

Abstract
Background:
Cystic fibrosis (CF) related diabetes is the most common comorbidity for CF patients and associated with islet dysfunction. Exocrine pancreas remodeling in CF alters the microenvironment in which islets reside. Since CFTR is mainly expressed in pancreatic ductal epithelium, we hypothesized altered CF ductal secretions could impact islet function through paracrine signals.
Method:
We evaluated the secretome and cellular proteome of polarized WT and CF ferret ductal epithelia using quantitative ratiometric mass spectrometry. Differentially secreted proteins (DSPs) or expressed cellular proteins were used to mine pathways, upstream regulators and CFTR interactomes to map candidate CF-associated alterations in ductal signaling and phenotype. Candidate DSPs were evaluated for their in vivo pancreatic expression patterns and their functional impact on islet hormone secretion.
Results:
The secretome and cellular proteome of CF ductal epithelia was significantly altered relative to WT and implicated dysregulated TGFb, WNT, and BMP signaling pathways. Cognate receptors of DSPs from CF epithelia were equally distributed among endocrine, exocrine, and stromal pancreatic cell types. IGFBP7 was a downregulated DSP in CF ductal epithelia in vitro and exhibited reduced CF ductal expression in vivo. IGFBP7 also altered WT islet insulin secretion in response to glucose. Many CFTR-associated proteins, including SLC9A3R1, were differentially expressed in the CF cellular proteome. Upstream regulators of the differential CF ductal proteome included TGFb, PDX1, AKT/PTEN, and INSR signaling. Data is available via ProteomeXchange with identifier PXD025126.
Conclusion:
These findings provide a proteomic roadmap for elucidating disturbances in autocrine and paracrine signals from CF pancreatic ducts and how they may alter islet function and maintenance.
Keywords: Cystic Fibrosis, Cystic Fibrosis Related Diabetes, pancreas, ductal cells, secretome, proteome
Introduction
Cystic Fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes a Cl− and HCO3− channel (1). While lung disease is the most life-threatening component of CF, damage to the pancreas causes significant morbidity and worsens the progression of lung disease. In this context, acinar cell loss in the CF pancreas leads to pancreatic insufficiency and poor nutritional status (2), and islet dysfunction leads to the development of CF related diabetes (CFRD) (3)—both worsen CF pulmonary function and life expectancy. Approximately 50% of adult CF patients develop CFRD, making it the most common co-morbidity. However, factors that contribute to the incidence and progression of CFRD remain poorly understood (4).
Cystic fibrosis pancreatic disease initiates very early in life and progresses rapidly, leading to pancreatic insufficiency in 75% of children by 1–4 years of age (4), accompanied by abnormal glucose tolerance (5). However, islet loss in adult CFRD patients (~25–30%) is relatively minor and cannot account for the severity of insulin insufficiency and glucose intolerance (4), suggesting that other factors likely contribute to islet dysfunction. Remodeling of the CF pancreas is accompanied by changes in its cellular composition, including a decline in acinar cells and expansion of adipocytes, fibroblasts, pancreatic stellate cells, and ductal cells (4). This process of pancreatic remodeling in turn alters the microenvironment of islets, which cluster around cystic ducts within fibrotic regions of the CF human and CF ferret pancreata (4, 6, 7). These changes to the cellular anatomy of the CF pancreas have been proposed to alter islet function through the gain and/or loss of paracrine signals (4, 8, 9).
CFTR knockout (CFTR-KO) ferrets have been instrumental in studying CF exocrine pancreatic damage and remodeling and its overall impact in the pathogenesis of CFRD. CFTR-KO ferrets are born with relatively minor pathology in the exocrine pancreas, but the damage rapidly progresses over the first two months of life (6, 7, 10). Despite minimal exocrine pancreatic involvement at birth, CFTR-KO (CF) kits demonstrate abnormal glucose tolerance, poorly regulated blood glucose, and lack of first-phase insulin secretion (7, 10). The inflammation and fibrosis during the first two months of life mark a transient period of spontaneous glycemic instability in CFTR-KO ferrets accompanied by reductions in the number of INS and GCG expression islet beta- and alpha-cells (7). This “glycemic crisis” is rapidly followed by an adipogenic program marked by an increase in PDX1 and PPARγ expression and transient glycemic stabilization (7). Notably, early impaired glucose tolerance is also observed at a higher frequency in CF children at 2–4 years of age (5), as compared to children in their teens (4), suggesting that the CF ferret model reproduces pathologic components in CF humans. As CF ferrets become adults (5–6 months of age), they develop glucose intolerance more similar to CFRD studied in humans (4), with insulin insufficiency (7) and islet coalescence within fibrotic regions surrounding ducts (6, 7).
Isolated neonatal CF ferret islets retain abnormalities in glucose-stimulated insulin secretion (GSIS) and have lower insulin content than WT islets (9, 10). In static assays, CF ferret islets secrete a larger fraction of insulin than WT controls at low concentrations of glucose, but lack a GSIS response (10). This heightened sensitivity of CF islets to glucose appears to be due to greater K-ATP channel inhibition and elevated basal intracellular Ca2+ in the resting CF islet (9). Notably, CF ferret islets had impaired first phase and second phase insulin secretion under perifusion conditions, as compared to WT controls, suggesting that a diffusible factor within CF islets impact β-cell function (9). A screen of inflammatory cytokines demonstrated that CFTR-KO islets secrete elevated levels of IL-6 and treatment of WT islets with IL-6 phenocopied the CF islet state (9). This study also demonstrated that the major CFTR-expressing cell type within both human and ferret islets were KRT7 positive ductal cells and that CFTR expression was absent in endocrine cells of the islet. Given that CFTR expression was limited to ductal cells associated with islets, this study proposed that the altered inflammatory state of CF ductal impacts β-cell function through paracrine signals (i.e., enhanced IL-6 secretion) (9).
With the hypothesis that CFTR impacts ductal cell secretions and in turn influences islet function, we sought to map the differences between secretomes and intracellular proteomes of CF and WT ferret ductal epithelium under stimulatory conditions. Differentially secreted and expressed proteomes where then subjected to informatic analysis to determine biologic pathways, CFTR-interacting proteins, and upstream regulators that might impact the CFTR-dependent alterations. Finally, we evaluated several proteins identified in the CF ductal epithelial secretome for their impact on islet INS and glucagon GCG secretion. Our findings from these studies implicate inflammatory and pancreatic development pathways that are significantly altered in CF ductal epithelium and provide a roadmap for elucidating how duct-to-endocrine paracrine signaling may alter islet function in CF.
Methods
Pancreatic ductal cell culture, propagation, and polarization.
