

Abstract
Characterizing the molecular mechanisms regulating gene expression is crucial for understanding the regulatory processes underlying physiological responses to environmental and developmental signals in eukaryotes. The covalent modification of histones contributes to the compaction levels of chromatin, as well as the recruitment of the transcriptional machinery to specific loci, facilitating metastable changes in gene activity. ChIP-seq (Chromatin Immunoprecipitation followed by sequencing) has become the gold standard method for determining histone modification profiles among different organisms, tissues, and genotypes. In the current protocol, we describe a highly robust method for performing ChIP-seq of histone modifications in Arabidopsis thaliana plantlets. Besides its robustness, this method uses in-house-prepared buffers for chromatin extraction, immunoprecipitation, washing, and elusion, making it cost-effective in contrast to commercial kits.
Keywords: Chromatin, Immunoprecipitation, ChIP-seq, Histone, Arabidopsis thaliana
Background
In eukaryotes, particular histone modifications can be associated with specific transcriptional status (Dong and Weng, 2013). Thus, integrating data on chromatin modification patterns with the transcriptional landscape of specific genotypes and tissues, as well as in response to treatments, can contribute with valuable information for the prediction of transcriptional networks regulating a plethora of physiological processes. ChIP-seq exploits the high specificity of immunoassays with the high-throughput nature of next-generation sequencing (NGS), representing a robust, replicable, and cost-effective technique. This method has been equally adapted for the identification of genomic targets of chromatin-interacting proteins, including transcription factors, allowing expanding its applications to practically any research field of biological sciences.
Materials and Reagents
50 ml Falcon tubes
Plastic film strips (6 × 2.5 cm)
100 μm filters
1.5 ml tube-adapted rotator mixer
-
ChIP-compatible antibodies (references)
Anti-H3K27me3 (Millipore, catalog number: 07-449)
Anti-H3K27me1 (Millipore, catalog number: 07-448)
Anti-H3K9ac (Millipore, catalog number: 07-352)
Anti-H3K14ac (Millipore, catalog number: 07-353)
Anti-H3K4me3 (Millipore, catalog number: 07-473)
Formaldehyde solution 37% (Sigma-Aldrich, catalog number: 252549)
Glycine (Sigma-Aldrich, catalog number: 410225)
Sucrose ≥99.5% (GC) (Sigma-Aldrich, catalog number: S9378)
Tris-HCl (Sigma-Aldrich, catalog number: T3253)
MgCl2 (Sigma-Aldrich, catalog number: M2670)
2-Mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
cOmpleteTM, EDTA-free Protease Inhibitor Cocktail (Roche, Sigma-Aldrich, catalog number: COEDTAF-RO)
TritonTM X-100 (Sigma-Aldrich, catalog number: X100)
Ethylenediaminetetraacetic acid (EDTA) 0.5 M solution (Sigma-Aldrich, catalog number: 03690)
