

ABSTRACT
Phased siRNAs (phasiRNAs) are a class of small interfering RNAs (siRNAs) which play essential roles in plant development and defence. However, only a few phasiRNAs have been extensively studied due to the difficulties in identifying and characterizing plant phasiRNAs by plant biologists. Herein, we describe a comprehensive and multi-functional web server termed PhasiRNAnalyzer, which is able to identify all crucial components in plant phasiRNA’s regulatory pathway (phase-initiator→PHAS gene→phasiRNA cluster→target gene). Currently, PhasiRNAnalyzer exhibits the following advantages: I) It is the most comprehensive platform which hosts 170 plant species with 256 genome data, 438 cDNA data and 271 degradome data. II) It can identify all crucial components in phasiRNA’s regulatory pathway, and verify the interactions between phasiRNAs and their target genes based on degradome data. III) It can perform differential expression analysis of phasiRNAs on each PHAS gene locus between different samples conveniently. IV) It provides the user-friendly interfaces and introduces several improvements, primarily by making more accurate and efficient analysis when dealing with deep sequencing data. In summary, PhasiRNAnalyzer is a comprehensive and systemic phasiRNA analysis server with high sensitivity and efficiency. It can be freely accessed at https://cbi.njau.edu.cn/PPSA/.
KEYWORDS: Plant, phased siRNA, phase-initiator, pathway, degradome, web server
Introduction
PhasiRNAs are phased, secondary, small interfering RNAs that play crucial roles in a variety of biological processes in plants, including growth and development, stress responses and genome stability [1,2]. Till now, there are numerous studies have uncovered that phasiRNAs are widely existed in plants and they can regulate their target genes by cleaving mRNAs at the post-transcriptional level [3], or directing DNA methylation at transcriptional level [4].
As shown in Figure S1, there are a large number of studies have proven that MIRNA (MIR) genes, encoded by plant genomes, are transcribed by DNA-dependent RNA Polymerase II (Pol II) [5] and then undergo a series of transcriptional modifications to form primary miRNA (pri-miRNA) [6]. Pri-miRNA is mainly cleaved by Dicer-like 1 (DCL1) to generate precursor miRNA (pre-miRNA), and the processing of the pre-miRNA yields an imperfect miRNA/miRNA* duplex [7]. This duplex is methylated by the small RNA methyltransferase HUA Enhancer 1 (HEN1) [8], and miRNA* is then degraded, leaving only the mature miRNA. The resulting mature miRNA can regulate gene expression via incorporation into Argonaute 1 (AGO1) and other proteins to form the RNA induced silencing complex (RISC) [9]. In some cases, the mature miRNA can trigger the production of secondary phasiRNAs from its target, and these secondary phasiRNAs can enhance the initial silencing signal, or affect other target genes [10,11]. A phase-initiator (usually a 22-nt miRNA) cleaves PHAS gene by binding the different AGOs and triggers two reported pathways [12] of ‘one-hit’ and ‘two-hit’. Then, RNA-dependent RNA-polymerase 6 (RDR6) and Suppressor of Gene Silencing 3 (SGS3) process the cleavage products into double-stranded RNAs (dsRNAs), which are cleaved by DCL4/DCL5 to generate 21-/24-nt phasiRNA clusters, respectively [13]. Finally, the 21-nt phasiRNAs direct the cleavages of target mRNAs [14], and 24-nt phasiRNAs usually direct DNA methylation [15]. In rice, ‘one-hit’ pathway is typified by a single target site of osa-miR2118/osa-miR2275 that results in processing downstream of the target transcript into 21-nt/24-nt phasiRNAs [16,17] and ‘two-hit’ pathway is typified by two target sites of osa-miR390 that results in processing upstream of the 3ʹ site to generate 21-nt phasiRNAs [18].