Primary ferret ductal cells were generated by enzymatic digestion of newborn ferret WT and CFTR-KO (CF) pancreata and propagated in PneumaCult EX+ media (Stem Cell Technologies) to obtain a uniform population of SOX9 and KRT7 expressing ductal cells by passage 8–10. Ductal cells were polarized at an air-liquid interface in Pneumacult ALI media (Stem Cell Technologies). See Supplemental Methods for details.
Short-circuit current (Isc) and surface fluid pH measurements.
CFTR-mediated Cl− transport by polarized WT and CFTR-KO ductal epithelial cultures was assessed by Ussing chambers measurements. The apical pH of polarized ductal cultures was assessed using SNARF-1 fluorescent ratiometric imaging and calibrated against pH standard curves. See Supplemental Methods for details.
Single molecule in situ fluorescent hybridization (smFISH) detection of mRNA.
ViewRNA ISH kits (Thermo Fisher) were used on paraffin embedded sections of WT and CFTR-KO ferret pancreata for in situ analysis of IGFBP7 and LGALS3 mRNA expression. See Supplemental Methods for details.
Mass spectrometry analysis of pancreatic ductal epithelial secretions and whole cell proteomes.
Apical and basolateral secretions from polarized CF and WT pancreatic ductal epithelial cultures were collected following a 1 hr with IBMX (100 μM) and forskolin (10 μM). Ten μg of denatured protein from each sample was digested with a Trypsin/Lys-C protease mix (ThermoFisher Scientific) and the digested WT and CF peptides were subjected to “light” or “heavy” stable dimethyl isotope labeling, respectively, for relative quantitation by MS. Paired WT and CF samples were then combined into one tube, fractionated by ion exchange to decrease the complexity of peptides, desalted, and then run through a Thermo QEx HF High Resolution MS Instrument (ThermoFisher Scientific). The ratio of heavy/light labeled peptides was then used to calculate the ratio (CF/WT) for each protein using Scaffold software. Whole cell proteomics of polarized pancreatic ductal epithelial cultures utilized the same ratiometric MS approach from WT and CF whole cell lysates in RIPA buffer. See Supplemental Methods for details on mass spectrometry data analysis and quantification. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025126 and 10.6019/PXD025126.
Isolation of adult ferret islets and static glucose stimulated insulin and glucagon secretion assays.
Freshly harvested adult WT ferret islets were subjected to static insulin and glucagon secretion assays as previously described (9). The secretory index for each hormone was calculated as the fraction secreted at low and high glucose relative to the total amount of hormone in each sample. When evaluating the impact of candidate ductal secreted proteins on islets hormone secretion, islets were continuously exposed to 500 nM of purified IGFBP7, RNASE1, LGALS3, or A2M protein. Supplemental Methods for details.
Results
CF pancreatic ductal epithelia have an altered cAMP-stimulated secretome.
Our method to generate pancreatic ductal epithelial cells from WT and CF neonatal ferret pancreata utilized long-term passaged cultures, which led to the expansion of a uniform population of cells expressing ductal markers SOX9 and KRT7 (Fig. 1A). Following polarization, WT ductal epithelium gave rise to cAMP-inducible and GlyH101-inhibited CFTR-dependent currents that were absent in CF epithelia (Fig. 1B,C). As expected, the pH of the apical surface of CF ductal epithelia was lower than that of WT (Fig.1D), consistent with a major role for CFTR in bicarbonate transport of pancreatic ducts (11). To evaluate potential alterations to the secretome of CF ductal epithelium, we collected IBMX/Frsk (I/F) stimulated secretions from the apical and basolateral surfaces of these polarized cultures, as well as whole cell lysates from unstimulated cultures, and performed quantitative mass spectrometry (Fig. 1E). The approach utilized heavy isotope labelled CF peptides and light isotope labelled WT peptides to calculate a fold change (CF/WT) in protein abundance of genotypically paired samples (Fig.1F).