NaCl (Sigma-Aldrich, catalog number: S7653)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 436143)
LiCl (Sigma-Aldrich, catalog number: 310468)
IGEPAL® CA-630 (Supelco, Sigma-Aldrich, catalog number: 56741)
NaHCO3 (Sigma-Aldrich, catalog number: S8875)
Phenol:Chloroform:Isoamyl alcohol (Sigma-Aldrich, catalog number: P2069)
Sodium Acetate Solution 3 M (Sigma-Aldrich, catalog number: 71196)
Dynabeads Protein A or G (ThermoFisher, Invitrogen, catalog number: 10001D/10003D)
GlycoBlueTM Coprecipitant (ThermoFisher, Invitrogen, catalog number: AM9516)
Proteinase K Solution (ThermoFisher, Invitrogen, catalog number: 25530049)
RNase CocktailTM Enzyme Mix (ThermoFisher, Invitrogen, catalog number: AM2286)
QubitTM dsDNA HS Assay Kit (ThermoFisher, Invitrogen, catalog number: Q32851)
Glycerol
Liquid nitrogen
100% ethanol, absolute
Sterile Milli-Q water
Extraction Buffer 1 (see Recipes)
Extraction Buffer 2 (see Recipes)
Extraction Buffer 3 (see Recipes)
Nuclei Lysis Buffer (see Recipes)
ChIP Dilution Buffer (see Recipes)
Low Salt Wash Buffer (see Recipes)
High Salt Wash Buffer (see Recipes)
LiCl Wash Buffer (see Recipes)
TE Buffer (see Recipes)
-
Elution Buffer (see Recipes)
Solutions for buffer preparation (see Recipes)
Glycine 2 M (autoclave)
Tris-HCl pH 8 1 M (autoclave)
Tris-HCl pH 6.5 1 M (autoclave)
Sucrose 2 M (autoclave)
MgCl2 1 M (autoclave)
NaCl 5 M (autoclave)
LiCl 4 M (autoclave)
Igepal CA-630 10% (do not autoclave)
Triton X-100 20% (do not autoclave)
Glycerol 30% (do not autoclave)
Equipment
Vacuum Desiccator
Mortars and pestles
Funnel
Refrigerated Microcentrifuge
Small paintbrush
Focused-Ultrasonicator (Covaris, model: S220)
Safe-Lock microcentrifuge tubes (Eppendorf®)
Water bath or heat block
Agarose for electrophoresis gel
Magnetic rack for 1.5 ml tubes
Mini spin centrifuge
Qubit Fluorometer (ThermoFisher, Invitrogen)
Software
The pipeline used for ChIP-seq analysis is presented in the original paper where this method has been used (Antunez- Sanchez et al., 2020 : https://elifesciences.org/articles/58533).
Procedure
A schematic diagram depicting the whole experimental procedure is shown in Figure 1 .
Figure 1. Experimental overview. A.
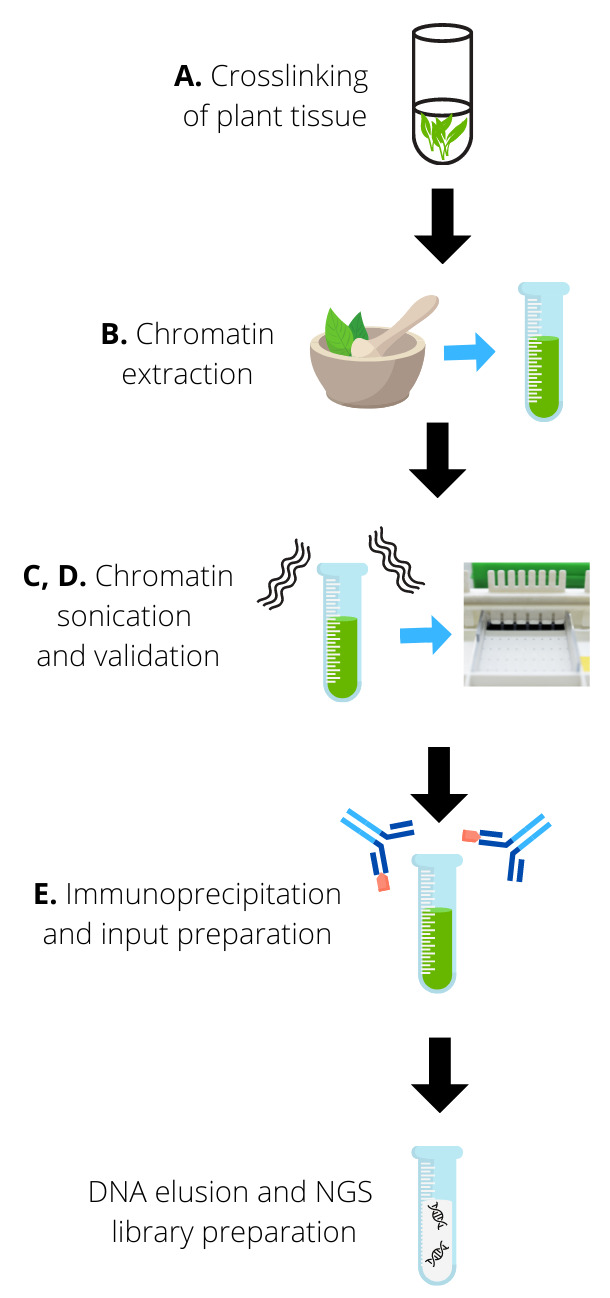
Plant tissue is crosslinked to preserve protein-DNA interactions. B. After crosslinking, chromatin is extracted from the tissue through grinding and using a series of extraction buffers. C-D. Extracted chromatin is sonicated to obtain DNA fragments of the desired size (150-500 bp). Sonication is validated by visualizing the DNA in an agarose gel. E. Sonicated chromatin is immunoprecipitated with the chosen antibodies and non-enriched chromatin washed with a series of washing buffers. The input is prepared. Finally, immunoprecipitated DNA is de-crosslinked from interacted proteins and eluted. Eluted DNA can be used for NGS library preparation.
