. R. rickettsii actin tails (phalloidin stained) were quantified by the percentage of bacteria that had evident actin filaments emanating from the bacteria per field of view (6,000 total R. rickettsii bacteria were assessed). Fragmentation of the TGN was assessed by whether TGN46 fluorescence was concentrated in discrete structures in close proximity to the nucleus or fluorescence was diffused throughout the cytosol, as described previously (over 600 total cells were counted) (18).
Statistical analyses.
Numerical data were graphed with GraphPad Prism v8.4.3.686. Statistical significance was assessed with ordinary one- or two-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
We thank Adam Nock, Nicholas Chamberlain, and Zoe Dimond for their helpful comments, suggestions, and discussions.
Contributor Information
Ted Hackstadt, Email: thackstadt@niaid.nih.gov.
Craig R. Roy, Yale University School of Medicine
REFERENCES
- 1.Paddock CD, Denison AM, Lash RR, Liu L, Bollweg BC, Dahlgren FS, Kanamura CT, Angerami RN, Pereira dos Santos FC, Brasil Martines R, Karpathy SE. 2014. Phylogeograpy of Rickettsia rickettsii genotypes associated with fatal Rocky Mountain spotted fever. Am J Trop Med Hyg 91:589–597. 10.4269/ajtmh.14-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdan C. 2015. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 36:161–178. 10.1016/j.it.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya ST, Bayakly N, Lloyd R, Benson MT, Davenport J, Fitzgerald ME, Rothschild M, Lamoreaux WJ, Coons LB. 2000. Nitric oxide synthase and cGMP activity in the salivary glands of the American dog tick Dermacentor variabilis. Exp Parasitol 94:111–120. 10.1006/expr.1999.4477. [DOI] [PubMed] [Google Scholar]
- 4.Porrini C, Ramarao N, Tran SL. 2020. Dr. NO and Mr. Toxic: the versatile role of nitric oxide. Biol Chem 401:547–572. 10.1515/hsz-2019-0368. [DOI] [PubMed] [Google Scholar]
- 5.Cinelli MA, Do HT, Miley GP, Silverman RB. 2020. Inducible nitric oxide synthase: regulation, structure, and inhibition. Med Res Rev 40:158–189. 10.1002/med.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogdan C. 2001. Nitric oxide and the immune response. Nat Immunol 2:907–916. 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 7.Bhatt KH, Sodhi A, Chakraborty R. 2011. Role of mitogen-activated protein kinases in peptidoglycan-induced expression of inducible nitric oxide synthase and nitric oxide in mouse peritoneal macrophages: extracellular signal-related kinase, a negative regulator. Clin Vaccine Immunol 18:994–1001. 10.1128/CVI.00541-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang FC, Vázquez-Torres A. 2019. Reactive nitrogen species in host-bacterial interactions. Curr Opin Immunol 60:96–102. 10.1016/j.coi.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henard CA, Vázquez-Torres A. 2011. Nitric oxide and Salmonella pathogenesis. Front Microbiol 2:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schick J, Etschel P, Bailo R, Ott L, Bhatt A, Lepenies B, Kirschning C, Burkovski A, Lang R. 2017. Toll-like receptor 2 and Mincle cooperatively sense corynebacterial cell wall glycolipids. Infect Immun 85:e00075-17. 10.1128/IAI.00075-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawakami T, Kawamura K, Fujimori K, Koike A, Amano F. 2016. Influence of the culture medium on the production of nitric oxide and expression of inducible nitric oxide synthase by activated macrophages in vitro. Biochem Biophys Rep 5:328–334. 10.1016/j.bbrep.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogdan C. 1997. Of microbes, macrophages and nitric oxide. Behring Inst Mitt 58–72. [PubMed] [Google Scholar]