Fig. 1. Mass spectrometry evaluation of pancreatic ductal secretions and whole cell proteome.
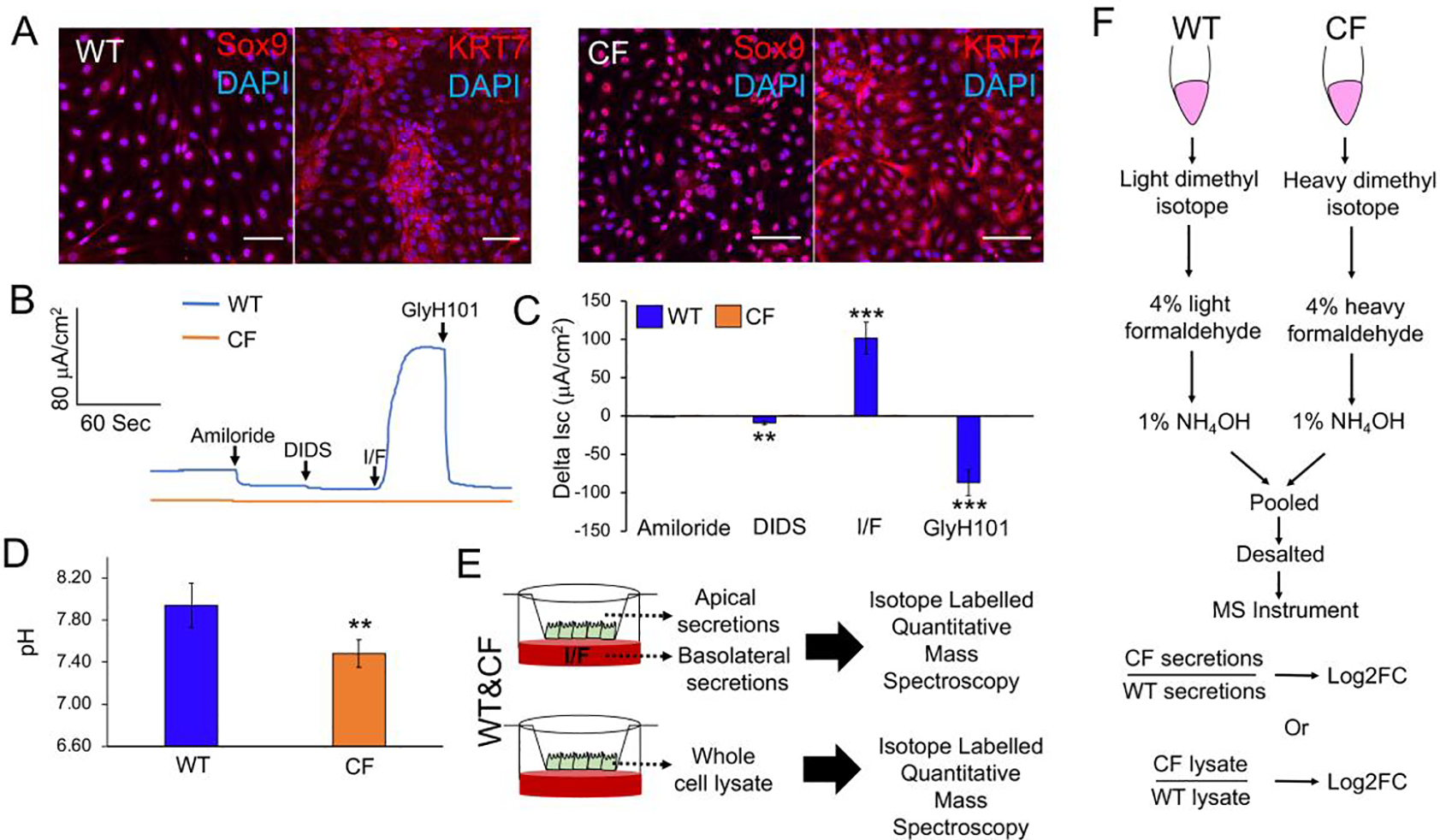
(A) Expression of pancreatic ductal cell-specific markers Sox9 and KRT7 in P10 WT and CF ferret ductal cells by immunofluorescence. Scale bars, 50 μm. (B,C) Short circuit current (Isc) analysis of CFTR-mediated currents in polarized WT and CF ductal epithelia. Cultures were sequentially treated with amiloride (to inhibit ENaC), DIDS (to inhibit non-CFTR chloride channels), IBMX/Forskolin (I/F, to activate CFTR), and GlyH101 (to inhibit CFTR). Shown is a representative Isc tracing (B) and the average change in response to each antagonist or agonist (C). Data depicts the Mean+/−SEM for N=6 transwells per genotype (6 WT and 6 CF donors). (D) Baseline apical surface pH measurements on WT and CF polarized duct epithelia. Data depicts the Mean+/−SEM for N=12 transwells per genotype (4 WT and 4 CF donors). (E) Schematic protocol for the mass spectrometry analysis of the IBMX/forskolin (I/F)-stimulated secretome and unstimulated whole cell proteome of WT and CF pancreatic duct epithelia. (F) Schematic protocol for quantitative mass spectrometry using isotope labelling to calculate the fold change (FC) of proteins in the secretome and whole cell proteome in CF relative to WT (CF/WT). Statistical analysis in (C, D) was performed using a two-tailed Student’s t-test, **p<0.01, ***p<0.001.
Apical and basolateral secretions from CF and WT epithelia contained 28 and 7 differentially secreted proteins (DSPs) (p<0.05) out of 135 and 32 total detected proteins, respectively (Fig. 2A; Table S1A,B). Proteins with Log2FC>1.5 were considered upregulated and log2FC<−1.5 were considered downregulated. Among these were proteins such as SPARC (Log2FC=−11.00), which is known to enhance islet insulin secretion (12), and PEBP1 (Log2FC=−11.79), which inhibits β-cell proliferation (13). Additionally, ~45% of the DSPs had previously reported roles in endocrine cell functions and/or epithelial to mesenchymal transition (Table S1C). Ingenuity Pathway Analysis (IPA) of upstream regulators was also evaluated to discern potential signaling pathways responsible for differential secretion. This analysis identified TGFβ, BMP, FGF, Wnt, and Notch pathways as potential upstream regulators of the DSPs by CF duct epithelium (Fig. 2B, Table S2A,B). Spatiotemporal regulation of these pathways has been shown to be necessary for endocrine lineage specification during pancreatic development (14).
Fig 2. Predicted alteration to upstream regulators and paracrine signaling in CF pancreatic ductal epithelia.
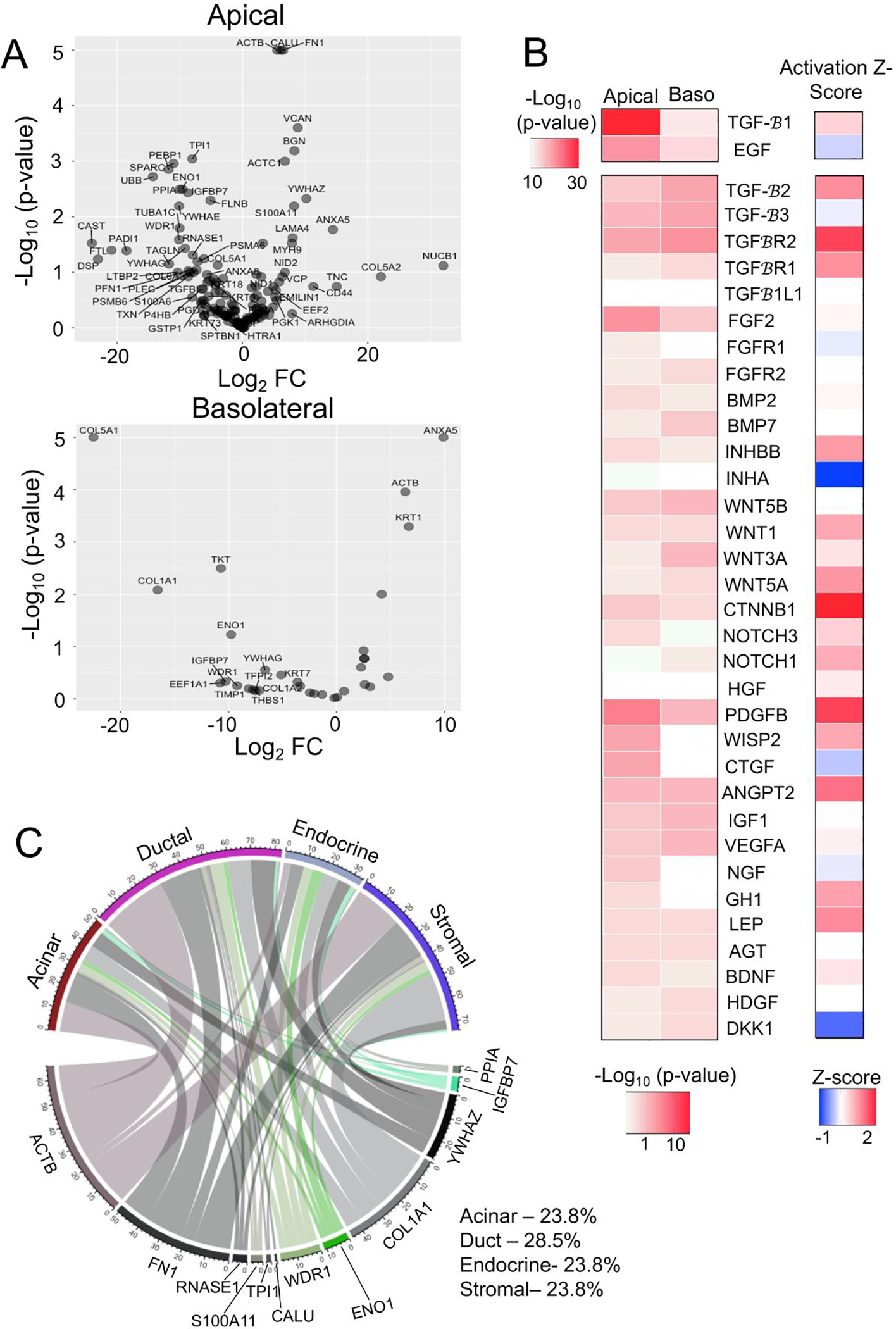
(A) Volcano plot showing cAMP-stimulated differentially secreted proteins (DSPs) from the apical or basolateral surface of WT (N=4 donors) and CF (N=4 donors) pancreatic duct epithelia. Proteins labelled in the plot have a Log2FC>5 (p<0.5) or Log2FC<−5 (p<0.05). (B) Heatmap of predicted upstream regulators of the differential secretomes from the basolateral (Baso) and apical surface (p<0.05) of CF and WT pancreatic duct epithelium. The activation Z-score for each of the upstream regulators are shown in the heatmap to the right. (C) Chordplot showing putative functional interaction between the DSPs from the CF duct epithelium with exocrine, endocrine, and stromal cell types of the pancreas.