-
Crosslinking of plant tissue (perform under extraction hood)
Collect 14-day-old plantlets grown in ½ MS. Add approximately 3 g of tissue to a 50 ml Falcon tube containing 36 ml of water (divide samples in different tubes if necessary).
Add 1 ml of 37% Formaldehyde stock to each Falcon tube (for a final concentration of 1%).
Close tubes and invert several times to mix.
Remove tube lids. Add one filmstrip to each tube, pushing it down until it touches the top of the solution (Figure 2). This prevents the plants from floating out of the liquid during the vacuum infiltration.
Rapidly proceed to vacuum infiltrate the samples for 15 min.
After 15 min, add 2.5 ml of the glycine 2 M solution, close the tubes, and mix by inversion.
Open the tubes and vacuum infiltrate for an additional 5 min (to quench the formaldehyde).
Strain the samples to remove the crosslinking solution, disposing of it in the appropriate chemical waste container.
Rinse with running distilled water until the crosslinking solution is completely washed away.
Remove the excess water from each sample by placing it on absorbing paper and pressing it gently.
Place the crosslinked tissue on an aluminum foil square and fold it to create a pouch. Label each sample and snap freeze them in liquid nitrogen.
If desired, the experiment can be stopped at this step and samples stored at -80°C. Otherwise, continue to chromatin extraction.
-
Chromatin extraction (perform on ice)
Cool-down Falcon- and Eppendorf-adapted centrifuges to 4°C.
Prepare Extraction Buffer 1, Extraction Buffer 2, and Extraction Buffer 3 (these buffers can be stored at 4°C prior to the addition of β-mercaptoethanol and protease inhibitors). Prepare Nuclei Lysis buffer (prepare fresh each time).
Add β-mercaptoethanol and protease inhibitors to Extraction Buffer 1, Extraction Buffer 2, and Extraction Buffer 3 to obtain the desired final concentrations (see materials and reagents).
Prepare and label a 50 ml Falcon tube per sample, remove the cap, and place it on ice.
Add liquid nitrogen to a mortar to cool it down. Grind samples thoroughly with mortar and pestle, adding liquid nitrogen constantly to avoid thawing. As soon as the sample is completely ground, place the powdered tissue in the labeled Falcon tube, close the tube, and place it in liquid nitrogen.
As soon as all samples are ground, place falcons with powdered tissue on ice and add 25 ml of Extraction Buffer 1 to each tube. Mix samples slowly by inversion until the solution is homogeneous; open the tubes several times during this step to avoid gas accumulation.
Prepare and label a new set of 50 ml Falcon tubes on ice. Filter each sample into a new Falcon using 100 μm-metal filters placed in a funnel (rinse the funnel and filter between each sample with distilled water).
Centrifuge filtrates at 1,500 × g for 15 min in a pre-cooled centrifuge.
Gently remove the supernatant with a 25 ml pipette or by slowly inverting the tubes on the appropriate chemical waste container. Be careful not to lose pellet material.
Carefully add 20 ml of cold Extraction Buffer 2 to each tube and resuspend the pellet gently using a paintbrush. After most of the pellet is resuspended, close the Falcon tubes, invert slowly, and incubate on ice for 5-10 min. Wash the paintbrush between samples.
Centrifuge at 1,500 × g for 10 min in a pre-cooled centrifuge (4°C).
Gently remove supernatant using a 25 ml pipette without disturbing the pellet.
Resuspend the pellet in 500 μl of Extraction Buffer 3 (stir with a paintbrush) and gently pipet up and down a few times, avoiding creating air bubbles (samples are delicate). If samples are too thick after resuspending them, add another 500 μl of Extraction Buffer 3.
Prepare two 1.5 ml Eppendorf tubes per sample and place them on ice. Add 500 μl of Extraction Buffer 3 on each tube (if an extra volume of Extraction Buffer 3 has been added on Step B13, prepare one extra Eppendorf tube/sample).