- 13.Akaberi D, Krambrich J, Ling J, Luni C, Hedenstierna G, Järhult JD, Lennerstrand J, Lundkvist Å. 2020. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol 37:101734. 10.1016/j.redox.2020.101734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vázquez-Torres A, Balish E. 1997. Macrophages in resistance to candidiasis. Microbiol Mol Biol Rev 61:170–192. 10.1128/mmbr.61.2.170-192.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng HM, Walker DH. 2000. Mechanisms of intracellular killing of Rickettsia conorii in infected human endothelial cells, hepatocytes, and macrophages. Infect Immun 68:6729–6736. 10.1128/IAI.68.12.6729-6736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turco J, Liu H, Gottlieb SF, Winkler HH. 1998. Nitric oxide-mediated inhibition of the ability of Rickettsia prowazekii to infect mouse fibroblasts and mouse macrophagelike cells. Infect Immun 66:558–566. 10.1128/IAI.66.2.558-566.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleba B, Clark TR, Lutter EI, Ellison DW, Hackstadt T. 2010. Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect Immun 78:2240–2247. 10.1128/IAI.00100-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aistleitner K, Clark T, Dooley C, Hackstadt T. 2020. Selective fragmentation of the trans-Golgi apparatus by Rickettsia rickettsii. PLoS Pathog 16:e1008582. 10.1371/journal.ppat.1008582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husain M, Bourret TJ, McCollister BD, Jones-Carson J, Laughlin J, Vázquez-Torres A. 2008. Nitric oxide evokes an adaptive response to oxidative stress by arresting respiration. J Biol Chem 283:7682–7689. 10.1074/jbc.M708845200. [DOI] [PubMed] [Google Scholar]
- 20.Forte E, Borisov VB, Konstantinov AA, Brunori M, Giuffrè A, Sarti P. 2007. Cytochrome bd, a key oxidase in bacterial survival and tolerance to nitrosative stress. Ital J Biochem 56:265–269. [PubMed] [Google Scholar]
- 21.Driscoll TP, Verhoeve VI, Guillotte ML, Lehman SS, Rennoll SA, Beier-Sexton M, Rahman MS, Azad AF, Gillespie JJ. 2017. Wholly Rickettsia! Reconstructed metabolic profile of the quintessential bacterial parasite of eukaryotic cells. mBio 8:e00859-17. 10.1128/mBio.00859-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams JC, Weiss E. 1978. Energy metabolism of Rickettsia typhi: pools of adenine nucleotides and energy charge in the presence and absence of glutamate. J Bacteriol 134:884–892. 10.1128/jb.134.3.884-892.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bovarnick MR, Allen EG. 1957. Reversible inactivation of typhus rickettsiae at 0 C. J Bacteriol 73:56–62. 10.1128/jb.73.1.56-62.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, Gabbard JL, Gerdes S, Guard A, Kenyon RW, Machi D, Mao C, Murphy-Olson D, Nguyen M, Nordberg EK, Olsen GJ, Olson RD, Overbeek JC, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomas C, VanOeffelen M, Vonstein V, Warren AS, Xia F, Xie D, Yoo H, Stevens R. 2020. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48:D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomason JA, III, Stevens R, Vonstein V, Wattam AR, Xia F. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williamson LR, Piano GV, Winkler HH, Krause DC, Wood DO. 1989. Nucleotide sequence of the Rickettsia prowazekii ATP/ADP translocase-encoding gene. Gene 80:269–278. 10.1016/0378-1119(89)90291-6. [DOI] [PubMed] [Google Scholar]
- 27.Winkler HH. 1976. Rickettsial permeability: an ADP-ATP transport system. J Biol Chem 251:389–396. 10.1016/S0021-9258(17)33891-7. [DOI] [PubMed] [Google Scholar]
- 28.Ramm LE, Winkler HH. 1973. Rickettsial hemolysis: effect of metabolic inhibitors upon hemolysis and adsorption. Infect Immun 7:550–555. 10.1128/iai.7.4.550-555.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker TS, Winkler HH. 1978. Penetration of cultured mouse fibroblasts (L cells) by Rickettsia prowazeki. Infect Immun 22:200–208. 10.1128/iai.22.1.200-208.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis KM, Mohammadi S, Isberg RR. 2015. Community behavior and spatial regulation within a bacterial microcolony in deep tissue sites serves to protect against host attack. Cell Host Microbe 17:21–31. 10.1016/j.chom.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Policastro PF, Hackstadt T. 1994. Differential activity of Rickettsia rickettsii opmA and ompB promoter regions in a heterologous reporter gene system. Microbiology (Reading) 140:2941–2949. 10.1099/13500872-140-11-2941. [DOI] [PubMed] [Google Scholar]