To date, several classic regulatory pathways for plant phasiRNAs which we introduced above have been well demonstrated in many studies [19–21]. However, the significance of most phasiRNAs is still unclear due to the difficulties in identifying and characterizing phasiRNAs, especially for plant biologists without bioinformatics experiences. Since phasiRNAs are critical in plants, it is urgent to develop a fast and convenient genome-wide phasiRNA’s regulatory pathway identification strategy to get a better understanding of phasiRNAs. To our knowledge, pssRNAMiner [22] is the only web server for phasiRNA cluster prediction. However, it cannot provide accurate downstream analysis for putative phasiRNAs and the algorithm is not optimized for the deep sequencing data. Although PhaseTank [23] and PHASIS [24] can analyse phasiRNAs systemically, programming skills are still required to successfully run standalone programs. Moreover, data collection and pre-processing are laborious and time-consuming, and the results are somehow uninformative. TasiRNAdb [25] is the only database for retrieving phasiRNA’s regulatory pathways, but it is limited by the number of plant species and it cannot perform customized analysis. Compared to these existing tools, PhasiRNAnalyzer can not only identify all important components in phasiRNA’s regulatory pathway and verifies the interactions between phasiRNAs and their target genes (PTIs) by using all available degradomes at a time, but also perform the differential expression analysis of phasiRNAs on each PHAS gene locus and make the phasiRNA’s regulatory networks (PRNs). Moreover, it also provides the sequence and graphical visualization for the analysis results. Currently, PhasiRNAnalyzer supports 170 plant species with 256 genome data, 438 cDNA data and 271 degradome data, which is the most comprehensive web server for phasiRNA’s regulatory pathway.
Results
Workflow and user interfaces
As shown in the Fig. 1, PhasiRNAnalyzer provides the flexible workflow composed of three stages, including I) Prediction of phasiRNA clusters, PHAS genes and phase-initiators (optional) by user-submitted small RNA sequences. II) Prediction and verification of phasiRNAs and their target genes by user-submitted or pre-loaded degradome data. III) Downloading and visualization function for the analysis results. Furthermore, the ‘Retrieve’ module allows users to trace their results with a unique job ID at a later time and the results will be retained in our back-end database for 30 days. In the ‘Scripts’ module, we also provided the standalone programs of PhasiRNAnalyzer for offline usage.
Figure 1.
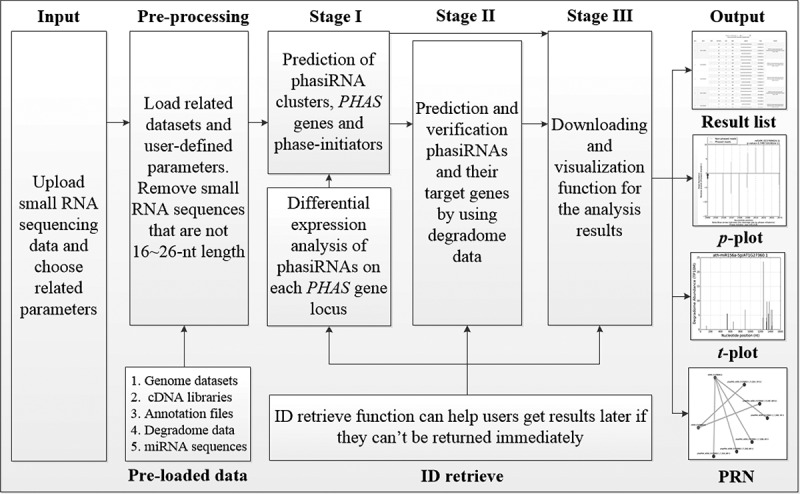
The architecture of PhasiRNAnalyzer
As shown in Fig. 2A,B, PhasiRNAnalyzer takes small RNA sequences as input and output a comprehensive list of predicted PHAS genes, phasiRNA clusters and phase-initiators (optional). Fig. 2C shows that PhasiRNAnalyzer uses degradome data to verify the PTIs and outputs an informative list as well. For the output data, we presented an example of visualization function in PhasiRNAnalyzer, where phasiRNA-plot (p-plot) figure (Fig. 2D) presents the alignment of phasiRNA clusters with their phase-initiator cleavage sites on the putative PHAS genes; sequence alignment figure (Fig. 2E) shows the details of sequence alignment between phasiRNAs and PHAS genes; degradome-based phasiRNA’s regulatory network (Fig. 2F) presents the reliable networks of verified PTIs based on degradome data; target-plot (t-plot) figure (Fig. 2G) visualizes the valid cleavage sites between phasiRNAs and their target genes. Thus, PhasiRNAnalyzer can graphically visualize the results to help researchers observe the phasiRNA’s regulatory pathway in a convenient way.
Figure 2.