Delicately place each sample from step 13 on top of the two (or three) tubes prepared on step 14 (containing 500 μl of Extraction Buffer 3). The buffer will be used to create a sugar gradient that will allow filtering the samples.
Centrifuge at 16,000 × g for 1 h in a pre-cooled centrifuge (4°C).
Prepare fresh Nuclei Lysis (can be kept ON or for a few days in the fridge) and ChIP dilution Buffer (can be kept a few weeks in the fridge).
Discard the supernatant from Step B16.
Resuspend each pellet in 300 μl of Nuclei Lysis Buffer (if the sample is too viscous, add 100 μl extra).
Regroup all the samples for the same genotype/treatment in a single cold 1.5 ml Eppendorf tube. If the final volume of chromatin per sample is less than 1 ml, complete to this volume with Nuclei Lysis Buffer.
If desired, the experiment can be stopped at this step and samples stored at -20°C. Otherwise, continue directly to chromatin sonication.
-
Chromatin sonication (perform all steps on ice)
Turn on Covaris Sonicator (it takes 20-30 min to cool down and degas).
Transfer 1 ml of chromatin in Covaris 1 ml tubes with AFA fiber.
Sonicate samples (program: Peak Incident Power = 175 W, Duty Factor = 20, cycle/burst = 200, and duration = 5 min).
Keep samples in Covaris tubes on ice and proceed to validate the chromatin sonication. Sonicated chromatin with debris can be stored at -20°C.
-
Validation of sonication quality (perform all steps on ice)
Place 60 μl of each sonicated chromatin into 1.5 ml Safe-Lock Eppendorf tubes and centrifuge for 5 min (16,000 × g, 4°C).
Transfer 50 μl of each supernatant into a new Eppendorf tube and add 1 μl of RNase A + T1. Incubate at 37°C for 20 min.
Add 2 μl of 5M NaCl + 1 μl of 0.5 M EDTA + 2 μl of 1 M Tris-HCl pH 6.5 + 1 μl Proteinase K to each sample. Incubate at 65°C for 1 h.
Spin down each sample briefly.
Add 114 μl of Phenol/Chloroform/IAA to each sample (under the laminar hood), close tightly, and vortex for 30 s.
Centrifuge at 13,000 × g for 5 min (room temperature).
Transfer the supernatant (~ 50 μl) to a new 1.5 ml tube and add an extra 1 μl of RNase A + T1 to each sample to completely eliminate RNAs. Incubate at 37°C for 20 min.
Mix 25 μl of each sample from Step D7 with 6 μl of 30% glycerol and load it on a 1% agarose gel, using a 100-1,000 bp ladder. (Glycerol replaces loading buffer).
Run the gel at 100 V for 20 min.
Visualize gel and check that DNA size ranges from 150-500 bp (Figure 3A-3C). If sonication worked as expected, proceed to prepare inputs and to the immunoprecipitation. If sonicated DNA size is larger than needed (Figure 3D, 3E), re-sonicate chromatin samples from Step C4 and repeat the validation of sonication.
-
Immunoprecipitation and Input preparation
Prepare ChIP Dilution Buffer, Low Salt Wash Buffer, High Salt Buffer, LiCl wash Salt Buffer, and TE buffer (can be stored at 4°C prior to the addition of protease inhibitors).
Transfer chromatin from the Covaris tube to 1.5 ml tubes and centrifuge at 16,000 × g 5 min at 4°C.
Transfer supernatant with chromatin into new tubes.
Transfer 10 μl of each sample of sonicated chromatin to be used as the input (store at -20°C).
For each immunoprecipitation, add 200 μl of sonicated chromatin to a 1.5 ml Safe-Lock tube and add 600 μl of cold ChIP dilution buffer. On average, three immunoprecipitations are set for every chromatin sample.
Add desired antibodies to each immunoprecipitation tube (the general standard for antibodies against histone modifications is 3 μg/reaction). Close lid well, invert tube several times to mix, and leave mixing ON in a rotator mixer at 4°C.
Next day, prepare 40 μl of Dynabeads (A for rabbit and G for mouse antibodies) per each immunoprecipitation sample in a 1.5 ml Eppendorf tube. Mix, spin quickly, and place the tube on the magnetic rack to separate beads from the supernatant. Remove supernatant while the tube is on the rack and repeat for a total of three washes with ChIP Dilution Buffer.