To evaluate the potential impact of the altered CF ductal secretome on paracrine signaling in the pancreas, we mapped DSPs to their cognate receptors found within pancreatic cell types of single cell RNAseq datasets (8) (Table S2C). Of the apically DSPs, targets of TGFβ signaling IGFBP7, COL1A1 and FN1 accounted for 42.3% of the total differential paracrine signaling pairs (Fig. 2C). Targets of WNT signaling ENO1, WDR1 and TPI1 were associated with 12.4% and the BMP targets RNASE1 and S100A11 were associated with 4.5% of the altered paracrine signaling in CF pancreas (Fig. 2C). Of the total altered signals originating from CF ducts, ~29% were autocrine signals, and paracrine signals included ~24% to each of stromal, endocrine, and acinar cell compartments (Fig. 2C). These findings suggest that altered properties of CF ductal epithelium could impact paracrine signaling to both exocrine and endocrine pancreatic compartments.
IGFBP7 alters glucose-stimulated islet insulin secretion.
Pathway analysis suggested that TGFβ signaling was altered in CF ductal epithelium and its target insulin-like growth factor-binding protein 7 (IGFBP7), which can bind to insulin to alter its action, was downregulated in secretions from CF ductal epithelium (Log2(FC)=−8.68)(Table S1A). We therefore evaluated the expression of IGFBP7 mRNA in CF and WT pancreata and compared its expression pattern to a control mRNA (LGALS3) for which its protein product was not differentially secreted by polarized CF and WT ductal epithelium (Fig. 3). Single molecule in situ hybridization (smFISH) demonstrated that ductal expression of IGFBP7 was absent in neonatal CF pancreata, whereas IGFBP7 was expressed at high levels in ducts of WT controls (Fig. 3A). Notably, mesenchymal cells surrounding the CF ducts and islets, previously characterized as activated myofibroblasts and stellate cells (9), showed increased expression of IGFBP7, whereas in WT pancreata IGFBP7 expression was limited to ductal epithelium (Fig. 3A). By contrast, ductal LGALS3 localization showed no discernable differences in expression between WT and CF pancreata (Fig. 3B).
Fig. 3. IGFBP7 expression is altered in the CF pancreas and treatment of islets with IGFBP7 alters glucose-stimulated insulin secretion.
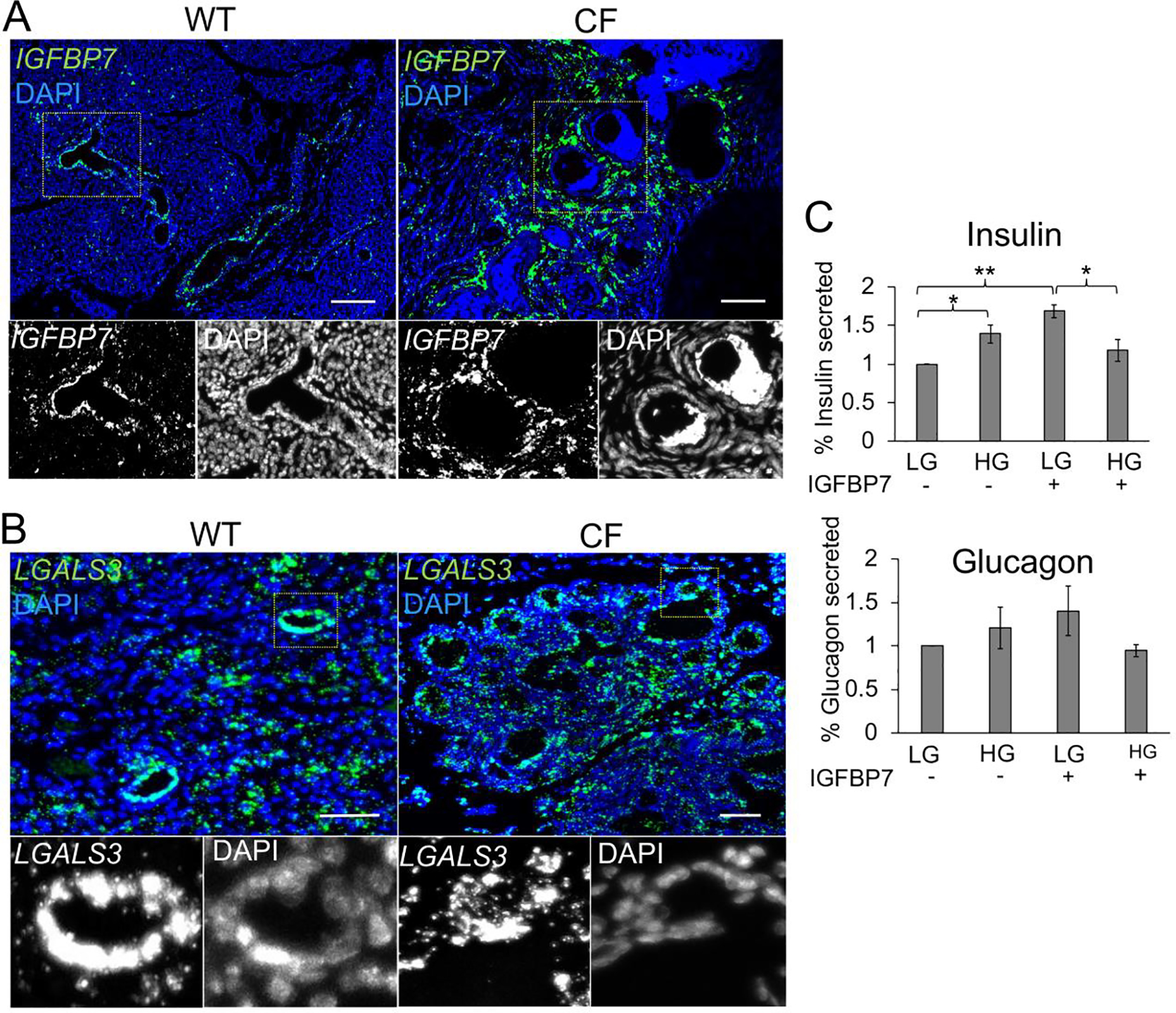
(A,B) smFISH localization of IGFBP7 (A) and LGALS3 (B) mRNA in neonate WT and CF ferret pancreata. Boxed regions in the main panels are shown below as enlarged single grey channels images. Micron bars, 50 μm. (C) Percent insulin and glucagon secretion from WT islets in low glucose and high glucose in the presence (+) and absence (−) of IGFBP7 exposure. Data shows the Mean+/−SEM for N=3 measurements from three independent donor islet preparations normalized to the mean hormone secreted at low glucose. Statistical analysis in (C) was performed using a two-tailed Student’s t-test, *p<0.05, **p<0.01.
