

Summary
We developed a metatranscriptomics method that can simultaneously capture the respiratory virome, microbiome, and host response directly from low biomass samples. Using nasal swab samples, we capture RNA virome with sufficient sequencing depth required to assemble complete genomes. We find a surprisingly high frequency of respiratory syncytial virus (RSV) and coronavirus (CoV) in healthy children, and a high frequency of RSV-A and RSV-B co-detections in children with symptomatic RSV. In addition, we have identified commensal and pathogenic bacteria and fungi at the species level. Functional analysis revealed that H. influenzae was highly active in symptomatic RSV subjects. The host nasal transcriptome reveled upregulation of the innate immune system, anti-viral response and inflammasome pathway, and downregulation of fatty acid pathways in children with symptomatic RSV. Overall, we demonstrate that our method is broadly applicable to infer the transcriptome landscape of an infected system, surveil respiratory infections, and to sequence RNA viruses directly from clinical samples.
Keywords: metatranscriptomics, virome, microbiome, next-generation sequencing, RNA-seq, acute respiratory infection, ARI, respiratory syncytial virus, RSV, coronavirus
Graphical abstract
Highlights
-
•
Develop metatranscriptomics to characterize virome, microbiome, and host response
-
•
High prevalence of RSV and coronavirus is observed in healthy children
-
•
RSV-A and RSV-B are co-detected in 56% of children with symptomatic RSV
-
•
H. influenzae is highly active in children with symptomatic RSV
Motivation
Metatranscriptomics is a powerful method to study the entire transcriptome landscape; however, the feasibility of using this approach to describe the entire respiratory RNA virome, microbiome, and host response from low biomass clinical samples remains elusive. Here, we present an optimized metatranscriptomics method and an accompanying computational workflow to simultaneously characterize the respiratory virome, microbiome, and host response directly from nasal samples. Although this method is optimized for nasal swab samples, further optimization of sample preservation and RNA extraction might be needed to obtain high-quality RNA for other clinical samples.
Rajagopala et al. develop a metatranscriptomic approach for simultaneously and directly characterizing the respiratory virome, microbiome, and host response from low biomass clinical samples such as nasal swabs.
Introduction
Respiratory RNA viruses (e.g., human rhinovirus [HRV], respiratory syncytial virus [RSV], influenza A virus [IAV], and coronaviruses [CoVs]) are a leading cause of morbidity, mortality, school and work absenteeism, and increased health care expenses worldwide (Iwane et al., 2004; Madhi and Klugman, 2006; Nichols et al., 2008; Tang et al., 2017). Both HRV and RSV infect ∼100% of children by the age of 3 years and are the most common etiologies of upper and lower acute respiratory infections (ARIs) in the pediatric population, respectively (Rossi and Colin, 2015). In addition to causing seasonal epidemics or localized outbreaks that affect both children and adults, IAV and CoVs can lead to pandemics with tremendous health, social, political, and economic consequences, such as the ongoing SARS-CoV-2 pandemic (Petersen et al., 2020).
Improving our knowledge of how respiratory RNA viruses interact with each other, with other upper airway microbes (such as bacteria or fungi), and with the host's immune response is important as these interactions are likely to impact the onset, severity, and progression of their diseases and can shed light on new therapeutic approaches (Chiu and Miller, 2019; Pallen, 2014; Rascovan et al., 2016; Rosas-Salazar et al., 2016a, 2016b; Shilts et al., 2020). To date, our understanding of these interactions has been limited, at least in part, due to a lack of methods that can accurately, comprehensively, and simultaneously characterize the whole respiratory virome, as well as the microbial and host's gene expression patterns, directly from clinical samples. Recent next-generation sequencing (NGS)-based methods allow for unbiased detection of multiple pathogens, including novel pathogens, and can also separately characterize the whole microbiome (i.e., the viral, bacterial, and fungal communities), as well as microbial and the host's gene expression patterns (Chiu and Miller, 2019; Chu et al., 2016, 2020; Mariani et al., 2017; Pallen, 2014; Rascovan et al., 2016; Shifman et al., 2019). However, these current NGS-based methods involve independent sample preparation, sequencing, and analytical approaches, which is labor intensive, can be cost prohibitive, and requires the availability of large sample volumes (Chiu and Miller, 2019). Furthermore, most of these NGS-based methods use targeted multiplexed amplicon sequencing of conserved regions of respiratory viral pathogens or metagenomic sequencing of DNA, which do not capture RNA viruses (Rascovan et al., 2016).
Metatranscriptomics is a powerful method that is commonly used to study the composition and functions of the microbiome (Abu-Ali et al., 2018), identify parasites in low-intensity infections (Galen et al., 2020), and characterize active antibiotic resistance genes and host-microbiome interactions (Zhang et al., 2020). However, the feasibility of implementing metatranscriptomics to describe the entire respiratory RNA virome, microbiome, and host response from low biomass samples remains elusive. In this study, we optimized the metatranscriptomics method and computational workflow to simultaneously characterize the respiratory virome, microbiome, and host response directly from low biomass clinical samples. By implementing this method on nasal swab clinical samples collected from young children, we were able to characterize the respiratory viruses' genomes, virus and bacterial co-infections, as well as bacterial and fungal communities present in healthy and RSV-ARI samples. Furthermore, our method was able to concurrently assess microbial and nasal mucosal host gene expression patterns, which revealed upregulation of the host innate immune system, interferon signaling, and anti-viral response during RSV-ARI.
Results
Pediatric cohort, workflow, and protocol optimization
We processed the nasal swab samples collected from healthy controls (HC) (n = 22), and RSV-positive children (RSV-ARI) (n = 43), based on clinical diagnosis, collected between November 2018 and February 2019 from children aged <3 years (see Table 1 and online methods for demography and clinical metadata). A total of 19 (86%) HC and 39 (90%) RSV-ARI samples passed sequencing library quality control. A schematic of the method is graphically represented in Figure 1A, and also detailed in the STAR Methods. There was an average of 46.7 million paired-end reads and 1.3 million single-end reads per sample after quality trimming. After removal of human rRNA, mitochondrial RNA, and bacterial rRNA, reads were partitioned into two bins: a human transcript bin with an average of 36.9 million reads and a microbial bin with an average of 3.8 million reads, which contained viral, bacterial, and fungal, as well as unclassified reads (see STAR Methods). Rarefaction analysis performed on five samples by using microbial reads showed that the number of unique bacterial species identified plateaus at around 4.5 million reads (Figure S1B).
Table 1.
Demographic and clinical characteristics of participants
| Healthy controls (N = 22) | Acute respiratory infection (N = 43) |
|||
|---|---|---|---|---|
| RSV-mild (N = 17) | RSV-severe (N = 26) | |||
| Demographics | ||||
| Age (months)a | 11.7 (4.6–21.2) | 8.8 (2.2–13) | 6.6 (2.75–16.4) | |
| Genderb | ||||
| Male | 14 (64) | 10 (59) | 16 (62) | |
| Female | 8 (36) | 7 (41) | 10 (38) | |
| Ethnicityb | ||||
| Caucasian | 6 (27) | 11 (65) | 21 (81) | |
| African American | 14 (64) | 6 (35) | 3 (11) | |
| Other | 2 (9) | 2 (8) | ||
| Clinical symptoms | ||||
| Length of hospitalization (days)a | N/A | 0 (0–1) | 4 (3–5.7) | |
N/A, not applicable.
Median (interquartile range).
Number (percentage).
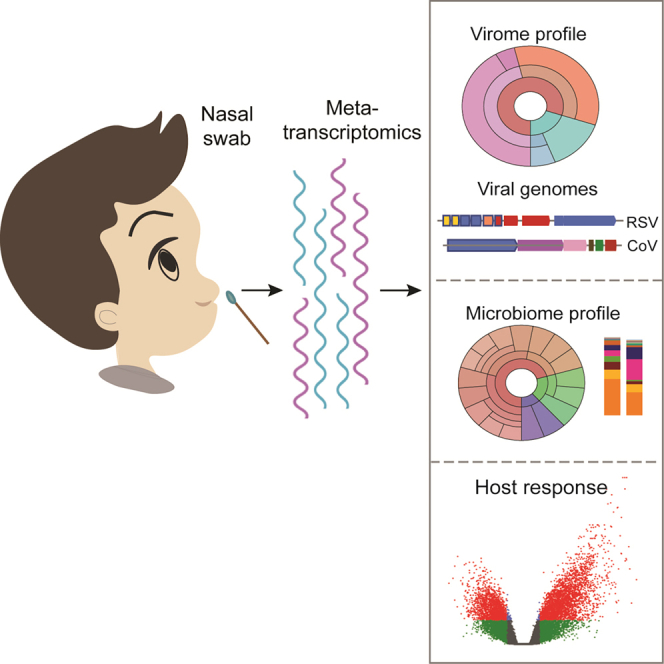
Figure 1.

Metatranscriptomics workflow and respiratory virome profile
(A) Schematic overview of metatranscriptomics sample preparation and data analysis. Total RNA was extracted from nasal samples, and human rRNA was depleted before library preparation and sequencing. The sequencing reads were used to profile virome, bacteriome, and host transcriptional response.
(B) Heatmap showing the virome profile. Each row represents a sample and each column represents the percentage of a virus genome recovered. The samples are grouped into healthy and RSV-ARI-positive samples; RSV samples are further split into RSV-severe and RSV-mild groups based on days of hospitalization. Complete genomes of common RNA respiratory viruses, such as RSV, coronavirus, rhinovirus, and influenza were recovered. In addition, a DNA virus, Bocavirus, was recovered in one sample. Plant viruses and phages were also recovered in both the HC and RSV samples.
Metatranscriptome captured complete genomes of respiratory RNA viruses
The depth of sequence coverage obtained from this method was sufficient for assembling complete genomes of the dominant and co-detecting RNA viruses. We assembled a total of 60 RSV genomes that were either complete or partial (minimum 90% of genome). These include 22 complete and 15 partial RSV-A genomes, and 12 complete and 2 partial RSV-B genomes from RSV-ARI samples; 4 complete and 4 partial RSV-A genomes, and one partial RSV-B genome from HC samples. In addition to complete coding regions, annotation of all the full-length RSV genomes we recovered 5′ and 3′ UTRs of the viruses (Table S1). Overall genome coverage depth for RSV-ARI samples were generally >1,000×, which is usually sufficient to capture the intra-host variability profile of the virus. We were also able to assemble coding complete genomes of several other RNA viruses, i.e., 4 CoVs, 1 influenza H3N2 virus, 11 HRVs (including HRV C and A), 1 EV, 1 ssDNA virus, and 1 BV (Table 2).