Resuspend the beads in 40 μl of ChIP dilution buffer per sample.
Add 40 μl of washed beads to each immunoprecipitated sample. Invert tubes to mix and place samples back in rotation at 4°C for at least 2 h in a lab rotator mixer.
Quickly spin the samples and place tubes on the magnetic rack until beads and supernatant are separated.
Remove supernatant by pipetting while the tubes stand on the magnetic rack.
Add 1 ml of Low Salt Wash Buffer to each sample, close tubes, remove from the magnetic rack, and invert to mix. Quickly spin the tubes and place them back on the magnetic rack until beads and supernatant are separated. Remove supernatant using a pipette while keeping the tubes on the rack.
Add 1 ml of Low Salt Wash Buffer to each sample, close tubes, and remove from the magnetic rack. Incubate 5 min at 4°C on a rotating wheel. Quickly spin the tubes and place them back on the magnetic rack until beads and supernatant are separated. Remove supernatant with a pipette while keeping the tubes on the rack.
Repeat Steps E12 and E13 with 1 ml of High Salt Buffer.
Repeat Steps E12 and E13 with 1 ml of LiCl wash Salt Buffer.
Repeat Steps E12 and E13 with 1 ml of TE buffer. Remove supernatant with pipette, and remove tubes from the magnetic rack.
Prepare Elution Buffer.
Add 200 μl of Elution Buffer, vortex, and incubate at 65°C for 15 min (vortex every 2-3 min).
Quickly spin and place the tubes on a magnetic rack until the beads and supernatant are completely separated.
Transfer supernatants into new 1.5 ml Eppendorf tubes while keeping the tubes with the beads on the magnetic rack.
Repeat Steps E18 and E19, and combine the supernatants into a single 1.5 ml Eppendorf tube for a final volume of 400 μl.
Take input samples from the freezer (10 μl) and add 390 μl of Elution Buffer. From this step, process inputs the same way as immunoprecipitations.
Add 16 μl of 5M NaCl and incubate at 65°C overnight to reverse crosslink.
Quickly spin and add 1 μl of RNase A+T1. Incubate at 37°C for 20 min.
Add 8 μl of 0.5 M EDTA + 16 μl of 1 M Tris-HCl pH 6.5 + 4 μl Proteinase K to each sample. Incubate at 65°C for 3 h.
Spin down briefly in a bench spin centrifuge to collect samples on the bottom of the tubes.
Add 450 μl of Phenol/Chloroform/IAA to each sample (under the laminar hood), close tightly, and vortex for 30 s (use Safelock tubes).
Centrifuge at 13,000 × g for 10 min (room temperature).
Transfer the supernatant (approximately 400 μl) of each sample into new tubes. Add 40 μl of 3M sodium acetate pH 5.5 + 1 μl Glycoblue. Mix by inverting and quickly spin. Add 1 ml of cold 100% ethanol, mix by inversion, and place at -20°C ON to precipitate DNA.
Centrifuge at 14,000 × g for 20 min (4°C).
Remove and discard the supernatant without disturbing the blue pellet. Wash pellet with 700 μl of 70% ethanol.
Centrifuge at maximum speed for 10 min (4°C).
Carefully remove the ethanol without disturbing the pellet. Allow the pellet to dry either on the bench (10 min) or under vacuum (5 min).
Resuspend the pellet in 10-20 μl of water (depending on the number of immunoprecipitations performed for each sample and taking into account that the maximum volume that can be used to produce DNA libraries with a NEBNext Kit is 50 μl).
Pull together the immunoprecipitations corresponding to the same sample.
Quantify the ChIPed DNA for each sample and input with the Qubit dsDNA High Sensitivity Assay Kit. For ChIP-seq, each library should be prepared from 5-10 ng of DNA (ideally 10 ng), using the NEBNext Ultra II DNA Library Prep Kit or equivalent (10-12 cycles on the PCR step). If ChIPed DNA is lower than 10 ng per sample, it is recommended to repeat the procedure from Step E5 with an additional 200 μl of chromatin and pool the resulting IPs together.
Figure 2. Schematic representation of the vacuum infiltration setup for crosslinking.