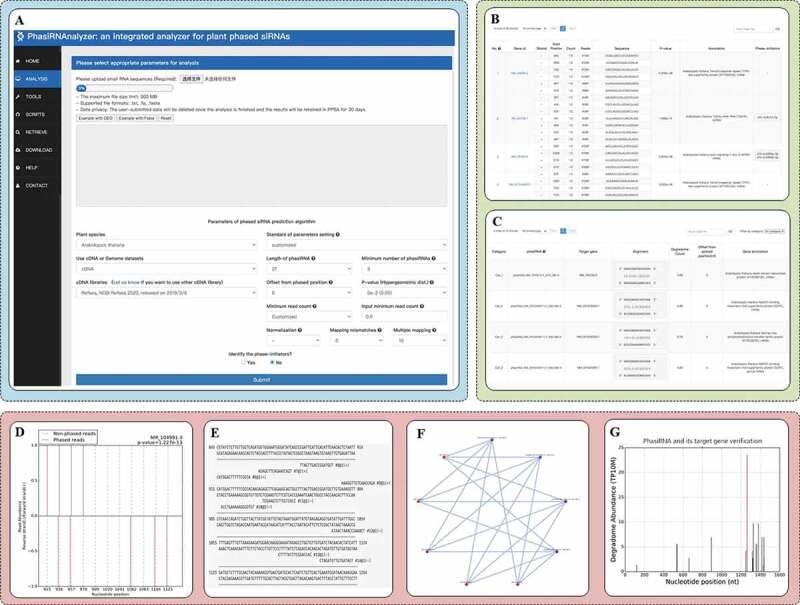
The user-interfaces of PhasiRNAnalyzer
A. Analysis function in PhasiRNAnalyzer. B. The result list of phasiRNA, PHAS gene prediction and phase-initiator. C. The result list of phasiRNA and its target gene. D.-G. Examples of phase-plot figure, sequence alignment figure, degradome-based phasiRNA’s regulatory network and target-plot figure.
Performance
To evaluate the performance of PhasiRNAnalyzer, we collected six representative Oryza sativa’s small RNA-seq datasets across various tissues with different sequencing depths from NCBI GEO database [26] (Table S1), and compared the performance of PhasiRNAnalyzer with pssRNAMiner [22] comprehensively. As shown in Table S2, the performance comparison demonstrated that PhasiRNAnalyzer significantly reduced the running time of predictions. It is worth mentioning that we manually filtered the reads by removing the lowly expressed reads (Counts Per Million<1) before submitting to pssRNAMiner, otherwise, the running time of pssRNAMiner will be much longer. For example, pssRNAMiner costed ~60 hours to identify phase-initiators based on GSM562947, while PhasiRNAnalyzer decreased the running time to ~1 hour by using high-performance computational pipelines; we also found that pssRNAMiner could not handle the deep sequencing small RNA-seq datasets (e.g. GSM686040 and GSM913525) due to the maximum file size and sequence number limit. Several improvements have been applied into PhasiRNAnalyzer (Table S3), including flexible thresholds for data pre-processing, flexible selections of cDNA datasets and friendly size limit API for data uploading, which makes PhasiRNAnalyzer suitable for deep sequencing small RNA sequences.
As shown in Table S2, we compared the number of predicted phasiRNAs, PHAS genes and phase-initiators between PhasiRNAnalyzer and pssRNAMiner based on rice MSU7.0 cDNA data. The detection rate of pssRNAMiner seemed higher than PhasiRNAnalyzer. In fact, the cut-off setting of p-values and the algorithm of dealing with the redundant phasiRNAs largely affects the number of predicted phasiRNAs. Since we introduced a stricter method to evaluate p-value and an efficient way to merge the redundant phasiRNAs, the detection accuracy of PhasiRNAnalyzer is much increased. In addition, as compared to rice cDNA data MSU7.0, we found that PhasiRNAnalyzer achieves better detection rate based on NCBI RefSeq cDNA data (Table S2), which is, however, not supported by pssRNAMiner. It is suggested that NCBI RefSeq cDNA data may be better for phasiRNA analysis in plants.
Identification of phasiRNA’s regulatory pathway
To identify 21-/24-nt phasiRNA’s regulatory pathway in Oryza sativa, we also used these 6 representative Oryza sativa’s small RNA-seq datasets (Table S1) based on NCBI RefSeq data. The details of parameters setting can be found in Table 1Table 2.
Table 1.
Identification of phasiRNA’s regulatory pathways in rice
| Accession ID |
Tissue | 21-nt phasiRNA |
24-nt phasiRNA |
||||
|---|---|---|---|---|---|---|---|
| #phasiRNA | #PHAS | Initiator | #phasiRNA | #PHAS | Initiator | ||
| GSM562947 | Panicle | 2,284 | 163 | miR156k, miR156l-5p, miR390-5p, miR1432-3p, miR1858a/b miR2118a-r, miR2876-3p, miR2925, miR6254 |
85 | 12 | miR167d-j, miR2275a, miR2275b, miR2275d |
| GSM686040 | Callus | 58 | 5 | miR390-5p | 0 | 0 | N/A |
| GSM816693 | Seedling | 57 | 6 | miR390-5p | 4 | 1 | N/A |
| GSM816705 | Root | 54 | 7 | miR390-5p | 0 | 0 | N/A |
| GSM816725 | Shoot | 64 | 6 | miR390-5p | 0 | 0 | N/A |
| GSM913525 | Leaf | 53 | 9 | N/A | 26 | 4 | N/A |
Table 2.