Table 2.
Complete genomes of RSV and other respiratory viruses assembled and submitted to GenBank
| Virus | Genomes from ARI samples | Genomes from healthy control samples | Total |
|---|---|---|---|
| RSV-A | 22 | 4 | 26 |
| RSV-B | 12 | 12 | |
| Coronavirus | 4 | 4 | |
| Rhinovirus | 9 | 2 | 11 |
| Influenza | 1 | 1 | |
| Bocavirus | 1 | 1 | |
| Rehmannia mosaic virus | 1 | 1 | |
| Tobacco mosaic virus | 1 | 1 | |
| Tomato mosaic virus | 1 | 1 | |
| Parechovirus | 1 | 1 | |
| Tomato brown rugose fruit virus | 1 | 1 |
The respiratory virome of healthy children is comprised of a high frequency of RSV and CoV
Using a cutoff of >40% genome coverage, we identified 10 unique RNA viruses, and 3 unique bacteriophages/prophages that constituted the virome community of HC cohort (Figure 1B; Table S1). All samples had at least one virus, and a maximum of three human RNA viruses were identified (average two viruses/sample), which included, for example, RSV, CoV, HRV, enteroviruses (EVs), and influenza. Interestingly, plant viruses were also identified in almost all samples, which has been reported before in human and mice lungs (Balique et al., 2013; Nakamura et al., 2009). With a stringent cutoff of >90% genome coverage, and minimum 5× coverage, we identified two plant viruses, rehmannia mosaic virus NcaS (ReMV-NcaS) and tobacco mosaic virus (Figure 1B; Table S2). In addition, bacteriophage/prophage RNA transcripts were also recovered from many of the samples, albeit with lower genome coverage.
Surprisingly, 79% (15 of 19 samples) of the HC samples had either RSV-A or RSV-B genomes, and 31% of the HC samples had both RSV-A and RSV-B genomes (Figure 1B). We assembled four coding complete and eight partial RSV-A genomes, and one partial RSV-B genome from HC samples (Table S1). To exclude the possibility of internal cross-contamination, we performed pairwise distancing and phylogenetic analyses of the assembled RSV genomes, and also compared the virome profiles between these samples, which strongly suggest the samples were not cross-contaminated (Figure S2).
Three different strains of CoVs (229E, OC43, and NL63) were identified in HC samples (Figure 2A). Of note, 68% (13 out of 19) of the HC samples had CoVs with a >25% genome coverage cutoff. CoV-NL63 was found in 11 samples, whereas CoV-229E and CoV-OC43 were found in one sample each. Complete coding sequences were assembled for two of the CoV-NL63 genomes, and both the CoV-229E and CoV-OC43 genomes (Figure 2A). Unlike RSV, multiple CoV types were not identified in any sample. Along with RSV, almost every sample had other known pathogenic viruses co-detected. HRVs were found in 36% (7 out of 19) of samples, EV-D68 reads were found in 26% (5 out of 19) of samples, and influenza reads were found in 5% (1 out of 19) of samples. We also found a significant amount of rotavirus reads, covering 80% (with an average read depth of 65×) of the rotavirus genome, in the respiratory sample from a 7-month-old infant; sequence analysis showed 100% similarity with the RotaTeq vaccine strain.
Figure 2.

Respiratory virome in healthy and children with RSV-ARI
(A) Read coverage maps showing three different coronavirus strains (NL63, OC43, and 229E) identified. The reference genomes were used to map the sequence reads to show that the complete genome sequences were recovered from the metatranscriptome approach.
(B) Bar plots showing number of samples with RSV-A, RSV-B, or co-detected by the metatranscriptomics method, in RSV-ARI samples. RSV-ARI samples were sub-grouped into RSV-severe and RSV-mild based on clinical presentations.
(C) Read coverage map showing four conditions of RSV presence. RSV-A (reference genome JX627336) and RSV-B (reference genome KM517573) genome sequences were concatenated and used as the reference. Mapping reads to this reference shows the presence of RSV-A in example 1, RSV-B in example 2, both in example 3 with RSV-A being dominant, and both in example 4 with RSV-B being dominant.
(D) Each row represents a sample and the columns represent results for each virus from the RPP (pink) followed by the metatranscriptomics method (purple). RPP results show the presence or absence of a virus, whereas metatranscriptomics results show the percentage of genome recovered for each virus. All viruses detected by RPP in at least one sample are shown here. The samples are grouped into healthy and RSV-ARI. The RSV-ARI samples are grouped into mild and severe.
The respiratory virome during RSV-ARI shows a high frequency of RSV-A and B co-detections
Using a very stringent threshold for virus identification (>90% genome coverage and an average read depth of 5×), we found RSV (either A or B subtype) in 38 out of 39 RSV-positive samples sequenced. Twenty-four samples had only RSV-A, 1 sample had only RSV-B, and 13 samples had co-infection of RSV-A and RSV-B (Figure 1B; Table S2). In the one remaining RSV-positive sample, 86% of the RSV-A genome was recovered. We co-detected RSV-A and RSV-B in 22 (56%) RSV-ARI samples by using a cutoff of >40% genome coverage (Figure 2B); in all cases, one of the RSV subtypes was found to be dominant (Figure 2C). Mapping the sequencing reads to reference genomes revealed four different patterns: samples positive for RSV-A (example 1); positive for RSV-B (example 2); positive for both RSV-A and RSV-B where RSV-A is dominant (example 3); and positive for both RSV-A and RSV-B where RSV-B is dominant (example 4). RSV-B was dominant in 12 subjects and RSV-A was dominant in 10 subjects. Strikingly, the read coverage depth of the dominant subtype was exponentially higher in RSV-A and RSV-B co-detected samples. For example, the RSV-B dominant co-detected samples show an average read coverage depth of 1,773× for RSV-B and 27× for RSV-A. The RSV-ARI samples were portioned into RSV-mild (outpatient/emergency department or under observation) and RSV-severe (hospitalized more than 1 day) groups. We observed a higher frequency of RSV-A and RSV-B co-detection in the RSV-severe group (69.6%) compared with the RSV-mild group (37.5%, p = 0.058, Fisher's exact test) (Figure 2B). In addition, as in the HC samples, several commensal or co-infecting viruses were identified in RSV-ARI samples: HRV was identified in nine samples and Bocavirus (BV) was identified in one RSV-ARI sample. Similar to HC samples, we also found plant viruses and bacteriophages/prophage transcripts with very low genome coverage (Figure 1B).
Comparison of the virome between healthy and RSV-infected children
We observed no difference in overall composition of the virome (number and types of viruses identified) between HC and RSV-ARI samples (Figure 1B). However, none of the RSV-ARI samples had CoVs, which was in sharp contrast to the HC samples, where CoVs (with >40% genome coverage) were identified in 47% of the samples (p = 1.788 × 10−5, chi-square test) (Figure 1B; Table S1). Another key difference between RSV-ARI and HC was that the sequencing read depth for RSV in HC samples was very shallow, with an average depth of 17× for RSV-A and 3.4× for RSV-B, compared with an average read depth of 1,525× for RSV-A and 1,054× for RSV-B in RSV-ARI samples.
Metatranscriptome is superior for RNA virus detection compared with multiplex panels
To evaluate our metatranscriptomics method, we compared the respiratory pathogens identified by metatranscriptomics with results from a clinical diagnostics panel, the NxTAG Respiratory Pathogen Panel (RPP) (Luminex) (Jassal et al., 2020). The comparison between these two methods is shown in Figure 2D and Table S3. In summary, for RNA viruses, we were able to match: all of the 22 RSV-A and 13 RSV-B; 3 out of 4 CoVs; 14 out of 15 HRV/EV; 1 out of 1 influenza; 1 out of 1 HMPV; and 0 out of 1 PIV4 identifications by RPP. However, in one instance where the panel identified CoV OC43 as the strain type, our method identifies it as the NL63 type with medium confidence (>50% genome recovered with average read depth >3×). In addition, we were able to identify OC43 in a sample and recover 100% of the genome with over 23× average read coverage, which the panel did not detect.
We note that the 35 RSV identifications by RPP originated from 34 samples, as it detected both RSV types in only 1 sample. This is in sharp contrast with our approach where we were able to co-detect in 13 out of the same 34 samples with high confidence (>95% of genome of each subtype recovered). In the one sample where RPP detected both RSV subtypes, we recovered 98% of the RSV-A genome and 63% of the RSV-B genome. The RPP was not capable of distinguishing between HRV and EV types (14 instances), whereas our sequencing approach was able to identify the strains of the 13 instances of HRV and 1 EV. These results suggest that our approach is more sensitive than RPP. However, a future independent study is required to validate the superiority of our method. In addition, we note that it has limitations in detecting DNA viruses, as our method was able to identify only 1 (out of 9) BV identified by RPP and it did not detect adenoviruses that were identified by the RPP. Furthermore, we were able to match the identification of one instance of the bacterial pathogen, Mycoplasma pneumoniae (Figure 2D).
Species-level respiratory microbiome profiling from transcripts
To profile the active respiratory bacteria and fungi with high confidence, we implemented a custom approach that includes k-mer-based taxonomic classification as the first step. Species-level identification was achieved by using a combination of metatranscriptomics assemblies, sequence homology searches, and stringent filtering of putative species, as described in STAR Methods. The high-confidence profiling resulted in a total of 88 bacterial species and 3 fungal species (Figure 3; Table S4) identified across all the samples. The top 5 most abundant bacterial species, Moraxella catarrhalis, Streptococcus pneumoniae, Streptococcus mitis, Haemophilus influenzae, and Cutibacterium acnes were identified in 60% of the samples. Of the three fungal species identified, Malassezia restricta was detected in 41% of samples, and Candida dubliniensis and Saccharomyces cerevisiae were detected in two (3.4%) samples each. Our high-confidence profiling approach detected an average of 9 (1 minimum and 28 maximum) bacterial species per sample, with up to 90% of the transcriptome recovered for highly abundant bacterial species (Figure 3). In the one instance where RPP identified a Mycoplasma pneumoniae co-infection, we were able to detect it with high confidence and also captured the entire coding complement of the M. pneumoniae genome (Figure 4A). Similarly, several pathogenic bacteria were identified in both RSV-ARI and HC samples with high confidence. For example, H. influenzae was identified in 27 (69%) RSV-ARI samples and 9 (47%) HCs; Streptococcus pneumoniae was identified in 26 (67%) RSV-ARI samples and 11 (58%) HCs; and Staphylococcus aureus was identified in 6 (15%) RSV-ARI samples and 2 (10%) HCs (Figure 3).