To evaluate whether IGFBP7 might impact islet function, we treated WT adult ferret islets with purified IGFBP7 protein and analyzed glucose-stimulated insulin and glucagon secretion (Fig. 3C). IGFBP7 treatment enhanced insulin secretion at low glucose and impaired insulin secretion at high glucose (Fig. 3C). By contrast, IGFBP7 treatment did not significantly alter glucagon secretion by islets (Fig. 3B). While the functional consequences of these findings in vivo remain unclear, since CF pancreata demonstrated enhanced IGFBP7 expression in non-ductal cell compartments, they provide a working example of how CF pancreatic remodeling of ductal cell phenotypes may impact islet function.
The CFTR interactome is altered in CF ductal epithelium.
We next investigated CF-associated changes in the cellular proteome using stable isotope labelled mass spectrometry of whole cell lysates from WT and CF duct epithelium (Fig. 1E,F). Of the 303 significantly altered proteins (p<0.05), 31 were downregulated (Log2FC<−1.5, p<0.05) and 24 were upregulated (Log2FC>1.5, p<0.05) (Table S3A). CFTR is an anion channel with seven transmembrane domains, an extracellular N-terminus, and an intracellular C-terminus with annotated functional interaction with cytoplasmic, extracellular and membranous proteins (Fig. 4A). In order to characterize changes in the cellular proteome that might result from a loss of direct interactions with CFTR, we cross referenced the CFTR-interactome obtained from IPA (Table S4A) with the proteins that were differentially expressed in CF ductal epithelia (Table S4B). Of the differentially expressed CFTR-interactome proteins (p<0.05), ~77% were cytoplasmic proteins, 9.4% were membrane proteins, and 3.5% were extracellular proteins (Fig. 4B) (Table S4B). Out of these, 8 proteins were upregulated (Log2FC>1.5, p<0.05) and 3 proteins were downregulated (Log2FC<−1.5, p<0.05). The largest increase in relative expression was observed in two cytoplasmic proteins: actin binding CAPZB (Log2FC=11.36, p=0.034) and proteasome complex constituent PSMD11 (Log2FC=8.48, p=0.037) (Fig. 4B; Table S4B). An additional upregulated CFTR-interactome protein included the sodium/hydrogen exchanger regulatory cofactor (SLC9A3R1/NHERF1) that regulates CFTR function (Log2FC=0.74, p<0.0001) (15).
Fig 4. CF pancreatic duct epithelia have altered expression of proteins that interact with CFTR.
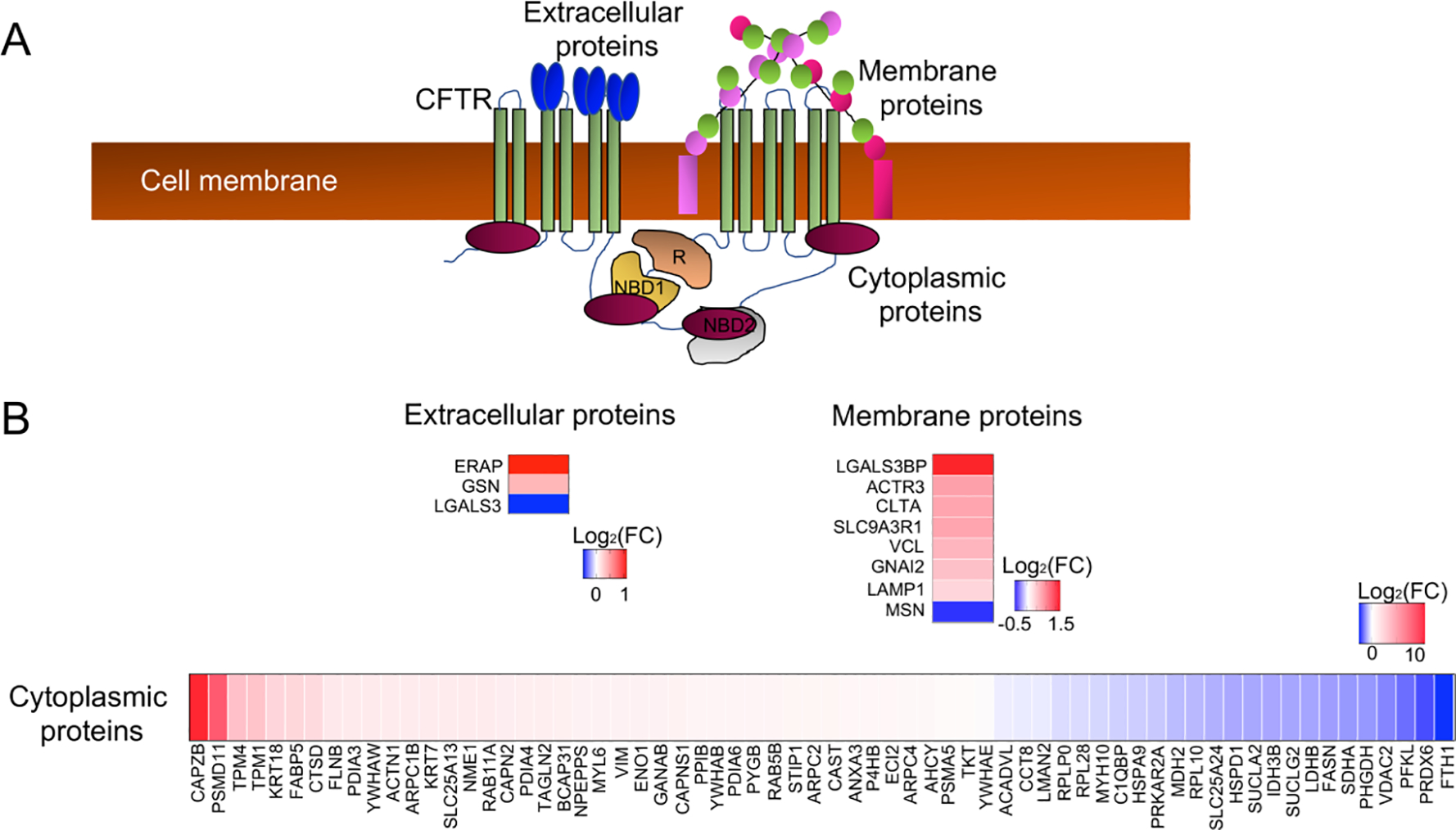
(A) Schematic of the CFTR interactome in the cytoplasmic, cell membrane, and extracellular compartments of epithelial cells. (B) Differentially expressed proteins from whole cell proteomic analysis of CF and WT pancreatic ductal epithelia cross-referenced to the CFTR interactome. Shown are heatmaps of the Log2(FC) for differentially expressed proteins (CF/WT) within the various domains of interaction with CFTR.