Setting of parameters
| Stage | Name of parameters | Value |
|---|---|---|
| PhasiRNA prediction | Length of phasiRNA | 21-/24-nt |
| Minimum number of phasiRNAs | 4 | |
| P-value | 1E-3 | |
| Offset of phased positions | 0 | |
| Mapping mismatches | 0 | |
| Minimum read count of small RNA-seq | ≥1 CPM | |
| Phase-initiator prediction | Mismatches in seed region | 0 |
| Mismatches in total region | 0 | |
| Expectation | 6.0 | |
| Cleavage site | 9–11th | |
| Distance from start site (multiple of 21-nt) | 1–5 | |
| Source of phase-initiator | 738 mature miRNAs from miRBase |
As shown in Table 1, we found that 21- and 24-nt phasiRNAs are much more abundant in panicle than other tissues in rice, suggesting that the productions of phasiRNAs are generally less in vegetative organs and they are predominantly expressed in reproductive organs, including panicle, inflorescence and anther, etc, which is consisted with the previous reports [14,16,27]. Moreover, we noticed that 21-nt phasiRNAs are more enriched in rice compared to 24-nt phasiRNAs across various rice tissues. Similar to miRNAs, the 21-nt phasiRNAs negatively regulate their target genes by cleaving mRNAs in complementary region at the post-transcriptional level [14,28]. Thus, this is a general gene regulation mechanism across different rice tissues, especially in reproductive organs. However, 24-nt phasiRNAs, which are a hallmark of RNA-directed DNA methylation in plants, usually function in specific tissues and stages [29], suggesting that 21-nt phasiRNAs are more widely present compared to 24-nt phasiRNAs in rice. Moreover, the phase-initiator osa-miR2118 family members [14] for producing 21-nt phasiRNAs and osa-miR2775 family members for producing 24-nt phasiRNAs [27] are both specifically identified in panicle, suggesting that osa-miR2118 and osa-miR2775 families play specific regulatory functions in development of reproductive organs by initiating secondary phasiRNAs in rice [29, 30].
To reveal the functions of identified phasiRNAs, we further investigated the target genes of 21-nt phasiRNAs by using three panicle samples including GSM562947, GSM648141 and GSM816732 (Table S1). We noticed that ≥85% identified PHAS genes are long non-coding RNAs (lncRNAs) and ~90 high reliable verified target genes of 21-nt phasiRNAs have been commonly identified based on 11 panicle degradome data (Table S4). These verified target genes are mainly involved in plant growth, defence response, flowering and floral morphogenesis, suggesting that 21-nt phasiRNAs are involved in diverse biological processes in rice. Furthermore, we also identified several reliable phasiRNA’s regulatory pathways to confirm our findings. For examples, osa-miR2118 family members directly cleave lncRNA XR_003239927.1, leading to the productions of 21-nt phasiRNAs, and these 21-nt phasiRNAs were confirmed by multiple degradome data to target XM_015775000.2 gene involving plant heat shock stress response; osa-miR2118 family members are recruited to initiate 21-nt phasiRNA productions from lncRNA XR_003239861.1 and XR_003239936.1, and these 21-nt phasiRNAs regulate the target genes XM_015779296.2 and XM_015789512.2, which encode MADS-box [31] and F-box [32] proteins involving floral morphogenesis.
As shown in Fig. 3, we also compared the expression levels (Counts Per Million≥1) of phasiRNAs with miRNAs and identified non-phased siRNAs (non-phasiRNAs) based on six samples across different rice tissues. The results showed that the expression levels of phasiRNAs were general lower than those of miRNAs, but higher than non-phasiRNAs in most samples, suggesting that 21-nt phasiRNAs, similar to miRNAs, are an important regulatory component in rice.
Figure 3.

Expression of miRNAs and 21-nt phasiRNAs among six samples
Differential expression analysis of phasiRNAs
The most distinctive feature in the ‘Tools’ module is that users can make differential expression analysis of phasiRNAs on each PHAS gene locus between different samples conveniently according to the job IDs. In this function, PhasiRNAnalyzer generated the count matrix of phasiRNAs according to the analysis results and integrated three reliable packages, including GFOLD [33], DESeq2 [34] and edgeR [35] for differential expression analysis based on different samples with/without biological replicates. Moreover, the count matrix of phasiRNAs and non-redundant phasiRNAs’ sequences can be downloaded for further analysis in the result page. We also released the source code for post-processing in the ‘Tools’ module, and it helps users merge the redundant phasiRNAs according to the analysis results.