Figure 3.

High-confidence nasal microbiome
Heatmap showing percentage of transcriptome recovered for each bacterial and fungal species identified. The columns represent each sample, which have been color coded to identify healthy controls (green), and RSV-mild (blue) and RSV-severe (pink) groups.
Figure 4.

Nasal microbiome abundance and diversity
(A) Read coverage along the full genome of Mycoplasma pneumoniae (reference genome: NC000912), which was recovered in a sample co-infected with RSV.
(B) Nasal microbiome relative abundance in HC and children with RSV-ARI. A color-coded bar plot shows the relative abundance of the nasal microbiome at the species level. The samples are portioned into HC and RSV-ARI groups. The RSV-ARI samples were portioned into RSV-mild and RSV-severe groups. Only the top 20 most abundant species are shown here.
(C) Differentially abundant nasal species in children with RSV-ARI and HC groups. All displayed values were calculated within the DESeq2 package, where we compared species abundance. On the x axis is displayed the q value for the tested species; only significant species with q < 0.05 are shown. On the y axis is displayed the log2 fold abundance change for that species. Error bars show the standard error of the log2 fold change. Log2 fold changes >0 indicate that a species was more abundant in RSV-ARI children compared with HC.
(D) Similar to (C), the nasal microbiome of children with severe RSV-severe was compared with the RSV-mild group. Log2 fold changes >0 indicate that a species was more abundant in RSV-severe children compared with the RSV-mild group. Malassezia globosa was less abundant and Staphylococcus aureus was more abundant in the RSV-severe group compared with RSV-mild group.
(E) Richness and alpha diversity of the nasal microbiome. Alpha diversity (measured by Shannon index) and richness (measured by S.chao1) are compared between the HC, RSV-severe, and RSV-mild groups. The richness was highest in the RSV-mild group compared with the HC and RSV-severe groups. The differences were significant between the RSV-mild and RSV-severe groups and the RSV-mild and HC groups. Differences in alpha diversity between the groups were not significant.
Respiratory microbiome abundance and diversity profile shows increased abundance and gene expression of H. influenzae during RSV-ARI
The respiratory microbiome of both HC and RSV-ARI groups were dominated by members of the Moraxella sp. (Moraxella catarrhalis, M. lincolnii, and M. nonliquefaciens), Corynebacterium sp. (Corynebacterium propinquum, Corynebacterium pseudodiphtheriticum, and Corynebacterium sp.), and Dolosigranulum pigrum, with these species having mean abundances of 40.8%, 22.4%, and 10.8%, respectively, in the HC group, and 23%, 21.2%, and 3.4%, respectively, in the RSV-ARI group (Figure 4B). Although the HC and RSV-ARI groups shared many low abundant taxa in common, differences were observed between the groups in bacterial Haemophilus sp. and Delftia sp. and a fungi Malassezia sp. (Figure 4B). The members of Haemophilus sp. (H. influenzae/aegyptius) and Delftia sp. were more highly abundant in the RSV-ARI group, with mean abundances of 21.4% (p = 0.056, Kruskal-Wallis) and 3.5% (p = 2.644 × 10−5), respectively, compared with the HC group, with mean abundances of 10.8% and 0.5%, respectively. The Malassezia sp. were more highly abundant in HC groups, with a mean abundance of 5.6% (p = 0.12) compared with the RSV-ARI group, with a mean abundance of 1.2%. Strikingly, the mean abundance of Haemophilus sp. was higher in the RSV-severe group with 26.7% compared with RSV-mild group with 13.7% (p = 0.76). The mean abundance of Malassezia sp. was lower in the RSV-severe group 0.034% (p < 0.001) compared with the RSV-mild group 2.9% (Figure 4B). These results did not change after removing seven RSV-ARI subjects who had taken antibiotics <1 month before sample collection. (Figure S3). To compare the bacterial signatures identified by the metatranscriptomics method, we profiled microbial communities by using 16S rRNA marker gene sequencing. Interestingly, the nasal bacteria and their abundance profiled by 16S and metatranscriptomics were highly comparable (Figure S4).
We further investigated whether specific taxa were differentially abundant using the DESeq2 package (Love et al., 2014). A comparison between the HC and RSV-ARI groups revealed that RSV-ARI children had a significantly higher relative abundance of H. influenzae (log2 fold change = 4.5, q = 0.01), Delftia sp. (log2 fold change = 3.41, q = 0.0029), and Cutibacterium acnes (log2 fold change = 2.5, q = 0.01) (Figure 4C). A comparison between the RSV-severe and RSV-mild groups showed a significantly higher relative abundance of Staphylococcus aureus (log2 fold change = 25.7, q = 3.56E-10) and a significantly lower abundance of Malassezia globosa (log2 fold change = −25.6, q = 3.56E-10) in the RSV-severe group (Figure 4D). Further analysis of bacterial species contributions to metabolic pathways showed 13 pathways that were significantly different (q < 0.05) between the HC and RSV-ARI groups (Table S5). Among these pathways, H. influenzae and Delftia sp. were major contributing species linked with the functional attributes (Figure S5), which further confirms their increased activity in RSV-ARI samples.
The mOTUs counts at the species level were used to compute the alpha diversity of the microbial communities by using the Shannon and Chao1 indices. The alpha diversity calculations (mean Shannon index) showed no significant difference between the HC, RSV-mild, and RSV-severe groups. The richness calculations (mean Chao1 index) reveal the microbiota alpha diversity of the RSV-mild group to be higher than that of the HC and RSV-severe groups, with this difference being statistically significant (p value for RSV-mild versus HC, 0.0115; p value for RSV-severe versus RSV-mild, 0.027, Wilcoxon rank-sum test). There was no significant difference between the HC versus RSV-severe groups (Figure 4E).
Comparison of nasal mucosal cells transcriptome between HC and RSV-ARI
Sequence reads that mapped to human transcripts were used to analyze host response to RSV-ARI. After estimating the library size by using the median ratio method, the samples with a low number of human transcripts (n = 13) and an ARI sample negative for RSV were removed from the analysis. RSV-ARI (n = 32) and HC (n = 12) samples were used for gene expression analysisby using DESeq2 (Love et al., 2014), which revealed that 2,878 genes were upregulated and 1,746 genes were downregulated in RSV-ARI subjects (Figure 5A; Table S6). Most of the overexpressed genes are involved in immune response during RSV infection, specifically interferon response genes, IFIT1, IFIT2, IFIT3, IFI6, and anti-viral genes, such as IFITM3 (Zani and Yount, 2018), IFITM1 (Smith et al., 2019), and MX1 (Villenave et al., 2015), as well as chemokines CCL4, CCL8, and CXCL10 (Steinbusch et al., 2019). We also identified ACOD1 as being significantly upregulated in RSV-ARI samples. The long non-coding RNA of ACOD1 is known to promote viral replication by modulating cellular metabolism (Wang et al., 2017). To confirm the significance and robustness of the observed differences in gene expression between RSV-ARI and HC, we performed pathway enrichment analysis (Kuleshov et al., 2016), which identified up- and downregulation of 69 and 2 human Reactome pathways, respectively (Jassal et al., 2020) (with an adjusted p < 0.05) (Table S7). The majority of upregulated pathways belonged to anti-viral response; e.g., interferon signaling, interleukin signaling, chemokine signaling, inflammasome pathway, and Toll-like receptor (TLR) signaling (Figure 5B). For example, we found 57% (39 out of 68) and 34% (31 out of 92) of the genes were significantly upregulated from interferon alpha/beta and MYD88 signaling, respectively, in RSV-ARI samples (Figure S6). Similarly, 9 out of 17 genes from the inflammasome pathway were found to be upregulated; NLRP3, the key driver of inflammasome activity (Yang et al., 2019), was significantly higher in RSV-ARI samples than HC (Figures 5B and 5C). Fatty acid synthesis and phase 1 functionalization of compounds were the two pathways enriched in the downregulated genes (Figure 5B). We identified 9 out of 15 genes from the fatty acid pathway were down modulated compared with HC (Figure 5D). Many of these enriched pathways have been found to be modulated upon RSV infection in human and animal models, including in cotton rat lung tissue (Dapat and Oshitani, 2016; Rajagopala et al., 2018).
Figure 5.

Host transcriptional response
(A) Volcano plot showing differentially expressed host genes between RSV-ARI and HC children. We use a threshold of log2 fold change >1 and adjusted p < 0.05 to call the genes that are up- or downregulated. The genes that satisfy the threshold are shown in red dots. Non-significant genes are shown in gray dots. The genes that pass the adjusted p value threshold but not log2 fold change are shown in blue dots. The genes that pass the log2 fold change threshold but not adjusted p value are shown in green dots.
(B) Enriched Reactome human pathways from the differential gene expression analysis. Upregulated pathways are shown in red and downregulated pathways are shown in blue. On the y axis is displayed the pathway name and number of genes in the pathway. On the x axis is displayed the percentage of genes upregulated in that pathway; only a subset of significant pathway enrichment with q < 0.05 are shown.
(C) Plot showing the Reactome (Jassal et al., 2020) inflammasome pathway (R-HSA-622312) genes that are significantly upregulated in the RSV-ARI group compared with the HC group. On the x axis is displayed the q value for the upregulated genes with q < 0.05. On the y axis is displayed the log2 fold change for those genes. The size of the dots represents base mean, which is the mean of normalized counts of all samples.
(D) Similar to (C), a plot showing the Reactome fatty acid synthesis pathway (R-HSA-211935) genes that are significantly downregulated in the RSV-ARI group compared with the HC group.