- 32.Heinzen RA, Hayes SF, Peacock MG, Hackstadt T. 1993. Directional actin polymerization associated with spotted fever group rickettsia infection of VERO cells. Infect Immun 61:1926–1935. 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teysseire N, Chiche-Portiche C, Raoult D. 1992. Intracellular movements of Rickettsia conorii and R. typhi based on actin polymerization. Res Microbiol 143:821–829. 10.1016/0923-2508(92)90069-Z. [DOI] [PubMed] [Google Scholar]
- 34.Fitzsimmons LF, Liu L, Kim JS, Jones-Carson J, Vázquez-Torres A. 2018. Salmonella reprograms nucleotide metabolism in its adaptation to nitrosative stress. mBio 9:e00211-18. 10.1128/mBio.00211-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Groote MA, Granger D, Xu Y, Campbell G, Prince R, Fang FC. 1995. Genetic and redox determinants of nitric oxide cytotoxicity in a Salmonella typhimurium model. Proc Natl Acad Sci USA 92:6399–6403. 10.1073/pnas.92.14.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bourret TJ, Boylan JA, Lawrence KA, Gherardini FC. 2011. Nitrosative damage to free and zinc-bound cysteine thiols underlies nitric oxide toxicity in wild-type Borrelia burgdorferi. Mol Microbiol 81:259–273. 10.1111/j.1365-2958.2011.07691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson AR, Dunman PM, Fang FC. 2006. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol 61:927–939. 10.1111/j.1365-2958.2006.05290.x. [DOI] [PubMed] [Google Scholar]
- 38.Jones-Carson J, Laughlin J, Hamad MA, Stewart AL, Voskuil MI, Vazquez-Torres A. 2008. Inactivation of [Fe-S] metalloproteins mediates nitric oxide-dependent killing of Burkholderia mallei. PLoS One 3:e1976. 10.1371/journal.pone.0001976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones-Carson J, Laughlin JR, Stewart AL, Voskuil MI, Vázquez-Torres A. 2012. Nitric oxide-dependent killing of aerobic, anaerobic and persistent Burkholderia pseudomallei. Nitric Oxide 27:25–31. 10.1016/j.niox.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croen KD. 1993. Evidence for antiviral effect of nitric oxide: inhibition of herpes simplex virus type 1 replication. J Clin Invest 91:2446–2452. 10.1172/JCI116479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klingström J, Akerström S, Hardestam J, Stoltz M, Simon M, Falk KI, Mirazimi A, Rottenberg M, Lundkvist A. 2006. Nitric oxide and peroxynitrite have different antiviral effects against hantavirus replication and free mature virions. Eur J Immunol 36:2649–2657. 10.1002/eji.200535587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM, Lowenstein CJ. 1999. An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 10:21–28. 10.1016/S1074-7613(00)80003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennedy MC, Antholine WE, Beinert H. 1997. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem 272:20340–20347. 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- 44.Ding H, Demple B. 2000. Direct nitric oxide signal transduction via nitrosylation of iron-sulfur centers in the SoxR transcription activator. Proc Natl Acad Sci USA 97:5146–5150. 10.1073/pnas.97.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yukl ET, Elbaz MA, Nakano MM, Moënne-Loccoz P. 2008. Transcription factor NsrR from Bacillus subtilis senses nitric oxide with a 4Fe-4S cluster. Biochemistry 47:13084–13092. 10.1021/bi801342x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duan X, Yang J, Ren B, Tan G, Ding H. 2009. Reactivity of nitric oxide with the [4Fe-4S] cluster of dihydroxyacid dehydratase from Escherichia coli. Biochem J 417:783–789. 10.1042/BJ20081423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henard CA, Tapscott T, Crawford MA, Husain M, Doulias PT, Porwollik S, Liu L, McClelland M, Ischiropoulos H, Vázquez-Torres A. 2014. The 4-cysteine zinc-finger motif of the RNA polymerase regulator DksA serves as a thiol switch for sensing oxidative and nitrosative stress. Mol Microbiol 91:790–804. 