The CF ductal epithelial proteome predicts disturbances in mitochondrial function and inflammatory and PTEN/AKT signaling.
Ingenuity Pathway Analysis (IPA) of differentially expressed cellular proteins returned numerous disease-associated pathways including Inflammatory Response (p=1.93E-43), Endocrine System Disorders (p=1.81E-26), Metabolism of Reactive Oxygen Species (p=1.7E-21), and Transmembrane Potential of Mitochondria (p=1.01E-13) (Fig. 5A) (Table S3C). Similarly, IPA canonical pathways included Mitochondrial Dysfunction (−Log10[p-value]=18.2), Oxidative Phosphorylation (−Log10[p-value]=10.8), TCA Cycle II (−Log10[p-value]=14.1), and Glutathione Redox Reactions I (−Log10[p-value]=5.6), suggesting alterations to the cellular redox state in the absence of CFTR (Fig. 5B; Table S3B). Other top canonical pathways were associated with redox-dependent inflammatory pathways that signal through PI3K/AKT/PTEN (16), including EIF2 Signaling (−Log10[p-value]=33.7), Regulation of eIF4 and p70S6K Signaling (−Log10[p-value]=13.1), and mTOR Signaling (−Log10[p-value]=10.6) (Fig.5B; Table S3B).
Fig. 5. Pathway analysis of differentially expressed proteins within CF ductal epithelia.
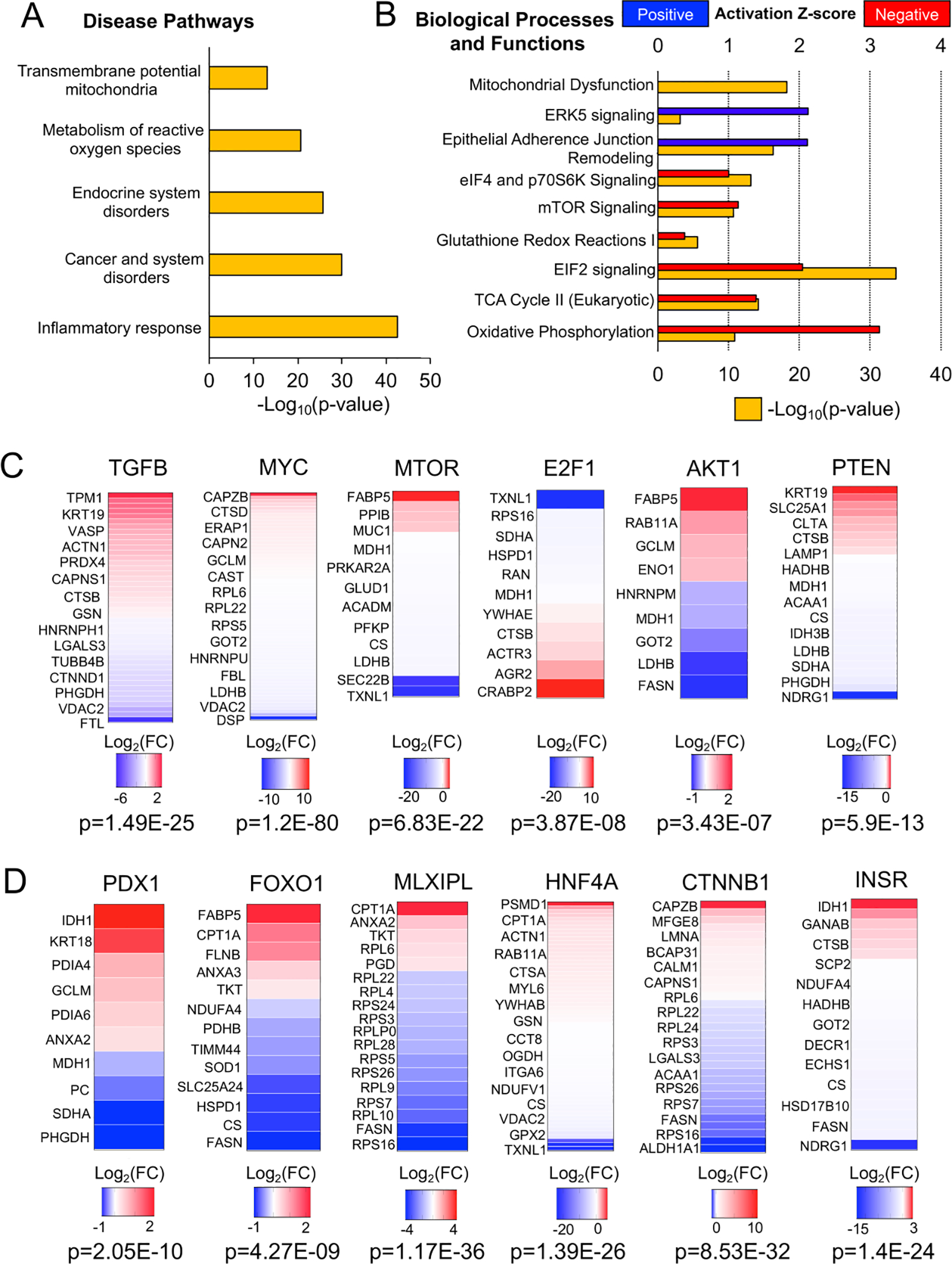
(A) IPA analysis of disease pathways associated with the differential CF ductal epithelial proteome. (B) IPA analysis of canonical biologic processes and functions associated with the differential CF ductal epithelial proteome. (C) Upstream regulators of the differential CF ductal epithelial proteome related to proinflammatory and redox-dependent signaling pathways. Heatmaps show the Log2(FC) of target molecules of each upstream regulator that were differentially expressed in CF ductal epithelia. (D) Upstream regulators of the differential CF ductal epithelial proteome that are known to influence specification of endocrine cells from ductal progenitors during pancreatic development. Heatmaps show the Log2(FC) of target molecules of each upstream regulator that were differentially expressed in CF ductal epithelia.