Discussion
A comprehensive understanding of viral ARIs, including the virome, microbiome (bacterial and fungal), and the host response, is essential to understand virus-host and virus-microbiome interactions and to model viral diseases. To date, there have been only a few studies focused on characterizing the entire human respiratory virome during health and/or disease (Abbas et al., 2019; Langelier et al., 2018; Li et al., 2019; Mitchell et al., 2016; Noell and Kolls, 2019; Wylie, 2017). This is mostly due to the inherent extensive sequence diversity of virus genetic material (for example, it is challenging to simultaneously sequence both DNA and RNA viruses) and lack of methods to capture complete genomes of RNA viruses directly from clinical samples. Although RNA viruses are a major threat to human health, there have been limited studies to characterize the entire RNA virome because of the higher cost and time associated with preserving, enriching, and sequencing RNA viruses from low biomass clinical samples. Current high-throughput genome sequencing methods for RNA viruses include cDNA synthesis followed by amplicon sequencing (Djikeng and Spiro, 2009; Geoghegan et al., 2015; Houldcroft et al., 2017; Ladner et al., 2014; Nelson et al., 2016; Tan et al., 2016). However, these methods suffer from limitations inherent to PCR amplification, such as polymerase errors, jackpot effects, uneven sequence coverage (or lack of coverage in specific regions of the genome), and they require an independent sequencing approach for each pathogen (Bustin and Nolan, 2004; Gu et al., 2019). In this study we have demonstrated that the metatranscriptomics method is suitable for complete genome sequencing of the dominant and co-infecting RNA viruses with sufficient depth. This approach also captures the majority of both the 3′ and 5′ UTRs that are usually not captured by an amplicon-based sequencing approach. These regions are known to be important for viral replication and immune modulation (Dreher, 1999; Guo et al., 2001). Comparing the results of our metatranscriptomics method with RPP demonstrates that our approach captures the respiratory RNA viruses, at a strain-level resolution, with high sensitivity and is agnostic to the pathogen of interest (Figure 2D). It was surprising that RSV was not detected by the NxTAG in HC, especially for the samples in which we recovered RSV complete genomes with >20× average coverage (Table S2). This suggests a limited sensitivity of detection for the current diagnostic method. By using our stringent approach, we could not only identify the viruses at the strain level, but at the genotype level, and could potentially assess intra-host variability of viruses with confidence, especially in samples that have >1,000× genome coverage. Although this method is clearly sensitive compared with existing panels in detecting RNA viruses, we note that it has limitations regarding detection of DNA viruses. However, in the one instance where we were able to detect BV, 97% of its genome sequence was assembled, suggesting that our method can detect active DNA viruses (Figure 1B).
We show that the respiratory RNA virome of children during health constitutes a wide array of human RNA viruses. Interestingly, although the presence of RSV, HRV, EV, and CoV in healthy children and adults has been reported previously (Wylie, 2017), the frequency at which our method detected RSV-A and RSV-B and CoV is substantially higher than previous reports. Even with a cutoff of >40% genome coverage, we identified RSV in 79% (although most RSV infections in children are thought to be symptomatic) and CoVs in 47% of HC samples (Table S1). Similarly, this study revealed a high frequency (56%) of RSV-A and RSV-B co-detections in RSV-ARI samples, whereas previous studies have shown RSV-A and RSV-B co-detections ranging from 0.1% to 0.4% of RSV-ARI (Bouzas et al., 2016; Gamino-Arroyo et al., 2017). The frequency of RSV co-detections was greater in the RSV-severe group (69.6%) compared with the RSV-mild group (37.5%) (Figure 2B).
In addition to capturing the virome, we have demonstrated the strength of our method in identifying the respiratory bacterial microbiome at the species level directly from low biomass clinical samples (Figure 3), as well as common fungi inhabiting the nasopharynx. We have identified a total of 88 unique bacterial species and 3 fungal species. Importantly, our method also captured the entire coding complement from the samples with a bacterial co-infection; for example, M. pneumoniae co-infection (Figure 3B). The microbial read depth was sufficient to assess the respiratory microbiome abundance and diversity in healthy and RSV-ARI-positive children. RSV-mild and with RSV-severe groups were positively associated with H. influenzae and Streptococcus abundance (de Steenhuijsen Piters et al., 2016). There was no significant alpha diversity difference between HC, RSV-mild, and RSV-severe groups. We observed higher richness (mean Chao1 index) in the RSV-mild group compare with the HC and RSV-severe groups; higher richness during an ARI might be protective against RSV-ARI disease severity (Figure 4E). However, these observations need to be further validated by a larger cohort study. Although several bacterial pathogens were detected in both RSV-ARI and HC samples, we detected S. pneumoniae more frequently in RSV-ARI (67% of RSV-ARI subjects). Previous studies have shown co-detection of RSV and S. pneumoniae in the nasopharynx was associated with more severe ARI (Brealey et al., 2018). Similarly, H. influenzae was detected more frequently (69%) in RSV-ARI samples compared with HC samples (47%). The relative abundance of H. influenzae was significantly higher (log2 fold change = 4.5, q = 0.01) in the RSV-ARI subjects (Figure 4D), which was further validated by the high metabolic activity of H. influenzae in the RSV-ARI subjects (Figure S5). To our knowledge, this is the first study to show that abundance of a fungus, Malassezia sp., is negatively associated with RSV-ARI infections. Malassezia sp. were significantly less abundant in RSV-ARI subjects, and their abundance was further reduced in the RSV-severe group (Figures 4B and 4E).
So far, there have only been a few studies describing the local mucosal immune response to RSV (de Steenhuijsen Piters et al., 2016; Ederveen et al., 2018; Shilts et al., 2020; Turi et al., 2018), as the majority of studies have so far focused on either adaptive or systemic transcriptional gene expression (de Steenhuijsen Piters et al., 2016; Russell et al., 2017; Sonawane et al., 2019). Our comparison of RSV-ARI with HC samples by using respiratory mucosal transcriptomics showed a massive anti-viral response, with upregulation of interferon signaling, interleukin signaling, chemokine signaling, inflammasome pathway, and TLR signaling in the RSV-ARI group. As expected, the majority of the interferon-stimulated genes and anti-viral genes were upregulated, i.e., IFITs, IFITMs, and MX dynamin-like GTPase (Smith et al., 2019; Villenave et al., 2015; Zani and Yount, 2018). We also found massive upregulation of the inflammasome pathway, where the key modulator is NLRP3 (Figure 5C). Of note, the NLRP3 inflammasome has been shown to be activated through the small hydrophobic protein of RSV viroporin, which induces membrane permeability to ions or small molecules (Kim and Lee, 2014; Triantafilou et al., 2013). Furthermore, Segovia et al. (2012) showed that NLRP3/ASC inflammasome activation was crucial for interleukin-1β production during RSV infection.
Several genes in fatty acid synthesis pathways were downregulated in RSV-ARI (Figure 5D); interestingly, it has been shown that short-chain fatty acids, specifically acetate, have anti-viral activity against RSV (Antunes et al., 2019) and influenza A virus (Sencio et al., 2020; Trompette et al., 2018). Furthermore, microbiota-derived acetate has been shown to protect against RSV infection through a GPR43-type 1 interferon response (Sencio et al., 2020). The most significant increase in gene expression was of ACOD1; ACOD1 is also called IRG1 and the long non-coding RNA of ACOD1 is involved in the inhibition of the inflammatory response (Luan and Medzhitov, 2016), and also serves as a negative regulator of the TLR-mediated inflammatory innate response by stimulating the tumor necrosis factor alpha-induced protein TNFAIP3 expression via reactive oxygen species in LPS-tolerized macrophages (Li et al., 2013). Furthermore, ACOD1-mediated itaconic acid production contributes to the antimicrobial activity of macrophages (Li et al., 2013; Michelucci et al., 2013), whereas the long non-coding RNA of ACOD1 is known to promote viral replication by modulating cellular metabolism (Runtsch and O'Neill, 2018; Wang et al., 2017).
Our study has considerable clinical and technological implications. To our knowledge, this is the first study to show the feasibility of capturing the entire respiratory RNA virome from low biomass sample. Importantly this method captures diverse viral complete genomes, including both coding and non-coding regions of the viruses, which could contribute to phylogenetic and phylodynamic analyses to understand principles of virus evolution, virus-virus interactions, and at the same time improve our understanding of intra-host variability and emergence. Furthermore, our method can simultaneously identify active bacterial and fungal co-infections.