In this work, we used two groups of samples for control and salt treatment (Table S5) to make the differential expression analysis of phasiRNAs by using PhasiRNAnalyzer. As shown in Figure S2, we found that osa-miR2118 family members directly cleave lncRNA XR_003239906.1 (LOC112937369), leading to the productions of 21-nt phasiRNAs. These 21-nt phasiRNAs are significantly accumulated (fold change>2, p-value<0.05) in control samples than salt-treated samples on the same PHAS gene loci. As shown in Table S6, two 21-nt phasiRNAs were then confirmed by multiple degradome datasets (Table S4) to target XM_015762600.2 (LOC4327387) and XM_015771917.2 (LOC4330345) genes, which encode Osmotin-like Proteins (OLPs) and histone H4. According to the published papers, we found that OLPs are kinds of proteins which were produced during plant adapting to the environmental stress and act as sentinels to confront salt, drought and other stress tolerance [36,37]. We also noticed that histone H4 acetylation is associated with transcriptional activation and is thought to be essential for the response to high salinity stress in plants [38]. Thus, the differential expression analysis in PhasiRNAnalyzer provides a convenient and efficient way to identify the crucial phasiRNAs in plant development and defence responses.
Implementation
We built PhasiRNAnalyzer in the ‘Linux-Apache2-PHP-PostgreSQL’ environment based on high-performance server. This web server has been tested and proven compatible with all major browser environments. The operating system has been installed all the software that necessary for PhasiRNAnalyzer, including Apache 2, PHP 7.0, PostgreSql-10, Biopython 1.7.6, Matplotlib 2.2.5, Numpy 1.16.6 and corresponding extension services. In the meanwhile, Python 2.7.15 and Perl 5.22.1 compilation environment has been installed as well. The user interfaces of PhasiRNAnalyzer is implemented by HTML, CSS, JAVASCRIPT and shell commands are used to execute related scripts in the back-end. PostgreSql-10 is responsible for storing and presenting all the analysis results online. The algorithms of PhasiRNAnalyzer are primarily written in Python and Perl. The Bowtie 1.0.0 [39] and Fasta-36.3.8 g was used for sequence alignment. We also applied the Ajax technology based on JQurey to make the upload function, and implied the file slice technology to improve the upload speed of large files.
Data privacy policy
Once the analysis is accomplished, the web server will delete user-submitted data immediately and we will not keep any user-submitted data or user information. The analysis result will be retained in our back-end database for 30 days for users to retrieve or download, and the scheduled task in our server will clean up the back-end database every 30 days.
Conclusion
In summary, we built a ‘one-click’ web server to identify all important components in phasiRNA’s regulatory pathway. We used many plant resources from different public databases and integrated a variety of functions in PhasiRNAnalyzer. Based on the significance test, a useful tool has been integrated into PhasiRNAnalyzer to conveniently analyse the differentially expressed phasiRNAs on each PHAS gene locus between different samples. Several methods were also proposed to improve the accuracy and efficiency when dealing with deep sequencing data. Moreover, the flexible analysis parameters and visualization function in PhasiRNAnalyzer help users study plant phasiRNAs in a more accurate and convenient way. Together, PhasiRNAnalyzer can be easily used by plant biologists without any bioinformatics experiences and we believe it can significantly accelerate the studies of phasiRNAs in plant community.
Materials and methods
Data collection
As shown in Table S7, all the data pre-loaded in PhasiRNAnalyzer, including genome datasets, cDNA datasets, degradome data (also referred as parallel analysis of RNA ends, PARE) and miRNA sequences were obtained from the reliable and published databases, including NCBI GenBank [40], NCBI RefSeq [41], EnsemblPlants [42], psRNATarget [43], Phytozome [44] and miRBase [45]. Currently, PhasiRNAnalyzer contains 170 plant species with 256 genome data, 438 cDNA data and 271 degradome data. The small RNA-seq data and degradome sequencing data used in this work can be found in Table S1, Table S4 and Table S5.
Parameters of phasiRNA’s regulatory pathway identification
To get reliable predictions, we only kept the small RNA reads with counts per million (CPM) more than 1 and required at least four phasiRNAs in each phasiRNA cluster (Table 2). We also set cut-off of p-value as 1E-3 (0.001) to evaluate all possible phasiRNA clusters.