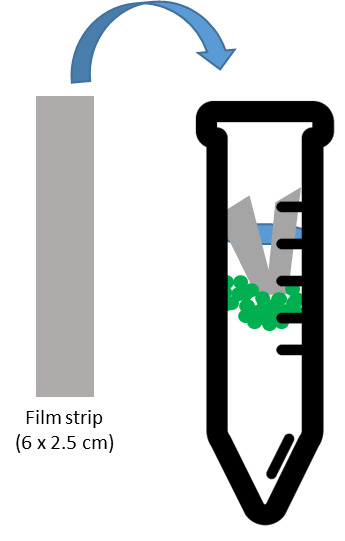
A plastic filmstrip is added to each tube to ensure that the plant tissue stays submerged during vacuum infiltration.
Figure 3. Agarose gel of sonicated Arabidopsis chromatin.
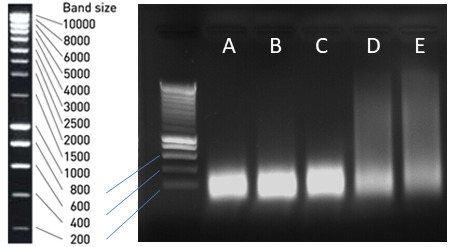
Samples in lanes A, B, and C are sufficiently sonicated. Samples in lanes D and E need further sonication before immunoprecipitation.
Data analysis
The pipeline used for ChIP-seq analysis is presented in the original paper where this method has been used (Antunez- Sanchez et al., 2020 : https://elifesciences.org/articles/58533).
Recipes
Stock Solutions:
-
Glycine 2 M (autoclave)
Composition For 100 ml Glycine 15 g H2O Complete to 100 ml -
Tris-HCl pH 8, 1 M (autoclave)
Composition For 100 ml Tris-HCl 12.114 g H2O Complete to 80 ml, adjust pH to 8 with NaOH, and then complete to 100 ml -
Tris-HCl pH 6.5, 1 M (autoclave)
Composition For 100 ml Tris-HCl 12.114 g H2O Complete to 80 ml, adjust pH to 6.5 with NaOH, and then complete to 100 ml -
Sucrose, 2 M (autoclave)
Composition For 500 ml Sucrose 342.2965 g H2O Complete to 500 ml -
MgCl2, 1 M (autoclave)
Composition For 100 ml MgCl2 9.5211 g (or 20.33 g if hexahydrate) H2O Complete to 100 ml -
NaCl, 5 M (autoclave)
Composition For 100 ml NaCl 29.22 g H2O Complete to 100 ml -
LiCl, 4 M (autoclave)
Composition For 50 ml LiCl 8.5 g H2O Complete to 50 ml -
Igepal CA-630 10% (do not autoclave)
Composition For 50 ml Igepal CA-630 5 ml H2O 45 ml -
Triton X-100 20% (do not autoclave)
Composition For 50 ml Triton X-100 10 ml H2O 40 ml -
Glycerol 30% (do not autoclave)
Composition For 10 ml Glycerol 3.33 ml H2O 6.66 ml -
Extraction Buffer 1
Note: It can be stored at 4°C before adding protease inhibitors and 2- mercaptoethanol.
Composition For 100 ml 0.4 M Sucrose 20 ml of 2 M solution 10 mM Tris-HCl pH 8 1 ml of 1 M solution 10 mM MgCl2 1 ml of 1 M solution 5 mM 2-mercaptoethanol 35 μl of 14.4 M solution Protease Inhibitors 200 μl of 1 tablet/ml solution H2O 77.8 ml -
Extraction Buffer 2
Note: It can be stored at 4°C before adding protease inhibitors and 2-mercaptoethanol.
Composition For 100 ml 0.25 M Sucrose 12.5 ml of 2 M solution 10 mM Tris-HCl pH 8 1 ml of 1 M solution 10 mM MgCl2 1 ml of 1 M solution 1% Triton X-100 5 ml of 20% solution 5 mM 2-mercaptoethanol 35 μl of 14.4 M solution Protease Inhibitors 200 μl of 1 tablet/ml solution H2O 80.3 ml -
Extraction Buffer 3
Note: It can be stored at 4°C before adding protease inhibitors and 2-mercaptoethanol.