Limitations of the study
We should also acknowledge several limitations. First, the library preparation method was not efficient in removing the bacterial rRNA (off-target molecules) because of lack of appropriate commercial reagents. Mitigating this step would further improve the resolution of microbial and host transcriptome data. Furthermore, all our samples were collected only during the cold and flu season, and thus it remains to be known if children with no symptoms of RSV or CoV are persistently infected all year long or only during the cold and flu season. Third, although our method is far superior than the multiplex diagnostic panels, it is expensive and computationally intensive. However, rapid reduction in sequencing costs, expanding pathogen sequence databases, and better data analysis tools can enable routine use and implementation in clinical settings. Despite these limitations, we have shown the feasibility of our metatranscriptomics method to capture multi-dimensional genomics data to study viral phylodynamics and virus-host and virus-microbial interactions; and, with reduction of sequencing costs and a robust automated analytic pipeline, this method could become a powerful tool for clinical and translational research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | PROVIDER | CATALOG NUMBER |
|---|---|---|
| Antibodies | ||
| N/A | N/A | N/A |
| Bacterial and virus strains | ||
| N/A | N/A | N/A |
| Biological samples | ||
| Nasal swabs | DNA Genotek | OMR-110 |
| Chemicals, reagents | ||
| QIAzol | Qiagen | 79306 |
| 2.0 mm zirconium oxide beads | Next Advance, Inc. | ZROB05 |
| Chloroform | Milipore Sigma | 288306 |
| Critical commercial assays | ||
| NxTAG® Respiratory Pathogen Panel (RUO) | Luminex Corporation | X051C0451 |
| RNeasy Plus Universal Mini Kit | Qiagen | 73404 |
| Agilent RNA 6000 Nano kit | Agilent | p/n 5067-1511 |
| Agilent RNA 6000 Pico kit | Agilent | p/n 5067-151 |
| Agilent High Sensitivity DNA kit | Agilent | p/n 5067-4627 |
| NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) | NEB | E7400X |
| NEBNext Ultra II RNA Library Prep Kit | NEB | E7770L |
| Deposited data | ||
| Sequence data | SRA Bioproject | PRJNA671738 |
| Experimental models: Cell lines | ||
| N/A | N/A | N/A |
| Experimental models: Organisms/strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| 16S primer - Forward primer binding site: GTGCCAGCMGCCGCGGTAA | IDT | Cite: (Kozich et al., 2013) |
| 16S prime - Reverse primer binding site: GGACTACHVGGGTWTCTAAT | IDT | Cite: (Kozich et al., 2013) |
| NEBNext Multiplex | NEB | E7600S |
| Recombinant DNA | ||
| N/A | N/A | N/A |
| Software and algorithms | ||
| Trimmomatic v0.39 | http://www.usadellab.org/cms/?page=trimmomatic | (Bolger et al., 2014) |
| Bbtools | http://jgi.doe.gov/data-and-tools/bbtools/ | (Bushnell et al., 2017) |
| KrakenUniq | https://github.com/fbreitwieser/krakenuniq | (Breitwieser et al., 2018) |
| VAPiD annotation tool | https://github.com/rcs333/VAPiD | (Shean et al., 2019) |
| mOTUs2 tool | https://github.com/motu-tool/mOTUs | (Milanese et al., 2019) |
| Phyloseq R package version 1.30.0 | https://bioconductor.org/packages/3.13/bioc/html/phyloseq.html | (McMurdie and Holmes, 2013) |
| DESeq2 | https://bioconductor.org/packages/3.13/bioc/html/DESeq2.html | (Love et al., 2014) |
| HUMAnN2 pipeline | http://huttenhower.sph.harvard.edu/humann2 | (Franzosa et al., 2018) |
| HISAT2 | http://daehwankimlab.github.io/hisat2 | (Kim et al., 2015) |
| HTSeq | https://github.com/simon-anders/htseq | (Anders et al., 2015) |
| Enrichr | https://CRAN.R-project.org/package=enrichR | (Kuleshov et al., 2016) |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Suman Das (suman.r.das@vumc.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
For this study, we enrolled 65 children, aged 0–36 months, at Vanderbilt University Medical Center (VUMC), Nashville, TN between November 2018 and February 2019. Forty-three of these children presented symptoms at the time of the hospital visit. Based on the Respiratory Pathogen Panel (RUO, Luminex Corporation) they were confirmed to be infected with Respiratory Syncytial Virus. Samples collected from these children are referred to in this study as “RSV-ARI” samples. The remaining 22 children were enrolled into the study when they visited the hospital for well-child visits. Samples collected from these children are referred in this study as Healthy Control (HC) samples. The demographic and clinical characteristics of participants are summarized in the Table 1. The RSV-ARI group included pediatric patients presenting to VUMC emergency department (ED) visit only (discharged home from ED) or admitted and seen in the ED during the same visit (Inpatient). Children were enrolled if they were less than 3-years-old and had no prior medical comorbidities including congenital cardiac or pulmonary disease. Children less than 1-month-old were excluded if premature (gestational age less than 37 weeks) or were newborns who had never been discharged home. All children were excluded if neutropenic (Absolute Neutrophil Counts [ANC] less than 600) and if they received antibiotics within the last 24 hours. HCs were excluded if they had symptoms of an infection, including rhinorrhea or congestion, within the past 3 days.
Ethics statement
Subject recruitment and study procedures were approved by and carried out in accordance with the Institutional Review Board of Vanderbilt University Medical Center (IRB number:111296). In compliance with the IRB approval, informed consent was obtained from study participants’ parents or guardian before the initial sample collection.
Method details
Sample collection and RNA extraction
Two flocked (type) swabs were used in each nostril with adequate swabbing to collect nasal epithelial cell samples. The samples were stored in the MMB collection tube containing RNA/DNA stabilizing liquid for microbiome (Genotek OMR-110). The samples were stored at room temperature until transported to the lab, then kept at −80°C for long term storage. The nasal swab samples in the MMB collection tube were vortexed for 2 minutes, then an aliquot of 250μL nasal swap sample was used for RNA extraction. The 250μL aliquot from the nasal swab sample was homogenized in 600μL QIAzol (Qiagen) and 500μL of 2.0 mm zirconium oxide beads (Next Advance, Inc. Cat: ZROB05) using a Bullet Blender homogenizer (BB24-AU, Next Advance, Inc). While homogenizing the samples, temperature was maintained at or near 4°C by using the dry ice cooling system in the Bullet Blender. The homogenate was treated with 100μL of genomic DNA Eliminator solution (Qiagen) to remove the genomic DNA. Next, 180μL of chloroform was added to the samples for phase separation. The total RNA in the aqueous phase was then purified using RNeasy Mini spin columns as recommend by the Qiagen RNeasy protocol. RNA integrity and RNA quantification were assessed using an Agilent Bioanalyzer RNA 6000 Nano/Pico Chip (Agilent Technologies, Palo Alto, California). Our RNA extraction approach resulted in high quality RNA with an average RIN value of ∼4.1 (Figure S1A).
Respiratory pathogen panel
Both RSV-ARI and HC samples were subjected to respiratory pathogen detection using the NxTAG Respiratory Pathogen Panel (RUO) (Luminex Corporation, Part Number: X051C0451). This assay simultaneously detects 22 respiratory pathogens, including 19 respiratory viruses (Influenza A, Influenza A - H1 subtype, Influenza A - H3 subtype, Influenza A 2009 H1N1 subtype, Influenza B, Respiratory Syncytial Virus A, Respiratory Syncytial Virus B, Parainfluenza 1, Parainfluenza 2, Parainfluenza 3, Parainfluenza 4, Human Bocavirus, Human Metapneumovirus, Rhinovirus/Enterovirus, Adenovirus, Coronavirus HKU1, Coronavirus NL63, Coronavirus OC43, and Coronavirus 229E), and three bacterial pathogens (Chlamydophila pneumoniae, Legionella pneumophila, and Mycoplasma pneumoniae). RNA extractions and pathogen detection assays were performed using the standard protocols provided by the reagent’s manufacturer.
Ribosomal RNA depletion, metatranscriptomic library preparation and sequencing
Eukaryotic ribosomal RNAs (rRNA) were depleted using the NEBNext rRNA Depletion Kit (Human/Mouse/Rat, Cat: E6310X). After rRNA depletion, the samples were checked by Agilent Bioanalyzer RNA 6000 Nano/Pico Chip to ensure depletion of the 18S and 28S ribosomal peaks. Next, Illumina sequencing libraries were made using the NEBNext Ultra II RNA Library Prep Kit (NEB #E7775). The quality of the libraries was assessed using an Agilent Bioanalyzer DNA High Sensitivity chip. The libraries were then sequenced on an Illumina NovaSeq6000 platform (S4 flow cells run) with 2x150 base pair reads, with a sequencing depth of 45-50 million paired-end reads per sample.
Quantification and statistical analysis
Preprocessing and quality control of NGS data
Adapter removal and quality-based trimming of the raw reads were performed using Trimmomatic v0.39 (Bolger et al., 2014) using default parameters. Trimmed reads shorter than 50nt were discarded. Low complexity reads were discarded using bbduk from bbtools (Bushnell et al., 2017) with entropy set at 0.7 (BBMap – Bushnell B. – sourceforge.net/projects/bbmap/).
Read binning
Reads were mapped to human rRNA and the human mitochondrial genome using bbmap from bbtools with default parameters. Mapped reads were discarded. The remaining reads were binned into human genome, bacterial rRNA, and a bin that contains all microbiome reads, using seal, from bbtools, with default parameters. The human genome (GRCh38) and SILVA bacterial rRNA database were used as references. Binning resulted in an average of 36,905,002 human transcript reads (32,506,155 – median) and an average of 3,815,821 microbiome reads (2,930,555 – median). The microbiome reads bin contains viral, bacterial, fungal as well as unclassified reads. In the microbial reads bin the distribution of viral reads range from ∼2 to 7%, bacterial reads range from 53 to 63% and fungal reads range from ∼2 to 7%.
Taxonomic classification of reads
Reads from the microbiome bin were subjected to taxonomic classification using KrakenUniq (Breitwieser et al., 2018) with default parameters. The reference NCBI nt database was installed via kraken2-build script.
Rarefaction analysis of microbiome reads
We performed rarefaction analysis on the microbiome reads from five samples. Two were HC samples (with 4,495,493 and 5,182,313 PE reads respectively) and three were RSV-ARI samples (5,093,637; 4,917,227 and 7,245,926 PE reads respectively). Each set of reads were sub-sampled five times at 1M, 2M, 3M, 4M, 5M, 6M, and 7M reads as possible. KrakenUniq was run on each sub-sampled read set, and reads were taxonomically classified. Species with at least 100 reads and a kmer to reads ratio of 7 were counted for rarefaction analysis. Number of species were plotted against reads per million after averaging across the five runs. Results show plateauing of the curve begins at around 4.5M reads (Figure S1B).
Virome profiling
To produce a high confidence virome profile, we developed a method that first produces de novo transcriptome assemblies, followed by putative virome identification using BLAST searches, and finally high confidence virome profiling based on read mapping to reference virus genomes. This workflow was implemented in a bash script. First, reads that were classified as viral by KrakenUniq were extracted using the script krakenuniq extract-reads, with taxonID 10239 (superkingdom, viruses). If more than 100,000 reads were extracted, they were first normalized to a target depth of 100 using bbnorm from bbtools. Reads were assembled using the metaSPAdes assembler. Resulting contigs were filtered for length, using reformat from bbtools, and only contigs that were at least 300bp were retained. Nucleotide BLAST (blastn) searches were performed on the resulting contigs, against the NCBI nt database with -max_target_seqs and -max_hsps set to 1. From the blast results, a list of subjects was compiled and their genome sequences (fasta) were extracted from the nt blast database using the blastdbcmd from BLAST. Each of those genome sequences were used as a reference and all the virome reads were mapped using bowtie2. Genome coverage and average read depth statistics were extracted from this mapping using samtools (Li et al., 2009). The high confidence virome profile was constructed using these coverage statistics.
Annotation of viral genomes
Assembled sequences where the full-length genome (coding complete) was recovered, were reverse complemented if needed, and annotated using the VAPiD annotation tool (Shean et al., 2019). In order to resolve the subtypes of RSV (A and B) genomes, a different approach was employed as described below.