Upstream regulator analysis of differentially expressed cellular proteins suggested involvement of TGFβ, MYC, MTOR, E2F1, AKT and PTEN signaling pathways (p<0.05) (Fig. 5C; Table S5A). The finding of TGFβ as a candidate upstream regulator is consistent with the observed genotypically altered IPA Inflammatory Response. IL2, which was also a significant upstream regulator (p=0.00213), is known to regulate PTEN/PI3K/AKT/MTOR/MYC signaling in the control of mitochondrial oxidation (17), also consistent with IPA findings of an altered redox state. EIF2 also regulates AKT/PTEN redox-sensitive activity through MTOR (16). In support of these findings, the PTEN target NDRG1, which is linked to the PTEN/AKT/MTOR signaling networks and also regulates TGFβ and Wnt signaling (18), was significantly downregulated (Log2FC=−18.67, p<0.05) in CF pancreatic duct epithelium.
Additional upstream regulators of differentially expressed cellular proteins were linked to pancreatic development, including PDX1 and MLXIPL, transcription factors important in bipotent ductal progenitor cell specification during pancreatic development (19), and HNF4A, a transcription factor also important in pancreatic development for which mutations lead to impair insulin secretion and diabetes (Table S5A) (20). The insulin receptor (INSR) was also identified as an inhibited upstream regulator (Z-score −3.629) (Table S5A) that signals through AKT/PTEN (also with negative Z-scores) and this correlated with downregulation of the AKT/PTEN target NDRG1 (Log2FC=−18.67, p<0.05). Notably, NDRG1 has known function in maintaining E-cadherin and β-catenin at membrane adherens junctions (21) and β-catenin (CTNNB1) was another upstream regulator identified (Fig. 4D) and also associates with CFTR (Table S4A). Furthermore, IPA canonical pathway Remodeling of Epithelial Adherens Junctions (−Log10[p-value]=16.2) was within the top five pathways.
Discussion
Paracrine signals between cell types in the pancreas are undoubtedly important in controlling collective organ functions (8). In the CF pancreas, certain cell types are depleted (i.e., acinar cells) and others are structurally reorganized (i.e., ductal cells and islets). However, the link between structure and function in the pancreas remain poorly studied, underscoring the knowledge gaps in understanding how disease-associated alterations in cellular anatomy contribute to the pathogenesis of pancreatogenic diabetes (4). Here we sought to investigate how the lack of CFTR impacts ductal cell phenotype and how CF-associated alterations in paracrine signals might impact islet function. Utilizing primary pancreatic ductal cell cultures from neonatal WT and CF ferrets, we demonstrate significant alterations to the secretome and cellular proteome between genotypes. Given that ductal epithelial cells were isolated from newborn CF and WT ferret pancreata at a very early stage of CF disease, our finding suggest that the CF-associated alterations in ductal cell phenotype likely initiate in utero and could have long-term consequences to organ function.
Differentially expressed cellular proteomes implicated genotypic differences in inflammatory pathway activation that were also reflected in the differentially secreted proteins. For example, YWHAZ secretion from CF ductal epithelia was enhanced (Log2FC=10.21, p=0.0047) and is known to associate with CFTR (Table S4A). YWHAZ suppresses the production of pro-inflammatory cytokines from macrophages in chronic inflammation (22), but it also reduces islet glucose responsiveness by inhibiting the GLP-1 receptor (23). Likewise, SPARC secretion from CF ductal epithelia was reduced (Log2FC=−11, p=0.0011) and was recently shown to enhance glucose-stimulated insulin secretion by β-cells (24). These findings inform the potential impact of an altered CF ductal secretome on islet function.
IPA analysis of alterations in the CF ductal epithelial cellular proteome suggested substantial changes to the cellular redox state. These pathways emphasized inhibition of Oxidative Phosphorylation (Z-score=−3.13) and Mitochondrial Dysfunction, Glutathione Redox Reaction, among others. CFTR has been shown to transport glutathione (GSH) (25) and thus can affect cellular redox state and mitochondrial redox metabolism (26, 27). Consistent with altered cellular redox balance were significant changes in expression of four peroxiredoxin PRDX3, 4, 5 and 6 (from −4 to +2 Log2FC), many of which reside in the mitochondria and utilize GHS as a substrate to degrade H202 and/or modify disulfide bonds in proteins. The findings of redox-sensitive AKT/PTEN/MTOR as upstream regulators is consistent with an altered redox state (16). The significant repression of NDRG1 expression in CF ductal epithelium is notable, since it is known to inhibit pro-inflammatory NFκB activation and PTEN/AKT/MTOR signaling, but also regulates Wnt and TGFβ signaling (18), and each of these factors were identified as significant upstream regulators.
Disruption of CFTR function and/or its association with other proteins could also be responsible for phenotypic changes observed in CF ductal epithelium. For example, the identified differentially activated upstream regulator PTEN (Z-score=−1.07 in CF ductal epithelium) has been shown to associate with CFTR to control mitochondrial metabolism (28) and was consistent with CF-associated pathway alterations in Oxidative Phosphorylation and Mitochondrial Dysfunction. CFTR serves as a scaffold for PTEN and reduced CFTR at the membrane leads to AKT activation and increased mitochondrial activity and reactive oxygen species (28). Notably, ablation of PTEN during pancreatic development leads to hyperplastic PDX1-expressing ducts and removal of acinar cells (29), a phenotype very similar to the CF pancreas and our findings of PDX1 as a differentially activated upstream regulator in CF ductal epithelium (Z-score=2.9). Additionally, in the absence of bicarbonate transport through CFTR, mouse intestinal stem cells have enhanced Wnt/CTNNB1(β-catenin) activation caused by a reduction in intracellular pH which leads to increased membrane localization of disheveled (30). β-catenin is also a member of the CFTR interactome and thus its absence at the membrane may act in concert with differentially expressed NDRG1 to regulate β-catenin availability (21).