Composition For 100 ml 1.7 M Sucrose 85 ml of 2 M solution 10 mM Tris-HCl pH 8 1 ml of 1 M solution 2 mM MgCl2 200 μl of 1 M solution 0.15% Triton X-100 750 μl of 20% solution 5 mM 2-mercaptoethanol 35 μl of 14.4 M solution Protease Inhibitors 200 μl of 1 tablet/ml solution H2O 12.8 ml -
Nuclei Lysis Buffer (prepare fresh each time)
Composition For 5 ml 50 mM Tris-HCl pH 8 250 μl of 1M solution 10 mM EDTA 100 μl of 0.5M solution 0.1% SDS 25 μl of 20% solution Protease inhibitors 100 μl of 1 tablet/ml solution H2O 4.525 ml -
ChIP Dilution Buffer
Note: It can be stored at 4°C before adding protease inhibitors.
Composition For 100 ml 1.1% Triton X-100 5.5 ml of 20% solution 1.2 mM EDTA 240 μl of 0.5 M solution 16.7 mM Tris-HCl pH 8 1.67 ml of 1 M solution 167 mM NaCl 3.34 ml of 5 M solution Protease inhibitors 200 μl of 1 tablet/ml solution H2O 89.05 ml -
Low Salt Wash Buffer
Note: It can be stored at 4°C.
Composition For 100 ml 150 mM NaCl 1.5 ml of 5M solution 0.1% SDS 250 μl of 20% solution 1% Triton X-100 2.5 ml of 20% solution 2 mM EDTA 200 μl of 0.5 M solution 20 mM Tris-HCl pH 8 1 ml of 1 M solution H2O 44.3 ml -
High Salt Wash Buffer
Note: It can be stored at 4°C.
Composition For 100 ml 500 mM NaCl 5 ml of 5 M solution 0.1% SDS 250 μl of 20% solution 1% Triton X-100 2.5 ml of 20% solution 2 mM EDTA 200 μl of 0.5 M solution 20 mM Tris-HCl pH 8 1 ml of 1 M solution H2O 41.05 ml -
LiCl Wash Buffer
Note: It can be stored at 4°C.
Composition For 50 ml 0.25 M LiCl 3.125 ml of 4 M solution 1% Igepal CA630 5 ml of 10% solution 1 mM EDTA 100 μl of 0.5 M solution 10 mM Tris-HCl pH 8 500 μl of 1 M solution H2O 41.25 ml -
TE Buffer
Note: It can be stored at 4°C.
Composition For 50 ml 1 mM EDTA 100 μl of 0.5 M solution 10 mM Tris-HCl pH8 500 μl of 0.5 M solution H2O 49.4 ml -
Elution Buffer (prepare fresh each time)
Composition For 20 ml 1% SDS 1 ml of 20% solution 0.1 M NaHCO3 168 g H2O 19 ml
Acknowledgments
This work was funded by the Agence National de la Recherche ANR (3DWheat project ANR-19-CE20-0001-01) and by the Institut Universitaire de France (IUF).
This protocol was derived from previously published research articles (Ramirez- Prado et al., 2019 ; Antunez- Sanchez et al., 2020 ; Concia et al., 2020 ).
Competing interests
The authors declare that no competing interests exist.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Antunez-Sanchez J., Naish M., Ramirez-Prado J. S., Ohno S., Huang Y., Dawson A., Opassathian K., Manza-Mianza D., Ariel F., Raynaud C., et al.(2020). A new role for histone demethylases in the maintenance of plant genome integrity. Elife 9: e58533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Concia L., Veluchamy A., Ramirez-Prado J. S., Martin-Ramirez A., Huang Y., Perez M., Domenichini S., Rodriguez Granados N. Y., Kim S., Blein T., et al.(2020). Wheat chromatin architecture is organized in genome territories and transcription factories. Genome Biol 21(1): 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong X. and Weng Z.(2013). The correlation between histone modifications and gene expression. Epigenomics 5(2): 113-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramirez-Prado J. S., Latrasse D., Rodriguez-Granados N. Y., Huang Y., Manza-Mianza D., Brik-Chaouche R., Jaouannet M., Citerne S., Bendahmane A., Hirt H., et al.(2019). The Polycomb protein LHP1 regulates Arabidopsis thaliana stress responses through the repression of the MYC2-dependent branch of immunity . Plant J 100(6): 1118-1131. [DOI] [PubMed] [Google Scholar]