Detection, assembly, and annotation of RSV
Human orthopneumovirus genomes, NC_038235.1 (Human orthopneumovirus Subgroup A, complete cds) and NC_001781.1 (Human orthopneumovirus Subgroup B, complete genome), were collected from RefSeq (Brister et al., 2015). They were merged into one fasta file with 100 “N”s in between, and used as the reference. Virome reads were mapped to this reference file using bbmap with default parameters. In its default setting, the reads with multiple top-scoring mapping locations are placed at the first best site. Since the overall identity between RSV subtypes A and B is only 81%, it is expected that bbmap has enough information for each paired read (2 x 150 bp) to be accurately placed. To confirm this, we have extracted reads mapped to the two genome sequences into two sets, and assembled each set independently using SPAdes (Bankevich et al., 2012). Blast searches were performed on the resulting contigs. The contigs hit the expected subtypes with greater than 99% identity. Assembled sequences where the full-length genome (coding complete) was recovered, were reverse complemented if needed, and annotated using the VAPiD annotation tool (Shean et al., 2019).
RSV co-infections and subtype identification
The nucleotide sequence identity between RSV-A (NC_038235) and RSV-B (NC_001781) reference subtypes is 81%. To resolve the subtypes of RSV in our samples, a merged reference genome of the two subtypes was used as a reference, and the viral reads were mapped to this reference using a high identity cutoff (95%). Since the overall identity between RSV subtypes is only 81%, it is expected that each paired read has enough information to be accurately placed. To further confirm this, we extracted reads that mapped to the two genomes, and assembled each set independently (see STAR Methods). BLAST searches of the resulting distinct assemblies, against the nucleotide database at NCBI, showed matches to RSV-A and RSV-B genomes with >95% identity.
RSV phylogenetic tree
We analyzed 28 complete genome sequences of RSV type A, and 12 complete genome sequences of RSV type B, assembled from samples in this study. Untranslated regions before the start of the first coding sequence (gene NS1) and after the end of the last coding sequence (gene L) were discarded. The remaining sequences, varying in length between 14,958 nt and 14,961 nt were used for pairwise comparison and phylogenetic analysis. Sequences were aligned using CLC Genomics Workbench (version 11.0.1). Pairwise comparisons of the sequences were performed using the “Maximum Likelihood (ML) Phylogeny” module in CLC Genomics Workbench was used to generate ML trees for RSV type A and RSV type B, using Neighbor Joining for construction method, Jukes Cantor for Nucleotide substitution model, and bootstrap analysis was performed with 1000 replicates. The trees were imported in FigTree (version 1.4.4) and annotated with metadata to highlight the sequences obtained from RSV-ARI samples and HCs.
Excluding possibility of cross-contamination of samples
To exclude the possibility of internal cross-contamination, we extracted complete coding sequences from 28 RSV-A genomes and 12 RSV-B genomes that were assembled, and performed pairwise comparison and phylogenetic analyses (Figures S2A and S2B). Nucleotide differences between the sequences were observed in all but five RSV-A genomes, where two sub-groups of two (S195 and S205 in Figure S2C) and three (S194, S213, S217) sequences were observed to be identical. Two of the three identical sequences were from HC samples (S213, S217). We then reviewed the virome profiles of these samples and observed that they are different, as other unique viruses were recovered. Near complete genome sequences for Influenza (in S217) and RSV-B (in S213) were recovered. 75% of the Rhinovirus A genome was recovered in the S205 sample. We have also performed read mapping to RefSeq RSV-A reference genome for all the healthy control samples, in which we detected RSV-A, followed by generation of SNP tree. The results from the SNP tree agree with pairwise comparison and phylogenetic analyses described above that the RSV genomes from the HC samples are distinct (data not shown).
Profiling high-confidence bacteria and fungi
We developed the following method to produce high confidence bacterial profiles. Taxonomy classification reports produced by KrakenUniq were parsed to retain entries at genus level. These entries were filtered to retain only the ones that have at least 100 reads and a Kmers to reads ratio of at least 7 or a minimum of 1000 Kmers. For each of the retained genera, reads were extracted and normalized to a target depth of 100 using bbnorm from bbtools, and assembled using metaSPAdes. BLAST searches were performed on the resulting contigs that are at least 300 nt long, using the NCBI nt database as reference (the NCBI nt database installed via kraken2-build script), and only the best hit for each contig was recorded. Only the contigs with at least 97% identical matches to a genome are retained for further analysis. The resulting file was parsed and a summary table was created with a list of species identified, along with the number of contigs and cumulative contig length for each species. Only species with a minimum of 10 contigs or a minimum cumulative contig length of 10,000 nt were included in the high confidence profile.
Microbiome abundance, diversity and functional profiling
We used the mOTUs2 tool (Milanese et al., 2019) to profile microbial abundance and the active microbiota. The mOTUs package uses 10 marker genes (MGs) to taxonomically profile and to quantify metabolically active members in metatranscriptome data. The microbial reads were subjected to mOTU analysis to obtain the relative abundance for each mOTU. Similarly, read counts for each mOTU were obtained at species and genus levels using mOTU profile reports. We tested for differences in proportions (relative abundance) of bacteria of interest using a Kruskal-Wallis test (Wallis, 1952). The Phyloseq R package version 1.30.0 (McMurdie and Holmes, 2013) was used to analyze microbial richness and alpha diversity metrics. The Shannon and Chao1 diversity indices were calculated from the mOTU counts in the samples to assess the alpha diversity of the microbial communities they represent. The Wilcoxon Rank Sum test was used to test for significant differences in microbial richness or alpha diversity between sample groups.
Significant associations between HC and RSV-ARI groups with bacterial taxa at mOTUs species and mOTUs genus levels were assessed using the R package DESeq2 (Love et al., 2014). Reported p-adjust values are the result of a Wald test with the Benjamini-Hochberg correction (Benjamini, 1995) applied to adjust for multiple comparisons.
To profile the abundance of microbial pathways in the metatranscriptomic sequencing data we used the HUMAnN2 pipeline (Franzosa et al., 2018), which involves a nucleotide search by mapping the reads to ChocoPhlAn and a translated search against Uniref90. First, the fungal reads were removed from the microbial reads bin. Then, paired-end reads were merged using bbmerg. Merged, unmerged, and single-end reads were combined into a single fastq file for each sample. All these samples were subjected to HumanN2 analysis. Each sample HUMAnN2 report was associated with a corresponding sample group using the humann2_associate script. The pathway abundance plots were produced using humann2_barplot, where the pathways are broken down into per-organism contributions, with the total read abundance consisting of pathways assigned to organisms.
Methods to analyze the bacterial microbiome with 16S rRNA gene amplicon sequencing
DNA was extracted with the DNeasy PowerSoil Kit (Qiagen). Mechanical lysis of bacterial cell walls was performed by shaking the samples on a TissueLyser II (Qiagen) for 20 minutes total. Dual-indexed universal primers appended with Illumina-compatible adapters were used to amplify the hypervariable V4 region of the bacterial 16S rRNA gene (Kozich et al., 2013). The PCR mix for each library contained 12.5 μl of MyTaq Mix (Bioline), 0.75 μl DMSO, 1 μl of forward primer, 1 μl of reverse primer, 7 μl of sample, and PCR Certified water (Teknova) was added to achieve a final volume of 25.25 μl. DNA was denatured at 95°C for 2 min, and then 30 cycles of 95°C for 20 seconds, 55°C for 15 seconds, and 72°C were performed. Samples were then incubated at 72°C for 10 min, and samples were held at 4°C until removal from the thermocycler. Each sample was run on a 1% agarose gel to verify reaction success. Libraries were cleaned and normalized with the Invitrogen SequalPrep Kit. After normalization to 1-2 ng/μl, 10 μl of each sample was combined to create the sequencing pool. The pool was cleaned with 1X AMPure XP beads (Beckman Coulter, Brea, California). Libraries were sequenced on an Illumina MiSeq with 2x250 bp reads. A mock community control (ZYMOBiomics) and extraction and PCR negative controls were run concurrently along with the samples to assess data quality and levels of background contamination.
We processed the 16S rRNA sequences using the dada2 pipeline by following its standard operating procedure (available at: https://benjjneb.github.io/dada2/tutorial.html, as of November 18, 2019) (Callahan et al., 2016). To this end, sequences were grouped into amplicon sequence variants (ASVs) and taxonomy was assigned using the SILVA reference database (Pruesse et al., 2007). Sequences were subsequently processed through the R package decontam (Davis et al., 2018) to remove any suspected contaminants that were found in the negative control samples. Potential contaminants were detected with the “prevalence” method, in which presence/absence of sequences in negative controls is compared to that of real samples. The R package phyloseq (McMurdie and Holmes, 2013) was used to facilitate data processing. The abundance counts were normalized to simple proportions within each sample to compare matched data to the bacterial microbiome profiles derived from the metatranscriptomics method.
Host response to RSV infection
The reads identified as originating from human transcripts were mapped to the human genome (hg19) using HISAT2 (Kim et al., 2015). The read counts for genomic features were quantified using HTSeq (Anders et al., 2015). The feature counts of all the samples were combined into a single matrix using a custom R script. Differential expression analysis was performed by comparing RSV-ARI and HC group samples using the DESeq2 package (Love et al., 2014). Genes with a significant log2 fold change with an adjusted p-value <0.05 were treated as differentially expressed. The lists of differentially expressed genes for each group were analyzed for enrichment of Reactome Human Pathways using Enrichr (Kuleshov et al., 2016), and were deemed significant when FDR < 0.05.
Acknowledgment
This work was supported by funds from the National Institute of Allergy and Infectious Diseases (under award nos. 1R21AI149262, 1R21AI154016, 1R21AI142321, R21AI142321-01A1S1, U19AI095227, and U19AI110819), and the National Heart, Lung, and Blood Institute (1R01HL146401), the Edward P. Evans Foundation, the Vanderbilt Institute for Clinical and Translational Research (grant support from the National Center for Advancing Translational Sciences under award no. UL1TR000445), start-up funds from Vanderbilt University Medical Center awarded to S.R.D., and the Vanderbilt Technologies for Advanced Genomics Core (grant support from the National Institutes of Health under award nos. UL1RR024975, P30CA68485, P30EY08126, and G20 RR030956).