These studies have created a roadmap for understanding phenotypic differences in the biology of CF ductal epithelium and their potential impact on pancreatic function and progression to a diabetic state. Several mechanisms could explain the observed phenotypic changes to CFTR-KO ductal epithelium: 1) in vivo epigenetic changes resulting from the inflammatory environment from which CF ductal cells were derived, 2) a cell-intrinsic requirement for CFTR protein, or 3) changes in luminal pH due to the lack of CFTR-mediated bicarbonate secretion. The use of a new CFTR-G551D ferret model, which is responsive to the CFTR modulator VX-770, may assist in addressing these hypotheses. While the mechanisms for disease associated changes in the CF ferret model remain speculative, the alterations in CF ductal epithelial secretome and cellular proteome were observed following long-term propagation in cultures after removal from the hostile environment of the CF pancreas, suggesting that these alterations may be epigenetically imprinted early during disease progression. Understanding the genesis of these changes and the functional implications on disease state may provide opportunities to develop therapies.
Supplementary Material
Highlights:
Mechanisms of islet impairment caused by CF exocrine pancreatic insufficiency are unknown.
Loss of CFTR alters the CF ductal epithelial secretome and proteome.
Alterations in CF ductal epithelial secretions implicate TGFβ, WNT, and BMP signaling.
IGFBP7 secretion by CF ductal epithelia is reduced and IGFBP7 alters glucose-stimulated islet insulin secretion.
Loss of ductal cell CFTR may impact islet function via paracrine signals.
TGFβ, PDX1, and PTEN/AKT signaling were upstream regulators implicated in the CF ductal phenotype.
Acknowledgements
We gratefully acknowledge contributions of the Proteomics Core Facility within the Carver College of Medicine at the University of Iowa, and an Endowment from the Carver Family Foundation which supports its operations.
Funding Sources
This work was supported by NIH grant RC2 DK124207, P30 DK054759 and contract 75N92019R0014 (to J.F.E.), R01 DK097820 (to A.U. & A.W.N.), the Carver Chair in Molecular Medicine (to J.F.E).
Footnotes
Conflict of Interest Statement
The authors declare no existing conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–31. [DOI] [PubMed] [Google Scholar]
- 2.Staab D Cystic fibrosis -- therapeutic challenge in cystic fibrosis children. Eur J Endocrinol. 2004;151 Suppl 1:S77–80. [DOI] [PubMed] [Google Scholar]
- 3.Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32(9):1626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norris AW, Ode KL, Merjaneh L, Sanda S, Yi Y, Sun X, et al. Survival in a bad neighborhood: pancreatic islets in cystic fibrosis. J Endocrinol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi Y, Norris AW, Wang K, Sun X, Uc A, Moran A, et al. Abnormal Glucose Tolerance in Infants and Young Children with Cystic Fibrosis. Am J Respir Crit Care Med. 2016;194(8):974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rotti PG, Xie W, Poudel A, Yi Y, Sun X, Tyler SR, et al. Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. Am J Pathol. 2018;188(4):876–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yi Y, Sun X, Gibson-Corley K, Xie W, Liang B, He N, et al. A Transient Metabolic Recovery from Early Life Glucose Intolerance in Cystic Fibrosis Ferrets Occurs During Pancreatic Remodeling. Endocrinology. 2016;157(5):1852–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyler SR, Rotti PG, Sun X, Yi Y, Xie W, Winter MC, et al. PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet scRNA-Seq. Cell Rep. 2019;26(7):1951–64 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun X, Yi Y, Xie W, Liang B, Winter MC, He N, et al. CFTR Influences Beta Cell Function and Insulin Secretion Through Non-Cell Autonomous Exocrine-Derived Factors. Endocrinology. 2017;158(10):3325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest. 2012;122(10):3755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilschanski M, Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med. 2013;3(5):a009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harries LW, McCulloch LJ, Holley JE, Rawling TJ, Welters HJ, Kos K. A role for SPARC in the moderation of human insulin secretion. PLoS One. 2013;8(6):e68253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardo FN, Altirriba J, Pradas-Juni M, Garcia A, Ahlgren U, Barbera A, et al. The role of Raf-1 kinase inhibitor protein in the regulation of pancreatic beta cell proliferation in mice. Diabetologia. 2012;55(12):3331–40. [DOI] [PubMed] [Google Scholar]
- 14.Bastidas-Ponce A, Scheibner K, Lickert H, Bakhti M. Cellular and molecular mechanisms coordinating pancreas development. Development. 2017;144(16):2873–88. [DOI] [PubMed] [Google Scholar]
- 15.Singh AK, Riederer B, Krabbenhoft A, Rausch B, Bonhagen J, Lehmann U, et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J Clin Invest. 2009;119(3):540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajesh K, Krishnamoorthy J, Kazimierczak U, Tenkerian C, Papadakis AI, Wang S, et al. Phosphorylation of the translation initiation factor eIF2alpha at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 2015;6:e1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity. 2015;43(4):690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Zhang D, Bae DH, Sahni S, Jansson P, Zheng Y, et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis. 2013;34(9):1943–54. [DOI] [PubMed] [Google Scholar]
- 19.Sharon N, Chawla R, Mueller J, Vanderhooft J, Whitehorn LJ, Rosenthal B, et al. A Peninsular Structure Coordinates Asynchronous Differentiation with Morphogenesis to Generate Pancreatic Islets. Cell. 2019;176(4):790–804 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maestro MA, Cardalda C, Boj SF, Luco RF, Servitja JM, Ferrer J. Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocr Dev. 2007;12:33–45. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z, Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. 2012;287(21):17016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong JS, Park JH, Yoo SA, Kim KM, Bae YJ, Park YJ, et al. Dynamic transcriptome analysis unveils key proresolving factors of chronic inflammatory arthritis. J Clin Invest. 2020;130(8):3974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim GE, Piske M, Lulo JE, Ramshaw HS, Lopez AF, Johnson JD. Ywhaz/14–3-3zeta Deletion Improves Glucose Tolerance Through a GLP-1-Dependent Mechanism. Endocrinology. 2016;157(7):2649–59. [DOI] [PubMed] [Google Scholar]
- 24.Hu L, He F, Huang M, Zhao Q, Cheng L, Said N, et al. SPARC promotes insulin secretion through down-regulation of RGS4 protein in pancreatic beta cells. Sci Rep 2020;10(1):17581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, et al. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003;22(9):1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valdivieso AG, Santa-Coloma TA. CFTR activity and mitochondrial function. Redox Biol. 2013;1:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Favia M, de Bari L, Bobba A, Atlante A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J Clin Med. 2019;8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riquelme SA, Lozano C, Moustafa AM, Liimatta K, Tomlinson KL, Britto C, et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci Transl Med. 2019;11(499). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–95. [DOI] [PubMed] [Google Scholar]
- 30.Strubberg AM, Liu J, Walker NM, Stefanski CD, MacLeod RJ, Magness ST, et al. Cftr Modulates Wnt/beta-Catenin Signaling and Stem Cell Proliferation in Murine Intestine. Cell Mol Gastroenterol Hepatol. 2018;5(3):253–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.