Pre-processing of small RNA sequences
The hypergeometric distribution method can identify small RNA sequences within putative PHAS genes, but it does not consider the number of small RNA reads in each position. When the depth of sequencing reaches a certain level, small RNA reads can be potentially mapped to many positions of a putative PHAS gene, which may lead to a high false-negative rate. To ensure the accuracy of prediction when dealing with user-submitted deep-sequencing small RNA sequences, we provided flexible thresholds to filter ‘background reads’. For example, the parameter of minimum read count of user-submitted small RNA sequences in stage I contains following options, including 1) Customized: small RNA reads are removed if less than a given number (0 means keep all the reads); 2) The lowest count of mapped reads: small RNA reads are removed if less than the lowest count of unmapped reads; 3) Median count of unmapped reads: small RNA reads are removed if less than the median count of unmapped reads; 4) Median count of mapped reads: small RNA reads are removed if less than the median count of mapped reads; 5) More than 1 after counts per million normalization: small RNA reads are removed if less than the 1 after counts per million normalization.
Identification of PHAS genes and phasiRNAs
To mimic the duplex of phasiRNAs, we established a double-strand structure by multiplying the phasing interval size (21-nt/24-nt) with 11 for calculating small RNA sequences phased in 21-nt or 24-nt intervals [46] (Fig. 4). Due to cleavage of DCLs, we incorporated a 2-bp shift between sense and antisense strand in this structure, and assumed phased positions are ranked on both strands in a phasing manner (every 21- or 24-nt). The double-stranded structure contains 2*m-1 phased positions on the mimic structure, and we define other positions as non-phased positions. We calculated the number of small RNAs which can map to this structure and the number of small RNAs which can align to the phased positions by custom python scripts. We also allowed 1-bp offset in both directions of phased positions.
Figure 4.
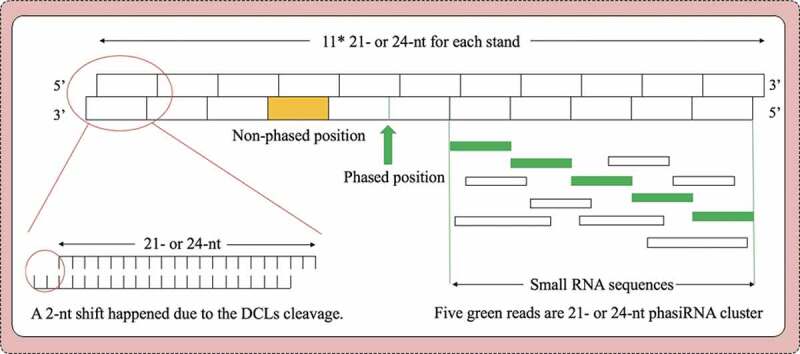
The schematic of phasiRNA and PHAS gene prediction
Moreover, we applied the previously described method of evaluating the p-value of hypergeometric distribution model [46] to evaluate all possible phasiRNA clusters.
where iis the increment of phasiRNA, i= 21 or 24; jis the length of single strand of phasiRNA, j= i *m, m= 11; n is the number of total positions having small RNA hits in j-bp region; k is the number of phased positions having small RNA hits in j-bp region; p is the number of phased positions, p= 21 (p= 2*m-1). (j*2-1)-p is number of non-phased position; j*2-1 is the total length of duplexes phasiRNA; s is offset from phased position.
Prediction of phase-initiators
We introduced the prediction algorithm with improved plant-specific penalty mechanism based on the previous studies [43,47,48] to identify whether the input candidate phase-initiators are the possible triggers of PHAS genes. The seed region is designed between 2 ~ 13-bp and we built a reasonable penalty mechanism and classified the bases paring into 1) perfect match, 2) G:U match, 3) mismatch and 4) insertion or deletion. The expectation value will be calculated according to the penalty rule and a lower expectation value means more similarity between phasiRNAs and the target mRNAs.
In this work, we designed 3 ~ 7 multiples extension (±) relative to start site of predicted phasiRNA cluster and simulated all possible phased positions ranked on the putative PHAS gene. By comparing consensus of cleavage sites and phased positions, we can obtain all valid cleavage sites and identify reliable phase-initiators (Fig. 5). We also allowed 1-nt offset from the cleavages position of candidate phase-initiators in complementary regions.
Figure 5.