Author contributions
N.H. and N.G.B. managed the study participants’ consent and sample collection. S.V.R. and M.H.S. developed and optimized the lab protocols. S.V.R., A.M., H.H.B., and R.McH. performed the experiments. S.B.P. and S.V.R. developed virome and bacterial profiling computational pipeline with inputs from S.R.D. and S.Y. S.V.R., S.B.P., M.H.S., and S.Y. performed computational analysis. S.V.R., S.B.P., and S.R.D. wrote the manuscript with input from S.Y., M.H.S., N.G.B., C.R.-S., and N.H. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100091.
Contributor Information
Seesandra V. Rajagopala, Email: s.v.rajagopala@vumc.org.
Suman R. Das, Email: suman.r.das@vumc.org.
Supplemental information
Data and code availability
-
•
The assembled viral genomes are submitted to GenBank, and the raw reads are submitted to SRA, which will be accessible via BioProject accession number PRJNA671738 upon acceptance of the manuscript for publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abbas A.A., Taylor L.J., Dothard M.I., Leiby J.S., Fitzgerald A.S., Khatib L.A., Collman R.G., Bushman F.D. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract associated with periodontitis and critical illness. Cell Host Microbe. 2019;25:719–729 e714. doi: 10.1016/j.chom.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Ali G.S., Mehta R.S., Lloyd-Price J., Mallick H., Branck T., Ivey K.L., Drew D.A., DuLong C., Rimm E., Izard J., et al. Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat. Microbiol. 2018;3:356–366. doi: 10.1038/s41564-017-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes K.H., Fachi J.L., de Paula R., da Silva E.F., Pral L.P., Dos Santos A.A., Dias G.B.M., Vargas J.E., Puga R., Mayer F.Q., et al. Microbiota-derived acetate protects against respiratory syncytial virus infection through a GPR43-type 1 interferon response. Nat. Commun. 2019;10:3273. doi: 10.1038/s41467-019-11152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balique F., Colson P., Barry A.O., Nappez C., Ferretti A., Moussawi K.A., Ngounga T., Lepidi H., Ghigo E., Mege J.L., et al. Tobacco mosaic virus in the lungs of mice following intra-tracheal inoculation. PLoS One. 2013;8:e54993. doi: 10.1371/journal.pone.0054993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y.a.H.Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995;57:289–300. doi: 10.2307/2346101. [DOI] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzas M.L., Oliveira J.R., Fukutani K.F., Borges I.C., Barral A., Van der Gucht W., Wollants E., Van Ranst M., de Oliveira C.I., Van Weyenbergh J., et al. Respiratory syncytial virus a and b display different temporal patterns in a 4-year prospective cross-sectional study among children with acute respiratory infection in a tropical city. Medicine (Baltimore) 2016;95:e5142. doi: 10.1097/MD.0000000000005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brealey J.C., Chappell K.J., Galbraith S., Fantino E., Gaydon J., Tozer S., Young P.R., Holt P.G., Sly P.D. Streptococcus pneumoniae colonization of the nasopharynx is associated with increased severity during respiratory syncytial virus infection in young children. Respirology. 2018;23:220–227. doi: 10.1111/resp.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser F.P., Baker D.N., Salzberg S.L. KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018;19:198. doi: 10.1186/s13059-018-1568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister J.R., Ako-Adjei D., Bao Y., Blinkova O. NCBI viral genomes resource. Nucleic Acids Res. 2015;43:D571–D577. doi: 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B., Rood J., Singer E. BBMerge—accurate paired shotgun read merging via overlap. PLoS One. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin S.A., Nolan T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004;15:155–166. [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J., Holmes S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C.Y., Miller S.A. Clinical metagenomics. Nat. Rev. Genet. 2019;20:341–355. doi: 10.1038/s41576-019-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.Y., Qiu X., McCall M.N., Wang L., Corbett A., Holden-Wiltse J., Slaunwhite C., Grier A., Gill S.R., Pryhuber G.S., et al. Airway gene expression correlates of RSV disease severity and microbiome composition in infants. J. Infect. Dis. 2020;223:1639–1649. doi: 10.1093/infdis/jiaa576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.Y., Qiu X., Wang L., Bhattacharya S., Lofthus G., Corbett A., Holden-Wiltse J., Grier A., Tesini B., Gill S.R., et al. The healthy infant nasal transcriptome: a benchmark study. Sci. Rep. 2016;6:33994. doi: 10.1038/srep33994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapat C., Oshitani H. Novel insights into human respiratory syncytial virus-host factor interactions through integrated proteomics and transcriptomics analysis. Expert Rev. Anti Infect. Ther. 2016;14:285–297. doi: 10.1586/14787210.2016.1141676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis N.M., Proctor D.M., Holmes S.P., Relman D.A., Callahan B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Steenhuijsen Piters W.A., Heinonen S., Hasrat R., Bunsow E., Smith B., Suarez-Arrabal M.C., Chaussabel D., Cohen D.M., Sanders E.A., Ramilo O., et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 2016;194:1104–1115. doi: 10.1164/rccm.201602-0220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Spiro D. Advancing full length genome sequencing for human RNA viral pathogens. Future Virol. 2009;4:47–53. doi: 10.2217/17460794.4.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher T.W. Functions of the 3’-untranslated regions of positive strand RNA viral genomes. Annu. Rev. Phytopathol. 1999;37:151–174. doi: 10.1146/annurev.phyto.37.1.151. [DOI] [PubMed] [Google Scholar]
- Ederveen T.H.A., Ferwerda G., Ahout I.M., Vissers M., de Groot R., Boekhorst J., Timmerman H.M., Huynen M.A., van Hijum S., de Jonge M.I. Haemophilus is overrepresented in the nasopharynx of infants hospitalized with RSV infection and associated with increased viral load and enhanced mucosal CXCL8 responses. Microbiome. 2018;6:10. doi: 10.1186/s40168-017-0395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa E.A., McIver L.J., Rahnavard G., Thompson L.R., Schirmer M., Weingart G., Lipson K.S., Knight R., Caporaso J.G., Segata N., et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods. 2018;15:962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galen S.C., Borner J., Williamson J.L., Witt C.C., Perkins S.L. Metatranscriptomics yields new genomic resources and sensitive detection of infections for diverse blood parasites. Mol. Ecol. Resour. 2020;20:14–28. doi: 10.1111/1755-0998.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamino-Arroyo A.E., Moreno-Espinosa S., Llamosas-Gallardo B., Ortiz-Hernandez A.A., Guerrero M.L., Galindo-Fraga A., Galan-Herrera J.F., Prado-Galbarro F.J., Beigel J.H., Ruiz-Palacios G.M., et al. Epidemiology and clinical characteristics of respiratory syncytial virus infections among children and adults in Mexico. Influenza Other Respir. Viruses. 2017;11:48–56. doi: 10.1111/irv.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J.L., Tan le V., Kuhnert D., Halpin R.A., Lin X., Simenauer A., Akopov A., Das S.R., Stockwell T.B., Shrivastava S., et al. Phylodynamics of enterovirus A71-associated hand, foot, and mouth disease in Viet Nam. J. Virol. 2015;89:8871–8879. doi: 10.1128/JVI.00706-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Miller S., Chiu C.Y. Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Allen E.M., Miller W.A. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol. Cell. 2001;7:1103–1109. doi: 10.1016/s1097-2765(01)00252-0. [DOI] [PubMed] [Google Scholar]
- Houldcroft C.J., Beale M.A., Breuer J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017;15:183–192. doi: 10.1038/nrmicro.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwane M.K., Edwards K.M., Szilagyi P.G., Walker F.J., Griffin M.R., Weinberg G.A., Coulen C., Poehling K.A., Shone L.P., Balter S., et al. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus, and parainfluenza viruses among young children. Pediatrics. 2004;113:1758–1764. doi: 10.1542/peds.113.6.1758. [DOI] [PubMed] [Google Scholar]
- Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. doi: 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.H., Lee H.K. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. 2014;47:184–191. doi: 10.5483/BMBRep.2014.47.4.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich J.J., Westcott S.L., Baxter N.T., Highlander S.K., Schloss P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner J.T., Beitzel B., Chain P.S., Davenport M.G., Donaldson E.F., Frieman M., Kugelman J.R., Kuhn J.H., O'Rear J., Sabeti P.C., et al. Standards for sequencing viral genomes in the era of high-throughput sequencing. mBio. 2014;5 doi: 10.1128/mBio.01360-14. e01360–01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier C., Kalantar K.L., Moazed F., Wilson M.R., Crawford E.D., Deiss T., Belzer A., Bolourchi S., Caldera S., Fung M., et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc. Natl. Acad. Sci. U S A. 2018;115:E12353–E12362. doi: 10.1073/pnas.1809700115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Fu X., Ma J., Zhang J., Hu Y., Dong W., Wan Z., Li Q., Kuang Y.Q., Lan K., et al. Altered respiratory virome and serum cytokine profile associated with recurrent respiratory tract infections in children. Nat. Commun. 2019;10:2288. doi: 10.1038/s41467-019-10294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhang P., Wang C., Han C., Meng J., Liu X., Xu S., Li N., Wang Q., Shi X., et al. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J. Biol. Chem. 2013;288:16225–16234. doi: 10.1074/jbc.M113.454538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan H.H., Medzhitov R. Food fight: role of itaconate and other metabolites in antimicrobial defense. Cell Metab. 2016;24:379–387. doi: 10.1016/j.cmet.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhi S.A., Klugman K.P. In: Disease and Mortality in Sub-saharan Africa. 2nd. Jamison D.T., Feachem R.G., Makgoba M.W., Bos E.R., Baingana F.K., Hofman K.J., Rogo K.O., editors. The International Bank for Reconstruction and Development / The World Bank; Washington, DC: 2006. Acute respiratory infections; pp. 149–162. [PubMed] [Google Scholar]