10.1111/mmi.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crawford MA, Tapscott T, Fitzsimmons LF, Liu L, Reyes AM, Libby SJ, Trujillo M, Fang FC, Radi R, Vázquez-Torres A. 2016. Redox-active sensing by bacterial DksA transcription factors is determined by cysteine and zinc content. mBio 7:e02161-15. 10.1128/mBio.02161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fitzsimmons L, Liu L, Porwollik S, Chakraborty S, Desai P, Tapscott T, Henard C, McClelland M, Vazquez-Torres A. 2018. Zinc-dependent substrate-level phosphorylation powers Salmonella growth under nitrosative stress of the innate host response. PLoS Pathog 14:e1007388. 10.1371/journal.ppat.1007388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mason MG, Shepherd M, Nicholls P, Dobbin PS, Dodsworth KS, Poole RK, Cooper CE. 2009. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat Chem Biol 5:94–96. 10.1038/nchembio.135. [DOI] [PubMed] [Google Scholar]
- 51.Jones-Carson J, Zweifel AE, Tapscott T, Austin C, Brown JM, Jones KL, Voskuil MI, Vázquez-Torres A. 2014. Nitric oxide from IFNγ-primed macrophages modulates the antimicrobial activity of β-lactams against the intracellular pathogens Burkholderia pseudomallei and nontyphoidal Salmonella. PLoS Negl Trop Dis 8:e3079. 10.1371/journal.pntd.0003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vitko NP, Spahich NA, Richardson AR. 2015. Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. mBio 6:e00045-15. 10.1128/mBio.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stern AM, Zhu J. 2014. An introduction to nitric oxide sensing and response in bacteria. Adv Appl Microbiol 87:187–220. 10.1016/B978-0-12-800261-2.00005-0. [DOI] [PubMed] [Google Scholar]
- 54.Seth D, Hausladen A, Stamler JS. 2020. Anaerobic transcription by OxyR: a novel paradigm for nitrosative stress. Antioxid Redox Signal 32:803–816. 10.1089/ars.2019.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alam MS, Zaki MH, Yoshitake J, Akuta T, Ezaki T, Akaike T. 2006. Involvement of Salmonella enterica serovar Typhi RpoS in resistance to NO-mediated host defense against serovar Typhi infection. Microb Pathog 40:116–125. 10.1016/j.micpath.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 56.Thompson KM, Rhodius VA, Gottesman S. 2007. SigmaE regulates and is regulated by a small RNA in Escherichia coli. J Bacteriol 189:4243–4256. 10.1128/JB.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tucker NP, Ghosh T, Bush M, Zhang X, Dixon R. 2010. Essential roles of three enhancer sites in sigma54-dependent transcription by the nitric oxide sensing regulatory protein NorR. Nucleic Acids Res 38:1182–1194. 10.1093/nar/gkp1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mehta HH, Liu Y, Zhang MQ, Spiro S. 2015. Genome-wide analysis of the response to nitric oxide in uropathogenic Escherichia coli CFT073. Microb Genom 1:e000031. 10.1099/mgen.0.000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poole RK. 2020. Flavohaemoglobin: the pre-eminent nitric oxide-detoxifying machine of microorganisms. F1000Res 9:F1000 Faculty Rev-7. 10.12688/f1000research.20563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillespie JJ, Ammerman NC, Dreher-Lesnick SM, Rahman MS, Worley MJ, Setubal JC, Sobral BS, Azad AF. 2009. An anomalous type IV secretion system in Rickettsia is evolutionarily conserved. PLoS One 4:e4833. 10.1371/journal.pone.0004833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cory J, Yunker CE, Ormsbee RA, Peacock M, Meibos H, Tallent G. 1974. Plaque assay of rickettsiae in a mammalian cell line. Appl Microbiol 27:1157–1161. 10.1128/am.27.6.1157-1161.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anacker RL, Mann RE, Gonzales C. 1987. Reactivity of monoclonal antibodies to Rickettsia rickettsii with spotted fever and typhus group rickettsiae. J Clin Microbiol 25:167–171. 10.1128/jcm.25.1.167-171.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]