The schematic of phase-initiator prediction
Verification of interactions between phasiRNAs and their targets
Since degradome data can provide experimental proofs for identifying verified PTIs [49], we quantify the abundances of degradome fragments mapping on a phasiRNA's target gene [47,50]. After comparing with the flanking regions, if the abundance of a degradome fragmentwhich exactly locates at splicing site (usually 10–11th nucleotides relative to the 5ʹend of phasiRNAs) has an obvious peak (Category I, Cat 1) or is greater than the median abundance value (Category II, Cat 2) of other degradome reads mapped on the target gene, this PTI is considered reliable and PhasiRNAnalyzer can present t-plot (Fig. 2G) according to verified PTI. We also integrated degradome-based validation algorithms into PhasiRNAnalyzer to verify the PTIs efficiently.
Supplementary Material
Acknowledgments
The authors thank the supports from Prof. Scott G. Kennedy and Dr. Jenny Yan from Harvard Medical School and Prof. Yufeng Wu from Center for Bioinformatics, Nanjing Agricultural University.
Funding Statement
This work was supported by the Fundamental Research Funds for the Central Universities [JCQY201901], Jiangsu Collaborative Innovation Center for Modern Crop Production, the Doctoral Thesis Innovation Project and the Cyrus Tang Crop Seed Innovation Center, Nanjing Agricultural University.
Author contributions
Ji Huang designed the study. Yuhan Fei developed the web server. Jiejie Feng, Rui Wang and Baoyi Zhang participated in data processing. Hongsheng Zhang supervised the study. Yuhan Fei and Ji Huang wrote the article. In addition, Jiejie Feng, Rui Wang and Baoyi Zhang contributed equally to this work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Deng P, Muhammad S, Cao M, et al. Biogenesis and regulatory hierarchy of phased small interfering RNAs in plants. Plant Biotechnol J. 2018;16(5):965–975. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu Y, Teng C, Xia R, et al. PhasiRNAs in plants: their biogenesis, genic sources, and roles in stress responses, development, and reproduction. Plant Cell. 2020;32(10):3059–3080. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang YC, Lei MQ, Zhou YF, et al. Reproductive phasiRNAs regulate reprogramming of gene expression and meiotic progression in rice. Nat Commun. 2020;11(1):6031. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dukowic-Schulze S, Sundararajan A, Ramaraj T, et al. Novel meiotic miRNAs and indications for a role of PhasiRNAs in meiosis. Front Plant Sci. 2016;7(1):762. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang J, Mei J, Ren G.. Plant microRNAs: biogenesis, homeostasis, and degradation. Front Plant Sci. 2019;10(1):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11(7):537–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wahid F, Shehzad A, Khan T, et al. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta. 2010;1803(11):1231–1243. . [DOI] [PubMed] [Google Scholar]
- [8].Ji L, Chen X. Regulation of small RNA stability: methylation and beyond. Cell Res. 2012;22(4):624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Diederichs S, Haber DA. Dual role for argonautes in MicroRNA processing and posttranscriptional Regulation of MicroRNA expression. Cell. 2007;131(6):1097–1098. [DOI] [PubMed] [Google Scholar]
- [10].Felippes FFD. Gene regulation mediated by microRNA-Triggered secondary small RNAs in plants. Plants. 2019;8(5):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vazquez F, Hohn T. Biogenesis and biological activity of secondary siRNAs in plant. Scientifica (Cairo). 2013;2013(3):783253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fei Q, Xia R, Meyers BC, et al. Small interfering RNAs in posttranscriptional regulatory networks. Plant Cell. 2013;25(7):2400–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Song X, Li P, Zhai J, et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012;69(3):462–474. . [DOI] [PubMed] [Google Scholar]
- [14].Jiang P, Lian B, Liu C, et al. 21-nt phasiRNAs direct target mRNA cleavage in rice male germ cells. Nature Commun. 2020;11(1):5191. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Atul K, S M M, Kun H, et al. Plant 24-nt reproductive phasiRNAs from intramolecular duplex mRNAs in diverse monocots. Genome Res. 2018;28(9):1333–1344. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Araki S, N T L, Koizumi K, et al. miR2118-dependent U-rich phasiRNA production in rice anther wall development. Nat Commun. 2020;11(1):3115. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fei Q, Yang L, Liang W, et al. Dynamic changes of small RNAs in rice spikelet development reveal specialized reproductive phasiRNA pathways. J Exp Bot. 2016;67(21):6037–6049. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Axtell MJ, Jan C, Rajagopalan R, et al. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127(3):565–577. . [DOI] [PubMed] [Google Scholar]
- [19].Allen E, Xie Z, A M G, et al. microRNA-directed phasing during trans-acting siRNA bio-genesis in plants. Cell. 2005;122(3):207–221. . [DOI] [PubMed] [Google Scholar]