- Mariani T.J., Qiu X., Chu C., Wang L., Thakar J., Holden-Wiltse J., Corbett A., Topham D.J., Falsey A.R., Caserta M.T., et al. Association of dynamic changes in the CD4 T-cell transcriptome with disease severity during primary respiratory syncytial virus infection in young infants. J. Infect. Dis. 2017;216:1027–1037. doi: 10.1093/infdis/jix400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A., Cordes T., Ghelfi J., Pailot A., Reiling N., Goldmann O., Binz T., Wegner A., Tallam A., Rausell A., et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. U S A. 2013;110:7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese A., Mende D.R., Paoli L., Salazar G., Ruscheweyh H.J., Cuenca M., Hingamp P., Alves R., Costea P.I., Coelho L.P., et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019;10:1014. doi: 10.1038/s41467-019-08844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A.B., Oliver B.G., Glanville A.R. Translational aspects of the human respiratory virome. Am. J. Respir. Crit. Care Med. 2016;194:1458–1464. doi: 10.1164/rccm.201606-1278CI. [DOI] [PubMed] [Google Scholar]
- Nakamura S., Yang C.S., Sakon N., Ueda M., Tougan T., Yamashita A., Goto N., Takahashi K., Yasunaga T., Ikuta K., et al. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS One. 2009;4:e4219. doi: 10.1371/journal.pone.0004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.I., Wentworth D.E., Das S.R., Sreevatsan S., Killian M.L., Nolting J.M., Slemons R.D., Bowman A.S. Evolutionary dynamics of influenza A viruses in US exhibition swine. J. Infect. Dis. 2016;213:173–182. doi: 10.1093/infdis/jiv399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols W.G., Peck Campbell A.J., Boeckh M. Respiratory viruses other than influenza virus: impact and therapeutic advances. Clin. Microbiol. Rev. 2008;21:274–290. doi: 10.1128/CMR.00045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noell K., Kolls J.K. Further defining the human virome using NGS: identification of redondoviridae. Cell Host Microbe. 2019;25:634–635. doi: 10.1016/j.chom.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallen M.J. Diagnostic metagenomics: potential applications to bacterial, viral and parasitic infections. Parasitology. 2014;141:1856–1862. doi: 10.1017/S0031182014000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen E., Koopmans M., Go U., Hamer D.H., Petrosillo N., Castelli F., Storgaard M., Al Khalili S., Simonsen L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020;20:e238–e244. doi: 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E., Quast C., Knittel K., Fuchs B.M., Ludwig W., Peplies J., Glockner F.O. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala S.V., Singh H., Patel M.C., Wang W., Tan Y., Shilts M.H., Hartert T.V., Boukhvalova M.S., Blanco J.C.G., Das S.R. Cotton rat lung transcriptome reveals host immune response to respiratory syncytial virus infection. Sci. Rep. 2018;8:11318. doi: 10.1038/s41598-018-29374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovan N., Duraisamy R., Desnues C. Metagenomics and the human virome in asymptomatic individuals. Annu. Rev. Microbiol. 2016;70:125–141. doi: 10.1146/annurev-micro-102215-095431. [DOI] [PubMed] [Google Scholar]
- Rosas-Salazar C., Shilts M.H., Tovchigrechko A., Chappell J.D., Larkin E.K., Nelson K.E., Moore M.L., Anderson L.J., Das S.R., Hartert T.V. Nasopharyngeal microbiome in respiratory syncytial virus resembles profile associated with increased childhood asthma risk. Am. J. Respir. Crit. Care Med. 2016;193:1180–1183. doi: 10.1164/rccm.201512-2350LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Salazar C., Shilts M.H., Tovchigrechko A., Schobel S., Chappell J.D., Larkin E.K., Shankar J., Yooseph S., Nelson K.E., Halpin R.A., et al. Differences in the nasopharyngeal microbiome during acute respiratory tract infection with human rhinovirus and respiratory syncytial virus in infancy. J. Infect. Dis. 2016;214:1924–1928. doi: 10.1093/infdis/jiw456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G.A., Colin A.A. Infantile respiratory syncytial virus and human rhinovirus infections: respective role in inception and persistence of wheezing. Eur. Respir. J. 2015;45:774–789. doi: 10.1183/09031936.00062714. [DOI] [PubMed] [Google Scholar]
- Runtsch M.C., O'Neill L.A. GOTcha: lncRNA-ACOD1 targets metabolism during viral infection. Cell Res. 2018;28:137–138. doi: 10.1038/cr.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell C.D., Unger S.A., Walton M., Schwarze J. The human immune response to respiratory syncytial virus infection. Clin. Microbiol. Rev. 2017;30:481–502. doi: 10.1128/CMR.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia J., Sabbah A., Mgbemena V., Tsai S.Y., Chang T.H., Berton M.T., Morris I.R., Allen I.C., Ting J.P., Bose S. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One. 2012;7:e29695. doi: 10.1371/journal.pone.0029695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencio V., Barthelemy A., Tavares L.P., Machado M.G., Soulard D., Cuinat C., Queiroz-Junior C.M., Noordine M.L., Salome-Desnoulez S., Deryuter L., et al. Gut dysbiosis during influenza contributes to pulmonary pneumococcal superinfection through altered short-chain fatty acid production. Cell Rep. 2020;30:2934–2947 e2936. doi: 10.1016/j.celrep.2020.02.013. [DOI] [PubMed] [Google Scholar]
- Shean R.C., Makhsous N., Stoddard G.D., Lin M.J., Greninger A.L. VAPiD: a lightweight cross-platform viral annotation pipeline and identification tool to facilitate virus genome submissions to NCBI GenBank. BMC Bioinformatics. 2019;20:48. doi: 10.1186/s12859-019-2606-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman O., Cohen-Gihon I., Beth-Din A., Zvi A., Laskar O., Paran N., Epstein E., Stein D., Dorozko M., Wolf D., et al. Identification and genetic characterization of a novel orthobunyavirus species by a straightforward high-throughput sequencing-based approach. Sci. Rep. 2019;9:3398. doi: 10.1038/s41598-019-40036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilts M.H., Rosas-Salazar C., Turi K.N., Rajan D., Rajagopala S.V., Patterson M.F., Gebretsadik T., Anderson L.J., Peebles R.S., Jr., Hartert T.V., et al. Nasopharyngeal Haemophilus and local immune response during infant respiratory syncytial virus infection. J. Allergy Clin. Immunol. 2020;147:1097–1101.e6. doi: 10.1016/j.jaci.2020.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.E., Busse D.C., Binter S., Weston S., Diaz Soria C., Laksono B.M., Clare S., Van Nieuwkoop S., Van den Hoogen B.G., Clement M., et al. Interferon-induced transmembrane protein 1 restricts replication of viruses that enter cells via the plasma membrane. J. Virol. 2019;93 doi: 10.1128/JVI.02003-18. e02003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane A.R., Tian L., Chu C.Y., Qiu X., Wang L., Holden-Wiltse J., Grier A., Gill S.R., Caserta M.T., Falsey A.R., et al. Microbiome-transcriptome interactions related to severity of respiratory syncytial virus infection. Sci. Rep. 2019;9:13824. doi: 10.1038/s41598-019-50217-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbusch M.M.F., Caron M.M.J., Surtel D.A.M., van den Akker G.G.H., van Dijk P.J., Friedrich F., Zabel B., van Rhijn L.W., Peffers M.J., Welting T.J.M. The antiviral protein viperin regulates chondrogenic differentiation via CXCL10 protein secretion. J. Biol. Chem. 2019;294:5121–5136. doi: 10.1074/jbc.RA119.007356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y., Hassan F., Schuster J.E., Simenauer A., Selvarangan R., Halpin R.A., Lin X., Fedorova N., Stockwell T.B., Lam T.T., et al. Molecular evolution and intraclade recombination of enterovirus D68 during the 2014 outbreak in the United States. J. Virol. 2016;90:1997–2007. doi: 10.1128/JVI.02418-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.W., Lam T.T., Zaraket H., Lipkin W.I., Drews S.J., Hatchette T.F., Heraud J.M., Koopmans M.P., investigators I. Global epidemiology of non-influenza RNA respiratory viruses: data gaps and a growing need for surveillance. Lancet Infect. Dis. 2017;17:e320–e326. doi: 10.1016/S1473-3099(17)30238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K., Kar S., Vakakis E., Kotecha S., Triantafilou M. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax. 2013;68:66–75. doi: 10.1136/thoraxjnl-2012-202182. [DOI] [PubMed] [Google Scholar]
- Trompette A., Gollwitzer E.S., Pattaroni C., Lopez-Mejia I.C., Riva E., Pernot J., Ubags N., Fajas L., Nicod L.P., Marsland B.J. Dietary fiber confers protection against flu by shaping Ly6c(–) patrolling monocyte hematopoiesis and CD8(+) T cell metabolism. Immunity. 2018;48:992–1005 e1008. doi: 10.1016/j.immuni.2018.04.022. [DOI] [PubMed] [Google Scholar]
- Turi K.N., Shankar J., Anderson L.J., Rajan D., Gaston K., Gebretsadik T., Das S.R., Stone C., Larkin E.K., Rosas-Salazar C., et al. Infant viral respiratory infection nasal immune-response patterns and their association with subsequent childhood recurrent wheeze. Am. J. Respir. Crit. Care Med. 2018;198:1064–1073. doi: 10.1164/rccm.201711-2348OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villenave R., Broadbent L., Douglas I., Lyons J.D., Coyle P.V., Teng M.N., Tripp R.A., Heaney L.G., Shields M.D., Power U.F. Induction and antagonism of antiviral responses in respiratory syncytial virus-infected pediatric airway epithelium. J. Virol. 2015;89:12309–12318. doi: 10.1128/JVI.02119-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis W.H.K.W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952;47:583–621. [Google Scholar]
- Wang P., Xu J., Wang Y., Cao X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science. 2017;358:1051–1055. doi: 10.1126/science.aao0409. [DOI] [PubMed] [Google Scholar]
- Wylie K.M. The virome of the human respiratory tract. Clin. Chest Med. 2017;38:11–19. doi: 10.1016/j.ccm.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Wang H., Kouadir M., Song H., Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10:128. doi: 10.1038/s41419-019-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zani A., Yount J.S. Antiviral protection by IFITM3 in vivo. Curr. Clin. Microbiol. Rep. 2018;5:229–237. doi: 10.1007/s40588-018-0103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Forst C.V., Gordon A., Gussin G., Geber A.B., Fernandez P.J., Ding T., Lashua L., Wang M., Balmaseda A., et al. Characterization of antibiotic resistance and host-microbiome interactions in the human upper respiratory tract during influenza infection. Microbiome. 2020;8:39. doi: 10.1186/s40168-020-00803-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The assembled viral genomes are submitted to GenBank, and the raw reads are submitted to SRA, which will be accessible via BioProject accession number PRJNA671738 upon acceptance of the manuscript for publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.