- [20].Yoshikawa M, Peragine A, Park MY, et al. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005;19(18):2164–2175. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vazquez F, Vaucheret H, Rajagopalan R, et al. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell. 2004;16(1):69–79. . [DOI] [PubMed] [Google Scholar]
- [22].Dai X, Zhao PX. pssRNAMiner: a plant short small RNA regulatory cascade analysis server. Nucleic Acids Res. 2008;36(Web Server issue):W114–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guo Q, Qu X, Jin W. PhaseTank: genome-wide computational identification of phasiRNAs and their regulatory cascades. Bioinformatics. 2015;31(2):284–286. [DOI] [PubMed] [Google Scholar]
- [24].Kakrana A, Li P, Patel P, et al. PHASIS: a computational suite for de novo discovery and characterization of phased, siRNA-generating loci and their miRNA triggers. bioRxiv. 2017;158832. [Google Scholar]
- [25].Zhang C, Li G, Zhu S, et al. tasiRNAdb: a database of ta-siRNA regulatory pathways. Bioinformatics. 2014;30(7):1045–1046. . [DOI] [PubMed] [Google Scholar]
- [26].Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41(Database issue):D991–995. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xia R, Chen C, Pokhrel S, et al. 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat Commun. 2019;10(1):627. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Komiya R. Biogenesis of diverse plant phasiRNAs involves an miRNA-trigger and Dicer-processing. J Plant Res. 2017;130(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li C, Xu H, Fu FF, et al. Genome-wide redistribution of 24-nt siRNAs in rice gametes. Genome Res. 2020;30(2):173–184. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ta KN, Sabot F, Adam H, et al. miR2118-triggered phased siRNAs are differentially expressed during the panicle development of wild and domesticated African rice species. Rice. 2016;9(1):10. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ng M, Yanofsky MF. Function and evolution of the plant MADS-box gene family. Nat Rev Genet. 2001;2(3):186–195. [DOI] [PubMed] [Google Scholar]
- [32].Xia R, Ye S, Liu Z, et al. Novel and recently evolved MicroRNA clusters regulate expansive F-BOX gene networks through phased small interfering RNAs in wild diploid strawberry. Plant Physiol. 2015;169(1):594–610. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Feng J, Meyer CA, Wang Q, et al. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics. 2012;28(21):2782–2788. . [DOI] [PubMed] [Google Scholar]
- [34].Love MI, Huber W, Anders S, et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550–571. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumar SA, Kumari PH, Jawahar G, et al. Beyond just being foot soldiers – osmotin like protein (OLP) and chitinase (Chi11) genes act as sentinels to confront salt, drought, and fungal stress tolerance in tomato. Environ Exp Bot. 2016;132(1):53–65. . [Google Scholar]
- [37].Wan Q, Hongbo S, Zhaolong X, et al. Salinity tolerance mechanism of osmotin and osmotin-like proteins: a promising candidate for enhancing plant salt tolerance[J]. Curr Genomics. 2017;18(6):553–556. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patanun O, Ueda M, Itouga M, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates salinity stress in cassava. Front Plant Sci. 2017;7(1):2039. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Langmead B. Aligning short sequencing reads with bowtie. Curr Protoc Bioinformatics. 2010;32(1):11.7.1–11.7.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sayers EW, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res. 2020;48(D1):D84–D86. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yates AD, Achuthan P, Akanni W, et al. Ensembl 2020. Nucleic Acids Res. 2020;48(D1):D682–D688. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dai X, Zhuang Z, Zhao PX. psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018;46(W1):W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Goodstein DM, Shu S, Howson R, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(D1):D1178–D1186. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen HM, Li YH, Wu SH. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A. 2007;104(9):3318–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fei Y, Mao Y, Shen C, et al. WPMIAS: whole-degradome-based plant MiRNA-target interaction analysis server. Bioinformatic. 2020;36(6):1937–1939. . [DOI] [PubMed] [Google Scholar]
- [48].Ma X, Liu C, Gu L, et al. TarHunter, a tool for predicting conserved microRNA targets and target mimics in plants. Bioinformatics. 2017;34(9):1574–1576. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Addo-Quaye C, Eshoo TW, Bartel DP, et al. Endogenous siRNA and miRNA targets identified by sequencing of the arabidopsis degradome. Curr Biol. 2008;18(10):758–762. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fei Y, Wang R, Li H, et al. DPMIND: degradome-based plant miRNA–target interaction and network database. Bioinformatics. 2018;34(9):1618–1620. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.